

Abstract
Carbon-carbon bonds are integral for pharmaceutical discovery and development. Frequently, C–C bond reactions utilize expensive catalyst/ligand combinations and/or are low yielding, which can increase time and expenditures in pharmaceutical development. To enhance C–C bond formation protocols, we developed a highly efficient, selective, and combinatorially applicable Friedel-Crafts acylation to acetylate the C-3 position of imidazo[1,2-a]pyridines. The reaction, catalyzed by aluminum chloride, is both cost effective and more combinatorial friendly compared to acetylation reactions requiring multiple, stoichiometric equivalents of AlCl3. The protocol has broad application in the construction of acetylated imidazo[1,2-a]pyridines with an extensive substrate scope. All starting materials are common and the reaction requires inexpensive, conventional heating methods for adaptation in any laboratory. Further, the synthesized compounds are predicted to possess GABA activity through a validated, GABA binding model. The developed method serves as a superior route to generate C-3 acetylated imidazo[1,2-a]pyridine building-blocks for combinatorial synthetic efforts.
Keywords: Imidazo[1,2-a]pyridine; Combinatorial chemistry; Friedel-Crafts acylation; Drug discovery; Library generation
1. Introduction
Several strategies have been utilized to expeditiously achieve the construction of chemical libraries for drug discovery. One of the most fundamental, sought after transformations is the creation of carbon-carbon bonds. As such, numerous strategies have been developed employing metal catalysis, which typically require electron-rich ligand counterparts, to construct novel drug-like libraries. Although operationally friendly, metal/ligand based carbon-carbon bond formation can be cost prohibitive and carries the burden of environmental impact. Therefore, combinatorial synthetic efforts can be unreasonably expensive generating large amounts of metal/ligand waste. Because of our interest in imidazo [1,2-a]pyridine chemistry [1–3] and the medicinal relevance of the imidazo[1,2-a]pyridine scaffold [4–14], we developed a high- yielding, Friedel-Crafts C-3 acetylation completed under neat conditions utilizing catalytic amounts of AlCl3. The acetylation, which is operationally simplistic, requires only conventional heating, and is exceptionally well-suited for combinatorial chemistry efforts.
The imidazo[1,2-a]pyridine scaffold has proved very significant in therapeutic development serving as the backbone in many marketed therapeutics including alpidem [4] (1) (anxiolytic), necopidem [5] (2) (anxiolytic), saripidem [7] (3) (anxiolytic), zolpidem [8] (4) (insomnia), olprinone [9] (5) (cardiotonic), minodronic acid [10] (6) (bisphosphonate), and zolimidine [11] (7) (antiulcer) (Fig. 1). Imidazo[1,2-a]pyridines also exhibit a wide range of biological activities and have been investigated as kinase inhibitors for PI3K12, CDK [13], IRAK [14], FLT3 [1], and RET [6]. Typically, the imidazo[1,2-a]pyridine occupies the same region as adenine from ATP at the kinase active site [1,6].
Fig. 1.
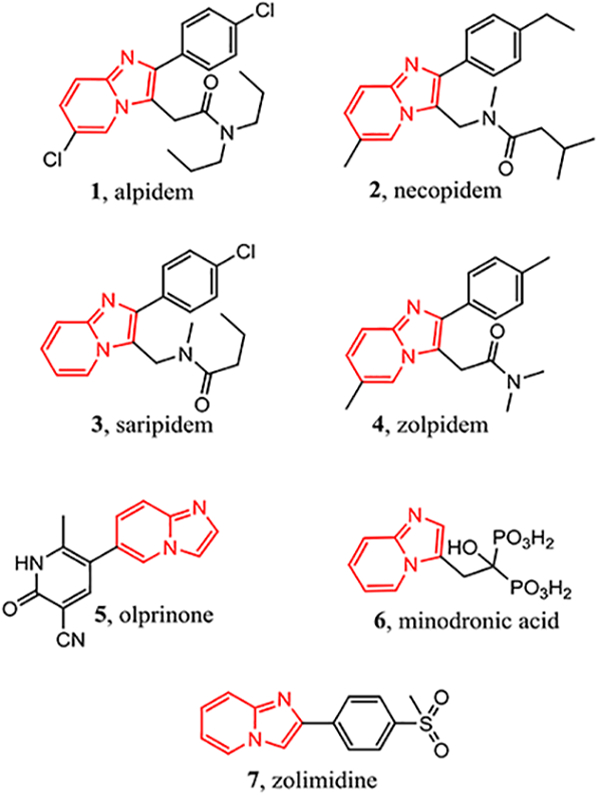
Marketed drugs bearing an imidazo[1,2-a]pyridine core.
Owing to the attractive biological properties of imidazo[1,2-a] pyridines, a variety of strategies to modify the core structure at the C-3 position have been developed [15–18]. Carbon-hydrogen activating, pseudo-Heck reactions that couple an aryl halide have been investigated with palladium metal and a ligand [19] and also in ligand free conditions [20–23]. Very recently, palladium and copper catalyzed direct C–H arylations of imidazo[1,2-a]pyridines at C-3 have been extensively studied [19–24]. Beyond carbon-carbon bond formation, synthetic advancement has been completed with diethyl azodicarboxylate (DEAD) as an amination agent [3] and nbromosuccinimide (NBS) as a halogenating agent [14]. Further, Vilsmeier-Haack reactions can selectively alkylate the C-3 position of imidazo[1,2-a]pyridine with DMF as the alkylation source, yet presenting with very low yields [25]. C-3 acetylations have also successfully been completed [26–36]. This has been accomplished in multiple steps, employing halogenated imidazo[1,2-a]pyridine C-3 derivatives with a strong base such as n-BuLi [37]. In another method, three molar equivalents of AlCl3, as well as one molar equivalent of carbon disulfide, are necessary to acetylate the C-3 position [38]. In general, known C-3 acetylation reactions of imidazo[1,2-a]pyridines are low yielding, must occur in multiple steps, and/or employ multiple molar equivalents of AlCl3 for reaction conversion.
Although arylations of imidazo[1,2-a]pyridines have been extensively investigated and optimized [1,2,12,17,18], the more simplistic C-3 acetylation is still low yielding, typically completed in step-wise fashion, and requires non-catalytic amounts of a Lewis acid [26,27,29–36], which is not optimal for combinatorial chemistry efforts. Therefore, it is important to develop an operationally-simplistic, inexpensive, high-yielding C-3 acetylation procedure, which employs a Lewis acid in catalytic amounts. The synthetic approach can be applied to the development of drug-like libraries, with more ecofriendly production that can further define the biological roles of imidazo[1,2-a]pyridine derivatives.
Combination of the C-3 directed imidazo[1,2-a]pyridine research, with further optimization for operational simplicity, we developed a one pot, single addition approach to acetylate imidazo [1,2-a]pyridine at the C-3 position requiring a catalytic amount of AlCl3 (Scheme 1). Herein, we report the successful employment of this methodology to the synthesis of 38 imidazo[1,2-a]pyridine derivatives. Investigation into the mechanism and a computational reaction coordinate diagram was completed to justify reaction conditions. Select derivatives were screened in a validated GABA receptor computational model and ligand/receptor binding was predicted. The acetylated derivatives can serve as drug-like building blocks or novel molecular probes to further define imidazo[1,2-a]pyridine biological properties.
Scheme 1.
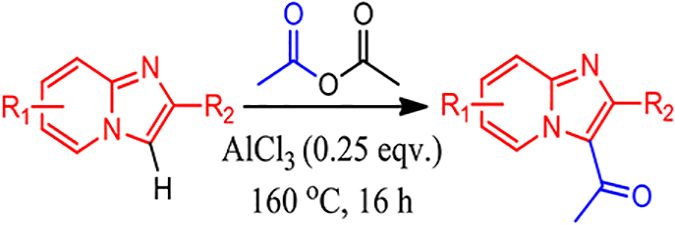
C-3 Friedel-Crafts Acetylation of Imidazo[1,2-a]pyridine.
2. Results and discussion
The identified method is able to effectively acetylate imidazo [1,2-a]pyridine and imidazo[1,2-a]pyridine analogues. The optimization of the procedure was focused on a simple set up, applicability to a wide range of imidazo[1,2-a]pyridine substrates, provision of high yield and purity, and catalytic requirement of a Lewis acid. We avoided reaction conditions that produced heterogeneous mixtures and incomplete conversion because, under parallel synthetic library production, complicated product mixtures can confound synthetic efficiency.
Initial evidence for the acetylation of imidazo[1,2-a]pyridine stemmed from the amination of imidazo[1,2-a]pyridine using DEAD [3]. Under the amination conditions, imidazo[1,2-a]pyridines can be successfully aminated at the C-3 position in aprotic solvents with gentle heating [3]. We utilized similar conditions in which DEAD was replaced with acetic anhydride under gentle heating to acetylate starting material 8 (Table 1). However, only trace amounts of conversion were observed (Table 1, Entry 1). To help progress the reaction, a variety of metal catalysts with carbonyl-activating properties were investigated (Table 1, Entry 2–8). Best conversion was observed with La(OTf)3 and AlCl3, yet conversion was very modest only producing the desired acetylation product in 32–40% yields. Because of laboratory availability and extremely minor cost, reaction optimization continued with AlCl3 as the Lewis acid catalyst.
Table 1.
Development and optimization for C-3 imidazo[1,2-a]pyridine acetylation.
| Entry | Catalyst | Eqv Cat | Eqv Ac2O | Solvent | T[°C] | Time (h) | Yield (%) |
|---|---|---|---|---|---|---|---|
| 1 | / | / | 2 | Acetonitrile | 80 | 6 | Trace |
| 2 | FeSO4 7H2O | 0.10 | 2 | Acetonitrile | 80 | 6 | NR |
| 3 | FeCl2 | 0.10 | 2 | Acetonitrile | 80 | 6 | NR |
| 4 | SeO2 | 0.10 | 2 | Acetonitrile | 80 | 6 | NR |
| 5 | La(NO3)3 6H2O | 0.10 | 2 | Acetonitrile | 80 | 6 | NR |
| 6 | ZrCl4 | 0.10 | 2 | Acetonitrile | 80 | 6 | 16 |
| 7 | La(0Tf)3 | 0.10 | 2 | Acetonitrile | 80 | 6 | 32 |
| 8 | A1C13 | 0.10 | 2 | Acetonitrile | 80 | 6 | 40 |
| 9 | A1C13 | 0.25 | 2 | Acetonitrile | 80 | 6 | 45 |
| 10 | A1C13 | 0.50 | 2 | Acetonitrile | 80 | 6 | 53 |
| 11 | A1C13 | 0.75 | 2 | Acetonitrile | 80 | 6 | 57 |
| 12 | A1C13 | 1.00 | 2 | Acetonitrile | 80 | 6 | 55 |
| 13 | A1C13 | 0.25 | 2 | Acetonitrile | 80 | 12 | 60 |
| 14 | A1C13 | 0.25 | 2 | Benzene | 80 | 12 | NR |
| 15 | A1C13 | 0.25 | 2 | Xylene | 80 | 12 | NR |
| 16 | A1C13 | 0.25 | 2 | Toluene | 80 | 12 | NR |
| 17 | A1C13 | 0.25 | 2 | DMA | 80 | 12 | Trace |
| 18 | A1C13 | 0.25 | 2 | DCE | 80 | 12 | NR |
| 19 | A1C13 | 0.25 | 2 | Dry DMF | 80 | 12 | NR |
| 20 | A1C13 | 0.25 | 2 | Dry THF | 80 | 12 | NR |
| 21 | A1C13 | 0.25 | 2 | Dry Dioxane | 80 | 12 | NR |
| 22 | A1C13 | 0.25 | 2 | Dry DMSO | 80 | 12 | NR |
| 23 | A1C13 | 0.25 | 2 | NMP | 80 | 12 | NR |
| 24 | A1C13 | 0.25 | Neata | Ac2O | 80 | 12 | 63 |
| 25 | A1C13 | 0.25 | Neata | Ac2O | 100 | 12 | 75 |
| 26 | A1C13 | 0.25 | Neata | Ac2O | 130 | 12 | 78 |
| 27 | A1C13 | 0.25 | Neata | Ac2O | 160 | 16 | 99 |
NR = No Reaction.
Completed in 0.33 M acetic anhydride solution.
Increasing catalytic load from 0.10 to 1.00 equivalent had a positive impact on yield, but the yield plateaued due to formation of undesired side products (Table 1, Entry 8–12). The AlCl3 load was decreased to 0.25 equivalence while the reaction duration was doubled, which furnished a desirable increase in yield (Table 1, Entry 9, 13). Various reaction solvents were investigated and acetonitrile provided the desired acetylation product in highest yields (Table 1, Entry 14–23). We concluded that reaction kinetics maybe important for conversion, and reactions were completed with acetic anhydride as solvent to increase reactant concentration (Table 1, Entry 24–27). An increase in yield was identified, especially when reaction temperature and duration were increased to 160 °C for 16 h (Table 1, Entry 17). With these conditions, the desired acetylated imidazo[1,2-a]pyridine product underwent 100% conversion and was isolated in 99% yield only requiring catalytic amounts of AlCl3 (Table 1, Entry 17).
With optimized reaction conditions in hand, substrate scope was examined on 38 different imidazo[1,2-a]pyridine analogues (Table 2). Conversion was generally high with a diverse set of electron withdrawing (EWG) and electron donating (EDG) groups, and the reaction appeared robust with low substrate specificity. Highest yields were observed with compounds 9, 11, 13, 17, 21, and 28 all of which contained EDG and EWG functionality (Table 2). Therefore, it can be concluded that this C-3 acetylation has a broad scope encompassing diverse electron donating and withdrawing substituents for product conversion. Yields were generally lower for imidazo[1,2-a]pyridine derivatives with substitution at the C-8 as halogen (24, 36, 37, 43), methyl (10, 12, 22, 30, 34) or ether (23) (Table 2). It is possible that substitution off C-8 can alter electron density in the imidazo[1,2-a]pyridine core, which can lower the nucleophilicity of C-3. Because C-8 is on the other end of the imidazo[1,2-a]pyridine compared to C-3, substitution is likely not lowering product conversion from steric effects. Substitution off the C-2 carbon had little overall effect on product conversion with a slight trend favoring electron donating groups as yields sequentially increased from examples 16 (−NO2, 70%), 15 (−CF3, 81%), 20 (−Cl, 85%) to 11 (−OCH3, 92%). The reaction was also tolerated with fluorinated, chlorinated, and brominated heteroaryl aryls, which can provide synthetic handles for further derivatization (Table 2). We further investigated alkyl (45) and no substitution (29) at the C-2 position of imidazo[1,2-a]pyridine, which both furnished high yields (Table 2). However, substitution with a chlorine at C-2 decreased yield to about 20%, displaying that halogens are not well tolerated at this position (46, Table 2). The reaction conditions were gentle enough for stability of a benzyl protecting group, as conversion was observed for compound 23 (Table 2). Nitrogenous heterocycles at the C-2 position were also tolerated as observed with compound 39 (Table 2). Surprisingly, despite using acetic anhydride at high temperatures with long reaction durations, the N-1 alkylation product was never detected. Overall, the developed acetylation protocol has a broad substrate scope, furnishes high isolated yields, and requires only catalytic amounts of AlCl3.
Table 2.
Synthesized drug-like library of acetylated imidazo[1,2-a]pyridines.
| Compd | Structure | Yielda (%) | Compd | Structure | Yielda (%) |
|---|---|---|---|---|---|
| 9 |  |
99 | 20 | 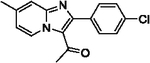 |
85 |
| 10 | 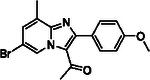 |
62 | 21 | 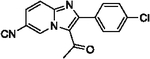 |
90 |
| 11 | 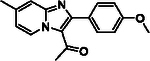 |
92 | 22 |  |
54 |
| 12 | 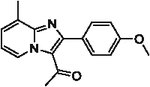 |
83 | 23 | 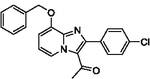 |
50 |
| 13 |  |
99 | 24 | 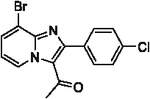 |
55 |
| 14 | 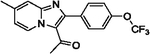 |
85 | 25 | 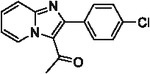 |
73 |
| 15 | 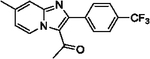 |
81 | 26 | 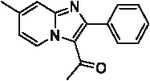 |
81 |
| 16 | 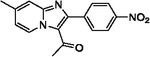 |
70 | 27 | 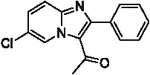 |
84 |
| 17 | 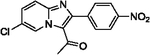 |
99 | 28 | 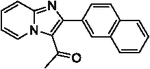 |
95 |
| 18 |  |
74 | 29 |  |
76 |
| 19 | 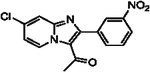 |
79 | 30 | 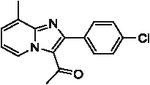 |
53 |
| 31 | 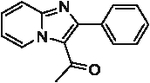 |
56 | 42 |  |
83 |
| 32 |  |
70 | 43 | 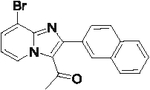 |
62 |
| 33 |  |
71 | 44 | 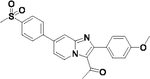 |
80 |
| 34 | 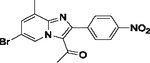 |
50 | 45 | 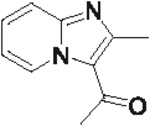 |
75 |
| 35 | 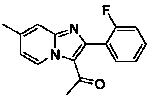 |
73 | 46 |  |
20 |
| 36 |  |
60 | |||
| 37 | 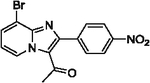 |
45b | |||
| 38 | 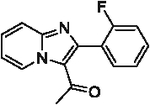 |
75 | |||
| 39 | 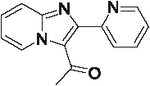 |
87 | |||
| 40 | 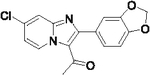 |
85 | |||
| 41 | 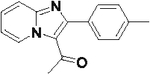 |
60 |
Isolated Yield.
Continued for 24 h.
A general, step-wise mechanism has been proposed for the acetylation of imidazo[1,2-a]pyridine using the developed conditions (Fig. 2, A). The mechanism follows the typical Friedel-Crafts acylation in which AlCl3 activates acetic anhydride by generating the acylium ion intermediate (Fig. 2 and 9a). It is postulated that the AlCl3(COO)− complex further activates the acylium ion acting as a strong electron-sink to break the resonance of the imidazo[1,2-a] pyridine core (Fig. 2 and 9b). The AlCl3(COO)− complex abstracts the C-3 proton from imidazo[1,2-a]pyridine restoring resonance, regenerating AlCl3, and also producing acetic acid (Fig. 2 and 9c).
Fig. 2.
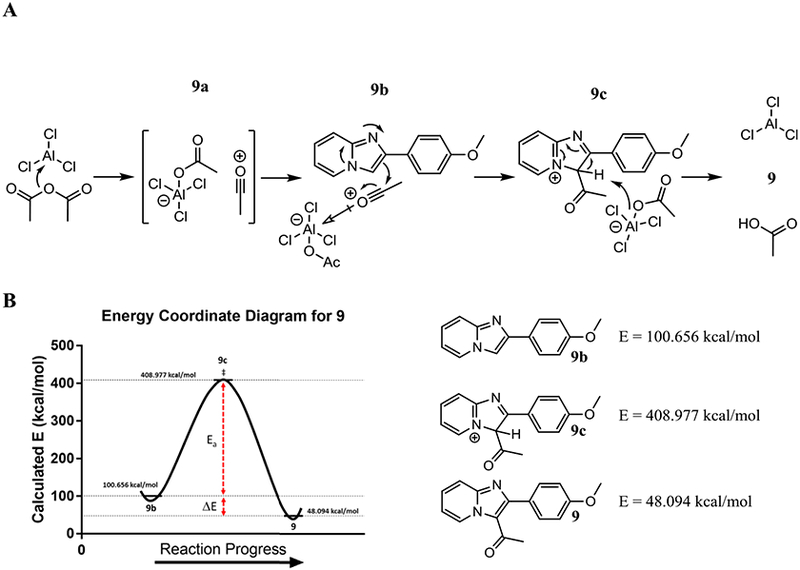
A) Proposed mechanism for the generation of acetylated imidazo[1,2-a]pyridines using the developed method. The mechanism is postulated to follow the typical Friedel-Crafts acylation. B) Calculated energy profiles for the starting material (9b), intermediate (9c), and product (9). In order to reach the transition state, a large activation energy barrier must be breached, and the generated product offers limited energetic payoffs. From the energy calculations, it appears reaching the transition state is rate limiting, which accounts for higher temperatures and reaction times necessary for complete product conversion.
To further study the reaction, computational energy profiles were generated for 9b, 9c, and 9 (Fig. 2, B). From the profiles, a reaction coordinate diagram was constructed, which displayed a large activation energy (Ea) barrier to reach the transition state of the reaction. In order to convert from the starting material (9b) to the transition state (9c), aromaticity within the imidazo[1,2-a] pyridine must be broken while simultaneously taking on a positive charge. This is not energetically favored and results in a large thermodynamic barrier that must be breached for product conversion. From calculations, the generated product offers a modest energy payoff and is likely not contributing significantly to reaction progression. Instead, breaching the Ea barrier is rate limiting and necessary for product generation. The computational results align well with the experimental results, as the large calculated Ea can account for the requirement of high heat and long reaction times for complete product conversion.
Acetylated imidazo[1,2-a]pyridines 9, 11, 13, 17, 21, and 28, which were isolated in varying yields (Table 2), were computationally modeled in the GABA receptor. The model was validated with zolpidem (4) (Fig. 3) and predicted zolpidem binding with reasonable affinity (Fig. 3, Table 3) [26,27,29,30,39]. Acetylated imidazo[1,2-a]pyridines were predicted to interact with the GABA receptor (4) (Table 3). Compounds 17 and 21 share a near-identical binding mode to that of zolpidem (4) (Fig. 3; Fig. 4). Compounds 9, 11, and 13 were also predicted to interact with GABA, but in a different conformation (Fig. 3; Fig. 4). Further, compounds 11, 13, 21, and 28 were predicted to have higher GABA affinity than zolpidem (4).
Fig. 3.
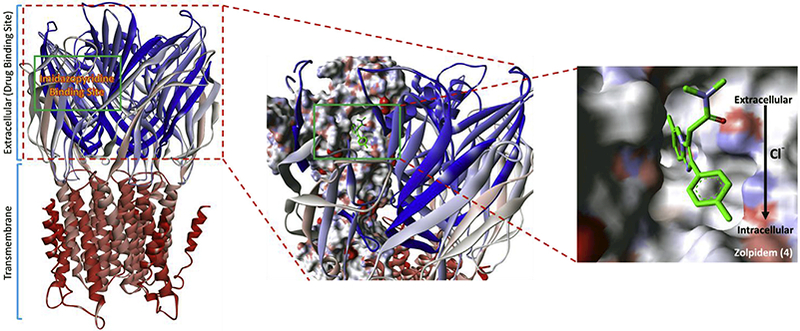
A) GABA ligand-gated ion channel structural overview. Imidazo[1,2-a]pyridine-based GABA drugs bind at the extracellular domain in a lipophilic pocket. The pocket is exposed to the central ion channel-core on the inside of the protein. Zolpidem (4) has been docked at the imidazo[1,2-a]pyridine binding site. Zolpidem (4) is an allosteric agonist of the GABA-A receptor. The amide and the toluene ring system point towards the ion channel, while the imidazo[1,2-a]pyridine is pushed into the pocket. Upon binding of zolpidem (4), the channel opens and relieves the chlorine membrane potential causing depolarization and depression of the neuron. PDB Accession Number: 2YOE [40].
Table 3.
GABA computational affinity of acetylated products.
| Compd | GABA-A ΔG (kcal/mol)a | Kd (μM)b |
|---|---|---|
| Zolpidem (4) | −7.6 | 2.6 |
| 9 | −7.4 | 3.7 |
| 11 | −7.7 | 2.2 |
| 13 | −8.1 | 1.1 |
| 17 | −7.4 | 3.7 |
| 21 | −7.7 | 2.2 |
| 28 | −8.0 | 1.3 |
Calculated with AutoDock Vina.
Calculated using Kd = e−ΔG/RT at 298 K.
Fig. 4.
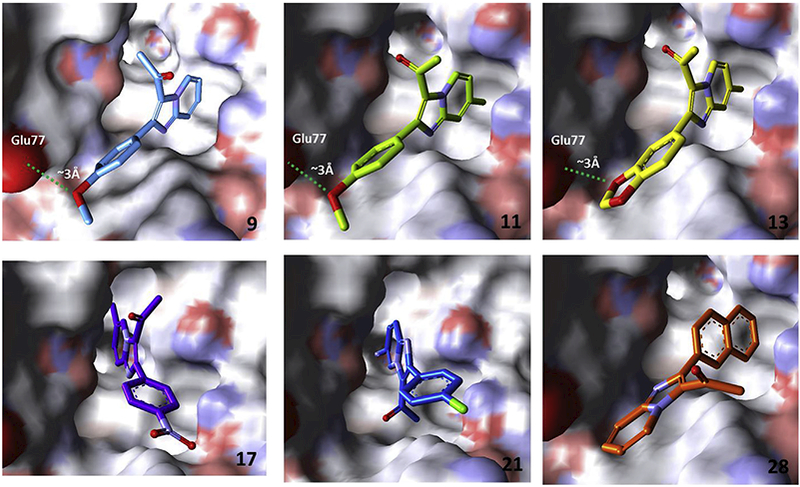
Computational binding models of acetylated imidazo[1,2-a]pyridines 9, 11, 13, 17, 21, and 28 in the GABA ion channel. PDB Accession Number: 2YOE [40].
Because of the high predicted affinity of compound 13 (Table 2), binding modes of 13 and zolpidem (4) were compared in detail (Fig. 5). Zolpidem (4) is predicted to accesses the back of a lipophilic channel, while 13 is predicted to enter a novel side-pocket. The methyl at the C-7 carbon on 13 seems necessary to fill the side-pocket (Fig. 5). The overlay of both 4 and 13 illustrate subtle differences in GABA binding (Fig. 5). However, both C-3 carbons from 4 and 13 are oriented upwards towards the opening of the ion channel (Fig. 5). The computational study supports the prospect of utilizing the acetylated imidazo[1,2-a]pyridine analogues as GABA binders. Further work is being completed to confirm the modeling results.
Fig. 5.
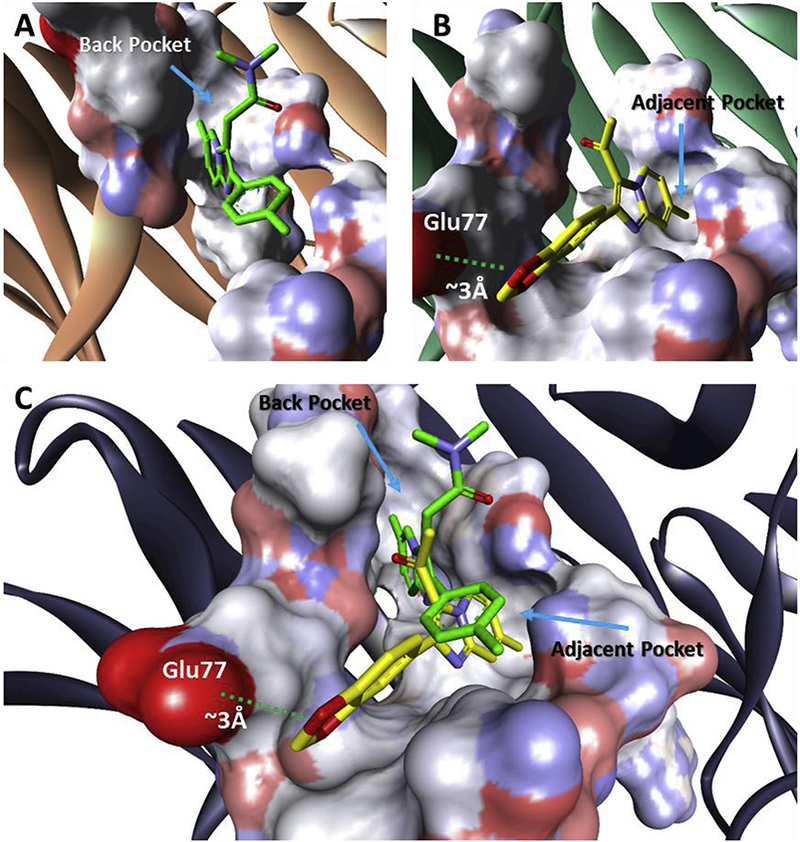
Comparison of predicted binding modes of zolpidem (4) and 13 in the GABA ion channel. PDB Accession Number: 2YOE [40].
In conclusion, we describe a selective C-3 Friedel-Crafts acylation of imidazo[1,2-a]pyridines that have predicted GABA binding properties. The method was built upon the Friedel-Crafts transformation and was completed neat under high heat to efficiently acetylate all starting material. The transformation was optimized for use in parallel synthesis to utilize (1) simple set up and basic lab-ware (conventional heating), (2) inexpensive and common reagents (acetic anhydride, AlCl3), (3) work-up free, extraction free, rapid silica-gel purification, and (4) catalytic amounts of AlCl3 for lower environmental impact. The optimized conditions permit facile synthesis of large, diverse imidazo[1,2-a]pyridine drug-libraries. Further, we presented modeling data that suggests the acetylated imidazo[1,2-a]pyridine analogues are plausible GABA binders. Understanding the important therapeutic impact of imidazo[1,2-a]pyridines [4–14], we believe that our developed acetylation method will be extremely useful for drug discovery as well as combinatorial efforts to expand the imidazo[1,2-a]pyridine scaffold.
3. Experimental procedures
All solvents were reagent grade or HPLC grade and all starting materials were obtained from commercial sources and used without further purification. Purity of final compounds was assessed using a Shimadzu ultra-high throughput LC/MS system (SIL-20A, LC-20AD, LC-MS 2020, Phenomenex® Onyx Monolithic C-18 Column) at variable wavelengths of 254 nM and 214 nM (Shimadzu PDA Detector, SPD-MN20A). The HPLC mobile phase consisted of a water-acetonitrile gradient buffered with 0.1% formic acid. 1H NMR spectra were recorded at 400 MHz and 13C spectra were recorded at 100 MHz, both completed on a Bruker 400 MHz instrument (Bruker Avance-III 400 MHz). Compound melting points were determined using differential scanning calorimetry (Q1000, TA Instruments). All compounds were purified using silicagel (0.035–0.070 mm, 60 Å) flash chromatography, unless otherwise noted.
3.1. General procedure for the synthesis of acetylated Imidazo[1,2-a] pyridines (9–44)
The imidazo[1,2-a]pyridine derivative (1.00 mmol) was placed into a glass vial affixed with a stir bar. Following, aluminum chloride (0.25 mmol) was added to the vial and acetic anhydride (3.0 mL) was added hitherto. The glass vial was securely sealed and heated to 160 °C in an oil bath. The reaction was heated for 16 h, at which point TLC analysis was completed to ensure complete product conversion. The crude reaction was diluted in DCM, transferred to a silica gel column and purified via flash chromatography. Depending on the polarity of the product, the reaction was either purified with a DCM/MeOH gradient (0% MeOH – 5% MeOH over 30 min) or a Hexanes/EtOAc (0% EtOAc – 50% EtOAc over 30 min) gradient. The purified product was collected, condensed, and dried in vacuo. Final acetylated products were isolated in yields from 21 to 99%.
3.2. Example experimental data of 9
White solid; yield, 99%; mp, 189.96 ± 1.22 °C; 1H NMR (400 MHz, CDCl3) δ 9.78 (d, J = 7.0 Hz, 1H), 7.81–7.66 (m, 1H), 7.65–7.43 (m, 3H), 7.09 (td, J = 6.9, 1.2 Hz, 1H), 7.05 (d, J 8.8 Hz, 2H), 3.91 (s, 3H), 2.26 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 189.21, 160.47, 155.27, 146.81, 131.20, 129.30, 129.06, 127.37, 121.36, 117.14, 114.71, 113.91, 55.37, 29.85; HRMS (ESI): m/z calculated for C16H14N2O2 [M+H]+: 267.10888; 267.11282 found.
3.3. General procedure for computational modeling studies
Computational modeling studies were completed using Auto-Dock Vina [41], AutoDock Tools, and Discovery Studio 3.5. Using AutoDock Tools, the GABA-A crystal structure [39] was prepared as follows: 1) All hydrogens were added as ‘Polar Only’ 2) A grid box at the binding site was created. Compounds to be computationally modeled were assigned appropriate rotatable bonds using Auto-Dock Tools (10) [1,6,42]. To computational model the compounds, AutoDock Vina [40] was employed. AutoDock Vina [41] provides docking scores in terms of ΔG values. After the modeling study, ΔG values were reported and binding modes were visualized and analyzed with Discovery Studio 3.5. ΔG values were converted into Kd values using the thermodynamic equation, Kd = e−ΔG/RT at 298 K with a gas constant value (R) of 1.987 10−3 kcal K−1 mol−1.
Acknowledgments
We would like to acknowledge the Biological Chemistry Program (BCP) at the University of Arizona for their continued scholarship support. We would also like to acknowledge the Therapeutic Sciences Program at the UAMS Cancer Institute.
Funding sources
This work was graciously supported by two training grants from The National Institutes of Health (3T32GM008804–10S1; T32 GM008804), University of Arizona startup funding, The Caldwell Health Sciences Research Fellowship, and the Yuma Friends Scholarship. This work was also supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number “P20 GM109005”. HL was supported by the grants (NIH 1R01CA194094–010 and 1R01CA197178–01A1).
Abbreviations
- GABA
gamma-aminobutyric acid
- C-3
carbon-3
- PI3K
phosphoinositide 3-kinase
- CDK
cyclin-dependent kinase
- IRAK
interleukin-1 receptor-associated kinase
- FLT3
fms-related tyrosine kinase 3
- RET
rearranged during transfection
- ATP
adenosine triphosphate
- C–H
carbon-hydrogen
- DEAD
diethyl azodicarboxylate
- NBS
N-Bromosuccinimide
- DMF
dimethylformamide
- La(OTf)3
lanthanum(III) trifluoromethanesulfonate
- AlCl3
aluminium chloride
- EWG
electron withdrawing group
- EDG
electron donating group
- C-2
carbon-2
- Glu
glutamic acid
- Å
angstrom
- C-7
carbon-7
- HPLC
high-performance liquid chromatography
- LC/MS
liquid-chromatography/mass spectrometry
- NMR
nuclear magnetic resonance
- MHz
megahertz
- mmol
millimole
- mL
milliliter
- TLC
thin layer chromatography
- DCM
dichloromethane
- MeOH
methanol
- EtOAc
ethylacetate
- mp
melting point
- CDCl3
deuterated chloroform
- ΔG
Gibbs free energy
- Kd
disassociation constant
- ACN
acetonitrile
- μM
micromolar
- K
Kelvin
- PDB
protein data bank
Footnotes
Notes
The Authors declare no competing financial interest.
Appendix A. Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.tet.2018.07.027.
References
- [1].Frett B, McConnell N, Smith CC, Wang Y, Shah NP, Li H, Eur. J. Med. Chem 94 (2015) 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wang Y, Frett B, Li H, Org. Lett 16 (2014) 3016–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wang Y, Frett B, McConnell N, Li H, Org. Biomol. Chem 13 (2015) 2958–2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Byrnes JJ, Greenblatt DJ, Miller LG, Brain Res. Bull 29 (1992) 905–908. [DOI] [PubMed] [Google Scholar]
- [5].Wang H, Wang Y, Liang D, Liu L, Zhang J, Zhu Q, Angew. Chem. Int. Ed 50 (2011) 5678–5681. [DOI] [PubMed] [Google Scholar]
- [6].Frett B, Moccia M, Carlomagno F, Santoro M, Li H, Eur. J. Med. Chem 86 (2014) 714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Renard S, Olivier A, Granger P, Avenet P, Graham D, Sevrin M, George P, Besnard F, J. Biol. Chem 274 (1999) 13370–13374. [DOI] [PubMed] [Google Scholar]
- [8].Depoortere H, Zivkovic B, Lloyd KG, Sanger DJ, Perrault G, Langer SZ, Bartholini G, J. Pharmacol. Exp. Therapeut 237 (1986) 649–658. [PubMed] [Google Scholar]
- [9].Mizushige K, Ueda T, Yukiiri K, Suzuki H, Cardiovasc. Drug Rev 20 (2002) 163–174. [DOI] [PubMed] [Google Scholar]
- [10].Yuasa T, Nogawa M, Kimura S, Yokota A, Sato K, Segawa H, Kuroda J, Maekawa T, Clin. Canc. Res 11 (2005) 853–859. [PubMed] [Google Scholar]
- [11].Zhang Y, Chen Z, Wu W, Zhang Y, Su W, J. Org. Chem 78 (2013) 12494–12504. [DOI] [PubMed] [Google Scholar]
- [12].Lee H, Kim SJ, Jung KH, Son MK, Yan HH, Hong S, Hong S-S, Oncol. Rep 30 (2013) 863–869. [DOI] [PubMed] [Google Scholar]
- [13].Hamdouchi C, Zhong B, Mendoza J, Collins E, Jaramillo C, De Diego JE, Robertson D, Spencer CD, Anderson BD, Watkins SA, Zhang F, Brooks HB, Bioorg. Med. Chem. Lett 15 (2005) 1943–1947. [DOI] [PubMed] [Google Scholar]
- [14].Buckley GM, Ceska TA, Fraser JL, Gowers L, Groom CR, Higueruelo AP, Jenkins K, Mack SR, Morgan T, Parry DM, Pitt WR, Rausch O, Richard MD, Sabin V, Bioorg. Med. Chem. Lett 18 (2008) 3291–3295. [DOI] [PubMed] [Google Scholar]
- [15].Pericherla K, Kaswan P, Pandey K, Kumar A, Synthesis (Stuttg) 47 (2015) 887–912. [Google Scholar]
- [16].Koubachi J, El Kazzouli S, Bousmina M, Guillaumet G, Eur. J. Org Chem (2014) 5119–5138. [DOI] [PubMed] [Google Scholar]
- [17].Enguehard-Gueiffier C, Gueiffier A, Mini Rev. Med. Chem 7 (2007) 888–899. [DOI] [PubMed] [Google Scholar]
- [18].Bagdi AK, Santra S, Monir K, Hajra A, Chem. Commun 51 (2015) 1555–1575. [DOI] [PubMed] [Google Scholar]
- [19].Koubachi J, El Kazzouli S, Berteina-Raboin S, Mouaddib A, Guillaumet G, Synlett 2006 (2006) 3237–3242. [Google Scholar]
- [20].Lee J, Chung J, Byun SM, Kim BM, Lee C, Tetrahedron 69 (2013) 5660–5664. [Google Scholar]
- [21].Fu HY, Chen L, Doucet H, J. Org. Chem 77 (2012) 4473–4478. [DOI] [PubMed] [Google Scholar]
- [22].Belkessam F, Derridj F, Zhao L, Elias A, Aidene M, Djebbar S, Doucet H, Tetrahedron Lett 54 (2013) 4888–4891. [Google Scholar]
- [23].Nair DK, Mobin SM, Namboothiri INN, Org. Lett 14 (2012) 4580–4583. [DOI] [PubMed] [Google Scholar]
- [24].Chernyak N, Gevorgyan V, Angew Chem. Int. Ed. Engl 49 (2010) 2743–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].McKenna CE, Kashemirov BA, K.M. Błaz_ewska, I. Mallard-Favier, C.A. Stewart, J. Rojas, M.W. Lundy, F.H. Ebetino, R.A. Baron, J.E. Dunford, M.L. Kirsten, M.C. Seabra, J.L. Bala, M.S. Marma, M.J. Rogers, F.P. Coxon, J. Med. Chem 53 (2010) 3454–3464. [DOI] [PubMed] [Google Scholar]
- [26].Byth KF, Cooper N, Culshaw JD, Heaton DW, Oakes SE, Minshull CA, Norman RA, Pauptit RA, Tucker JA, Breed J, Pannifer A, Rowsell S, Stanway JJ, Valentine AL, Thomas AP, Bioorg. Med. Chem. Lett 14 (2004) 2249–2252. [DOI] [PubMed] [Google Scholar]
- [27].Reddy BJ, Reddy MS, J. Chil. Chem. Soc 55 (2010) 483–485. [Google Scholar]
- [28].Burkholder C, Dolbier WR, Medebielle M, Ait-Mohand S, Tetrahedron Lett 42 (2001) 3077–3080. [Google Scholar]
- [29].Li R-D, Wang H-L, Li Y-B, Wang Z-Q, Wang X, Wang Y-T, Ge Z-M, Li R-T, Eur. J. Med. Chem 93 (2015) 381–391. [DOI] [PubMed] [Google Scholar]
- [30].Matsufuji T, Ikeda M, Naito A, Hirouchi M, Kanda S, Izumi M, Harada J, Shinozuka T, Bioorg. Med. Chem. Lett 23 (2013) 2560–2565. [DOI] [PubMed] [Google Scholar]
- [31].Melvin JR; Graupe M; Venkataramani C Fused Heterocyclic Inhibitor Compounds US 2010/0029638 A1, February 4, 2010.
- [32].Xiang D, et al. , Faming Zhuanli Shenqing, June 05, 2013, 103130792. [Google Scholar]
- [33].Berg S, Hellberg S, PCT Int. Appl (2002) WO/2002/065979.
- [34].Terauchi J, Koike T, Honda E, Nakamura M, Kajita Y, Hoashi Y, Morimoto S, Fujimoto T, PCT Int. Appl (2011) WO/2011/002067.
- [35].Melvin LS, Graupe M, Venkataramani C, PCT Int. Appl (2010) WO/2010/009155.
- [36].Thomas AP, Breault GA, Beattie JF, Jewsbury PJ, PCT Int. Appl (2001) WO/2001/014375.
- [37].Chezal JM, Moreau E, Chavignon O, Lartigue C, Blache Y, Teulade JC, Tetrahedron 59 (2003) 5869–5878. [Google Scholar]
- [38].Cupido T, Rack PG, Firestone AJ, Hyman JM, Han K, Sinha S, Ocasio CA, Chen JK, Angew. Chem. Int. Ed 48 (2009) 2321–2324. [DOI] [PubMed] [Google Scholar]
- [39].Kapur J, Macdonald RL, Mol. Pharmacol 50 (1996) 458–466. [PubMed] [Google Scholar]
- [40].Spurny R, Ramerstorfer J, Price K, Brams M, Ernst M, Nury H, Verheij M,Legrand P, Bertrand D, Bertrand S, Dougherty DA, de Esch IJP, Corringer P-J, Sieghart W, Lummis SCR, Ulens C, Proc. Natl. Acad. Sci 109 (2012) E3028–E3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Trott O, Olson AJ, J. Comput. Chem 31 (2010) 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Frett B, McConnell N, Wang Y, Xu Z, Ambrose A, Li H, Med. Chem. Commun 5 (2014) 1507–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]