

Abstract
Despite purines making up one of the largest classes of metabolites in a cell, little is known about the regulatory mechanisms that facilitate efficient purine production. Under conditions resulting in high purine demand, enzymes within the de novo purine biosynthetic pathway cluster into multienzyme assemblies called purinosomes. Purinosome formation has been linked to molecular chaperones HSP70 and HSP90; however, the involvement of these molecular chaperones in purinosome formation remains largely unknown. Here, we present a new-found biochemical mechanism for the regulation of de novo purine biosynthetic enzymes mediated through HSP90. HSP90-client protein interaction assays were employed to identify two enzymes within the de novo purine biosynthetic pathway, PPAT and FGAMS, as client proteins of HSP90. Inhibition of HSP90 by STA9090 abrogated these interactions and resulted in a decrease in the level of available soluble client protein while having no significant effect on their interactions with HSP70. These findings provide a mechanism to explain the dependence of purinosome assembly on HSP90 activity. The combined efforts of molecular chaperones in the maturation of PPAT and FGAMS result in purinosome formation and are likely essential for enhancing the rate of purine production to meet intracellular purine demand.
Purine metabolites are responsible for the maintenance of cellular homeostasis and growth by supplying the cell with the building blocks for the transfer of genetic information, the required energy, and the signaling molecules and cofactors for biochemical mechanisms. This class of metabolites is produced through the coordinated action of compensatory metabolic pathways, de novo and salvage processes, to meet the intracellular demand. The cell predominantly leverages the salvage pathway by recycling DNA bases and nucleosides back into its corresponding nucleotide.1 However, under cellular conditions that result in high purine demand, the resupply is met through the activation of the de novo purine biosynthetic pathway.2,3 In humans, this process transforms phosphoribosyl pyrophosphate into inosine 5’-monophosphate with the involvement of six enzymes. The regulatory mechanisms that modulate purine metabolism by either pathway are largely unknown.
One plausible mechanism for efficient purine metabolism is the spatial organization of sequential metabolic enzymes into multiprotein complexes called metabolons. Initial intracellular localization studies using fluorescent chimeras of de novo purine biosynthetic enzymes indicated formation of a metabolon only under purine-depleted growth conditions.4 These metabolons were termed purinosomes. Unlike most reported metabolons to date, purinosomes are non-membrane-bound, and the processes of complexation and enzyme recruitment are highly transient.5
Our understanding of the underlying mechanisms of purinosome formation is constantly evolving. Previously, we have shown that FGAMS, a purinosome marker, colocalizes with heat shock proteins 90 (HSP90) and 70 (HSP70) under cellular conditions that activate the de novo purine biosynthetic pathway, implicating an association between these two subcellular complexes.6 Purinosome assembly was severely impaired upon HSP90 inhibition with NVP-AUY922 and 17-AAG; however, we did not understand how these chaperones were responsible for its complexation. Up to this point, no one appears to have studied whether any enzyme involved in the de novo purine biosynthetic pathway interacts with and/or serves as a client of HSP90, or how this interaction might facilitate purinosome formation.
HSP90 is a highly abundant protein and is shown to interact with more than 20 co-chaperones that modulate its biochemical (chaperoning and ATPase) activities and, more recently, to have a role in selective client protein class recruitment.7,8 The list of reported HSP90 clients is ever growing, with the best characterized clients being hormone receptors and protein kinases. Although HSP90 is most often associated with the folding of an immature protein to its active state, clients can also be processed for membrane translocation or proteasome-mediated degradation.7,9 Here, we aim to further probe the mechanism of involvement of HSP90 in purinosome formation by first asking whether any of the de novo purine biosynthetic enzymes are clients of HSP90.
HSP90-client complexes are highly dynamic in nature. Because of the overall low expression level of many HSP90 clients and the likely prevalence of post-translational modifications, 293T cells were transiently expressed individually with one of the six de novo purine biosynthetic enzymes fused with a 3×FLAG tag. After expression for 48 h, HSP90-client interactions were assessed by co-immunoprecipitation with anti-FLAG resin, which revealed that of the six enzymes evaluated, two enzymes, PPAT and FGAMS, interact with HSP90 (Figure 1). Validation of these interactions was performed using a more quantitative luminescence-based mammalian interactome mapping (LUMIER) assay10 (Figure 2). This assay involves the expression of a 3×FLAG-tagged bait protein in a 293T cell line stably expressing the prey protein (HSP90AB1) fused to Renilla luciferase. The complex is then immunoprecipitated using anti-FLAG-coated plates, and the presence of prey and bait proteins is assessed by luminescence and enzyme-linked immunosorbent assays, respectively (Figure 2A). This assay platform has provided a more global understanding of how HSP90 promotes cellular protein homeostasis, including more recently a correlation between mutation-driven disease severity and enhanced interaction of the mutant client protein with HSP90.8,11
Figure 1.
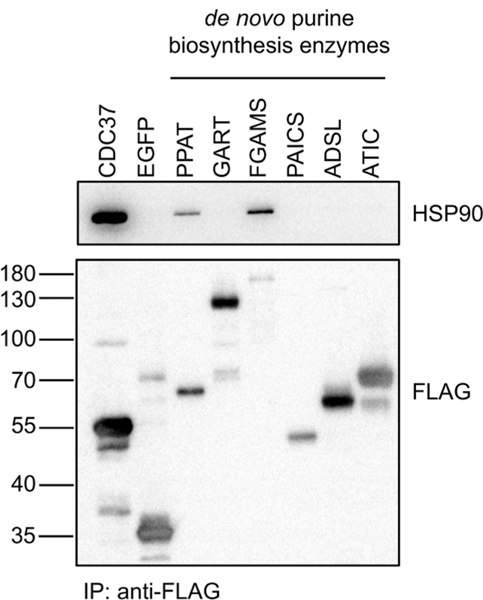
PPAT and FGAMS interact with HSP90 in 293T cells. Interactions between 3×FLAG-tagged enzymes within the de novo purine biosynthetic pathway and HSP90 were assessed by co-immunoprecipitation of 3×FLAG bait proteins with anti-FLAG resin and detection of HSP90 by Western blot. CDC37 and EGFP served as positive and negative controls, respectively.
Figure 2.
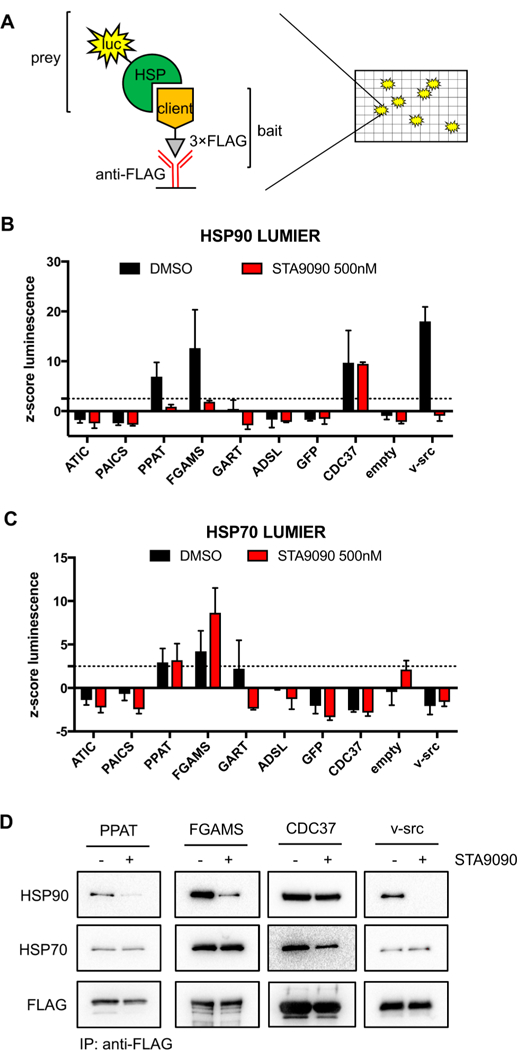
Effects of HSP90-client protein interactions upon HSP90 inhibition with STA9090 in 293T cells. (A) General scheme of the LUMIER assay previously developed, in which the luminescence signal is associated with Hsp90-client protein interactions. (B) A LUMIER assay confirmed PPAT and FGAMS as clients of HSP90 (black bars). Upon HSP90 inhibition with STA9090, a decrease in the level of Renilla luciferase—HSP90 (luc-HSP90AB1) interaction with the 3×FLAG-tagged client proteins was detected (red bars). The dotted line represents a z-score of 2.5, the threshold set for statistical significance for the LUMIER assay. (C) The decrease in the level of HSP90—client interactions had no effect on HSP70—client protein interactions (luc—HSPA8). Similar bait protein expression levels were obtained in the LUMIER assays in panels B and C (Figure S1). (D) Co-immunoprecipitation of 3×FLAG-tagged client proteins with anti-FLAG resin and detection of HSP90 and HSP70 by Western blot in the presence or absence of 500 nM STA9090 for 1 h. For all studies, CDC37 and v-src were used as a positive and negative controls, respectively, for STA9090-mediated inhibition of HSP90.
The two newly found HSP90 clients, PPAT and FGAMS, play pivotal roles in the de novo purine biosynthetic pathway. PPAT is the first enzyme in the pathway and catalyzed the conversion of phosphoribosyl pyrophosphate to 5-phosphoribosylamine and is considered to be rate-limiting. Allosteric modulation of PPAT activity through a feedback mechanism mediated by downstream purine nucleotides is hypothesized to serve as a switch between the de novo and salvage pathways.12 FGAMS, a protein encoded by the PFAS gene, is responsible for the generation of N-formylglycinamide ribonucleotide, the substrate needed to fulfill the nonsequential reactions catalyzed by GART in efficient purine biosynthesis.
We next asked whether PPAT and FGAMS behaved like traditional clients of HSP90, where HSP90 inhibition abrogates its interactions with client proteins. STA9090, a resorcinol-containing triazolone inhibitor that binds the N-terminal ATP binding site of HSP90, was selected for these studies.13 Transiently transfected 293T cells were treated with 500 nM STA9090 for 1 h prior to assessing the extent to which the interactions were retained by the developed LUMIER assay. Using v-src as a positive control, interactions of both PPAT and FGAMS with HSP90 were significantly hindered (Figure 2B). Similarly, CDC37 was used as a negative control, where HSP90 inhibition has minimal effects on its interaction. The disruption of HSP90-client interactions upon HSP90 inhibition suggests that both PPAT and FGAMS are clients of HSP90 (Figure 2).
To explore whether the inhibition of HSP90 also influenced HSP70 recruitment of the clients to HSP90, we used a modified LUMIER assay in which a stably transfected Renilla luciferase-HSP70 293T cell line was transiently transfected with 3×FLAG-tagged de novo purine biosynthetic enzymes. The extent of HSP70-client interactions was evaluated in the presence and absence of a 1 h treatment of 500 nM STA9090. HSP70 was shown to interact with PPAT and FGAMS, and the interaction was not significantly altered upon HSP90 inhibition (Figure 2C). Co-immunoprecipitation assays confirmed the results obtained from the LUMIER assays (Figure 2D). The lack of displacement of PPAT or FGAMS from HSP70 is consistent with a proposed model in which HSP70 recruits these clients to HSP90, and this event might sequester them in a state to protect them from degradation. However, it is also possible that HSP70 is working in a manner independent of HSP90 on these client proteins. At this time, our investigations do not definitively prove one model over the other.
Much like other HSP90 inhibitors, STA9090 leads to the degradation of various oncogenic client proteins such as kinases Akt, c-kit, and HER2/neu.13 We explored whether the same phenomenon holds true with PPAT and FGAMS. 293T cells were transiently transfected with either PPAT-3×FLAG or FGAMS-3×FLAG and allowed to express for 48 h prior to cycloheximide-mediated inhibition of protein translation in the presence or absence of STA9090. The level of expression of both client proteins decreased relative to the DMSO control samples, suggesting that HSP90 is stabilizing these clients in a specific state (Figure S2). When the cells were co-treated with STA9090 and proteasome inhibitor MG132, no decrease in the level of client expression was observed. Together, these results show that HSP90 inhibition results in the proteasome-mediated degradation of client proteins PPAT and FGAMS, so HSP90 must play an important role in promoting a stable form of these clients.
The remaining question is exactly how HSP90 is contributing to purinosome formation. To address whether HSP90 is acting as a platform to assist in purinosome stability or whether HSP90 is processing PPAT and FGAMS to prepare these enzymes for complexation, similar degradation studies were performed as previously described under purine-depleting growth conditions. Purine depletion results in the activation of the de novo purine biosynthetic pathway and has been shown to lead to enhanced purinosome formation in a wide variety of mammalian cells.4,14–16 If HSP90 is involved in the stabilization of the purinosome, then other enzymes that are comprised of the complex will become destabilized and likely degraded upon HSP90 inhibition. On the other hand, if no degradation is observed among the other enzymes in the purinosome, then the most probable explanation is that HSP90 is transforming these enzymes into a state that promotes complexation.
When 293T cells transiently expressing clients PPAT-3×FLAG or FGAMS-3×FLAG were cultured under purine-depleting growth conditions, levels of both PPAT and FGAMS expression decreased upon HSP90 inhibition with STA9090 (Figure 3). The decrease in the level of client expression was attributed to proteasome-mediated degradation. We next asked whether other pathway enzymes in the purinosome also showed a similar trend. Pathway enzymes within the purinosome are classified as either core or periphery as inferred through protein proximity and diffusion studies.17,18 The purinosome core consists of the first three enzymes within the pathway: PPAT, GART, and FGAMS. The other proteins in the pathway (PAICS, ADSL, and ATIC) have significantly weaker interprotein interactions. Given that PPAT and FGAMS are identified clients of HSP90, we first sought to determine whether the level of protein expression of the remaining core protein, GART, also decreased upon HSP90 inhibition. Purine-depleted 293T cells that transiently expressed GART-3×FLAG for 48 h were treated with cycloheximide in the presence and absence of STA9090 for 2 h. No change in protein expression was observed upon the inhibition of HSP90 (Figure 3A). Similarly, no peripheral enzyme showed a decrease in the level of protein expression 2 h post-HSP90 inhibition with STA9090 (Figure 3B). Combined, these results suggest that HSP90’s role in purinosome assembly is the processing of clients PPAT and FGAMS to promote complexation and not stabilizing either the core of the purinosome or the whole complex itself. Figure 4 shows our current mechanistic model of how HSP90 effectively couples the activity of the purinosome complex to even mild changes in purine supply.
Figure 3.
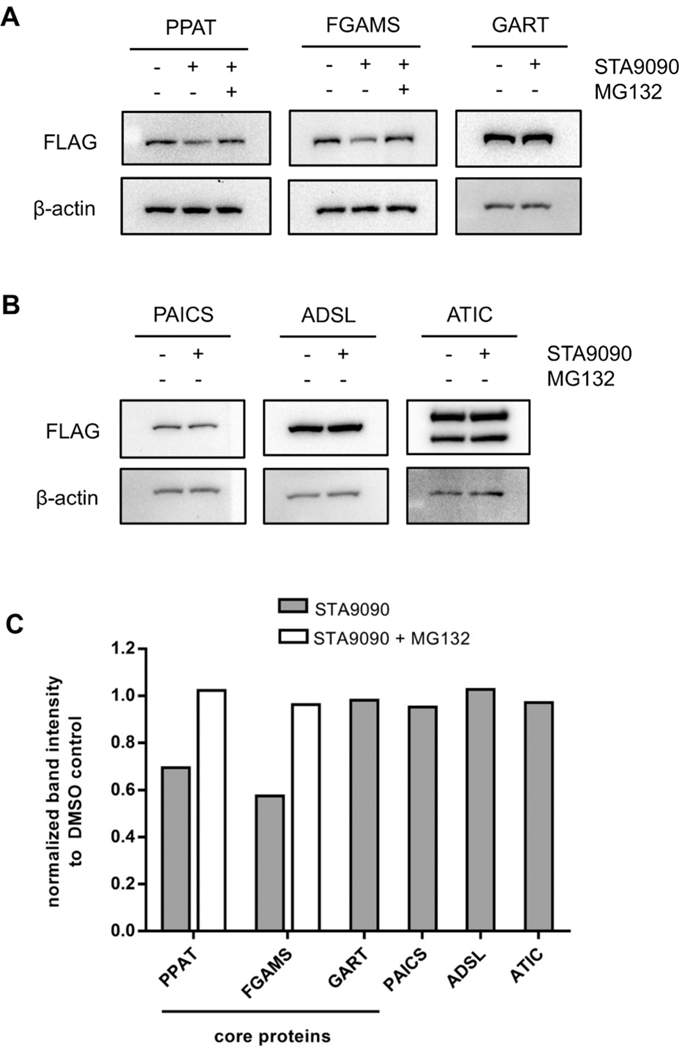
HSP90 protects purinosome core proteins, PPAT and FGAMS, from proteasome-mediated degradation. Purine-depleted 293T cells transiently expressing 3×FLAG-tagged purinosome enzymes were treated with 20 μg/mL cycloheximide in the presence of DMSO (control), 100 nM STA9090, or 100 nM STA9090 and 10 μM proteasome inhibitor MG132 for 2 h. Lysates were analyzed for the expression of purinosome (A) core and (B) periphery enzymes. (C) Band intensities were normalized to β-actin (loading control), and the normalized band intensity values were represented as fractions of the DMSO-treated control.
Figure 4.
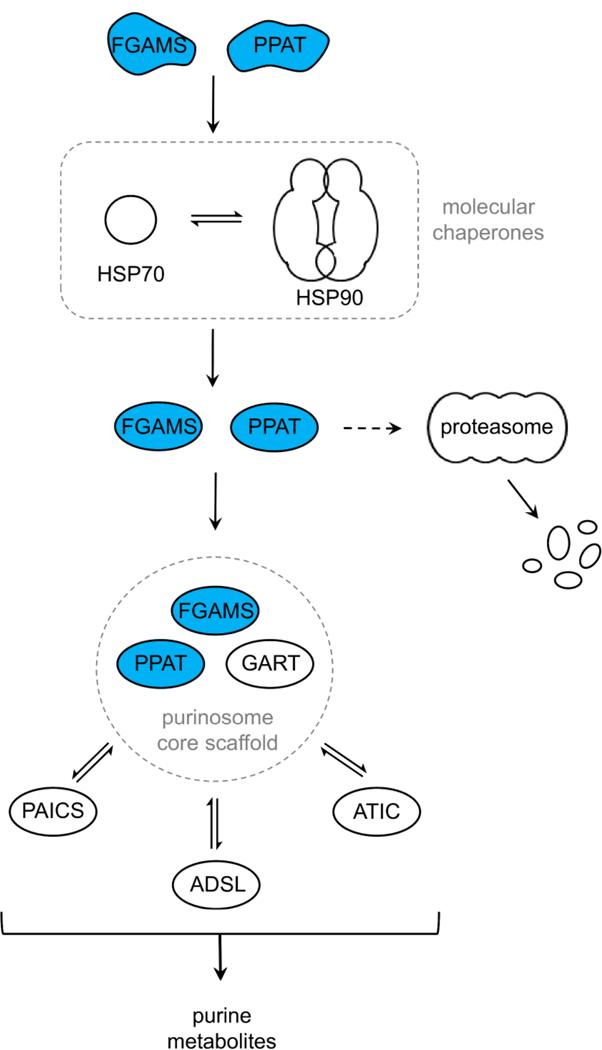
Our current mechanistic model of purinosome formation. Client proteins of HSP90 are colored blue. The dashed arrow is a side pathway induced upon HSP90 inhibition.
Here, we have further investigated the influence that the molecular chaperone HSP90 has on enzymes in the de novo purine biosynthetic pathway. Two pathway enzymes, PPAT and FGAMS, were shown to interact with HSP90 and, upon inhibition of HSP90 with STA9090, resulted in a decrease in the level of HSP90-client protein interactions, further validating these enzymes as bona fide clients of HSP90. HSP70-client protein interactions were not perturbed upon HSP90 inhibition, suggesting that HSP70 might sequester and stabilize a PPAT and FGAMS prior to delivering them to HSP90. While co-immunoprecipitation and LUMIER assays do not guarantee direct protein—protein interactions, the characteristics of these newly defined clients and HSP90 mimic those of other well-established HSP90 clients that have been shown to directly interact.8,10 The exact structural changes made by HSP90 to these clients remain hypothetical as do those molecular features that recruit these clients to HSP90. A previous proteomics study showed that FGAMS might be driven to HSP90 through an assortment of co-chaperones and ancillary proteins such as J-domain proteins, HOP, and p23.6 Although the exact composition of the molecular chaperone complex remains unknown, the activity of such a HSP90 complex is imperative in purinosome formation by providing core enzymes PPAT and FGAMS in a postprocessed state that promotes complexation.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Jack P. Boylan for his technical assistance in preparation of reagents used in this work.
Funding
Financial support for this study was provided by the U.S. National Institutes of Health (GM024129 to S.J.B.).
ABBREVIATIONS
- HSP90
heat shock protein 90 kDa
- HSP70
heat shock protein 70 kDa
- PPAT
phosphoribosylpyrophosphate amidotransferase
- FGAMS
phosphoribosyl formylglycinamide synthase
- LUMIER
luminescence-based mammalian interactome mapping
- STA9090
ganetespib
- IP
immunoprecipitation
- luc
Renilla luciferase
- DMSO
dimethyl sulfoxide
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.8b00140.
Materials and Methods and Supplemental Figures S1 and S2 (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Murray AW (1971) The biological significance of purine salvage. Annu. Rev. Biochem. 40, 811—826. [DOI] [PubMed] [Google Scholar]
- (2).Mayer D, Natsumeda Y, Ikegami T, Faderan M, Lui M, Emrani J, Reardon M, Olah E, and Weber G (1990) Expression of key enzymes of purine and pyrimidine metabolism in a hepatocyte- derived cell line at different phases of the growth cycle. J. Cancer Res. Clin. Oncol. 116, 251—258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Natsumeda Y, Prajda N, Donohue JP, Glover JL, and Weber G (1984) Enzymic capacities of purine de Novo and salvage pathways for nucleotide synthesis in normal and neoplastic tissues. Cancer Res. 44, 2475—2479. [PubMed] [Google Scholar]
- (4).An S, Kumar R, Sheets ED, and Benkovic SJ (2008) Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 320, 103—106. [DOI] [PubMed] [Google Scholar]
- (5).Pedley AM, and Benkovic SJ (2017) A New View into the Regulation of Purine Metabolism: The Purinosome. Trends Biochem. Sci 42, 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).French JB, Zhao H, An S, Niessen S, Deng Y, Cravatt BF, and Benkovic SJ (2013) Hsp70/Hsp90 chaperone machinery is involved in the assembly of the purinosome. Proc. Natl. Acad. Sci. U. S. A. 110, 2528–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Taipale M, Jarosz DF, and Lindquist S (2010) HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 11, 515–528. [DOI] [PubMed] [Google Scholar]
- (8).Taipale M, Tucker G, Peng J, Krykbaeva I, Lin ZY, Larsen,Choi, H., Berger, B., Gingras, A. C., and Lindquist, S. (2014) A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell 158, 434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Whitesell L, and Lindquist SL (2005) HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5, 761–772. [DOI] [PubMed] [Google Scholar]
- (10).Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, and Lindquist S (2012) Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 150, 987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Karras GI, Yi S, Sahni N, Fischer M, Xie J, Vidal M, D’Andrea AD, Whitesell L, and Lindquist S (2017) HSP90 Shapes the Consequences of Human Genetic Variation. Cell 168, 856–866.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Yamaoka T, Yano M, Kondo M, Sasaki H, Hino S, Katashima R, Moritani M, and Itakura M (2001) Feedback inhibition of amidophosphoribosyltransferase regulates the rate of cell growth via purine nucleotide, DNA, and protein syntheses. J. Biol.Chem. 276, 21285–21291. [DOI] [PubMed] [Google Scholar]
- (13).Wang Y, Trepel JB, Neckers LM, and Giaccone G (2010) STA-9090, a small-molecule Hsp90 inhibitor for the potential treatment of cancer. Curr. Opin Invest. Drugs 11, 1466–1476. [PubMed] [Google Scholar]
- (14).Baresova V, Skopova V, Sikora J, Patterson D, Sovova J, Zikanova M, and Kmoch S (2012) Mutations of ATIC and ADSL affect purinosome assembly in cultured skin fibroblasts from patients with AICA-ribosiduria and ADSL deficiency. Hum. Mol. Genet. 21, 1534–1543. [DOI] [PubMed] [Google Scholar]
- (15).Fu R, Sutcliffe D, Zhao H, Huang X, Schretlen DJ, Benkovic S, and Jinnah HA (2015) Clinical severity in Lesch- Nyhan disease: the role of residual enzyme and compensatory pathways. Mol. Genet. Metab. 114, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhao H, Chiaro CR, Zhang L, Smith PB, Chan CY, Pedley AM, Pugh RJ, French JB, Patterson AD, and Benkovic SJ (2015) Quantitative analysis of purine nucleotides indicates that purinosomes increase de novo purine biosynthesis. J. Biol. Chem. 290, 6705–6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Deng Y, Gam J, French JB, Zhao H, An S, and Benkovic SJ (2012) Mapping protein-protein proximity in the purinosome. J. Biol. Chem. 287, 36201–36207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kyoung M, Russell SJ, Kohnhorst CL, Esemoto NN, and An S (2015) Dynamic architecture of the purinosome involved in human de novo purine biosynthesis. Biochemistry 54, 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.