

Abstract
Background
WRKY transcription factors, so named because of the WRKYGQK heptapeptide at the N-terminal end, are widely distributed in plants and play an important role in physiological changes and response to biotic and abiotic stressors. Many previous studies have focused on the evolution of WRKY transcription factors in a given plant; however, little is known about WRKY evolution in legumes. The gene expression pattern of duplicated WRKY transcription factors remains unclear.
Results
We first identified the WRKY proteins in 12 legumes. We found that the WRKYGQK heptapeptide tended to mutate into WRKYGKK. The Q site in WRKYGQK preferentially mutated, while W, K, and Y were conserved. The phylogenetic tree shows that the WRKY proteins in legumes have multiple origins, especially group IIc. For example, WRKY64 from Lupinus angustifolius (LaWRKY64) contains three WRKY domains, of which the first two clustered together in the N-terminal WRKY domain of the group I WRKY protein, and the third WRKY domain grouped in the C-terminal WRKY domain of the group I WRKY protein. Orthologous WRKY genes have a faster evolutionary rate and are subject to constrained selective pressure, unlike paralogous WRKY genes. Different gene features were observed between duplicated WRKY genes and singleton WRKY genes. Duplicated Glycine max WRKY genes with similar gene features have gene expression divergence.
Conclusions
We analyzed the WRKY number and type in 12 legumes, concluding that the WRKY proteins have multiple origins. A novel WRKY protein, LaWRKY64, was found in L. angustifolius. The first two WRKY domains of LaWRKY64 have the same origin. The orthologous and paralogous WRKY proteins have different evolutionary rates. Duplicated WRKY genes have gene expression divergence under normal growth conditions in G. max. These results provide insight into understanding WRKY evolution and expression.
Electronic supplementary material
The online version of this article (10.1186/s12870-018-1467-2) contains supplementary material, which is available to authorized users.
Keywords: Evolutionary rate, Gene feature, Legume, Ortholog, Paralog, Phylogenetic relationship, WRKY
Background
The WRKY gene family comprises a class of important transcription factors involved in physiological change and response to biotic and abiotic stress [1–4]. WRKY transcription factors contain a WRKYGQK heptapeptide at the N-terminal end and a zinc-finger motif (CX4-5CX22-23HXH or CX7CX23HXC) at the C-terminal end [1, 2]. WRKY proteins can be classified into groups I–III based on the number of WRKY domains and the type of zinc-finger motif [1, 2]. Group I WRKY contains two WRKY domains and a zinc-finger motif [1, 2]. Group II WRKY contains a single WRKY domain and a CX4-5CX22-23HXH zinc-finger motif, and group II WRKY can be divided into the following five subgroups: IIa, IIb, IIc, IId, and IIe [1, 2]. Group III WRKY has a single WRKY domain and a CX7CX23HXC zinc-finger motif [1, 2]. WRKY can also be classified into two groups based on their intron type, R-type or V-type [2]. R-type introns are distributed in groups Ic, IIc, IId, and III, which contains an intron spliced immediately after the R (Arg) position [2, 5, 6]. V-type introns are distributed in groups IIa and IIb. One intron is located before the K (Val) position, which is at the sixth amino acid after the second C residue in the C2H2 zinc finger motif [2, 5, 6].
WRKY transcription factors can activate downstream genes, involving physiological change and response to biotic and abiotic stress by binding cis-acting elements [1, 2]. WRKY transcription factors are involved in seed development [7, 8], seed dormancy and germination [9–11], flowering [12, 13], senescence [14], metabolic pathways [7], morphogenesis of trichomes [15], and plant growth [16]. In addition, WRKY transcription factors, especially group III, can be involved in response to herbivores, pathogens, and nematodes [1, 17–21]. Furthermore, researchers have found that WRKY also aids plant resistance to abiotic stress such as high temperature, low temperature, salt and drought, H2O2, and UV radiation [22–30].
Many studies have focused on the evolution of the WRKY gene family, but there is a debate about the origin of each type of WRKY in various plant species. According to a phylogenetic tree, the evolutionary relationships proposed revealed that the WRKY gene family can be classified into four clades including groups I + IIc, groups IIa + IIb, group IId, and group IIe [6]. Based upon phylogenetic analyses, researchers have proposed that the group II and III WRKY domains are descendants that have originated from the C-terminal WRKY domain of group I [2, 6]. With the development of sequencing technology, an increasing number of complete genome-wide sequences have been reported for various plant species. Researchers have identified more WRKY gene families in various plants, obtaining results that contrasted the above conclusion. Specifically, Zhu et al. [31] found that the Triticum aestivum subgroup IIc WRKY domains originated from the N-terminal WRKY domain of group I. Wei et al. [32] demonstrated that group I WRKY proteins first appeared in monocotyledons, followed by groups III and II. Brand et al. [33] reported that group I and other WRKY proteins likely originated from subgroup IIc. Recently, Rinerson et al. [4] detected the number and type of WRKY gene families ranging from lower organisms to higher organisms without the use of phylogenetic trees. Rinerson et al. [4] proposed two alternative hypotheses of WRKY protein evolution: the “Group I Hypothesis” and the “IIa + b Separate Hypothesis” [4]. The “Group I Hypothesis” proposed that all WRKY proteins evolved from the C-terminal WRKY domains of group I proteins, whereas the “IIa + b Separate Hypothesis” suggested that groups IIa and IIb evolved directly from a single domain algal gene separated from a group I-derived lineage [4].
To date, genome-wide sequences have been reported for 12 legume species, and their phylogenetic relationships have been revealed based on genomic data [34–36] (Fig. 1). However, studies on the phylogenetic relationships of the WRKY gene family are limited in these legume species. In this study, we identified WRKY genes using the same method utilized by other studies and confirmed the number and type of WRKY genes in each genome. Then, we identified orthologs and paralogs and estimated their evolutionary rate. Further, we compared gene features between duplicated WRKY genes and singleton WRKY genes as well as the gene features and expression in WRKY paralogs. These results provide a greater depth of the understanding of WRKY evolution.
Fig. 1.

A phylogenetic tree of 12 legume species. The topology of the phylogenetic tree is revised based on references [34–36]
Methods
Identification of WRKY transcription factors in 12 legumes
A total of 12 legumes have reported genome sequences including Arachis duranensis (V14167.a1), Arachis ipaënsis (K30076.a1), Cajanus cajan (Cc 1.0), Cicer arietinum (cicar.CDCFrontier.v1.0), Glycine max (Wm82.a2), Lotus japonicus (Lj3.0), Lupinus angustifolius (La1.0), Medicago truncatula (Mt4.0), Phaseolus vulgaris (V10), Trifolium pratense (Tp2.1), Vigna angularis (Va3.0), and Vigna radiata (Vr1.0) [35–45]. These 12 legume genome sequences were downloaded from their sequencing websites (data downloaded on 2017-6-25).
There are many methods currently available to identify gene families in a genome. For example, the similarity-based method [46] and hidden Markov models (HMMs) method. Here, we used the HMMs method to identify the WRKY gene family in the 12 legumes, because this method has high accuracy and sensitivity. Additionally, this method has also been previously used to detect the WRKY gene family in A. duranensis, A. ipaënsis, and G. max [18, 29]. However, the WRKY gene family was identified in C. arietinum, L. japonicus, and M. truncatula using the similarity-based method [47–49]. In this study, we re-identified this gene family in the previously investigated genomes using the HMMs method. In addition, little is known about the WRKY gene family in C. cajan, L. angustifolius, P. vulgaris, T. pratense, V. angularis, and V. radiata. We also used the HMMs method to identify the WRKY gene family in these genomes. In brief, the HMM profile of the WRKY domain (PF03106) was downloaded from the Pfam protein family database (http://pfam.xfam.org/) [50] and was used to survey all proteins using the HMMER program [51]. To verify the reliability of the search results, each protein sequence was checked in the Pfam database. In this study, we excluded the WRKY sequences that had lost the WRKYGQK motif or the zinc finger motif.
Phylogenetic analyses
The multiple sequence alignment of all WRKY domains and groups I, II, and III domains were executed using MAFFT 7.0 [52]. The maximum likelihood (ML) estimate used the best-fitting model of sequence evolution as determined by ProtTest [53]. The phylogenetic trees were constructed using IQ-TREE [54] and were estimated using an SH-aLRT test with 1000 random addition replicates and ultrafast bootstrap approximation set to 10,000.
For Arachis, homologous genes were identified in the transcriptome assembly using the similarity-based method [37, 55]. Here, we used the same method to identify WRKY orthologs and paralogs in the 12 legumes. In brief, the multiple alignment of coding sequences (CDSs) was executed using each species with the local BLAST program [56]. The following evaluation criteria were used as thresholds to determine inclusion in subsequent analyses [55]: (1) length of aligned sequences > 80% of each sequence length; (2) identity > 80%; and (3) E-value ≤10− 10. MAFFT 7.0 [52] was used to align duplicated CDS and amino acid pairs. PAL2NAL [57] was used for the conversion of amino acid sequences into the corresponding CDSs. PAML 4.0 [58] was used to calculate the nonsynonymous substitution rates (Ka) and synonymous substitution rates (Ks). If the Ks value was less than 0.01 or more than 3, and the Ka value was nearly 0, these duplicated genes were excluded, because low sequence divergence could result in unknown estimates, and a high Ks value indicated potential sequence saturation [58, 59]. When Ka/Ks = 1 it indicated neutral selection, when it was > 1 it indicated positive selection, and when it was < 1 it indicated purifying selection.
Gene feature in homologs
To compare duplicated and singleton WRKY genes, we estimated the gene features between these genes. The gene features included polypeptide length, GC content at three codon positions (GC1, GC2, and GC3), and the frequency of optimal codons (Fop). The Fop and polypeptide length were calculated using the codon W program (version 1.4, http://codonw.sourceforge.net). The GC content was estimated using the in-house perl script.
Glycine max transcriptome data under normal growth conditions
The normalized data (Reads/Kb/Million, RPKM) for 14 G. max tissues collected during different growth periods, including young leaves, flowers, one cm pods, pod shell 10 days after flowering (DAF), pod shell 14 DAF, seeds 10 DAF, seeds 14 DAF, seeds 21 DAF, seeds 25 DAF, seeds 28 DAF, seeds 35 DAF, seeds 42 DAF, roots, and nodules, was reported by Severin et al. [60]. This data was downloaded from the SoyBase website [61]. The RPKM value was log2-transformed as gene expression.
Results
WRKY genes in 12 legume species
A total of 75 AdWRKY, 77 AiWRKY, and 178 GmWRKY proteins with both WRKYGQK heptapeptide and zinc-finger motifs were identified in A. duranensis, A. ipaënsis, and G. max genome sequences, respectively [18, 29]. Re-identification of WRKY proteins in C. arietinum, L. japonicus, and M. truncatula showed that 58 CaWRKY, 78 LjWRKY, and 98 MtWRKY proteins, respectively, were found using the HMMs method (Table 1). We detected 92 CcWRKY, 108 LaWRKY, 88 PvWRKY, 89 TpWRKY, 77 VaWRKY, and 76 VrWRKY proteins that contained both the WRKYGQK heptapeptide and zinc-finger motif using the HMMs method in C. cajan, L. angustifolius, P. vulgaris, T. pratense, V. angularis, and V. radiata genomes, respectively (Table 1). In addition, genomes with missing zinc-finger motifs and/or partial WRKY proteins are presented in Additional file 1: Table S1. We named WRKYs based on the order of genes located in chromosomes. CaWRKY, LjWRKY, and MtWRKY have been named in previous studies [47–49]. Newly detected WRKY genes from C. arietinum, L. japonicus, and M. truncatula were named based on the order of their location on the chromosome. If alternative splicing was observed for the WRKY genes, we retained the primary transcript. These WRKY genes can be classified into three groups: groups I, II, and III. A comparison of the number of WRKY genes among the 12 legumes showed that G. max contained the greatest number of WRKY genes, while C. arietinum contained the fewest WRKY genes.
Table 1.
The number and type of WRKY proteins in 12 legume species
| Group I | Group II | Group III | Total | |
|---|---|---|---|---|
| Arachis duranensis | 16 | 46 | 13 | 75 |
| Arachis ipaënsis | 14 | 48 | 15 | 77 |
| Cajanus cajan | 16 | 63 | 13 | 92 |
| Cicer arietinum | 12 | 39 | 7 | 58 |
| Glycine max | 30 | 124 | 24 | 178 |
| Lotus japonicus | 14 | 56 | 8 | 78 |
| Lupinus angustifolius | 24 | 72 | 12 | 108 |
| Medicago truncatula | 16 | 64 | 18 | 98 |
| Phaseolus vulgaris | 14 | 62 | 12 | 88 |
| Trifolium pratense | 14 | 60 | 15 | 89 |
| Vigna angularis | 15 | 53 | 9 | 77 |
| Vigna radiata | 16 | 48 | 12 | 76 |
WRKY genes can regulate downstream genes that are involved in physiological change and response to biotic and abiotic stress by WRKYGQK heptapeptide binding to the cis-acting element of the downstream gene [1]. In this study, we found that 139 WRKYGQK sequences were observed in at least one mutation site (Fig. 2). WRKYGKK tended to be mutated. Among these sequences, most WRKYGQK sequences from group II WRKY genes appeared to be mutated (Additional file 1: Table S1), indicating that the biological function of the group II WRKY genes was more diverse. Further, in this study, the Q in WRKYGQK preferentially mutated, while the W, K, and Y were conserved (Fig. 3). Previous studies have revealed that mutation of the K positions in WRKYGQK sequences disrupt protein-DNA interactions [62, 63]. In this study, some WRKYGQK sequences were observed with a K mutation, suggesting that the mutation influences protein-DNA interactions.
Fig. 2.

The mutated WRKY domain. The figure was constructed using the wordcloud package in R script. The font size indicates the number of mutations
Fig. 3.
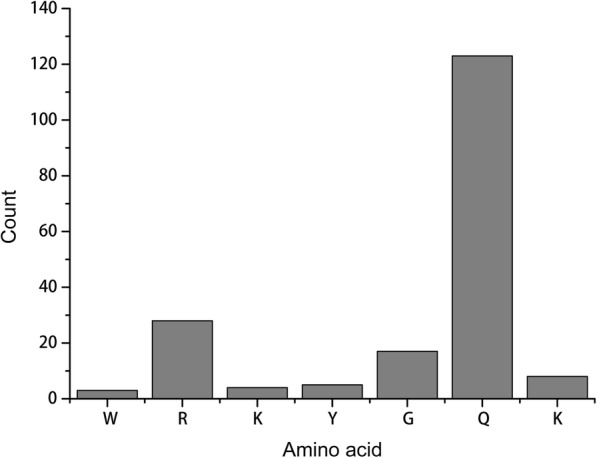
The number of amino acids of the WRKYGQK heptapeptide. The figure was constructed using Origin 9.0. The Y-axis indicates the count, and the X-axis indicates the amino acid
A WRKY protein can form a fusion protein with the nucleotide binding site-leucine-rich repeat (NBS-LRR) protein [1, 4], which is involved in plant response to pathogens [64]. We found two, one, and one WRKY-NBS fusion proteins in A. duranensis, A. ipaënsis, and G. max, respectively. We propose that there is no evolutionary relationship among WRKY proteins, NBS-LRR proteins, and WRKY-NBS fusion proteins due to the absence of WRKY-NBS proteins in close relatives to A. duranensis, A. ipaënsis, and G. max. WRKY-NBS fusion proteins can be classified into eight groups based on a phylogenetic tree [4]. As reported in a previous study, a G. max WRKY-NBS protein that belongs to the group RW4 was identified [4], but there is no record of the Arachis WRKY-NBS fusion protein. We constructed an ML phylogenetic tree using the JTT + I + G model using reported WRKY-NBS fusion proteins and Arachis WRKY-NBS fusion proteins. The phylogenetic tree showed that Arachis WRKY-NBS fusion proteins are a part of the group RW4 (Additional file 2: Figure S1). In addition, an ML phylogenetic tree using the JTT + G model based upon 12 legume WRKY domains was constructed (Fig. 4 and Additional file 3: Figure S2). GmWRKY, AdWRKY, AiWRKY, MtWRKY, LjWRKY, and CaWRKY have been reported in previous studies [18, 29, 47–49]. Accordingly, we used these WRKY genes as reference sequences to further classify other group II WRKY genes. The ML tree showed that the WRKY proteins can be classified into eight clusters: In, Ic, IIa, IIb, IIc, IId, IIe, and III (Fig. 4). However, we found that some clades contain different types of WRKY proteins. For example, some group II WRKY proteins clustered with group I WRKY proteins, and some group I WRKY proteins mixed with groups IIa, IIc, IIe, and III (Fig. 4). Further, two WRKY domains of group I grouped with IIa, IIc, and III, but only N-terminal WRKY domains of group I clustered in group IIe. In addition, the WRKY proteins in groups In, IIa, IIe, and III clustered into separate clades, but the other types of WRKY proteins formed multiple mixed clades (Fig. 4). These results indicated that WRKY proteins have multiple origins, especially group IIc.
Fig. 4.

A phylogenetic tree of the WRKY domains in 12 legumes. The phylogenetic tree was constructed using IQ-tree. The phylogenetic tree was estimated using maximum likelihood with the Jones-Taylor-Thornton (JTT) model, and branch support estimates are based on 1000 bootstrap replicates. The legume species included are Arachis duranensis (V14167.a1), Arachis ipaënsis (K30076.a1), Cajanus cajan (Cc 1.0), Cicer arietinum (cicar.CDCFrontier.v1.0), Glycine max (Wm82.a2), Lotus japonicus (Lj3.0), Lupinus angustifolius (La1.0), Medicago truncatula (Mt4.0), Phaseolus vulgaris (V10), Trifolium pratense (Tp2.1), Vigna angularis (Va3.0), and Vigna radiata (Vr1.0)
To reveal the phylogenetic relationships of group II WRKY proteins, we constructed an ML phylogenetic tree using the JTT + G model based on all group II WRKY proteins in the 12 legumes (Fig. 5 and Additional file 4: Figure S3). The topological structure of the phylogenetic tree is similar between using solely group II WRKY proteins and using all WRKY proteins. For example, groups IIa and IIe clustered in a clade, and other WRKY proteins grouped into multiple clades. However, compared with using all WRKY proteins to construct the phylogenetic tree, the number of clusters of group IIc WRKY proteins is relatively lower. We found a mixed clade, IIm, including group IIb and IIc WRKY proteins. Further, we found that members of the clade IIm were observed in group II, including MtWRKY99 and VaWRKY2, but were not classified (Fig. 5). These results indicated that the IIm WRKY proteins have multiple origins.
Fig. 5.

A phylogenetic tree of the WRKY group II domain in 12 legumes. The phylogenetic tree was constructed using IQ-tree. The phylogenetic tree was estimated using maximum likelihood with the Jones-Taylor-Thornton (JTT) model, and branch support estimates are based on 1000 bootstrap replicates. The legumes included are Arachis duranensis (V14167.a1), Arachis ipaënsis (K30076.a1), Cajanus cajan (Cc 1.0), Cicer arietinum (cicar.CDCFrontier.v1.0), Glycine max (Wm82.a2), Lotus japonicus (Lj3.0), Lupinus angustifolius (La1.0), Medicago truncatula (Mt4.0), Phaseolus vulgaris (V10), Trifolium pratense (Tp2.1), Vigna angularis (Va3.0), and Vigna radiata (Vr1.0)
The number of WRKY domains showed that LaWRKY64 contains three WRKY domains (Fig. 6), and we propose this newly identified sequence as a member of group I. In addition, LaWRKY96 and VrWRKY68 contain three WRKY domains, but the second and third WRKY domain of LaWRKY96 and VrWRKY68 are abnormal. We speculate that the original LaWRKY96 and VrWRKY68 contain two WRKY domains because there is a short sequence inserted into the second WRKY domain in LaWRKY96 and VrWRKY68 (Fig. 6). The phylogenetic tree showed that the first two WRKY domains of LaWRKY64 clustered together in In, and the third WRKY domain grouped in Ic (Fig. 4). Accordingly, we proposed that the second WRKY domain possibly originated from the first WRKY domain in LaWRKY64. To verify this hypothesis, we constructed a phylogenetic tree using group I WRKY proteins. The results showed that the sequences were highly homologous between the first and second WRKY domains (Additional file 5: Figure S4).
Fig. 6.
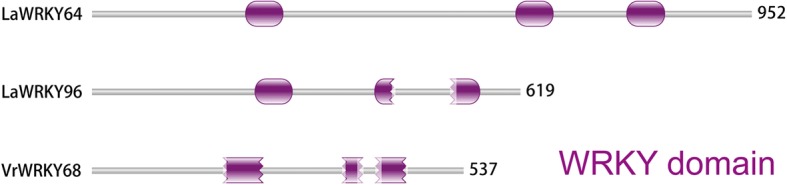
The WRKY domains in three WRKY proteins. The three amino acids are uploaded on the Pfam database (http://pfam.xfam.org/), and the domain structure was constructed in Pfam
Evolutionary rate and gene feature in homologous genes
In this study, we identified 317 orthologous pairs and 67 paralogous pairs in 12 legumes. We excluded one orthologous pair and six paralogous pairs due to the abnormality of the evolutionary rates of these seven homologous pairs. The evolutionary rate showed that the average Ka, Ks, and Ka/Ks of the paralogs was 0.06, 0.19, and 0.34, respectively. The average Ka, Ks, and Ka/Ks of the orthologs was 0.08, 0.34, and 0.28, respectively. A comparison of paralogs and orthologs revealed that the average Ka and Ks (a proxy for evolutionary rate) of the orthologs was higher than that of the paralogs, but the Ka/Ks (a proxy for selective pressure) for the orthologs was lower than that of the paralogs (Fig. 7). These results indicated that orthologs have a faster evolutionary rate and are subject to constrained selective pressure, unlike paralogs.
Fig. 7.
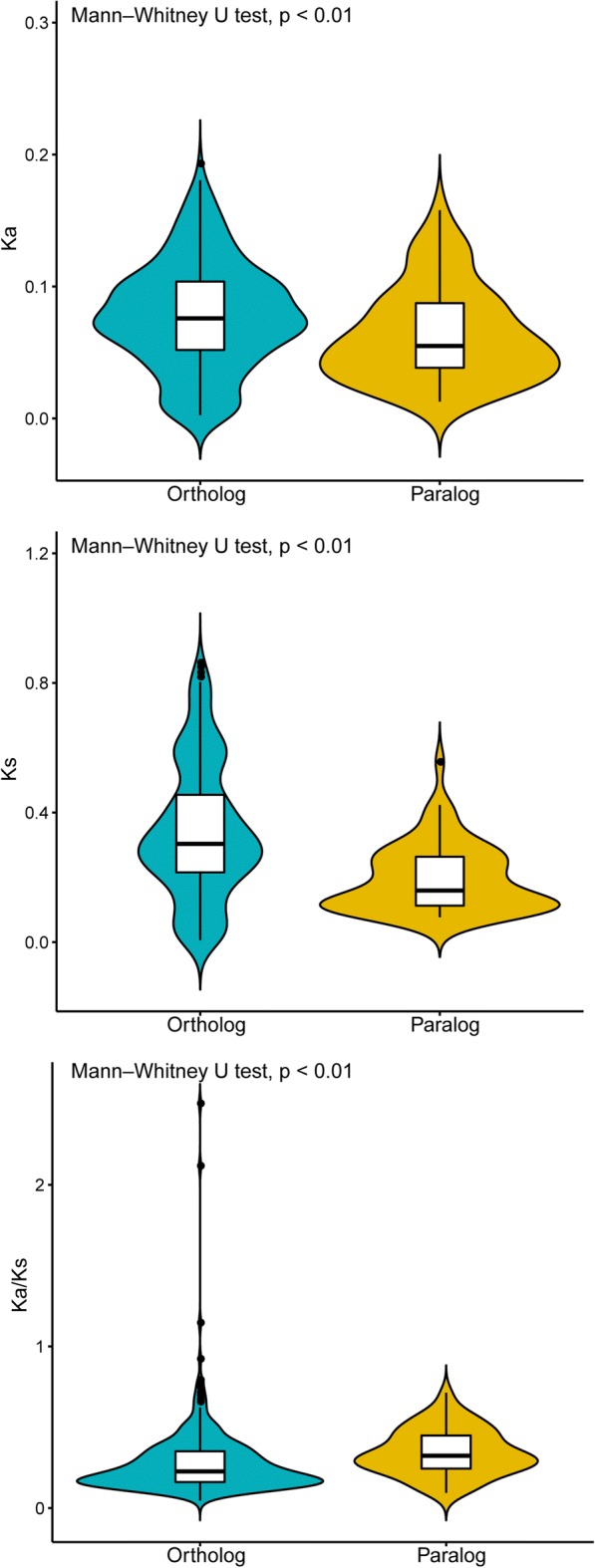
Comparison of the evolutionary rate between orthologs and paralogs. PAL2NAL was used to convert amino acid sequences into the corresponding nucleotide sequences. PAML 4.0 was used to calculate the nonsynonymous/synonymous substitution (Ka/Ks) rate. The figure was constructed using the ggpubr package in R script
The correlation analyses showed that Ka was positively correlated with Ks in paralogs and orthologs (paralog: r = 0.73, P < 0.01; ortholog: r = 0.58, P < 0.01). The Ks was negatively correlated with the Ka/Ks in paralogs and orthologs (paralog: r = − 0.27, P < 0.05; ortholog: r = − 0.42, P < 0.01). The Ka was not correlated with the Ka/Ks in orthologs (ortholog: r = 0.07, P > 0.05), but the Ka was significantly positively correlated with the Ka/Ks in paralogs (paralog: r = 0.38, P < 0.01). These results indicated that Ka influenced the Ka/Ks in paralogs, while the Ks determined the Ka/Ks in paralogs and orthologs.
Comparison of gene features between paralogs and orthologs showed that the average Fop and GC content of the orthologs was not significantly lower than that of paralogs (Additional file 6: Table S2).
Correlation of evolutionary rate and gene feature between orthologs and paralogs
There are correlations between evolutionary rate and gene feature [59, 65, 66]; however, different correlations have been found in various organisms. In this study, our results showed that the Ka was significantly negatively correlated with Fop, GC1, and GC2, and the Ka/Ks was significantly negatively correlated with Fop, polypeptide length, GC1, and GC2 in orthologs (Table 2). In paralogs, the Ka was significantly negatively correlated with Fop, GC2, and GC3, and the Ka/Ks was significantly negatively correlated with Fop, GC1, GC2, and GC3 (Table 2). These results indicated that both Fop and GC2 influenced in Ka, and Fop, GC1, and GC2 affected Ka/Ks in paralogs and orthologs.
Table 2.
The correlation of evolutionary rate and gene feature in WRKY orthologs and paralogs
| Fop | Polypeptide length | GC1 | GC2 | GC3 | |
|---|---|---|---|---|---|
| Ortholog | |||||
| Ks | 0.09026 | −0.0958 | − 0.10494 | − 0.0116 | 0.0672 |
| Ka | − 0.18988** | − 0.10997 | − 0.25635** | − 0.25051** | − 0.06667 |
| Ka/Ks | − 0.18418** | − 0.15026** | −0.23272** | − 0.23049** | −0.0526 |
| Paralog | |||||
| Ks | 0.10751 | 0.09914 | −0.08703 | 0.03401 | −0.10284 |
| Ka | −0.36815** | 0.11903 | −0.24856 | −0.36392** | − 0.37883** |
| Ka/Ks | −0.58738** | − 0.04473 | −0.2972* | − 0.54631** | −0.29905* |
Fop frequency of optimal codons, GC1 GC content at first codon positions, GC2 GC content at second codon positions, GC3 GC content at third codon positions, Ks synonymous substitution ratio, Ka nonsynonymous substitution ratio, Ka/Ks nonsynonymous to synonymous substitution ratio
*indicates significance at P < 0.05; **indicates significance at P < 0.01
Comparison of gene features between duplicated and singleton WRKY genes
Here, we addressed the gene features between duplicated WRKY genes and singleton WRKY genes in legumes. We found that the Fop (a proxy for codon usage bias) of duplicated WRKY genes was slightly higher than that of singleton WRKY genes (Table 3). The polypeptide length of duplicated WRKY genes was longer than that of singleton WRKY genes (Table 3). The GC content at the three codon positions of duplicated WRKY genes was higher than that of singleton WRKY genes (Table 3), but this is not statistically significant in GC1 content (Table 3). These results indicated that different gene features were observed between duplicated WRKY genes and singleton WRKY genes.
Table 3.
Comparison of gene features between duplicate and singleton WRKY genes
| aDuplicate | aSingleton | P value | |
|---|---|---|---|
| Fop | 0.3999 ± 0.0344 | 0.3910 ± 0.0443 | 0.014 |
| Polypeptide length | 398 ± 144 | 376 ± 164 | 0.014 |
| GC1 | 47.5282 ± 4.5553 | 46.6569 ± 4.3516 | 0.0981 |
| GC2 | 43.5792 ± 4.2996 | 42.1790 ± 4.5924 | 0.0189 |
| GC3 | 42.3161 ± 7.7402 | 37.2784 ± 7.6023 | 1.49E-10 |
Fop frequency of optimal codons, GC1 GC content at first codon positions, GC2 GC content at second codon positions, GC3 GC content at third codon positions
aMean ± SD
Glycine max WRKY paralogs
Compared with WRKY genes in other legumes, more duplicated GmWRKY (41 gene pairs) and LaWRKY (19 gene pairs) genes remained during the evolutionary process. Therefore, we investigated the gene features, evolutionary rates, and gene expression patterns in duplicated WRKY genes. To that end, we used duplicated GmWRKY because multiple tissue transcriptome datasets have been published for G. max, and it has the greatest number of duplicated WRKY genes. Our results showed that there is a slight difference in Fop (0–0.073), polypeptide length (0–14), GC1 (0–2.466), GC2 (0–3.087), and GC3 (0.015–4.664) among duplicated GmWRKY genes (Additional file 7: Table S3). However, the gene expression levels of 14 different tissues were observed to be largely different in duplicated GmWRKY genes. The differential gene expression of greater than 50% of the duplicated WRKY genes was observed as up to a two-fold difference, except for duplicated GmWRKY genes in seed tissue (Additional file 7: Table S3). These results indicated that the duplicated WRKY genes with similar gene features have gene expression divergence.
Negative correlations were found between the Ka/Ks and the gene expression level in 14 different tissues, but no statistical significance was found in young leaves, flowers, pod shells, and nodule tissues (Table 4). The Ka was significantly negatively correlated with the gene expression level in seeds 14 DAF and root tissues (Table 4). However, the Ks had a coefficient of irregularity with the gene expression level, but it was not statistically significant (Table 4). The correlation between gene features and gene expression level revealed that gene expression level was positively correlated with Fop, GC2, and GC3, but was not statistically significantly different in some tissues (Table 4). For example, the gene expression levels of leaves and pods 14 DAF were positively correlated with Fop, but were not statistically significantly different. There is correlation but no significance between the gene expression level of pods and GC2 (Table 4). In addition, the gene expression level was significantly positively correlated with GC3 in seeds 14 DAF, roots, and nodule tissues (Table 4). The correlations among gene expression level, polypeptide length, and GC1 were irregular and not statistically significant (Table 4).
Table 4.
The correlation between evolutionary rate, gene feature, and gene expression in Glycine max duplicates
| Young leaf | Flower | One cm pod | Pod shell 10DAF | Pod shell 14DAF | Seed 10DAF | Seed 14DAF | Seed 21DAF | Seed 25DAF | Seed 28DAF | Seed 35DAF | Seed 42DAF | Root | Nodule | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K s | 0.16331 | 0.0698 | −0.0229 | 0.10568 | 0.10625 | −0.06594 | 0.1175 | 0.19648 | 0.18418 | 0.15055 | 0.21299 | 0.2218 | −0.07009 | 0.03759 |
| K a | −0.06796 | −0.1692 | − 0.29357 | −0.14733 | − 0.08252 | −0.3319* | − 0.23597 | −0.11462 | − 0.21668 | −0.20247 | − 0.09324 | −0.1118 | − 0.4617** | −0.1818 |
| K a /K s | −0.20271 | −0.2411 | − 0.31353* | −0.27015 | − 0.12681 | −0.36376* | − 0.42364** | −0.32785* | − 0.46039** | −0.42072** | − 0.36024* | −0.4012** | − 0.52122** | −0.23178 |
| Fop | 0.29568 | 0.30819* | 0.34913* | 0.35181* | 0.26262 | 0.38268* | 0.39778* | 0.3624* | 0.49315** | 0.53284** | 0.44177** | 0.50804** | 0.36783* | 0.43526** |
| Polypeptide length | 0.06744 | 0.04395 | 0.08372 | −0.00281 | −0.07692 | 0.16338 | 0.12091 | 0.1345 | 0.18838 | 0.20758 | 0.22477 | 0.19481 | 0.09388 | 0.20409 |
| GC1 | 0.01633 | 0.05774 | 0.3434* | 0.00975 | −0.14317 | 0.38559* | 0.32063* | 0.32252* | 0.23863 | 0.2538 | 0.21561 | 0.22572 | 0.40488** | 0.22962 |
| GC2 | 0.32679* | 0.32974* | 0.42521** | 0.29401 | 0.21021 | 0.50622** | 0.52072** | 0.46359** | 0.57195** | 0.57169** | 0.48728** | 0.47168** | 0.65552** | 0.46916** |
| GC3 | 0.16546 | 0.20181 | 0.28002 | 0.16329 | 0.17571 | 0.25886 | 0.31821* | 0.30376 | 0.28938 | 0.28818 | 0.19329 | 0.21183 | 0.45942** | 0.31169* |
DAF day after flowering, Fop frequency of optimal codons, GC1 GC content at first codon positions, GC2 GC content at second codon positions, GC3 GC content at third codon positions, Ks synonymous substitution ratio, Ka nonsynonymous substitution ratio, Ka/Ks nonsynonymous to synonymous substitution ratio
*indicates significance at P < 0.05; **indicates significance at P < 0.01
Discussion
WRKY number, structure, and evolution in legumes
To date, genome-wide sequences obtained via different pipelines have been published for 12 legume species [35–45]. Accordingly, the sequencing depth of these legumes varies, making it difficult to compare the number of WRKY genes. However, in this study, the largest number of GmWRKY genes was detected for G. max compared with the 11 other legume species. This is because G. max is an autotetraploid [34], and more than three whole-genome duplication (WGD) events have been identified for G. max [67]. Our previous study revealed that the number of WRKY genes was not correlated with genome size but positively correlated with the number of WGD events [18]. This is also consistent with our other study, in which we found that the number of GmLOX genes is greater than in other legumes [68]. Unexpectedly, L. angustifolius, a diploid species, has more WRKY genes than other diploid legumes in this study. One reasonable explanation is the retention rate of duplicated WRKY genes is higher than in other legumes.
Normally, WRKY proteins contain one or two WRKY domains [1, 2]. However, Mohanta et al. [69] found that WRKY proteins contained three WRKY domains in Gossypium raimondii and Linum usitatissimum and four WRKY domains in Aquilegia coerulea and Setaria italic. We speculate that WRKY proteins containing three or four WRKY domains might have recently evolved because these types of WRKY proteins have only been observed in higher plant species, not in lower plant species [69]. We also identified a WRKY protein containing three domains in L. angustifolius. Our results indicated that the middle WRKY domain possibly produced the N-terminal WRKY domain, indicating that WRKY domains can be replicated using gene duplication.
Genomic rearrangement plays a pivotal force in forming WRKY-NBS fusion proteins [4], and NBS-LRR is a resistance gene involved in response to pathogens [64]. WRKY genes located in central positions mediating fast and efficient activation of defense programs [21]. Accordingly, we speculated that the fusion of WRKY proteins and NBS-LRR proteins can increase disease resistance in plants. The effector PopP2 and AvrRps4 can bind to the WRKY domain of the Arabidopsis WRKY-NBS fusion protein, which activates another NBS-LRR protein involved in response to bacterial pathogens in Nicotiana benthamiana and Nicotiana tabacum [70].
It is hard to explain the origin of the WRKY gene family using only one hypothesis, such as the “Group I Hypothesis” and “IIa + b Separate Hypothesis” [4]. This is because an increasing number of studies has revealed that the WRKY gene family has multiple origins. The “Group I Hypothesis” proposed that all WRKY proteins evolved from C-terminal WRKY domains of group I proteins, whereas the “IIa + b Separate Hypothesis” stated that groups IIa and IIb evolved directly from a single domain algal gene separated from the group I-derived lineage [4]. Our results are consistence with these two hypotheses. Based on our phylogenetic analyses, we found that WRKY genes from different groups clustered into a clade. Furthermore, our study fills a gap in the knowledge on the evolution of WRKY genes by comparing gene features and evolutionary rates between WRKY orthologs and paralogs. We concluded that (1) Ka influenced Ka/Ks in paralogs, while the Ks determined the Ka/Ks in paralogs and orthologs; (2) orthologs have a faster evolutionary rate and are subject to constrained selective pressure, unlike paralogs; and (3) both Fop and GC2 influenced Ka, and Fop, GC1, and GC2 affected Ka/Ks in paralogs and orthologs. However, more studies are required on legume WRKY genes because our current analysis does not clarify the evolutionary relationship among each group of WRKY genes. Therefore, further studies should not only focus on producing phylogenetic trees, but also work towards identifying novel WRKY genes, with a particular focus on WRKY proteins containing more than two domains or WRKY fusion proteins.
Gene expression in duplicated GmWRKY genes
The copy of duplicated genes will often be lost after WGD or small-scale duplication (SSD) [67]. However, many copies will be retained in the genome because they have novel molecular, structural, or adaptive traits functions [71]. Some researchers have proposed that duplicated genes with the same biological function will be lost due to fitness cost [72, 73]. Others researchers hold that duplication allows further adaptive changes to accumulate [71, 74, 75]. In addition to these contrasting proposals, four mechanisms can explain the retention of duplicates: gene dosage increase [72]; duplication, degeneration, and complementation (DDC) [76]; gene balance [77]; and paralog interference [74]. In this study, we found that the gene features of duplicated gene were similar, but the gene expression patterns of duplicated genes were different in 14 different tissues. The duplicated GmWRKY genes might have been retained because copies had the asymmetric expression pattern when following the explanation of the DDC mechanism. The DDC model proposed that the mutations that cause subfunctionalization are explicitly neutral [67, 76].
In this study, we found a negative correlation between Ka/Ks and gene expression level in 14 different tissues from G. max. This result is consistence with the expression-rate of sequence evolution anticorrelation model (E-R anticorrelation) [78]. The model proposed that the most highly expressed genes are also subject to the strongest selective constraint [79]. This can be explained by the expression cost hypothesis, the protein misfolding avoidance hypothesis, the protein misinteraction avoidance hypothesis, and the mRNA folding requirement hypothesis [80]. In addition, our results showed that Fop was positively correlated with gene expression levels, indicating that highly expressed genes have high codon usage bias. This is supported by natural selection in highly expressed genes preferentially using optimal codons [81]. Accordingly, we propose that natural selection plays a crucial role in codon usage bias of GmWRKY genes.
Conclusions
We identified the WRKY proteins in 12 different legume species, then we compared the gene number and type of WRKY proteins among the legumes. We found a novel WRKY protein, LaWRKY64, which contains three WRKY domains. The phylogenetic tree showed that the WRKY proteins in the 12 legumes have multiple origins. Duplicated and singleton WRKY genes have different gene features. Duplicated GmWRKY genes with similar gene features have gene expression divergence.
Additional files
Table S1. The name, chromosomal location, number, and type of WRKY in 12 legumes. aNull indicates that the protein lacks WRKY features. bThe bold font indicates an amino acid mutation. (XLSX 107 kb)
Figure S1. A phylogenetic tree of WRKY-NBS proteins. The phylogenetic tree was constructed using IQ-tree. The phylogenetic tree was estimated using maximum likelihood with the Jones-Taylor-Thornton (JTT) model, and branch support estimates are based on 1000 bootstrap replicates. (TIF 386 kb)
Figure S2. A phylogenetic tree of the WRKY domains in 12 legumes. The phylogenetic tree was constructed using IQ-tree. The phylogenetic tree was estimated using maximum likelihood with the Jones-Taylor-Thornton (JTT) model, and branch support estimates are based on 1000 bootstrap replicates. The legumes included are Arachis duranensis (V14167.a1), Arachis ipaënsis (K30076.a1), Cajanus cajan (Cc 1.0), Cicer arietinum (cicar.CDCFrontier.v1.0), Glycine max (Wm82.a2), Lotus japonicus (Lj3.0), Lupinus angustifolius (La1.0), Medicago truncatula (Mt4.0), Phaseolus vulgaris (V10), Trifolium pratense (Tp2.1), Vigna angularis (Va3.0), and Vigna radiata (Vr1.0). (PDF 1560 kb)
Figure S3. A phylogenetic tree of the WRKY group II domain in 12 legumes. The phylogenetic tree was constructed using IQ-tree. The phylogenetic tree was estimated using maximum likelihood with the Jones-Taylor-Thornton (JTT) model, and branch support estimates are based on 1000 bootstrap replicates. The legumes included are Arachis duranensis (V14167.a1), Arachis ipaënsis (K30076.a1), Cajanus cajan (Cc 1.0), Cicer arietinum (cicar.CDCFrontier.v1.0), Glycine max (Wm82.a2), Lotus japonicus (Lj3.0), Lupinus angustifolius (La1.0), Medicago truncatula (Mt4.0), Phaseolus vulgaris (V10), Trifolium pratense (Tp2.1), Vigna angularis (Va3.0), and Vigna radiata (Vr1.0). (PDF 990 kb)
Figure S4. A phylogenetic tree of the WRKY group I domain in 12 legumes. The phylogenetic tree was constructed using IQ-tree. The phylogenetic tree was estimated using maximum likelihood with the Jones-Taylor-Thornton (JTT) model, and branch support estimates are based on 1000 bootstrap replicates. The legumes included are Arachis duranensis (V14167.a1), Arachis ipaënsis (K30076.a1), Cajanus cajan (Cc 1.0), Cicer arietinum (cicar.CDCFrontier.v1.0), Glycine max (Wm82.a2), Lotus japonicus (Lj3.0), Lupinus angustifolius (La1.0), Medicago truncatula (Mt4.0), Phaseolus vulgaris (V10), Trifolium pratense (Tp2.1), Vigna angularis (Va3.0), and Vigna radiata (Vr1.0). The bold font indicates WRKY proteins with three WRKY domains. (TIF 5840 kb)
Table S2. Comparison of gene features between WRKY orthologs and paralogs. Fop: frequency of optimal codons; GC1: GC content at first codon positions; GC2: GC content at second codon positions; GC3: GC content at third codon positions. aMean ± SD (XLSX 10 kb)
Table S3. Absolute value of gene feature and gene expression between Glycine max WRKY duplicates. DAF: day after flowering; Fop: frequency of optimal codons; GC1: GC content at first codon positions; GC2: GC content at second codon positions; GC3: GC content at third codon positions. (XLSX 16 kb)
Acknowledgments
Funding
This study was supported by the Forage Industrial Innovation Team, Shandong Modern Agricultural Industrial and Technical System (SDAIT-23-01) and China Agriculture Research System (CARS-34).
Availability of data and materials
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CDS
Coding sequence
- DAF
Day after flowering
- DDC
Duplication, degeneration, and complementation
- E-R anticorrelation
Expression-rate of sequence evolution anticorrelation model
- Fop
Frequency of optimal codons
- GC1
GC content at first codon positions
- GC2
GC content at second codon positions
- GC3
GC content at third codon positions
- HMM
Hidden Markov model
- Ka
Nonsynonymous substitution ratio
- Ka/Ks
Nonsynonymous to synonymous substitution ratio
- Ks
Synonymous substitution ratio
- ML
Maximum likelihood
- NBS–LRR
Nucleotide binding site-leucine-rich repeat
- RPKM
Reads/Kb/Million
- SSD
Small-scale duplication
- WGD
Whole-genome duplication
Authors’ contributions
HS and GY conceived and designed this research. HS analyzed data and wrote the manuscript. HS and WS executed the data analyses. JS participated in the discussion of the results. GY contributed to the evaluation and discussion of the results and manuscript revisions. All authors have read and approved the final version.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hui Song, Email: biosonghui@outlook.com.
Weihong Sun, Email: 1083164023@qq.com.
Guofeng Yang, Email: yanggf@qau.edu.cn.
Juan Sun, Email: sunjuan@qau.edu.cn.
References
- 1.Rushton P, Somssich I, Ringler P, Shen Q. WRKY transcription factors. Trends Plant Sci. 2010;15(5):247–258. doi: 10.1016/j.tplants.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Eulgem T, Rushton P, Robatzek S, Somssich I. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000;5:199–206. doi: 10.1016/S1360-1385(00)01600-9. [DOI] [PubMed] [Google Scholar]
- 3.Riechmann JL, Heard J, Martin G, Reuber L, Jiang CZ, Keddie J, et al. Arabidopsis transcription factors:genome-wide comparative analysis among eukaryotes. Science. 2000;290:2105–2110. doi: 10.1126/science.290.5499.2105. [DOI] [PubMed] [Google Scholar]
- 4.Rinerson CI, Rabara RC, Tripathi QJ, Shen PJ, Rushton PJ. The evolution of WRKY transcription factors. BMC Plant Biol. 2015;15:66. doi: 10.1186/s12870-015-0456-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu K, Guo Z, Wang H, Li J. The WRKY family of transcription factors in rice and Arabidopsis and their origins. DNA Res. 2005;12:9–26. doi: 10.1093/dnares/12.1.9. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Wang L. The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evol Biol. 2005;5:1. doi: 10.1186/1471-2148-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun C, Palmqvist S, Olsson H, Borén M, Ahlandsberg S, Jansson C. A novel WRKY transcription factor, SUSIBA2, participates in sugar signaling in barley by binding to the sugar-responsive elements of the iso1 promoter. Plant Cell. 2003;15:20776–20792. doi: 10.1105/tpc.014597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luo M, Dennis E, Berger F, Peacock W, Chaudhury A. MINISEED3 (MINI3), a WRKY family gene, and HAIKU2 (IKU2), a leucine-rich repeat (LRR) KINASE gene, are regulators of seed size in Arabidopsis. Proc Natl Acad Sci U S A. 2005;102(48):17531–17536. doi: 10.1073/pnas.0508418102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Z, Xie Z, Zou X, Casaretto J, Ho T, Shen Q. A rice WRKY gene encodes a transcriptional repressor of the gibberellin signaling pathway in aleurone cells. Plant Physiol. 2004;134:1500–1513. doi: 10.1104/pp.103.034967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zou X, Neuman D, Shen Q. Interactions of two transcriptional repressors and two transcriptional activators in modulating gibberellin signaling in aleurone cells. Plant Physiol. 2008;148:176–186. doi: 10.1104/pp.108.123653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zentella R, Zhang Z, Park M, Thomas S, Endo A, Murase K, et al. Global analysis of della direct targets in early gibberellin signaling in Arabidopsis. Plant Cell. 2007;19:3037–3057. doi: 10.1105/tpc.107.054999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luo X, Sun X, Liu B, Zhu D, Bai X, Cai H, et al. Ectopic expression of a WRKY homolog from Glycine soja alters flowering time in Arabidopsis. PLoS One. 2013;8(8):e73295. doi: 10.1371/journal.pone.0073295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen L, Zhang L, Yu D. Transcription factor WRKY75 interacts with DELLA proteins to affect flowering. Plant Physiol. 2018;176(1):790–803. [DOI] [PMC free article] [PubMed]
- 14.Chen L, Xiang S, Chen Y, Li D, Yu D. Arabidopsis WRKY45 interacts with the DELLA protein RGL1 to positively regulate age-triggered leaf senescence. Mol Plant. 2017;10(9):1174–1189. doi: 10.1016/j.molp.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 15.Johnson C, Kolevski B, Smyth D. TRANSPARENT TESTA GLABRA2, a trichome and seed coat development gene of Arabidopsis, encodes a WRKY transcription factor. Plant Cell. 2002;14:1359–1375. doi: 10.1105/tpc.001404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen C, Chen Z. Potentiation of developmentally regulated plant defense response by AtWRKY18, a pathogen-induced Arabidopsis transcription factor. Plant Physiol. 2002;129:706–716. doi: 10.1104/pp.001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu X, Chen C, Fan B, Chen Z. Physical and functional interaction betwenn pathogen-induced Arabidosis WRKY18, WRKY40, and WRKY60 transcription factors. Plant Cell. 2006;18:1310–1326. doi: 10.1105/tpc.105.037523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song H, Wang P, Lin JY, Zhao C, Bi Y, Wang X. Genome-wide identification and characterization of WRKY gene family in peanut. Front Plant Sci. 2016;7(9):534. doi: 10.3389/fpls.2016.00534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skibbe M, Qu N, Galis I, Baldwin I. Induced plant defenses in the natural environment: Nicotiana attenuata WRKY3 and WRKY6 coordinate responses to herbivory. Plant Cell. 2008;20:1984–2000. doi: 10.1105/tpc.108.058594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grunewald W, Karimi M, Wieczorek K, Van de Cappelle E, Wischnitzki E, Grundler F, et al. A role for AtWRKY23 in feeding site establishment of plant-parasitic nematodes. Plant Physiol. 2008;148:358–368. doi: 10.1104/pp.108.119131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eulgem T, Somssich I. Networks of WRKY transcription factors in defense signaling. Curr Opin Plant Biol. 2007;10:366–371. doi: 10.1016/j.pbi.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 22.Rushton DL, Tripathi P, Rabara RC, Lin J, Ringler P, Boken AK, et al. WRKY transcription factors: key components in abscisic acid signalling. Plant Biotechnol J. 2012;10:2–11. doi: 10.1111/j.1467-7652.2011.00634.x. [DOI] [PubMed] [Google Scholar]
- 23.Chen L, Song Y, Li S, Zhang L, Zou C, Yu D. The role of WRKY transcription factors in plant abiotic stresses. BBA-Gene Regul Mech. 2012;1819:120–128. doi: 10.1016/j.bbagrm.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 24.Tripathi P, Rabara RC, Rushton PJ. A systems biology perspective on the role of WRKY transcription factors in drought responses in plants. Planta. 2014;239:255–266. doi: 10.1007/s00425-013-1985-y. [DOI] [PubMed] [Google Scholar]
- 25.Phukan UJ, Jeena GS, Shukla RK. WRKY transcription factors: molecular regulation and stress responses in plants. Front Plant Sci. 2016;7:760. doi: 10.3389/fpls.2016.00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang J, Ma S, Ye N, Jiang M, Cao J, Zhang J. WRKY transcription factors in plant responses to stresses. J Integr Plant Biol. 2017;59(2):86–101. doi: 10.1111/jipb.12513. [DOI] [PubMed] [Google Scholar]
- 27.Li S, Fu Q, Huang W, Yu D. Functional analysis of an Arabidopsis transcription factor WRKY25 in heat stress. Plant Cell Rep. 2009;28:683–693. doi: 10.1007/s00299-008-0666-y. [DOI] [PubMed] [Google Scholar]
- 28.Izaguirre M, Scopel A, Baldwin I, Ballaré C. Convergent responses to stress. Solar ultraviolet-B radiation and Manduca sexta herbivory elicit overlapping transcriptional responses in field-grown plants of Nicotiana longiflora. Plant Physiol. 2003;132:1755–1767. doi: 10.1104/pp.103.024323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song H, Wang P, Hou L, Zhao S, Zhao C, Xia H, et al. Global analysis of WRKY genes and their response to dehydration and salt stress in soybean. Front Plant Sci. 2016;7:9. doi: 10.3389/fpls.2016.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vandenabeele S, Van Der Kelen K, Dat J, Gadjev I, Boonefaes T, Morsa S, et al. A comprehensive analysis of hydrogen peroxide-induced gene expression in tobacco. Proc Natl Acad Sci U S A. 2003;100:16113–16118. doi: 10.1073/pnas.2136610100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu X, Liu S, Meng C, Qin L, Kong L, Xia G. WRKY transcription factors in wheat and their induction by biotic and abiotic stress. Plant Mol Biol Report. 2013;31:1053–1067. doi: 10.1007/s11105-013-0565-4. [DOI] [Google Scholar]
- 32.Wei K, Chen J, Chen Y, Wu L, Xie D. Multiple-strategy analyses of ZmWRKY subgroups and functional exploration of ZmWRKY genes in pathogen responses. Mol BioSyst. 2012;8:1940–1949. doi: 10.1039/c2mb05483c. [DOI] [PubMed] [Google Scholar]
- 33.Brand LH, Fischer NM, harter K, Kohlbacher O, Wanke D. Elucidating the evolutionary conserved DNA-binding specificities of WRKY transcription factors by molecular dynamics and in vitro binding assays. Nucleic Acids Res. 2013;41:9764–9778. doi: 10.1093/nar/gkt732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J, Sun P, Li Y, Liu Y, Yu J, Ma X, et al. Hierarchically aligning 10 legume genomes establishes a family-level genomics platform. Plant Physiol. 2017;174(1):284–300. doi: 10.1104/pp.16.01981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Vega JJ, Ayling S, Hegarty M, Kudrna D, Goicoechea JL, Ergon Å, et al. Red clover (Trifolium pratense L.) draft genome provides a platform for trait improvement. Sci Rep. 2015;5:17394. doi: 10.1038/srep17394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hane JK, Ming Y, Kamphuis LG, Nelson MN, Garg G, Atkins CA, et al. A reference genome sequence for lupin (Lupinus angustifolius), an emerging health food: insights into plant-microbe interactions and legume evolution. Plant Biotechnol J. 2017;15:318–330. doi: 10.1111/pbi.12615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bertioli DJ, Cannon SB, Froenicke L, Huang G, Farmer AD, Cannon EKS, et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat Genet. 2016;48:438–446. doi: 10.1038/ng.3517. [DOI] [PubMed] [Google Scholar]
- 38.Varshney RK, Chen W, Li Y, Bharti AK, Saxena RK, Schlueter JA, et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat Biotechnol. 2012;30(1):83–89. doi: 10.1038/nbt.2022. [DOI] [PubMed] [Google Scholar]
- 39.Jain M, Misra G, Patel RK, Priya P, Jhanwar S, Khan AW, et al. A draft genome sequence of the pulse crop chickpea (Cicer arietinum L.) Plant J. 2013;74:715–729. doi: 10.1111/tpj.12173. [DOI] [PubMed] [Google Scholar]
- 40.Sato S, Nakamura Y, Kaneko T, Asamizu E, Kato T, Nakao M, et al. Genome structure of the legume, Lotus japonicus. DNA Res. 2008;15(4):227–239. doi: 10.1093/dnares/dsn008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Young ND, Debellé F, Oldroyd GE, Geurts R, Cannon SB, Udvardi MK, et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature. 2011;480(7378):520–524. doi: 10.1038/nature10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, et al. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463(7278):178–183. doi: 10.1038/nature08670. [DOI] [PubMed] [Google Scholar]
- 43.Schmutz J, McClean PE, Mamidi S, Albert Wu G, Cannon SB, Grimwood J, et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat Genet. 2014;46:707–713. doi: 10.1038/ng.3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang K, Tian Z, Chen C, Luo L, Zhao B, Wang Z, et al. Genome sequencing of adzuki bean (Vigna angularis) provides insight into high starch and low fat accumulation and domestication. Proc Natl Acad Sci U S A. 2015;112(43):13213–13218. doi: 10.1073/pnas.1420949112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang YJ, Satyawan D, Shim S, Lee T, Lee J, Hwang WJ, et al. Draft genome sequence of adzuki bean, Vigna angularis. Sci Rep. 2015;5:8069. doi: 10.1038/srep08069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Remm M, Storm CE, Sonnhammer EL. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol. 2001;314(5):1041–1052. doi: 10.1006/jmbi.2000.5197. [DOI] [PubMed] [Google Scholar]
- 47.Kumar K, Srivastava V, Purayannur S, Kaladhar VC, Cheruvu PJ, Verma PK. WRKY domain-encoding genes of a crop legume chickpea (Cicer arietinum): comparative analysis with Medicago truncatula WRKY family and characterization of group-III gene(s) DNA Res. 2016;23(3):225–239. doi: 10.1093/dnares/dsw010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song H, Nan Z. Genome-wide indentification and analysis of WRKY transcription factors in Medicago truncatula. Hereditas (Beijing) 2014;36(2):152–168. doi: 10.3724/SP.J.1005.2014.00152. [DOI] [PubMed] [Google Scholar]
- 49.Song H, Wang P, Nan Z, Wang X. The WRKY transcription factor genes in Lotus japonicus. Int J Genomics. 2014;2014:420128. doi: 10.1155/2014/420128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finn RD, Mistry J, Schuster-Böckler B, Griffiths-Jones S, Hollich V, Lassmann T, et al. Pfam:clan, web tools and services. Nucleic Acids Res. 2006;34(suppl 1):247–251. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39(suppl 2):W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30(4):772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27(8):1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(1):268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clevenger J, Chu Y, Scheffler B, Ozias-Akins P. A developmental transcriptome map for allotetraploid Arachis hypogaea. Front Plant Sci. 2016;7:1446. doi: 10.3389/fpls.2016.01446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Altschul S, Madden T, Schäffer A, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suyama M, Torrents D, Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006;34(suppl 2):609–612. doi: 10.1093/nar/gkl315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 59.Song H, Gao H, Liu J, Tian P, Nan Z. Comprehensive analysis of correlations among codon usage bias, gene expression, and substitution rate in Arachis duranensis and Arachis ipaënsis orthologs. Sci Rep. 2017;7:14853. doi: 10.1038/s41598-017-13981-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Severin AJ, Woody JL, Bolon YT, Joseph B, Diers BW, Farmer AD, et al. RNA-Seq atlas of Glycine max: a guide to the soybean transcriptome. BMC Plant Biol. 2010;10:160. doi: 10.1186/1471-2229-10-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grant D, Nelson DT, Cannon SB, Shoemaker RC. SoyBase, the USDA-ARS soybean genetics and genomics database. Nucleic Acids Res. 2010;38(suppl 1):D843–D846. doi: 10.1093/nar/gkp798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Duan MR, Nan J, Liang YH, Mao P, Lu L, Li L, et al. DNA binding mechanism revealed by high resolution crystal structure of Arabidopsis thaliana WRKY1 protein. Nucleic Acids Res. 2007;35(4):1145–1154. doi: 10.1093/nar/gkm001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maeo K, Hayashi S, Kojima-Suzuki H, Morikami A, Nakamura K. Role of conserved residues of the WRKY domain in the DNA-binding of tobacco WRKY family proteins. Biosci Biotechnol Biochem. 2001;65(11):2428–2436. doi: 10.1271/bbb.65.2428. [DOI] [PubMed] [Google Scholar]
- 64.Dodds PN, Rathjen JP. Plant immunity: towards an integrated view of plant-pathogen interactions. Nat Rev Genet. 2010;11(8):539–548. doi: 10.1038/nrg2812. [DOI] [PubMed] [Google Scholar]
- 65.Song H, Zhang Q, Tian P, Nan Z. Differential evolutionary patterns and expression levels between sex-specific and somatic tissue-specific genes in peanut. Sci Rep. 2017;7:9016. doi: 10.1038/s41598-017-09905-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guo Y, Liu J, Zhang J, Liu S, Du J. Selective modes determine evolutionary rates, gene compactness and expression patterns in Brassica. Plant J. 2017;91:34–44. doi: 10.1111/tpj.13541. [DOI] [PubMed] [Google Scholar]
- 67.Conant GC, Wolfe KH. Turning a hobby into a job: how duplicated genes find new functions. Nat Rev Genet. 2008;9:938–950. doi: 10.1038/nrg2482. [DOI] [PubMed] [Google Scholar]
- 68.Song H, Wang P, Li C, Han S, Lopez-Baltazar J, Zhang X, et al. Identification of lipoxygenase (LOX) genes from legumes and their responses in wild type and cultivated peanut upon Aspergillus flavus infection. Sci Rep. 2016;6:35245. doi: 10.1038/srep35245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mohanta TK, Park YH, Bae H. Novel genomic and evolutionary insight of WRKY transcription factors in plant lineage. Sci Rep. 2016;6:37309. doi: 10.1038/srep37309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sarris PF, Duxbury Z, Huh SU, Ma Y, Segonzac C, Sklenar J, et al. A plant immune receptor detects pathogen effectors that target WRKY transcription factors. Cell. 2015;161(5):1089–1100. doi: 10.1016/j.cell.2015.04.024. [DOI] [PubMed] [Google Scholar]
- 71.Panchy N, Lehti-Shiu M, Shiu SH. Evolution of gene duplication in plants. Plant Physiol. 2016;171(4):2294–2316. doi: 10.1104/pp.16.00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ohno S. Evolution by gene duplication. New York, USA: Springer-Verlag; 1970. [Google Scholar]
- 73.Zhang JZ. Evolution by gene duplication: an update. Trends Ecol Evol. 2003;18(6):292–298. doi: 10.1016/S0169-5347(03)00033-8. [DOI] [Google Scholar]
- 74.Baker CR, Hanson-Smith V, Johnson AD. Following gene duplication, paralog, interference constrains transcriptional circuit evolution. Science. 2013;342(6154):104–108. doi: 10.1126/science.1240810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.De Smet R, Sabaghian E, Li Z, Saeys Y, Van de Peer Y. Coordinated functional divergence of genes after genome duplication in Arabidopsis thaliana. Plant Cell. 2017;29(11):2786–2800. doi: 10.1105/tpc.17.00531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151:1531–1545. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Freeling M, Thomas BC. Gene-balanced duplications, like tetraploisy, provide predictable drive to increase morphological complexity. Genome Res. 2006;16:805–814. doi: 10.1101/gr.3681406. [DOI] [PubMed] [Google Scholar]
- 78.Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH. Why highly expressed proteins evolve slowly. Proc Natl Acad Sci U S A. 2005;102(40):14338–14343. doi: 10.1073/pnas.0504070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Geiler-Samerotte KA, Dion MF, Budnik BA, Wang SM, Hartl DL, Drummond DA. Misfolded proteins impose a dosage-dependent fitness cost and trigger a cytosolic unfolded protein response in yeast. Proc Natl Acad Sci U S A. 2011;108(2):680–685. doi: 10.1073/pnas.1017570108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang J, Yang J. Determinants of the rate of protein sequence evolution. Nat Rev Genet. 2015;16:409–420. doi: 10.1038/nrg3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hershberg R, Petrov DA. Selection on codon bias. Annu Rev Genet. 2008;42:287–299. doi: 10.1146/annurev.genet.42.110807.091442. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The name, chromosomal location, number, and type of WRKY in 12 legumes. aNull indicates that the protein lacks WRKY features. bThe bold font indicates an amino acid mutation. (XLSX 107 kb)
Figure S1. A phylogenetic tree of WRKY-NBS proteins. The phylogenetic tree was constructed using IQ-tree. The phylogenetic tree was estimated using maximum likelihood with the Jones-Taylor-Thornton (JTT) model, and branch support estimates are based on 1000 bootstrap replicates. (TIF 386 kb)
Figure S2. A phylogenetic tree of the WRKY domains in 12 legumes. The phylogenetic tree was constructed using IQ-tree. The phylogenetic tree was estimated using maximum likelihood with the Jones-Taylor-Thornton (JTT) model, and branch support estimates are based on 1000 bootstrap replicates. The legumes included are Arachis duranensis (V14167.a1), Arachis ipaënsis (K30076.a1), Cajanus cajan (Cc 1.0), Cicer arietinum (cicar.CDCFrontier.v1.0), Glycine max (Wm82.a2), Lotus japonicus (Lj3.0), Lupinus angustifolius (La1.0), Medicago truncatula (Mt4.0), Phaseolus vulgaris (V10), Trifolium pratense (Tp2.1), Vigna angularis (Va3.0), and Vigna radiata (Vr1.0). (PDF 1560 kb)
Figure S3. A phylogenetic tree of the WRKY group II domain in 12 legumes. The phylogenetic tree was constructed using IQ-tree. The phylogenetic tree was estimated using maximum likelihood with the Jones-Taylor-Thornton (JTT) model, and branch support estimates are based on 1000 bootstrap replicates. The legumes included are Arachis duranensis (V14167.a1), Arachis ipaënsis (K30076.a1), Cajanus cajan (Cc 1.0), Cicer arietinum (cicar.CDCFrontier.v1.0), Glycine max (Wm82.a2), Lotus japonicus (Lj3.0), Lupinus angustifolius (La1.0), Medicago truncatula (Mt4.0), Phaseolus vulgaris (V10), Trifolium pratense (Tp2.1), Vigna angularis (Va3.0), and Vigna radiata (Vr1.0). (PDF 990 kb)
Figure S4. A phylogenetic tree of the WRKY group I domain in 12 legumes. The phylogenetic tree was constructed using IQ-tree. The phylogenetic tree was estimated using maximum likelihood with the Jones-Taylor-Thornton (JTT) model, and branch support estimates are based on 1000 bootstrap replicates. The legumes included are Arachis duranensis (V14167.a1), Arachis ipaënsis (K30076.a1), Cajanus cajan (Cc 1.0), Cicer arietinum (cicar.CDCFrontier.v1.0), Glycine max (Wm82.a2), Lotus japonicus (Lj3.0), Lupinus angustifolius (La1.0), Medicago truncatula (Mt4.0), Phaseolus vulgaris (V10), Trifolium pratense (Tp2.1), Vigna angularis (Va3.0), and Vigna radiata (Vr1.0). The bold font indicates WRKY proteins with three WRKY domains. (TIF 5840 kb)
Table S2. Comparison of gene features between WRKY orthologs and paralogs. Fop: frequency of optimal codons; GC1: GC content at first codon positions; GC2: GC content at second codon positions; GC3: GC content at third codon positions. aMean ± SD (XLSX 10 kb)
Table S3. Absolute value of gene feature and gene expression between Glycine max WRKY duplicates. DAF: day after flowering; Fop: frequency of optimal codons; GC1: GC content at first codon positions; GC2: GC content at second codon positions; GC3: GC content at third codon positions. (XLSX 16 kb)
Data Availability Statement
The datasets generated and analysed during the current study are available from the corresponding author on reasonable request.