

Abstract
Neisseria gonorrhoeae, an obligate human pathogen, is a leading cause of communicable diseases globally. Due to rapid development of drug resistance, the rate of successfully curing gonococcal infections is rapidly decreasing. Hence, research is being directed toward finding alternative drugs or drug targets to help eradicate these infections. 4-Hydroxy tetrahydrodipicolinate reductase (DapB), an important enzyme in the meso-diaminopimilate pathway, is a promising target for the development of new antibiotics. This manuscript describes the first structure of DapB from N gonorrhoeae determined at 1.85 Å. This enzyme uses NAD(P)H as cofactor. Details of the interactions of the enzyme with its cofactors and a substrate analog/inhibitor are discussed. A large scale bioinformatics analysis of DapBs’ sequences is also described.
Keywords: lysine biosynthesis; 4-hydroxy tetrahydrodipicolinate reductase; dihydrodipicolinate reductase; NADH; NADPH; 2,6-pyridine dicarboxylic acid
Introduction
Neisseria gonorrhoeae is an obligate human pathogen which is a leading cause of communicable diseases. It is a Gram negative, diplococcus, fastidious microorganism that grows optimally at 35.5°C, pH 7.4. These values are very close to the temperature and pH of human blood, which makes humans ideal hosts for this pathogen [1]. N. gonorrhoeae ranks second in terms of commonly reported notifiable disease in the United States with an infection rate of 145.8 cases per 100,000 population reported recently [2]. This rate in reality may be much higher due to underreporting and latent asymptomatic infections [3]. This bacterium commonly establishes infection in the urethra and the uterine cervix in men and women, respectively. Although it does not commonly infect other organs, it is known to cause arthritis-dermatitis syndrome, endocarditis, osteomyelitis, and meningitis in some patients [4]. In HIV infected individuals, N. gonorrhoeae induces an increase in the expression of viral RNA, thus aggravating the infection [3]. In addition to HIV susceptibility, the infection can result in pelvic inflammatory disease, which results in permanent fallopian tube scarring and blockage. Furthermore, it is associated with subsequent infertility and ectopic pregnancies in women [5]. Apart from adults, N. gonorrhoeae can also cause ophthalmia neonatorum, pharyngitis, rectal infections, and pneumonia in infants, if their mother is a carrier (symptomatic or asymptomatic infection) [6].
The first line of treatment for N. gonorrhoeae infection uses drugs like cefixime, ceftrioxane, ciprofloxacin or levofloxacin [6, 7]. Unfortunately, these antibiotics are proving inadequate in eradicating this pathogen due to its rapid development of drug resistance. Hence, the current approved medication for gonococcal infections is a combination of the above mentioned drugs with azithromycin [2]. However, there are several repots that indicate failure of combination therapy in many patients [8]. It is also being speculated that gonococcal infections will soon become untreatable [6–8]. This situation raises the need for development of new drugs and identification of new drug targets in the pathogen.
Regarding the identification of new drug targets in N. gonorrhoeae, the meso- diaminopimilate (lysine biosynthesis) pathway is a promising drug target due to its essentiality in microbial metabolism of lysine. Furthermore, this pathway is not found in mammals, permitting drugs targeting this pathway to be selective for pathogens [9–12]. The general pathway is outlined in Figure 1. 4-hydroxy-tetrahydrodipicolinate (HTPA) reductase (DapB; formerly dihydrodipicolinate reductase) is an important enzyme of this pathway that is critical for the production of lysine and meso-diaminopimelate. Microorganisms use these compounds to covalently link peptidoglycan in the cell wall [13]. Hence, blocking the production of these compounds will introduce defects in cell wall synthesis and protein production due to lysine deficiency [12]. DapB catalyzes the reduction of (2S,4S)-4-hydroxy-2,3,4,5- tetrahydrodipicolinate to 2,3,4,5-tetrahydrodipicolinate in an NADH/NADPH dependent reaction (Fig. 1). Apart from being a reductase, it is also speculated that DapB may act as a dehydratase [14].
Figure 1.

Lysine (meso-diaminopimilate ) biosynthesis through succinylase pathway (most commonly observed in Gram negative bacteria) [9]. The reaction catalyzed by DapB is highlighted in gray.
Structurally, all the bacterial homologs of the DapB enzyme have been reported to have homotetrameric assembly. Each monomer consists of a coenzyme binding and an oligomerization domain [15]. The coenzyme domain binds to nucleotide via the GXXGXXG motif that is conserved in most of the known DapB’s [16]. The oligomerization domain consists of conserved active site residues with a central lysine residue. This manuscript describes the first structure of 4-hydroxy-tetrahydrodipicolinate reductase from N. gonorrhoeae. Interactions of this enzyme with its cofactors NADH and NADPH, as well as 2,6-pyridine dicarboxylic acid (2,6- PDC) which is a substrate analog and an inhibitor are discussed. A detailed bioinformatics based analysis of DapB sequences is shown. This analysis reveals that DapBs can be segregated into three distinct groups, with one group lacking any representative structure.
Materials and Methods
Production of recombinant NgDapB
The NgDapB gene was codon optimized for expression in Escherichia coli, synthesized, and cloned into pJ411 plasmid vector system by ATUM, Inc. (Newark, CA). The plasmid contains an inducible T7 promotor, and kanamycin as selection marker. For ease of purification, a cleavable N-terminal hexahistidine tag was added to the NgDapB gene sequence [17]. The plasmid was co-expressed in E. coli BL21 (DE3) cells with pGro7 chaperone plasmid (groES- groEL chaperone system) (Takara Bio. Inc.). Co-expression experiments were done using arabinose (0.5 mg/L) as the inducer for pGro7 plasmid. Cultures were grown to an O.D. of 0.6 in Luria-Bertani (LB) at 37°C and then induced with 0.5 mM isopropyl β-D-1- thiogalactopyranoside (IPTG) and grown overnight at 16°C. Cell pellets were suspended in binding buffer (50 mM Tris-HCl, 500 mM NaCl, 10 mM imidazole, pH 8.0) supplemented with protease inhibitor tablets (Pierce), and lysed by sonication. Insoluble cellular debris was separated from supernatant by centrifugation. The supernatant was loaded onto Ni-NTA resin (Qiagen or Pierce) equilibrated with binding buffer. The protein was eluted using elution buffer (50 mM Tris-HCl, 500 mM NaCl, 300 mM imidazole, pH 8.0). Elution fractions containing protein were identified by SDS-PAGE, pooled, and dialyzed overnight using SnakeSkin Dialysis Tubing (MW cutoff 10,000 kDa) (ThermoFisher, Grand Island, NY) in dialysis buffer (10 mM Tris-HCl, 150 mM NaCl, pH 7.4). The dialyzed protein was concentrated and further purified by size exclusion chromatography using a GE Healthcare AKTA-Pure FPLC and HiLoad Superdex 200 column. The protein content of the concentrated elution fractions was determined by Bradford Assay [18] and purity determined by SDS-PAGE. Protein stability was evaluated using Differential Scanning Fluorimetry [19].
Crystallization, data collection and structure determination
Sitting drop vapor diffusion method and 96 well crystallization plates (Hampton Research, Aliso Viejo, CA) were used for all crystallization experiments. Different crystallization conditions and recombinant NgDapB (~10 mg/mL) were mixed in 1:1 ratio. The well diffracting crystal producing the data detailed herein was grown from the Hampton Research Index Screen condition of 0.1 M Bis-Tris pH 6.5 and 2.0 M ammonium sulfate at room temperature. The crystal was cryo-cooled in liquid nitrogen for the X-ray diffraction experiment.
Diffraction data was collected using South Regional Collaborative Access Team (SER- CAT) 22ID beamline at Advanced Photon Source (APS), Argonne National Laboratory (Lemont, IL). HKL-2000 software package was used to process diffraction images [20]. Data collection and refinement statistics are shown in Table 1. Structure was solved by molecular replacement using MOLREP [21] integrated with HKL-3000 [22]. E. coli DapB (PDB ID: 1ARZ) was used as a search model. The initial model was rebuilt using BUCCANEER [23]. REFMAC [24] and HKL-3000 were used for refinement. COOT [25] was used for model rebuilding and validation. MOLPROBITY was used as a final validation tool for the structure [26].The final coordinate file along with structure factors were deposited to the Protein Data Bank with accession code 6BDX.
Table 1.
Data collection, processing and refinement statistics. Parameters for the high resolution shell are in parenthesis.
| PDB ID | 6BDX |
| Diffraction source | APS Beamline 22ID |
| Wavelength (Å) | 1.000 |
| Temperature (K) | 100 |
| Space group | I222 |
| a, b, c (Å) | 57.8, 71.7, 138.4 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution range (Å) | 40.00–1.85 (1.88–1.85) |
| Solvent content (%) | 67 |
| No. of unique reflections | 25492 (1715) |
| Completeness (%) | 99.9 (100.0) |
| Redundancy | 7.9 (7.8) |
| 〈 I/σ(I)〉 | 48.1 (3.4) |
| Rmeasure | 0.067 (0.757) |
| Rp.i.m. | 0.024 (0.270) |
| Overall B factor from Wilson plot (Å2) | 31.6 |
| CC1/2 | 0.863 |
| Rcryst | 0.169 (0.216) |
| Rfree | 0.209 (0.267) |
| R.m.s. deviations | |
| Bonds (Å) | 0.021 |
| Angles (°) | 1.9 |
| Average B factors (Å2) | 39.8 |
| Ramachandran plot | |
| Favoured (%) | 98 |
| Allowed (%) | 100 |
Sequence similarity based clustering
The full length sequences of NgDapB homologs, corresponding to proteins from Pfam families 4-hydroxy-tetrahydrodipicolinate reductase N-terminus (DapB_N - PF01113; coenzyme binding domain) and 4-hydroxy-tetrahydrodipicolinate reductase C-terminus (DapB_C - PF05173; oligomerization domain), were downloaded from the Pfam database [27]. The sequences were merged, followed by the removal of redundant entries, rendering a final dataset of 4907 sequences. To visualize sequence similarities between subgroups of related proteins CLANS (CLuster ANalysis of Sequences) was used [28], where the P-values of highly-scoring segment pairs (HSPs) obtained from an N x N BLAST search, were used to compute attractive and repulsive forces between each sequence pair. The clustering was performed in 2D, using P- value threshold of 1e-6.
Isothermal titration calorimetry (ITC)
Binding of NgDapB with nucleotides (NADH and NADPH) and substrate analog 2,6- pyridine dicarboxylic acid (2,6-PDC) was studied using ITC. Binding of nucleotides was studied using a VP-ITC Micro Calorimeter (MicroCal, Northampton, MA) whereas the interactions with 2,6-PDC were analyzed using Afinity ITC (TA Instruments, New Castle, DE ). Purified enzyme was buffer exchanged into 25 mM HEPES, pH 7.5, using gel filtration chromatography as described above. 2 mM nucleotide and 2,6-PDC solutions were prepared in the same buffer. Prior to use, these solutions were degassed under vacuum using gentle stirring. For all the experiments with nucleotides, the final protein concentration in the sample cell was 75 μM. 4 μL of ligand was injected in 9.6 s for 25 injections with a spacing of 200 s between injections. The syringe was rotated at 310 rpm. Data analysis was done using ORIGIN software (OriginLab, Northampton, MA). For experiments with 2,6-PDC, 100 μM protein was used. 20 injections of 2 μL each with injection interval of 250 s and syringe rotation of 150 rpm was used for every run. For 2,6-PDC binding, protein with N-terminal hexahistidine tag cleaved was used. Hexahistidine tag was cleaved using previously described protocol [19]. The temperature was maintained at 25°C for all the experiments, and the experiments were done in triplicates.
Various Computational Calculations
Figures were prepared with Pymol [29] and Espript [30]. PDBePISA [31] was used to calculate areas between protein chains in the oligomeric assembly. SSM algorithm [32] as implemented in COOT was used for superposition of structures.
Results and Discussion
Overall structure of NgDapB
The recombinant protein was crystallized with the N-terminal hexahistidine tag attached to it. Crystal structure of apo-NgDapB was determined at 1.85 Å resolution. The protein crystallized with one chain in the asymmetric unit, however, the biological assembly corresponds to a tetramer. Despite extensive efforts, we were unable to obtain crystal structures of NgDapB with cofactors. The apo structure of NgDapB exhibits tetrameric arrangement which is a common characteristic of all bacterial homologs of DapB. Each protein chain consists of 267 residues that form coenzyme-binding (residues 1–126 and 237–268) and oligomerization (residues 127–236) domains. The coenzyme-binding domain adopts a Rossmann fold architecture where seven parallel β strands in the center are flanked by six α-helices. The oligomerization domain is composed of two a helices and four P strands. These β strands from each monomer interact with each other to form a central β barrel which forms the base for the tetrameric assembly (Fig. 2). In case of NgDapB, the interfaces between a single protein chain and two neighboring chains from the tetrameric assembly are large and there areas are 1460Å2 and 870Å2 respectively.
Figure 2.

Structure of NgDapB. A) Cartoon representation (top panel) and space filling model (bottom panel) of NgDapB monomer. Coenzyme binding domain and oligomerization domain are marked by dashed circles. B) Cartoon representation (top panel) and space filling model (bottom panel) of NgDapB monomer colored according to sequence conservation among bacterial homologs. Regions with identical residues are marked by red whereas highly conserved areas are indicated in blue. C) Tetrameric assembly of NgDapB. Four monomers are indicated in different colors.
Structural studies indicate that apo-DapBs exist predominantly in an open conformation that is also observed in the case of NgDapB. The open conformation of NgDapB is for example similar to those observed in DapB’s from E. coli (PDB ID: 1ARZ (chain A); 62% of sequence identity; rmsd 1.2Å over 248 superposed Ca atoms), P. aeruginosa (PDB ID: 4YWJ (chain A); 60% of sequence identity; rmsd 1.3Å over 250 superposed Cα atoms) and B. thailandensis (PDB ID: 4F3Y (chain A); 60% of sequence identity; rmsd 1.0Å over 249 superposed Cα atoms) (Fig. 3). However, after NADH/NADPH and HTPA binding, the protein conformation changes dramatically. The conformation change involves a large rotation of the coenzyme binding domain toward the oligomerization domain that leads to a closed conformation and allows for the proper positioning of both substrates [33].
Figure 3.
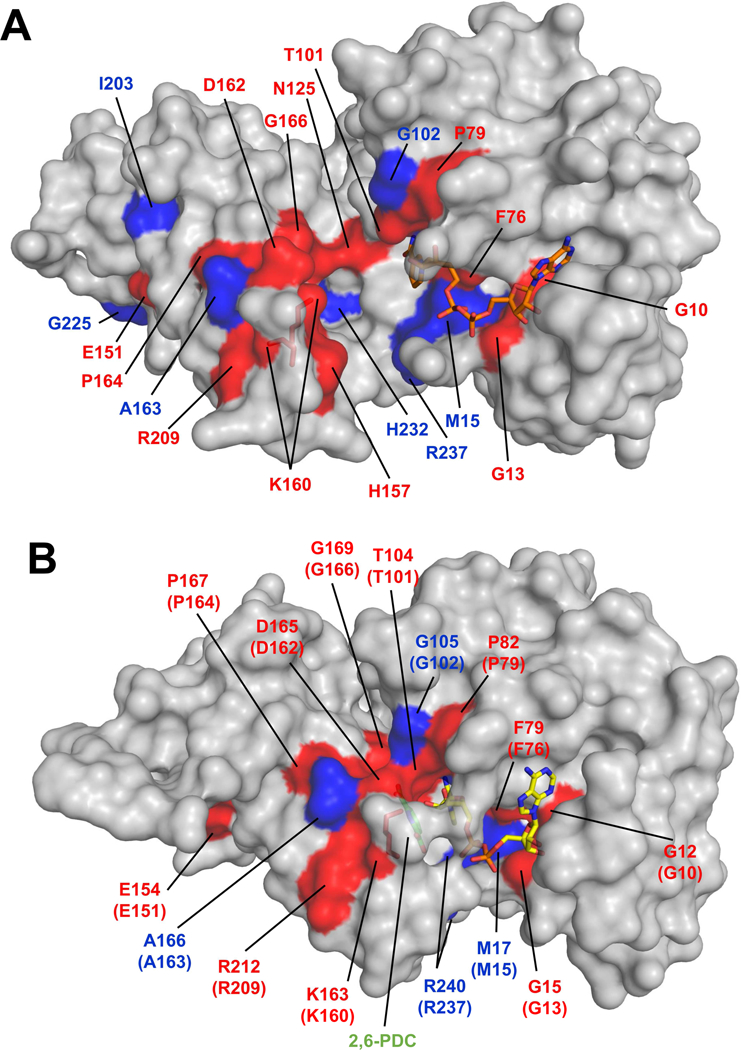
Structure of NgBapB (A) and EcDapB (B) with sequence conservation mapped on protein structure. The residues marked in red are completely conserved among DapBs that have their structure determined (see Supplemental Figure 1). The residues marked in blue are conserved in at least 10 out of 12 DapBs that have their structure determined. Coenzymes, 2,6- pyridine dicarboxylic acid (green) and the catalytic lysine residues are shown in stick representation. In case of NgDapB, the position of the coenzyme is modeled using structure of PaDapB (PDB ID: 4YWJ).
Sequence similarity based clustering
Domain architecture of NgDapB shows two domains classified in Pfam, namely 4- hydroxy-tetrahydrodipicolinate reductase N-terminus (DapB_N-PF01113; coenzyme binding domain) and 4-hydroxy-tetrahydrodipicolinate reductase C-terminus (DapB_C-PF05173; oligomerization domain). The domain organization, where both domains are present, is the most common architecture among NgDapB homologs (Fig. 2). The full-length sequences of all proteins classified to either of the two Pfam domains, which therefore share similarity with NgDapB, were used, and pair-wise sequence similarity-based clustering was carried out to visualize groups of more similar protein sequences within the analyzed dataset. The clustering results are summarized in Figure 4.
Figure 4.

Two-dimensional projection of the CLANS clustering results. Proteins are indicated by dots. Lines indicate sequence similarity detectable with BLAST and are colored by a spectrum of shades of gray according to the BLAST P-value. Sequences corresponding to structures in the PDB are indicated by blue dots, Sequence of NgDapB is indicated by red dot.
Two of the groups showing the lowest similarity to DapBs formed two satellite clusters. One is comprised of poorly characterized proteins with DapB_N domain and SAF domain. The second is composed of multi-domain proteins, annotated as various transcription regulators. These are characterized by the presence of DapB_N domain accompanied by several domains including Per-Arnt-Sim (PAS) domains, which are responsible for the binding of various proteins and small molecules, a regulatory Sigma-54 interaction domain, and a helix-turn-helix motif.
The cluster of glyceraldehyde-3-phosphate dehydrogenases is represented by several structurally characterized proteins (PDB IDs: 1CF2, 2YYY, 2CZC, 1B7G). Proteins in this group are characterized by the presence of DapB_N domain (PF01113) merged to the glyceraldehyde 3-phosphate dehydrogenase C-terminal domain (PF02800), This group shows the closest similarity to the cluster of meso-diaminopimelate D-dehydrogenases (PDB ID: 3WYB), where the DapB_N domain is accompanied by the diaminopimelic acid dehydrogenase C-terminal domain (PF16654).
There are four clusters of protein sequences annotated as DapB. The largest one (Fig. 4, cluster 4-hydroxy-tetrahydrodipicolinate reductase I) is comprised mostly of DapBs from Proteobacteria, including NgDapB and structurally characterized DapBs from Bartonella henselae, Burkholderia thailandensis, Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus and Thermotoga maritima (PDB IDs: 3IJP, 4F3Y, 1ARZ, 4YWJ, 3QY9, 1VM6).
The second group of DapBs (Fig. 4, cluster 4-hydroxy-tetrahydrodipicolinate reductase II) is predominantly composed of proteins from Bacteroidetes and has no structurally characterized representatives. Sequences in this cluster show high similarity to 4-hydroxy- tetrahydrodipicolinate reductase cluster I, but not III.
In the third group (Fig. 4, cluster 4-hydroxy-tetrahydrodipicolinate reductase III) two subgroups can be distinguished, where one is comprised of proteins from Actinobacteria represented by structurally characterized proteins from Corynebacterium glutamicum and Mycobacterium tuberculosis (PDB IDs: 5EER and 1C3V). The second is represented by DapB from Anabaena variabil (PDB ID: 5KT0), comprising of sequences from various bacteria, predominantly Firmicutes and Cyanobacteria, but also proteins from Viridiplantae chloroplasts.
In contrast to the three above-mentioned groups that have both DapB_N and DapB_C domains, proteins in the fourth cluster (Fig. 4, cluster 4-hydroxy-tetrahydrodipicolinate reductase IV) have only well-defined N-terminal DapB_N domain and unclassified C-terminal extension. These proteins show similarity to clusters I and II, and only the second subcluster of cluster III that includes DapB from Anabaena variabilis.
NADH and NADPH binding
In parallel with structural studies, nucleotide specificity of NgDapB was determined using ITC. Data analysis revealed that NgDapB can bind both the cofactors but has preference for NADH. The dissociation constants (Kd) were found to be 2.5 ± 0.5 (μM and 4.8 ± 0.6 (μM for NADH and NADPH respectively (Fig. S2). A lot of variation and different preferences for nucleotide binding has been reported for DapB homologs (Table 2). For example, E. coli DapB has greater affinity for NADH compared to NADPH [34, 35]. On the other hand, S. aureus DapB (MRSA) binds NADPH with higher affinity [36]. It has been speculated that the acidic and basic residues that are present around 20 amino acids down the conserved GXXGXXG motif play an important role in cofactor affinity and specificity of DapB enzyme [37]. In E. coli DapB, residues Glu38 and Arg39 are important for binding of NADH and NADPH [38]. In NgDapB, Glu36 is conserved but His37 is the counterpart for Arg39. This may be a contributing factor in the slightly lower affinity for NADH as compared to E. coli, even though both enzymes show preference for NADH over NADPH. Another possibility is that a different residue altogether plays a role in cofactor binding as seen in Methicillin resistant S. aureus [37]. However, these conjectures remain elusive due to a lack of crystal structures of NgDapB with its cofactors bound.
Table 2.
. Dissociation constants (Kd) for NADH or NADPH complexes with DapB’s from different bacteria as determined using ITC.
Interactions of NgDapB with 2,6-Pyridine dicarboxylic acid
2,6-Pyridine dicarboxylic acid (2,6-PDC) is a widely studied inhibitor of the DapB enzyme [39]. The unstable nature of the substrate for DapB makes it difficult to use for crystallization, and as a result, all the structures for this enzyme have been determined using 2,6- PDC in place of its substrate. It was reported that E. coli DapB follows an ordered sequential mechanism, in which the binding of cofactors (NAD(P)H) is followed by the binding of the substrate [40, 41]. However, later NMR studies of E. coli protein suggested that 2,6-PDC may bind to the enzyme both in the presence or absence of nucleotide (cofactor). In contrast to this, it was shown that enzyme from S. aureus cannot bind 2,6-PDC in absence of NADH [37]. Our results demonstrate that NgDapB can bind 2,6-PDC without cofactors, with a Kd of 21.2 ± 0.7 μM (Fig. S3). This observation suggests that the NgDapB catalyzed reaction follows a random sequential mechanism rather than the ordered sequential mechanism. Currently, there is no structure of any DapB in complex with 2,6-PDC alone, hence it is not clear what conformation the enzyme adopts upon inhibitor binding and which residues participate in 2,6-PDC binding when cofactor is not present. Interestingly, in the case of the E. coli protein, it was demonstrated that while 2,6-PDC binds cooperatively, NADH does not [42].
In summary, this study provides a structural background for investigating 4-hydroxy- tetrahydrodipicolinate reductase as a potential drug target in N. gonorrhoeae. Furthermore, we believe that the compounds directed against NgDapB may be tailored so they can target the enzyme’s various conformational states.
Supplementary Material
N gonorrhoeae is an obligate human pathogen which is a leading cause of communicable diseases.
4-Hydroxy-tetrahydrodipicolinate reductase from N. gonorrhoeae (NgDapB) is a new drug target, and its structure was determined.
Using Isothermal Titration Calorimetry (ITC) we have demonstrated and characterized binding of NADH, NADPH and an inhibitor (2,6-PDC) to NgDapB.
In the case of NgDapB, the ITC results suggest random sequential mechanism for substrate binding, as 2,6-PDC may be treated a substrate analog.
A large scale analysis of DapBs’ sequences was performed. This analysis reveals that DapBs can be segregated into three distinct groups, with one group lacking any representative structure.
Acknowledgements
Diffraction data were collected at Southeast Regional Collaborative Access Team beamline at APS, Argonne National Laboratory. Use of APS was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract Nos. DE-AC02–06CH11357 and W-31–109-Eng-38. This work was partially supported by an ASPIRE III grant from the Office of the Vice President of Research at the University of South Carolina and grant form National Institute of Allergy and Infectious Diseases (R01AI120987).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Ng L-K, Martin IE, The laboratory diagnosis of Neisseria gonorrhoeae, The Canadian Journal of Infectious Diseases & Medical Microbiology, 16 (2005) 15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Gonorrhea, Center for Disease Control and Prevention, 2017.
- [3].Satterwhite CL, Torrone E, Meites E, et al. , Sexually Transmitted Infections Among US Women and Men: Prevalence and Incidence Estimates, 2008, Sexually Transmitted Diseases, 40 (2013) 187–193. [DOI] [PubMed] [Google Scholar]
- [4].Lee MH, Byun J, Jung M, et al. , Disseminated Gonococcal Infection Presenting as Bacteremia and Liver Abscesses in a Healthy Adult, Infection & Chemotherapy, 47 (2015) 60–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Edwards JL, Apicella MA, The Molecular Mechanisms Used by Neisseria gonorrhoeae To Initiate Infection Differ between Men and Women, Clinical Microbiology Reviews, 17 (2004) 965–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Miller KE, Diagnosis and treatment of Neisseria gonorrhoeae infections, American Family Physician, 73 (2006) 1779–1784. [PubMed] [Google Scholar]
- [7].Patel AL, Chaudhry U, Sachdev D, et al. , An insight into the drug resistance profile & mechanism of drug resistance in Neisseria gonorrhoeae, The Indian Journal of Medical Research, 134 (2011) 419–431. [PMC free article] [PubMed] [Google Scholar]
- [8].Tapsall JW, Antibiotic Resistance in Neisseria gonorrhoeae, Clinical Infectious Diseases, 41 (2005) S263–S268. [DOI] [PubMed] [Google Scholar]
- [9].Velasco AM, Leguina JI, Lazcano A, Molecular Evolution of the Lysine Biosynthetic Pathways, Journal of Molecular Evolution, 55 (2002) 445–449. [DOI] [PubMed] [Google Scholar]
- [10].Cox RJ, The DAP pathway to lysine as a target for antimicrobial agents, Natural Product Reports, 13 (1996) 29–43. [DOI] [PubMed] [Google Scholar]
- [11].Cox RJ, Sutherland A, Vederas JC, Bacterial diaminopimelate metabolism as a target for antibiotic design, Bioorganic & Medicinal Chemistry, 8 (2000) 843–871. [DOI] [PubMed] [Google Scholar]
- [12].Hutton CA, Perugini MA, Gerrard JA, Inhibition of lysine biosynthesis: an evolving antibiotic strategy, Molecular BioSystems, 3 (2007) 458–465. [DOI] [PubMed] [Google Scholar]
- [13].Wehrmann A, Phillipp B, Sahm H, et al. , Different Modes of Diaminopimelate Synthesis and Their Role in Cell Wall Integrity: a Study with Corynebacterium glutamicum, Journal of Bacteriology, 180 (1998) 3159–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Devenish SRA, Blunt JW, Gerrard JA, NMR Studies Uncover Alternate Substrates for Dihydrodipicolinate Synthase and Suggest That Dihydrodipicolinate Reductase Is Also a Dehydratase, Journal of Medicinal Chemistry, 53 (2010) 4808–4812. [DOI] [PubMed] [Google Scholar]
- [15].Pearce FG, Sprissler C, Gerrard JA, Characterization of Dihydrodipicolinate Reductase from Thermotoga maritima Reveals Evolution of Substrate Binding Kinetics, The Journal of Biochemistry, 143 (2008) 617–623. [DOI] [PubMed] [Google Scholar]
- [16].Sagong HY KK, Structural Insight into Dihydrodipicolinate Reductase from Corybebacterium glutamicum for Lysine Biosynthesis, Journal of Microbiology and Biotechnology, 26 (2016). [DOI] [PubMed] [Google Scholar]
- [17].Stols L, Gu M, Dieckman L, et al. , A New Vector for High-Throughput, Ligation- Independent Cloning Encoding a Tobacco Etch Virus Protease Cleavage Site, Protein Expression and Purification, 25 (2002) 8–15. [DOI] [PubMed] [Google Scholar]
- [18].Bradford MM, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Analytical Biochemistry, 72 (1976) 248–254. [DOI] [PubMed] [Google Scholar]
- [19].Booth WT, Schlachter CR, Pote S, et al. , Impact of an N-terminal Polyhistidine Tag on Protein Thermal Stability, ACS Omega, 3 (2018) 760–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Otwinowski Z, Minor W, [20] Processing of X-ray diffraction data collected in oscillation mode, Methods in Enzymology, Academic Press; 1997, pp. 307–326. [DOI] [PubMed] [Google Scholar]
- [21].Vagin A, Teplyakov A, MOLREP: an Automated Program for Molecular Replacement, Journal of Applied Crystallography, 30 (1997) 1022–1025. [Google Scholar]
- [22].Minor W, Cymborowski M, Otwinowski Z, et al. , HKL-3000: the integration of data reduction and structure solution - from diffraction images to an initial model in minutes, Acta Crystallographica Section D, 62 (2006) 859–866. [DOI] [PubMed] [Google Scholar]
- [23].Cowtan K, The Buccaneer software for automated model building. 1. Tracing protein chains, Acta Crystallographica Section D, 62 (2006) 1002–1011. [DOI] [PubMed] [Google Scholar]
- [24].Murshudov GN, Skubak P, Lebedev AA, et al. , REFMAC5 for the refinement of macromolecular crystal structures, Acta Crystallographica Section D, 67 (2011) 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Emsley P, Cowtan K, Coot: model-building tools for molecular graphics, Acta Crystallographica Section D, 60 (2004) 2126–2132. [DOI] [PubMed] [Google Scholar]
- [26].Davis IW, Leaver-Fay A, Chen VB, et al. , MolProbity: all-atom contacts and structure validation for proteins and nucleic acids, Nucleic Acids Research, 35 (2007) W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Finn RD, Coggill P, Eberhardt RY, et al. , The Pfam protein families database: towards a more sustainable future, Nucleic Acids Research, 44 (2016) D279–D285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Frickey T, Lupas A, CLANS: a Java application for visualizing protein families based on pairwise similarity, Bioinformatics, 20 (2004) 3702–3704. [DOI] [PubMed] [Google Scholar]
- [29].DeLano WS, The PyMOL Molecular Graphics System; Schröddinger, LLC, 2002. [Google Scholar]
- [30].Robert X, Gouet P, Deciphering key features in protein structures with the new ENDscript server, Nucleic Acids Research, 42 (2014) W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Henrick E.K.a.K., Inference of macromolecular assemblies from crystalline state, J. Mol. Biol, 372 (2007) 774–797. [DOI] [PubMed] [Google Scholar]
- [32].Krissinel E, Henrick K, Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions, Acta Crystallographica Section D, 60 (2004) 2256–2268. [DOI] [PubMed] [Google Scholar]
- [33].Janowski R, Kefala G, Weiss MS, The structure of dihydrodipicolinate reductase (DapB) from Mycobacterium tuberculosis in three crystal forms, Acta Crystallographica Section D, 66 (2010) 61–72. [DOI] [PubMed] [Google Scholar]
- [34].Pearce FG, Sprissler C, Gerrard JA, Characterization of dihydrodipicolinate reductase from Thermotoga maritima reveals evolution of substrate binding kinetics, Journal of biochemistry, 143 (2008) 617–623. [DOI] [PubMed] [Google Scholar]
- [35].Reddy SG, Scapin G, Blanchard JS, Interaction of pyridine nucleotide substrates with Escherichia coli dihydrodipicolinate reductase: thermodynamic and structural analysis of binary complexes, Biochemistry, 35 (1996) 13294–13302. [DOI] [PubMed] [Google Scholar]
- [36].Girish TS, Navratna V, Gopal B, Structure and nucleotide specificity of Staphylococcus aureus dihydrodipicolinate reductase (DapB), FEBS letters, 585 (2011) 2561–2567. [DOI] [PubMed] [Google Scholar]
- [37].Dommaraju SR, Dogovski C, Czabotar PE, et al. , Catalytic mechanism and cofactor preference of dihydrodipicolinate reductase from methicillin-resistant Staphylococcus aureus, Archives of Biochemistry and Biophysics, 512 (2011) 167–174. [DOI] [PubMed] [Google Scholar]
- [38].Scapin G, Reddy SG, Zheng R, et al. , Three-Dimensional Structure of Escherichia coli Dihydrodipicolinate Reductase in Complex with NADH and the Inhibitor 2,6- Pyridinedicarboxylate, Biochemistry, 36 (1997) 15081–15088. [DOI] [PubMed] [Google Scholar]
- [39].Cirilli M, Zheng R, Scapin G, et al. , The Three-Dimensional Structures of the Mycobacterium tuberculosis Dihydrodipicolinate Reductase-NADH-2,6-PDC and -NADPH-2,6-PDC Complexes. Structural and Mutagenic Analysis of Relaxed Nucleotide Specificity, Biochemistry, 42 (2003) 10644–10650. [DOI] [PubMed] [Google Scholar]
- [40].Reddy SG, Sacchettini JC, Blanchard JS, Expression, purification, and characterization of Escherichia coli dihydrodipicolinate reductase, Biochemistry, 34 (1995) 3492–3501. [DOI] [PubMed] [Google Scholar]
- [41].Sagong HY, Kim KJ, Structural Insight into Dihydrodipicolinate Reductase from Corybebacterium glutamicum for Lysine Biosynthesis, J Microbiol Biotechnol, 26 (2016) 226–232. [DOI] [PubMed] [Google Scholar]
- [42].Ge X, Olson A, Cai S, et al. , Binding synergy and cooperativity in dihydrodipicolinate reductase: implications for mechanism and the design of biligand inhibitors, Biochemistry, 47 (2008) 9966–9980. [DOI] [PubMed] [Google Scholar]
- [43].Girish TS, Navratna V, Gopal B, Structure and nucleotide specificity of Staphylococcus aureus dihydrodipicolinate reductase (DapB), FEBS Letters, 585 (2011) 2561–2567. [DOI] [PubMed] [Google Scholar]
- [44].Lee CW, Park S-H, Lee SG, et al. , Crystal structure of dihydrodipicolinate reductase (PaDHDPR) from Paenisporosarcina sp. TG-14: structural basis for NADPH preference as a cofactor, Scientific Reports, 8 (2018) 7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.