

Abstract
A wealth of evidence has implicated inflammation in the development of depression. Yet, the heterogeneous nature of depression has impeded efforts to understand, prevent, and treat the disease. The purpose of this integrative review is to summarize the connections between inflammation and established core features of depression that exhibit more homogeneity than the syndrome itself: exaggerated reactivity to negative information, altered reward processing, decreased cognitive control, and somatic syndrome. For each core feature, we first provide a brief overview of its relevance to depression and neurobiological underpinnings, and then review evidence investigating a potential role of inflammation. We focus primarily on findings from experimental paradigms of exogenously-induced inflammation. We conclude that inflammation likely plays a role in exaggerated reactivity to negative information, altered reward reactivity, and somatic symptoms. There is less evidence supporting an effect of inflammation on cognitive control as assessed by standard neuropsychological measures. Finally, we discuss implications for future research and recommendations for how to test the role of inflammation in the pathogenesis of heterogeneous psychiatric disorders.
1.1. Introduction
An estimated 11–15% of the global population will experience a major depressive episode in their lifetime (Bromet et al., 2011; Kessler and Bromet, 2013). Only about half of affected individuals achieve remission under available treatments (Nemeroff, 2007), and depression remains the leading global cause of disability. (“WHO | Depression,” 2017). Identifying biological and behavioral processes underlying depression—and the links among them—that can be targeted by interventions is a global health priority. One of the more promising research areas in this vein involves inflammation. Substantial evidence across various populations and study designs has implicated inflammation in the etiology of depression (Dantzer et al., 2008; Irwin, 2002; Miller et al., 2009; Raison et al., 2006). However, depression is an exceedingly heterogeneous disorder as evidenced by the mixed symptom phenotypes that qualify for the disorder (Fried and Nesse, 2015; Young et al., 2014) and the mixed effectiveness of existing treatments (Biesheuvel-Leliefeld et al., 2015; Cipriani et al., 2018; Piet and Hougaard, 2011; Shea et al., 1992; Vittengl et al., 2007). Thus, it is likely that a better understanding of Major Depressive Disorder and its effective treatment will emerge by focusing on the role of inflammation in the onset and maintenance of more homogenous core features that are disrupted among depressed patients (Craske, 2012; Gottesman and Gould, 2003; Hasler et al., 2004).
The aim of the present review is to determine if exogenously-induced inflammation may cause core features of depression, or endophenotypes. Endophenotypes are quantitative traits that are heritable and reflect the functioning of a discrete biological system (Gottesman and Gould, 2003). Traits falling within the definition of an endophenotype are thought to more closely reflect the root cause of a given disease than the broad clinical phenotype represented by symptoms. First, we review the evidence that inflammation plays a role in depression, and underscore the value of an endophenotype approach to organizing the heterogeneity of psychopathology into treatment relevant, dimensional domains. Next, we identify and define several key endophenotypes for depression: reactivity to negative information, altered reward processing, cognitive control, and somatic syndrome. For each endophenotype, we review evidence supporting its relevance for depression and its neurobiological correlates, and then examine evidence that inflammation modulates the processes represented by the endophenotype. Evidence informing the role of inflammation in each endophenotype is drawn primarily from experimental studies employing one of three paradigms for inducing inflammation: typhoid vaccination, injection with endotoxin, or treatment with interferon (IFN)-α. The strength of limiting our review to studies using these three paradigms is that such studies can test a causal role of inflammation in each psychological construct. Finally, we propose future research directions aimed at understanding the role of inflammation in the pathogenesis of depression.
1.2. Mechanisms linking inflammation and depression
Inflammation is a key component of the innate immune system’s ability to clear infection and repair injured tissue. Inflammation results from the release of proinflammatory cytokines from innate immune cells. In addition to their effects in the periphery, cytokines can communicate with the brain and result in a host of emotional, cognitive, and behavioral changes collectively termed “sickness behaviors” (Dantzer et al., 2008). Of particular relevance for this review, peripheral inflammation has been shown to induce depressive-like behaviors in animal models, including anhedonia (e.g., reduced sucrose consumption), decreases in exploratory, novelty-seeking and social behaviors, reduced food intake; and sleep disturbance (Anisman and Matheson, 2005; De La Garza, 2005; Dunn et al., 2005; Larson and Dunn, 2001; Pecchi et al., 2009). These sickness behaviors are considered an adaptive response intended to reduce the spread of infection and promote healing. However, prolonged inflammatory signaling, such as when the inflammatory response is maintained by ongoing psychosocial stress, can have detrimental effects that include risk for depression and other psychiatric diseases (Dantzer et al., 2008; Miller et al., 2009; Miller and Raison, 2016; Slavich and Irwin, 2014).
Research conducted over the last several decades has elucidated the mechanisms by which peripheral inflammation can access the brain to influence neural processes relevant for depression, including neuroplasticity, neurotransmitter systems, and neuroendocrine function (see Banks and Erickson, 2010; Haroon et al., 2012 for comprehensive reviews on this topic). For example, inflammatory cytokines can alter neuroplasticity by decreasing expression of the brain-derived neuroprotective hormone BDNF (Calabrese et al., 2014). Inflammation can also lead to changes in dopaminergic systems, with relevance for depression (See Felger, 2017 for review). Indeed, single injections of high doses of LPS (5 mg/kg) lead to degeneration in dopaminergic system in the brain, particularly in the substantia nigra and the striatum (Qin et al., 2007; Reinert et al., 2014). Further, chronic treatment with IFN-α in non-human primates leads to decreases in the synthesis and availability of dopamine in the striatum, which are correlated with reductions in effort-dependent sucrose consumption (Felger et al., 2013). There is a wealth of experimental research in rodents demonstrating that decreases in dopamine release and binding to the dopamine receptor is closely linked with deficits in reward-motivated behavior in depression (Nestler and Carlezon, 2006).
In addition to effects on dopamine, inflammation can influence the serotonin system through alterations in tryptophan metabolism. Under normal, healthy conditions, approximately 5% of tryptophan is metabolized into serotonin while 95% is metabolized through the kynurenine pathway. Metabolism of tryptophan through the kynurenine pathway can either be converted to neurotoxic 3-hydroxykynurenin (OHK) and quinolinic acid (QUIN) or neuroprotective kynurenic acid. How tryptophan is metabolized is determined by the presence of indoleamine-2,3-dioxygenase (IDO). In preclinical models, activation of inflammation using LPS increases the activity of IDO thereby allocating more tryptophan metabolism toward the kynurenine pathway rather than serotonin synthesis (Zunszain et al., 2012), ultimately resulting in serotonin deficiency (See Dantzer et al., 2011; Nikkheslat et al., 2018; O’Farrell and Harkin, 2017 for a review of research on the kynurenine pathway). In preclinical models, LPS-induced depressive behavior is mediated by IDO activation (O’Connor et al., 2009). Of note, inflammation and IDO can both influence glutamate metabolism, which may also play a role in depression
Inflammation can impact almost all aspects of glutamate neurotransmission (See Haroon and Miller, 2017 for review). To summarize briefly, glutamate is released at a higher rate under conditions of stress (Goshen and Yirmiya, 2009), and excess presence of synaptic glutamate can lead to excitotoxicity and neuronal death (Ankarcrona et al., 1995). Control of synaptic glutamate concentrations depend upon release and clearance by neurons, astrocytes, microglia, and oligodendrocytes. Following the summary of the kynurenine pathway in the previous paragraph, QUIN and glutamate released by activated microglia operate synergistically to promote excitotoxicity (Dantzer and Walker, 2014). When synaptic glutamate is detected by glial cells via their glutamate receptors, they activate gliotransmission in order to maintain regulation of local excitatory activity. However, inflammatory cytokines such as TNF-α and IL-1ß interfere with the ability of glial cells to do this by altering their Ca2+ channels. The effect of inflammatory cytokines on calcium ion channels also leads to reverse efflux, which means that instead of glial cells taking in glutamate and detoxifying it, glutamate is released back into the extracellular space (Malarkey and Parpura, 2008).
Further, inflammation has potent effects on neuroendocrine systems. Inflammatory cytokines may indirectly upregulate glucocorticoids in the body by interfering with the functional capacity of the glucocorticoid receptor (Pace, Hu, & Miller, 2007; Raison & Miller, 2003), resulting in continued synthesis of corticotropin releasing hormone (CRH) and sustained activation of the HPA-axis which has long been implicated in the pathogenesis of depression (Lopez-Duran et al., 2009; Nikkheslat et al., 2018; Pariante and Lightman, 2008). This process can become self-perpetuating because chronically elevated glucocorticoids can stimulate production of neuroinflammation by activating microglia (Frank et al., 2013; Nair and Bonneau, 2006), the resident immune cells of the brain.
In summary, preclinical models have elegantly outlined the mechanisms through which inflammation can access the brain to influence neural processes relevant for depression, providing strong mechanistic support for this association. Next, we consider studies conducted in humans that have interrogated links between inflammation and depression, focusing on experimental paradigms.
1.4. Inflammation and depression in humans: experimental paradigms
There is a growing body of evidence in humans that inflammation plays a role in the pathogenesis of depression. Markers of inflammation are elevated in depressed individuals (Dowlati et al., 2010; Haapakoski et al., 2015; Howren et al., 2009) and predict increases in depressive symptoms in longitudinal studies (Gimeno et al., 2009; Valkanova et al., 2013; van den Biggelaar et al., 2007; Wium-Andersen et al., 2013). Similarly, recent data from a study of twin pairs—which holds constant shared genetic and early environmental factors—showed that the twin with higher CRP concentrations at baseline was more likely to develop depression five years later (Huang et al., 2017). Yet, there is also evidence that the inflammation-depression pathway is bidirectional as depressive symptoms predict increases in inflammation in some samples (Huang et al., 2017; Matthews et al., 2010; Stewart et al., 2009). Experimental paradigms, such as those using exogenously-induced inflammation, are needed to inform our understanding of the directional pathways between inflammation and depression.
Causal evidence in humans supporting a role for inflammation in core features of depression comes from studies administering an inflammatory challenge and examining subsequent changes in cognitive and behavioral functioning. Three predominant models have been used to this end: vaccination, endotoxin, and interferon-α (IFN-α) therapy. The results of studies using these paradigms will be the focus of the present review. Key features and evidence from these three models will be discussed below; Table 1 also provides a summary of the key characteristics of each of these models.
Table 1.
Examples of the three paradigms used to examine the link between inflammation and depression, and their key features
| Paradigm | Citation | Dosing schedule |
Actual or estimateda IL-6 increase (% increase) |
Time of IL-6 assessment |
Duration of increase |
Strengths | Limitations |
|---|---|---|---|---|---|---|---|
| IFN-α | Models effects of chronic, low/moderate levels of inflammation; allows for the investigation of temporal development of symptoms and relationships between symptoms |
Effects
of inflammation may be confounded by presence of pre-existing chronic disease. Limits generalizabilit y of findings. |
|||||
| Hepatitis C (3 MU/m2 dose) |
Dowell et al., 2016 |
3 days/wk, for 4 – 24 mos |
2.18 pg/mL −1.02 |
4
hrs following initial injection |
Sustained throughout treatment |
||
| Malignant melanoma (20 / 10 MU/m2 dose) |
Capuron et al., 2003 |
20 MU/m2 5 days/wk, for 1 month / then 10 MU/m2 3 days/wk for 12 mos |
46 pg/mL (1533%)a |
3
hrs following initial injection |
Sustained throughout treatment; however, at 8 wks, IL-6 increase to injection fell to 2.33 pg/mL (233% increase from baseline) |
||
|
Endotoxin 0.2 ng/kg of body weight dose |
Krabbe et al., 2005 |
1x |
4 pg/mL (400%)a |
3 hrs post- injection |
Resolves within 4.5 hours |
Models effects of acute exposure to high levels of inflammation. |
Can
cause illness symptoms. Elicits increases in |
| 0.8 ng/kg of body weight dose |
Eisenberge r et al., 2009 |
1x | 145 pg/mL (28900%)a |
3 hrs post- injection |
Resolves within 6 hrs |
Used in medically healthy samples to avoid confound of pre-existing disease |
inflammation higher than typically seen in individuals with depression. |
| 2.0 ng/kg of body weight dose |
Lasselin et al., 2017 |
1x | 1000 pg/mL (49900%)a |
3 hrs post- injection |
Did not resolve by last assessment (7 hours post- injection) |
||
|
Vaccination Typhoid vaccine Influenza vaccine |
Wright et al., 2005 Kuhlman et al., 2018 |
1x 1x |
0.87 pg/mL (106%) 0.33 pg/mL (29%) |
3 hrs post- injection 24 hours post- injection |
Resolves within 32 hrsb Resolves within 3–7 daysc |
Models effects of acute exposure to mild elevations in inflammation, comparable to elevations seen in individuals with depression. Is safe to use in diverse samples. Does not typically cause illness symptoms. |
May not be sufficient to elicit sustained increases in depressed mood or changes in behavior. |
Values are approximate, based on graphs.
Hingorani et al., 2000.
Some of the earliest evidence of a link between inflammation and depression came from clinical observations of patients with Hepatitis C or cancer, who commonly develop clinically significant depressive symptoms after the initiation of IFN-α therapy (Capuron and Miller, 2004; Raison et al., 2005). IFN-α is a cytokine released by the innate immune system, and stimulates the release of other proinflammatory cytokines such as IL-6 (Taylor and Grossberg, 1998). Studies of patients receiving this treatment have shown that IFN-α can induce symptoms of depressed mood, fatigue, sleep disturbance, loss of appetite, pain, anxiety, anhedonia, anger, hostility, cognitive impairment, and suicidal ideation (Capuron et al., 2002; Janssen et al., 1994; Lotrich et al., 2007). IFN-α treatment causes symptoms sufficient for a major depression diagnosis in up to 50% of patients (Musselman et al., 2001).
Dosing schedules for IFN-α therapy typically involve multiple treatments per week over a period of months. Thus, a key feature of the IFN-α model is that it reflects chronic inflammatory stimulation. Additionally, IFN-α treatment results in moderate to robust increases in inflammatory markers that are sustained over weeks, depending on the disease being treated and associated dosing schedule. For example, patients with Hepatitis C typically receive a relatively mild dose of IFN-α several days per week for at least 16 weeks; one study showed an IL-6 increase of 2.18 pg/mL (a 102% increase) at four hours following the initial injection (Dowell et al., 2016). Patients receiving IFN-α for malignant melanoma typically receive higher doses and a more rigorous dosing schedule; one study showed an increase in IL-6 levels of approximately 46 pg/mL (a 1533% increase) at three hours after the initial injection; by week 8 a 233% increase from pre-treatment levels persisted (Capuron et al., 2003). Thus, IFN-α studies model the effects of exposure to chronic inflammation at mild or moderate levels, depending on the indication and associated dosing regimen. Modeling chronic inflammatory exposure allows for examination of temporal manifestation of different symptoms across a time period that is similar to the development of depression, and the relationships between symptoms or symptom clusters. Yet, one limitation of this model is its use exclusively in the context of chronic disease (hepatitis C, cancer).
Experimental evidence that inflammation can contribute to the development of depression even among medically healthy individuals comes from studies in which bacterial endotoxin - which elicits a strong inflammatory response - is administered to healthy individuals. These studies have shown that endotoxin administration leads to symptoms of depression including negative mood, anhedonia, cognitive impairment, fatigue, reduced food intake, altered sleep, and social withdrawal, as well as anxiety symptoms (DellaGioia and Hannestad, 2010; Eisenberger et al., 2010a; Reichenberg et al., 2001). Additionally, the effects of endotoxin on mood and anxiety appear to manifest in a dose-dependent manner, with higher doses associated with degradations in mood and increases in anxiety (Grigoleit et al., 2011). Endotoxin results in a robust but short-term inflammatory response that ranges from 40 to 190 pg/mL (up to a 38000% increase) two hours after injection depending on the bacterial strain and dose (Andreasen et al., 2008; Grigoleit et al., 2011). Regardless of these factors, IL-6 increases following endotoxin appear to resolve within six hours of administration. Thus, the endotoxin model can inform our understanding of the acute effects of exposure to high levels of inflammation. In contrast to IFN-α, a strength of the endotoxin model is that it is typically carried out among medically healthy individuals. This reduces confounds associated with the presence of pre-existing disease; however, it can also limit the ability to generalize findings to groups of individuals with chronic diseases who are at particular risk for depression.
Inflammation can also be induced by vaccines (Carty et al., 2006; Christian et al., 2011; Paine et al., 2013;Tsai et al., 2005). A third model for studying the effects of inflammation on depression involves administration of a typhoid vaccine to elicit increases in inflammation among healthy individuals. Results from such studies have shown that typhoid vaccination can lead to symptoms relevant to depression, including negative mood, fatigue, and confusion (Harrison et al., 2009; Wright et al., 2005). Typhoid vaccination leads to a modest, short-term inflammatory response; for example, an average increase of 0.82 pg/mL (a 106% increase) in IL-6 at 3 hours post-vaccination (Wright et al., 2005) and increases in inflammation resolve within 24 hours following typhoid vaccination (Paine et al., 2013). Thus, typhoid vaccine studies model the effects of mild, acute increases in inflammation. A strength of these studies is that the mild increases elicited by typhoid vaccination are comparable to the elevations in inflammation seen in some individuals with depression (Dowlati et al., 2010; Raison and Miller, 2011). Additionally, unlike IFN-α treatment or endotoxin, vaccines do not typically lead to fever, nausea, or other physical illness symptoms. Therefore, any effects on mood and cognition are not simply attributable to illness symptoms, but rather to more direct effects of cytokines on the brain. As a result of the typhoid studies, the influenza vaccine is now emerging as a useful, mild inflammatory stimulus to further interrogate these within-subject associations in a wider range of populations (Kuhlman et al., 2018).
Together, these experimental models have been instrumental in providing causal evidence for an association between inflammation and depression. Here, we use this research to examine the effects of inflammation on specific features of depression, using an endophenotype approach.
1.5. Core features of depression: An endophenotype approach
Endophenotypes are measurable, intermediate components, including neurophysiological, biochemical, endocrine, neuroanatomical, cognitive, or neuropsychological, that exist “along the pathway between disease and distal genotype” (Gottesman and Gould, 2003; Gould and Gottesman, 2006). Endophenotypes were intended to aid in identification of genes that cause diseases; because endophenotypes are conceptualized to represent more proximal psychological and biological processes to the level of gene action, it follows that they may be informed by fewer (and thus easier to identify) genes than complex diagnostic categories (Gottesman and Gould, 2003; Gould and Gottesman, 2006). As researchers have realized that elementary depressogenic processes (rather than traditional diagnostic categories) can help identify not just genetic vulnerability factors but a myriad other biological, psychological, and environmental contributors, the endophenotype approach has outgrown its initial aim to identify solely genetic inputs (Miller and Rockstroh, 2013). Importantly, the endophenotype approach to understanding psychiatric disorders has informed the National Institute of Mental Health’s Research Domain Criteria (RDoC) initiative (Cuthbert and Insel, 2010; Sanislow et al., 2010), which emphasizes largely transdiagnostic intermediate phenomena and their links upstream to genetic, molecular, and cellular factors, and downstream to clinically relevant behavior (Insel et al., 2010).
Putative endophenotypes for depression include behavioral, biological, and neural processes (Goldstein and Klein, 2014; Hasler et al., 2006, 2004; Webb et al., 2016). The current review characterizes our current knowledge of the role of inflammation in four established cognitive and behavioral endophenotypes of depression: exaggerated reactivity to negative information, altered reward processing, deficits in cognitive control, and somatic syndrome. These four endophenotypes were selected for this review based on two criteria: 1) identified in the depression literature as an empirically-validated phenotype; and 2) examined in relation to inflammation in human research. With respect to the first criteria, a number of published studies have aimed to establish the empirical validity of endophenotypes within depression. For example, Hasler and colleagues (2004) identified 8 psychopathological endophenotypes: mood bias, impaired reward function, impaired learning and memory, direction of appetite change, diurnal variation, executive function, psychomotor change, and increased stress sensitivity. Among these, mood bias, increased stress sensitivity, impaired reward function, diurnal variation, and executive function were rated as having the most consistent evidence to support their role as endophenotypes. Webb and colleagues (2016) conducted a similar investigation proposing that neuroticism, blunted reward learning, and cognitive control deficits were depression endophenotypes by showing their distinct neural signature via EEG. Finally, Glahn and colleagues (2012) rank-ordered measures of behavioral endophenotypes and the top ranked measures included the Beck Depression Inventory II, neuroticism, working memory, facial memory, attentional control, psychomotor speed, and emotional recognition. Exaggerated reactivity to negative information, altered reward processing, deficits in cognitive control, and somatic syndrome were selected to broadly reflect the domains identified in these previous studies; importantly, these endophenotypes also meet the second criteria as they have been examined in studies of induced inflammation in humans. It should be noted that while these core features were identified from studies aimed specifically at identifying endophenotypes for depression, each of these constructs is largely transdiagnostic.
In the following sections, we review these four core features of depression in terms of their neurobiological and behavioral correlates, and the role of inflammation as determined in human studies that have used IFN-α, typhoid vaccination, and endotoxin administration to exogenously stimulate inflammatory processes.
2.0. Inflammation and core features of depression
2.1.1. Exaggerated reactivity to negative information in depression.
Individuals with depression exhibit increased reactivity to negative information (Gotlib and Joormann, 2010). For example, compared to non-depressed controls, individuals with depression exhibit more negative facial expressions in response to unpleasant images (Sloan et al., 1997), report greater perceived stress in response to a laboratory stress tasks (de Rooij et al., 2010), and display greater increases in negative affect in response to daily stressors (Myin-Germeys et al., 2003). Depressed individuals also display exaggerated physiological responses to laboratory stress tasks, including norepinephrine and epinephrine (Weinstein et al., 2010).
In addition to increased behavioral, affective, and physiological reactivity in depression, altered neural reactivity to negative emotional information has been observed among depressed individuals. Studies using event-related potentials (ERPs) to emotional stimuli have shown that healthy individuals exhibit greater slow-wave amplitudes to happy and neutral faces compared with sad whereas depressed individuals exhibit equivalent slow-wave amplitudes to sad, happy, and neutral faces (Deveney and Deldin, 2004). Studies using fMRI have elucidated neural regions involved in this altered responding. Among individuals with depression, processing of negative information is associated with stronger and longer lasting activation of the amygdala (Beevers et al., 2010; Drevets, 2001; Fu et al., 2004; Siegle et al., 2002; Stuhrmann et al., 2013), a region responsible for interpreting the emotional quality and salience of a given stimulus (Fu et al., 2004). Reactivity to negative information may be sustained due to deficits in left dlPFC inhibitory control over the amygdala (Disner et al., 2011; Drevets, 2001); depressed individuals tend to exhibit anatomical and functional abnormalities in the dlPFC, such as reduced gray matter volume, lower resting-state activity, and decreased activation to emotional stimuli (Gotlib and Hamilton, 2008).
Another region implicated in heightened processing of negative emotional information in depression is the anterior cingulate cortex (ACC). With ample connections to both the excitatory limbic regions and the inhibitory prefrontal cortex, the ACC is thought to play an important role in integration of the neural circuitry dedicated to emotional processing and affect regulation (Stevens et al., 2011). Two regions within the ACC that may be of particular relevance to depression and inflammation due to their key contributions to emotional processing are the dorsal anterior cingulate cortex (dACC) and the subgenual anterior cingulate cortex (sACC), (Etkin et al., 2011). The dACC is believed to be involved in the appraisal and evaluation of threat and negative emotions (Etkin et al., 2011). Along with the amygdala, anterior insula, and periaqueductal gray, the dACC has been posited as part of a ‘neural alarm system’ that detects threat and elicits responses to impending danger or harm (Eisenberger and Cole, 2012). The sACC has been characterized as central to the integration and processing of emotional information from the limbic system (Disner et al., 2011), and increased activation of this region has been consistently implicated in depression (Mayberg, 2003; Mayberg et al., 1999; Sacher et al., 2012, 2012; Tremblay et al., 2005).
2.1.2. Inflammation and exaggerated reactivity to negative information.
Inflammation may exaggerate affective and physiological reactivity to stressors, thus contributing to reactivity to negative stimuli as seen in depression. Typhoid vaccination caused greater negative mood following two stress tasks (a Stroop color-word task and a simulated public speaking exercise) compared to a placebo group; elevations in negative mood were driven by increases in fatigue and confusion (Brydon et al., 2009). Additionally, participants with larger vaccine-induced IL-6 responses exhibited elevated systolic blood pressure responses to stress tasks, as well as elevated post-stress cortisol and a marker of noradrenaline (Brydon et al., 2009). To examine the neural mechanisms by which inflammation may influence mood reactivity, a subset of participants underwent neuroimaging while completing an emotional face processing task following typhoid vaccination (Harrison et al., 2009). In this study, vaccine-induced negative mood was associated with enhanced sACC activation to emotional information (Harrison et al., 2009).
Endotoxin studies have also shown that inflammation can upregulate stress reactivity, particularly at the neural level. For example, although endotoxin did not increase self-reported affective reactivity to social stress, endotoxin did lead to exaggerated activation of threat-related neural circuits, including greater activation of the amygdala and dACC (Muscatell et al., 2016). Another study similarly found that endotoxin led to exaggerated amygdala responses to threatening stimuli, particularly socially threatening stimuli (e.g., fear faces versus guns) (Inagaki et al., 2012). Moreover, the exaggerated amygdala response to socially threatening images was associated with increased feelings of social disconnection (Inagaki et al., 2012); thus, inflammation may upregulate amygdala reactivity to social threats, resulting in the experience of social disconnection and withdrawal.
Overall, the existing literature, though limited in size, supports an effect of inflammation on exaggerated affective and neural reactivity to stressors. The emerging picture is that proinflammatory cytokines can signal the brain to effectively alter the functioning of threat-related neural circuitry leading to amplified neural responses to stressful information. Heightened neural activation in these regions may result in downstream increases in physiological arousal; indeed, regions including the dACC and amygdala modulate sympathetic nervous system and hypothalamic-pituitary axis (HPA) activity (Muscatell and Eisenberger, 2012). Given that heightened inflammation is often the result of illness or injury that leaves the organism in a vulnerable state, cytokine upregulation of threat-related neural circuitry has been postulated as an adaptive response in the service of increasing vigilance to further threats in the environment such as unfriendly strangers that could result in potential further harm (Inagaki et al., 2012; Muscatell et al., 2016; Slavich and Irwin, 2014). However, when this circuitry is chronically activated, the chronic expression of behavioral symptoms (including negative mood, fatigue, confusion, and feelings of social disconnection) may culminate into clinically significant depression (Miller et al., 2013).
2.2.0. Inflammation and altered reward processing
2.2.1. Altered reward processing in depression.
Anhedonia, the loss of pleasure or interest in previously rewarding stimuli, is a hallmark of depression and is one of the core diagnostic criteria for major depression (American Psychiatric Association, 2013). Anhedonia is a broad construct that includes deficits in anticipation of, motivation toward, and responsiveness to reward, as well as deficits in reward learning (ability to recall and make decisions based on past rewarding experiences) (Treadway and Zald, 2011) and is estimated to be 46% heritable in studies of monozygotic twins (Bogdan and Pizzagalli, 2009). Anhedonia is often conceptualized as the behavioral manifestation of altered reward processing in depressed patients (Beck, 2004; Nutt et al., 2007; Webb et al., 2016; Winer and Salem, 2015), and may underlie many of the profound social impairments that are central to depression (Kupferberg et al., 2016). Importantly, these alterations in reward processing precede and potentially contribute to the etiology of depression in at-risk groups (Gotlib et al., 2010), and number of prospective studies have shown that self-reported anhedonia (less frequent and intense positive affect) is a strong predictor of subsequent increases in depression severity (Morris et al., 2009).
In non-depressed individuals, the experience and maintenance of positive affect is related to activity in brain regions associated with reward and motivation (Nestler and Carlezon, 2006; Tremblay et al., 2005); these regions are modulated by top-down control by the PFC (Del Arco and Mora, 2008). Reward-related brain regions consist of neural structures receiving dopamine input from the ventral tegmental area, including the nucleus accumbens (NAc) of the ventral striatum, which plays a critical role in reward learning and the experience of pleasure. The ventromedial PFC and amygdala are also major dopaminergic targets that are involved in reward processes (Berridge and Kringelbach, 2008; Lieberman and Eisenberger, 2009).
Depressed individuals display attenuated subjective and neural reactivity to positive stimuli, including diminished emotional and affective responses to pleasant stimuli compared with non-depressed controls (Dunn et al., 2004; Sloan et al., 2001, 1997). Additionally, depressed individuals or those with trait-like anhedonia display attenuated reactivity of the NAc to pleasurable stimuli (Harvey et al., 2007; Keedwell et al., 2005), monetary rewards (Pizzagalli et al., 2009; Wacker et al., 2009), and positive words (Epstein et al., 2006).
In addition to reduced reactivity to reward, there is also evidence that individuals with depression have reduced capacity to sustain positive affect following a reward. Among individuals with depression, NAc and PFC activation has been shown to dissipate more quickly following the presentation of positive stimuli, compared to healthy controls (Epstein et al., 2006; Heller et al., 2009). In a study in which participants were explicitly asked to sustain their positive mood following the presentation of positive stimuli, depressed individuals failed to sustain NAc activation over time compared with controls; moreover, this reduced capacity was associated with lower self-reported positive affect (Heller et al., 2009).
2.2.2. Inflammation, altered reward processing, and anhedonia.
Evidence from IFN- α, endotoxin, and typhoid research supports an effect of induced inflammation on aspects of reward. For example, after 4–6 weeks of IFN-α therapy, patients with hepatitis C showed significantly attenuated ventral striatal activity (as measured by fMRI) to receipt of monetary rewards in a gambling task when compared with hepatitis C patients not receiving IFN-α (Capuron et al., 2012). Reduced ventral striatal activation was in turn significantly associated with more severe symptoms of anhedonia, depressed mood, and fatigue (Capuron et al., 2012). These changes may occur through the effect of proinflammatory cytokines on dopaminergic functioning in the ventral striatum (Capuron et al., 2012), as outlined in section 1.2. This possibility was investigated using positron emission tomography (PET) with radiolabeled 18-F fluorodopa (18F-dopa) in patients with Hepatitis C receiving IFN-α for 4–6 weeks. 18F-dopa is taken up by dopaminergic neurons and converted into dopamine, which is then stored and subsequently released. PET imaging using 18F-dopa can assess the uptake and release of 18F-dopa, which is likely a measure of presynaptic dopamine activity (Miller et al., 2013). Patients exhibited increased uptake and decreased turnover of 18F-dopa in the same ventral striatal regions shown to be effected by IFN-α in the previously mentioned fMRI study (Capuron et al., 2012). Thus, inflammatory cytokines may influence reward processing by specifically targeting the ventral striatum via dopaminergic functioning.
Inflammation has been shown not only to modulate responses to reward consumption, but also the anticipation of reward. In one report, endotoxin (vs. placebo) led to attenuated activation of the left ventral striatum to anticipation of monetary reward (Eisenberger et al., 2010a). Further, blunted ventral striatum activity to anticipated reward mediated the relationship between endotoxin exposure and increases in observer-rated (but not self-reported) depressed mood (Eisenberger et al., 2010a). Thus, inflammation may impact not only consummatory but also anticipatory reward processes, potentially leading to behavioral manifestations of depressed mood.
The effects of inflammation have also been examined in relation to novelty, a different kind of rewarding stimuli. Novel stimuli are high in intrinsic reward value, thereby motivating adaptive behaviors such as exploration and social approach (Kakade and Dayan, 2002). The hippocampus and substantia nigra (a basal ganglia structure) are central to novelty detection and reward evaluation of novelty (Bunzeck and Düzel, 2006; Lisman and Grace, 2005). Typhoid vaccination (vs. placebo) had no effect on hippocampal activation to novelty, but led to blunted substantia nigra reactivity to novel stimuli (Harrison et al., 2015). Moreover, analyses of the functional connectivity between the substantia nigra and hippocampus revealed that substantia nigra-hippocampus connectivity to novelty was increased in the placebo condition and attenuated in the vaccine condition (Harrison et al., 2015). Thus, inflammatory signaling may reduce basal ganglia (specifically substantia nigra) responsivity to novel stimuli, which is relevant to reductions in motivational and approach behavior seen in depression, and expands our understanding of the types of rewards and reward-related neural mechanisms impacted by inflammation.
Although there is some compelling evidence that inflammation may dampen neural reactivity to rewards, a few studies present more complex findings. For example, inflammation can lead to heightened ventral striatal reactivity to social rewards, specifically positive social feedback (Muscatell et al., 2016) and viewing images of close others (Inagaki et al., 2015). The potential facilitation effects of inflammation on reward may also extend to monetary rewards. For example, endotoxin led to increased motivation for monetary rewards using a behavioral measure of reward motivation, the Effort Expenditure for Rewards Task (EEfRT), but only when the probability of winning was high (Lasselin et al., 2016b). These, perhaps unexpected, findings may reflect that inflammation facilitates some adaptive behaviors during acute illness or infection, such as drawing an individual closer to potential caretakers and conserving energy by selectively pursuing rewards that are highly likely and valuable (Eisenberger et al., 2016; Lasselin et al., 2016b).
In summary, all three models of induced inflammation has demonstrated effects on neural responses to reward. Much of this work has shown attenuated reward-related neural activity following an inflammatory stimulus, which is generally consistent with preclinical research on inflammation and reward (See Swardfager et al., 2016 for review). From this work, we have begun to understand the neurobiological mechanisms of the effects of inflammation on reward processing, which may involve alterations in corticostriatal neural circuitry (including the ventral striatum) via modulation of dopamine availability and release (Felger and Treadway, 2016). However, effects of inflammation on reward are complex and may depend on the level of analysis (neural vs. behavioral responsiveness), the domain of reward processing (anticipation, motivation, consumption, and learning), the nature of the reward (social vs. monetary rewards; primary vs. secondary rewards), the level of reward value and probability of receiving the reward (for reward anticipation and motivation tasks), as well as inflammatory dose and duration. More research interrogating these possible moderators is needed. Importantly, novel behavioral tasks that tap into discrete aspects of reward are being developed, such as the EEfRT, which taps reward motivation, and the Probabilistic Reward Task, which taps implicit reward learning (Pizzagalli et al., 2008; Treadway et al., 2009). These and other tasks can help us understand how and when inflammation-induced changes in neural reactivity manifest into behavioral changes relevant to depression (e.g., Lasselin et al., 2016b), and to investigate effects on inflammation on distinct components of reward.
2.3.0. Inflammation and cognitive control
2.3.1. Cognitive control deficits in depression.
Deficits in cognitive control are among the most frequent complaints for individuals with depression (Simons et al., 2009). Cognitive control broadly describes the ability of higher-level cognitive processes to organize and regulate automatic cognitive functions (such as perception or motor responses) in order to guide behavior towards a goal (Alvarez and Emory, 2006; Banich, 2009). Cognitive control, often operationalized as executive functioning within neuropsychology, includes the ability to create, maintain, and switch between task goals; inhibition of automatic responses and distracting information; planning and decision-making; and selecting among competing options (Snyder, 2014). Impaired cognitive control is heritable and (Friedman et al., 2008) has been proposed as an important depression endophenotype within and across several units of analysis (Castaneda et al., 2008; Hammar and Ardal, 2009; Hasler et al., 2004; Webb et al., 2016). A meta-analysis of 113 studies found significant deficits in executive functioning among depressed individuals compared to non-depressed controls, including deficits in updating, shifting, inhibition, working memory, planning, and verbal fluency, with effect sizes ranging from d = 0.32 – 0.97 (Snyder, 2014). Cognitive control relies heavily on the prefrontal cortex, particularly the dorsolateral PFC (dlPFC), ventrolateral PFC (vlPFC) and anterior cingulate cortex (ACC), but also recruits broader neural networks including subcortical areas (Snyder, 2014). See Table 2 for definitions of these cognitive control domains and the tasks typically used to measure these components in the context of depression.
Table 2.
Domains of executive functioning commonly disrupted in depression and the tasks typically used to measure them
| Component | Definition | Tasks and descriptions |
|---|---|---|
| Updating | Ability to replace no longer
relevant information with new, relevant input |
N-back: Subjects
must indicate if a displayed stimulus (letter or number) matches a stimulus that was presented n (e.g., 4) items back. DVs: Accuracy and reaction time |
| Shifting | Ability to switch between task sets
or response rules |
Trail Making Test Part
B: Subjects must alternatively connect
letters and numbers; results are contrasted with part A, which doesn’t require shifting between letters and numbers. DV: Total time to complete task. Wisconsin Card Sorting Task: Subjects initially sort cards based on one characteristic (e.g., color), but must subsequently switch to sort by a different characteristic after receiving negative feedback. DVs: Number of perseverative errors (continuing to sort by old rule), successful switches achieved. |
|
Intradimensional/Extradimensional Shift
Task: Similar to Wisconsin Card Sorting Task, but shifts are initially intra-dimensional (switching to a previously non-rewarded stimulus) and subsequently extradimensional (switching to a different stimulus dimension). DVs: Number of perseverative errors, successful switches achieved. |
||
| Inhibition | Ability to suppress a
prepotent response and instead make a more effortful, task-relevant response |
Color-word
Stroop: Color words are presented in
incongruously colored ink; subjects must name the ink color of each word while ignoring the word meaning. Performance is compared to a neutral condition in participants name the ink color in the absence of conflicting color information. DVs: An interference score that measures the difference in completion time between incongruent and control conditions; a score that represents accuracy in the incongruent condition relative to the control condition. |
| Working Memory |
Ability to actively maintain
or manipulate (verbal, visuospatial) information across a short delay |
Forward and backward digit
span (verbal working memory):
Subjects hear a sequence of numbers and must repeat it back in forward or reverse order. DV: Length of longest sequence correctly repeated |
|
Block span test
(visuospatial working memory): Subjects watch a pattern of taps on arranged shapes and must repeat the taps in the same order. DV: Length of longest sequence correctly repeated. |
||
| Planning | Ability to identify and organize
a sequence of steps towards the completion of some larger goal |
Tower of London:
Subjects must move beads from a starting position to a target position in a minimum number of moves. Obtaining the target solution is believed to require the subject to visualize the solution several moves in advance. DVs: number of moves to reach target solution; number of targets reached in the minimum number of moves. |
| Verbal fluency | Ability to retrieve verbal
information from memory. |
Word generation
tasks: Subjects must generate as many words
as possible for a given category within a specific period of time. Categories can be semantic (e.g., animals, fruits) or phonemic (e.g., words that begin with the letter a). DV: Number of words generated within the time limit. |
2.3.2. Inflammation and cognitive control deficits.
There is some evidence that inflammation may elicit changes in the subjective experience of cognitive control. In particular, IFN-α treatment has been associated with self-reported cognitive deficits, such as concentration difficulties (Capuron et al., 2002). Typhoid vaccination increases subjective ratings of mental confusion and impaired concentration (Brydon et al., 2009; Harrison et al., 2009).
Evidence that inflammation leads to changes in objective measures of cognitive control is decidedly more mixed. Counter to the updating deficits commonly exhibited in individuals with depression, the only published study examining the role of inflammation in performance on behavioral tasks of updating found that moderate inflammation facilitated (rather than slowed) updating speed (Grigoleit et al., 2011). In their evaluation of low (0.4 ng/kg) and moderate (0.8 ng/kg) doses of endotoxin on updating via working memory using the n-back task, there was no significant effect of low-dose endotoxin on n-back task performance; however, the moderate-dose endotoxin group exhibited significantly faster reaction times on the n-back task compared to the placebo condition (with comparable levels of accuracy across the two groups) (Grigoleit et al., 2011).
A larger number of studies have examined effects of inflammation on shifting ability as measured with the Trail Making Test (TMT) part B. Leukemia patients receiving IFN-α treatment showed deficits on the TMT part B (Pavol et al., 1995), and hepatitis C patients who received 6 months of IFN-α performed significantly worse on the TMT part B when compared with hepatitis C patients who did not undergo IFN-α treatment (Hilsabeck et al., 2005). However, several other studies of IFN-α treated hepatitis C patients have found no significant performance deficits on the Trail Making Test part B (Amodio et al., 2005; Drozdz et al., 2008; Fontana et al., 2007), the Wisconsin Card Sorting task (Fontana et al., 2007), or on the Intradimensional/Extradimensional Shift task (Majer et al., 2008). Consistent with this, studies examining effects of both moderate- (0.8 ng/kg) (Reichenberg et al., 2001) and very low-dose (0.2 ng/kg) (Krabbe et al., 2005) endotoxin have failed to find significant effects on performance on the Trail Making Test part B. Thus, a role of inflammation in shifting ability impairments is mixed, with the majority of studies finding no effect of inflammation on shifting ability.
A number of studies have examined the effect of inflammation on inhibitory control, as assessed with the color-word Stroop task; most have found no effect. Stroop performance was not impaired after endotoxin administration at either a low dose (0.4 ng/mL) (Grigoleit et al., 2010) or high dose (2.0 ng/mL) (van den Boogaard et al., 2010). A study of hepatitis C patients found no change in Stroop performance over the course of IFN-α treatment (Amodio et al., 2005). Brydon et al. (2008) examined Stroop performance after typhoid vaccination, and found that individuals with higher vaccine-induced IL-6 levels had significantly slower reaction times across both incongruent and congruent trials. Thus, inflammation did not impair inhibitory control, but rather led to an overall psychomotor slowing and/or decrease in motivation (Brydon et al., 2008).
Various studies have examined the effect of inflammation on working memory, and have failed to support a role of inflammation in this aspect of executive functioning. Among the four studies that have been conducted, no significant effect of endotoxin exposure on digit span forward or backward performance was observed (Grigoleit et al., 2010; Krabbe et al., 2005; Reichenberg et al., 2001; van den Boogaard et al., 2010). Similarly, two studies among hepatitis C patients (Amodio et al., 2005; Fontana et al., 2007) and one study among melanoma patients (Caraceni et al., 1998) receiving IFN-α therapy found no effect of treatment on digit span performance. In addition, no significant effects of inflammation on visuospatial working memory were observed following either acute endotoxin (Grigoleit et al., 2010) nor following IFN-α therapy (Caraceni et al., 1998). Thus, the currently available data provide little evidence that inflammation impairs verbal or visuospatial working memory.
Only one study to our knowledge has examined the effects of an inflammatory stimulus on planning ability, and failed to find effects. This study used IFN-α (plus ribavirin) as an inflammatory stimulus and measured planning ability with the Stockings of Cambridge task, and no effect of inflammation on planning ability was observed (Majer et al., 2008).
The evidence is mixed with regard to the effect of inflammation on verbal fluency. Inflammation did not impact performance on a verbal fluency task following endotoxin administration (Reichenberg et al., 2001). Similarly, several studies found no effect of IFN-α treatment on verbal fluency (Amodio et al., 2005; Fontana et al., 2007; Pavol et al., 1995), although one study showed a significant reduction in verbal fluency after three months of IFN-α treatment (Lieb et al., 2006)
In summary, detrimental effects of inflammation have not been consistently observed for updating, inhibition, working memory, or planning ability, and in some cases facilitation effects have been observed in these domains. There is some evidence that inflammation impairs shifting ability and verbal fluency, yet even in these domains there are more studies reporting null findings than not. It is possible that the types of neuropsychological tasks typically used to assess executive functioning following an inflammatory challenge are not sensitive enough to detect the magnitude of changes triggered by inflammation. Many of the neuropsychological tasks that are commonly used and discussed in this section were developed to distinguish between normal and abnormal cognitive abilities following major insults or neurological disease and may not capture more subtle changes in executive function. Of note, inflammation has been associated with increases in subjective reports of cognitive difficulties, which may be more sensitive to small changes in cognitive function. Additionally, studies have not examined effects of inflammation on neural correlates of executive functioning that may not be manifest behaviorally.
2.4.0. Inflammation and somatic syndrome
Somatic symptoms in depression, often referred to as neurovegetative, include fatigue, sleep disturbance, appetite disturbance, and psychomotor changes. Up to 74% of individual differences in somatic symptoms are explained by genetic factors in twin studies (Kato et al., 2009; Kendler et al., 1992), which supports the co-occurrence of these somatic symptoms as a depression endophenotype.
2.4.1. Fatigue in depression.
Fatigue is a prevalent presenting symptom of major depressive disorder (Demyttenaere et al., 2005). Symptoms of physical fatigue include feelings of tiredness, low energy, reduced activity, decreased physical endurance, increased effort to perform physical tasks, general weakness, heaviness, and slowness or sluggishness (Arnold, 2008). A large epidemiological study of more than 1800 depressed patients found that 73% reported feeling tired, and that this was the second most commonly endorsed symptom, after low mood (Tylee et al., 1999).
Fatigue may also play a role in the pathogenesis of depression. Non-depressed individuals with fatigue are at a higher risk for developing MDD later in life compared with individuals without fatigue (Addington et al., 2001; Kroenke and Price, 1993; Walker et al., 1993). Persistent fatigue can lead to substantial impairment in fulfilling social, familial, and work roles, and patients with residual fatigue are more likely to experience depression recurrence despite continued prophylactic treatment (Targum and Fava, 2011). At the neural level, fatigue is associated with alterations in dopamine and associated frontostriatal regions (Dobryakova et al., 2015). In addition, conceptual models have suggested a role for the anterior insula and the dorsal ACC in fatigue, particularly in the context of inflammation (Dantzer et al., 2014; Karshikoff et al., 2017), while the basal ganglia and frontal lobes have been implicated in diseases such chronic fatigue syndrome (DeLuca et al., 2009).
2.4.2. Inflammation and fatigue.
Studies across the endotoxin, typhoid vaccination, and IFN-α paradigms have consistently shown that exogenous stimulation of proinflammatory cytokines leads to reports of increased fatigue (Brydon et al., 2008; Capuron et al., 2002; Dowell et al., 2016; Eisenberger et al., 2010a; Grigoleit et al., 2011; Harrison et al., 2009; Moieni et al., 2015) and decreases in ratings of vigor (Dowell et al., 2016; Wright et al., 2005). Indeed, increased fatigue may be the most consistently reported symptom across these paradigms. In IFN-α models, fatigue is the most commonly reported symptom, affecting an estimated 80% of patients (Capuron et al., 2002). Studies using typhoid vaccination to elicit modest increases in inflammation have shown that increases in mood disturbance are driven by increases in items assessing fatigue (as well as confusion), rather than increases in depressed mood (Brydon et al., 2009, 2008; Harrison et al., 2009).
Studies have begun to elucidate neurobiological mechanisms of inflammation-induced fatigue. IFN-α therapy led to reports of fatigue in a sample of patients with malignant melanoma that were positively correlated with increased glucose metabolism in the basal ganglia (the left putamen and left NAc) (Capuron et al., 2007). In another sample of hepatitis C patients, IFN-α treatment elicited increases in fatigue that began 4 hours after patients’ initial IFN-α injection, and continued to persist throughout the 6-month treatment period, peaking between 4–12 weeks (Dowell et al., 2016). They also observed changes in ventral striatal microstructure within 4 hours post-injection, which prospectively predicted fatigue experience at 4 weeks (Dowell et al., 2016). In a typhoid vaccination study, individual differences in inflammation-associated fatigue were predicted by activity changes within bilateral mid/posterior insula and left anterior cingulate during performance of the Stroop color-word task (Harrison et al., 2009).
2.4.3. Psychomotor slowing in depression.
Psychomotor retardation, defined as a slowing of motor activity and difficulty responding quickly and spontaneously to environmental cues, is a common symptom of depression (Taylor et al., 2006). Psychomotor speed can be assessed by self-report, clinician assessment, or behaviorally, using neuropsychological tests that assess reaction time, speech rate, motor speed, mental speed, and initiation and spontaneity of response.
Psychomotor slowing involves disruptions in dopamine functioning and the basal ganglia circuit; dopaminergic abnormalities underlie the psychomotor slowing seen in depression as they do in Parkinson’s disease (Rogers et al., 1987). Depressed individuals with psychomotor retardation may have a dopaminergic abnormality within the frontal-subcortical network that involves the basal ganglia and PFC. For example, psychomotor retardation is associated with reduced activity in higher-order prefrontal regions that connect to the basal ganglia and are associated with the initiation, planning, and control of movement, including the dlPFC and orbitofrontal cortex (Bench et al., 1993; Videbech et al., 2002). Moreover, depressed patients with psychomotor slowing (as assessed by a clinician) exhibit decreased cerebrospinal fluid levels of homovanillic acid, the primary metabolite of dopamine (van Praag and Korf, 1971), and decreased presynaptic dopamine function in the left caudate (Martinot et al., 2001). Further, decreased D2 binding among depressed patients was significantly correlated with reduced speech rate (FAS-Verbal Fluency test) and reduced speed of movement on a reaction time task (Shah et al., 1997).
2.4.4. Inflammation and psychomotor slowing.
IFN-α (plus ribavirin) therapy led to significant decreases in motor speed among patients with hepatitis C (Majer et al., 2008). Notably, decreased motor speed in this study was also associated with increased symptoms of depression and fatigue (Majer et al., 2008). Similarly, IFN-α treatment was associated with an increase in response latency on a reaction time task among cancer patients, especially when the cytokine was administered intravenously and when the task involved higher attentional resources (Capuron et al., 2001). Further, greater IL-6 following typhoid vaccination was associated with slower response times on all trials of a color-word Stroop task (both congruent and incongruent trials) (Brydon et al., 2008). Notably, increases in IL-6 and decreases in motor speed were both associated with altered activation patterns (enhanced activation) in the substantia nigra (Brydon et al., 2008).
2.4.5. Sleep changes in depression.
An estimated 50–90% of individuals with diagnosed depression complain of poor sleep quality (Casper et al., 1985; Hetta et al., 1985; Kupfer et al., 1969; Riemann et al., 2001). Common sleep alterations in depression include difficulty initiating sleep, frequent nocturnal awakenings, and early morning awakenings. Depressed subjects also show reductions in slow wave (stage 3/4) sleep, increases in rapid eye movement (REM) sleep, including a shorter latency to REM initiation, a protraction of the REM sleep period, and increases in REM density (Palagini et al., 2013; Riemann et al., 2001; Tsuno et al., 2005). Hypersomnia can also accompany depression, though it is somewhat less common (Garvey et al., 1984; Tsuno et al., 2005). Sleep deprivation appears to cause alterations to neural processes that underlie a wide range of cognitive tasks, including attentional control, memory, reward processing, and emotion discrimination which involve multiple brain regions and the connectivity between them (Krause et al., 2017). These neural regions include, but are not limited to the dlPFC, mPFC, amygdala, intraparietal sulcus, the hippocampus, the anterior Cingulate cortex, and instability within the default mode network (See Krause et al., 2017 for review).
2.4.6. Inflammation and sleep.
Proinflammatory cytokines are involved in normal regulation of sleep-wake cycles and the homeostatic drive for sleep in both animals and humans. The proinflammatory cytokines IL-1, TNF-α, and IL-6 display a circadian pattern of secretion with peak levels correlating with feelings of sleepiness and sleep onset (Imeri and Opp, 2009; Vgontzas et al., 2005). Indeed, proinflammatory cytokines can actively inhibit wake-promoting neurons and stimulate (a subset of) sleep-promoting neurons in the preoptic area and basal forebrain (Alam et al., 2004). Given that cytokines can regulate sleep, it follows that disturbances in cytokine levels would be associated with sleep dysregulation.
An IFN-α study in patients with hepatitis C showed that inflammation alters objective measures of sleep, and began to identify the specific components of sleep disrupted by inflammation using polysomnography (Raison et al., 2010). In particular, IFN-α treatment led to increased awakenings after sleep onset, decreases in stage 3/4 sleep and sleep efficiency, increases in REM latency, and decreases in stage 2 sleep over 12 weeks (Raison et al., 2010). Endotoxin studies have also provided causal evidence that cytokines can lead to acute sleep changes, though effects depend on dose. For example, very low doses of endotoxin (0.2 ng/kg) promoted sleep, whereas moderate doses (0.8 ng/kg) reduced total sleep time and increased sleep onset latency (Hermann et al., 1998; Korth et al., 1996; Mullington et al., 2000). Specifically, low-level immune activation leads to increased non-rapid eye movement (NREM) sleep, while decreasing or having no effect on REM sleep; in contrast, more robust immune activation decreased both NREM and REM sleep (Mullington et al., 2000).
Taken together, results suggest that inflammation can disrupt sleep in ways that mirror the sleep disruptions observed in depression, including reductions in sleep quality, increases in NREM sleep, and decreases in REM sleep latency following IFN-α and moderate doses of endotoxin. However, exogenously-induced inflammation using both endotoxin and IFN-α also led to a suppression of REM sleep (Mullington et al., 2000; Raison et al., 2010), which is the opposite of changes observed in depressed patients (Palagini et al., 2013; Riemann et al., 2001; Tsuno et al., 2005). Across the few existing studies, the effects of inflammation on components of sleep may depend on the magnitude of immune activation. Neuroimaging studies are lacking in this area; thus, the neurobiological mechanisms by which heightened inflammation affects sleep and its components are relatively unexplored, and represents an important avenue for future work.
3.0. Discussion
The current review considered four core features of depression and their links with inflammation: exaggerated reactivity to negative information, reward processes, cognitive control, and somatic symptoms. Here, we briefly summarize the results of the review, focusing on similarities and differences across the different models of cytokine induction. We then consider this body of research from a network perspective, and conclude with clinical implications and global suggestions for future research.
3.1. Summary and integration across models
Effects of inflammation on core features of depression have been interrogated using different stimuli to induce peripheral inflammatory activity. While the IFN-α paradigm models the effects of chronic exposure to moderate levels of inflammation, endotoxin and typhoid vaccination model the short-term effects of acute exposure at high and low levels, respectively. One important conclusion that can be drawn from this review is that the dose and sustained exposure to inflammation induced by each of these three different paradigms may matter. Table 3 provides a summary of findings by endophenotype by paradigm and level of analysis and Figure 1 provides a summary of results of experimental studies so far.
Table. 3.
Review of associations between exogenous immune activation and core features of depression by paradigm and level of analysis.
| Endophenotype | Paradigm | Subjective reports | Objective behavior | Neural activity |
|---|---|---|---|---|
|
Exaggerated reactivity to negative information |
Typhoid vaccination |
• greater negative
mood following stress tasks |
• elevated systolic
blood pressure, cortisol, and noradrenaline following stress tasks |
• enhanced sACC activation
to emotional information in subjects with negative mood |
| Endotoxin | • increased feelings
of social disconnection • did not increase self- reported affective reactivity to social stress |
• No data available | • greater activation of
the amygdala and dACC in subjects with greater IL-6 response |
|
| IFN-α therapy |
• No data available | • No data available | • No data available | |
|
Altered
reward processing |
Typhoid vaccination |
• No data available | • No data available | • blunted substantia
nigra reactivity to novel stimuli • attenuated substantia nigra- hippocampus connectivity to novelty |
| Endotoxin | • greater increases
in self-reported and observer-rated depressed mood |
•increased effort for
high probability monetary rewards |
•attenuated activation of
the left ventral striatum to anticipation of monetary reward • heightened ventral striatal reactivity to positive social feedback and viewing images of close others |
|
| IFN-α therapy |
• more
severe symptoms of anhedonia |
• No data available | • attenuated ventral
striatal activity to monetary rewards • increased uptake and decreased turnover of 18F dopa in ventral striatum |
|
|
Cognitive control deficits |
Typhoid vaccination |
• subjective ratings
of mental confusion and impaired concentration |
• No data available | • No data available |
| Endotoxin | • No data available | • facilitated updating
speed • faster reaction times on the n- back task (0.8 ng/kg dose only) • no effect on working memory or reaction time following low dose (0.4 ng/kg) • no effect on shifting ability • no effect on inhibitory control |
• No data available | |
| IFN-α | •
self-reported concentration difficulties |
• deficits in shifting
ability observed in 2 studies; no effect on shifting ability observed in 5 studies • no effect on working memory • no effect on planning • one study observed impaired verbal fluency, 3 studies observed no effect on verbal fluency |
• No data available | |
|
Somatic symptoms: fatigue, psychomotor retardation, sleep disturbance |
Typhoid vaccination |
• increased self- reported fatigue |
• slower reaction times | • enhanced activity in
the substantia nigra |
| Endotoxin | • increased
self- reported fatigue • decreased vigor |
• low dose endotoxin (0.2 ng/kg) promotes sleep via increased sleepiness, increased nighttime NREM sleep • moderate dose (0.8 ng/kg) reduced total sleep time (REM and NREM) and increased sleep onset latency |
• No data available | |
| IFN-α | • increased
self- reported fatigue • decreased vigor |
• decreased reaction
times • more frequent awakening after sleep onset, decreased stage ¾ sleep, decreased sleep efficiency, increases in REM latency, and decreases in stage 2 sleep |
• increased
glucose metabolism in the basal ganglia (the left putamen and left NAc) as assessed with PET neuroimaging with fluorine-18-labeled fluorodeoxyglucose • changes in ventral striatal microstructure |
Figure 1.
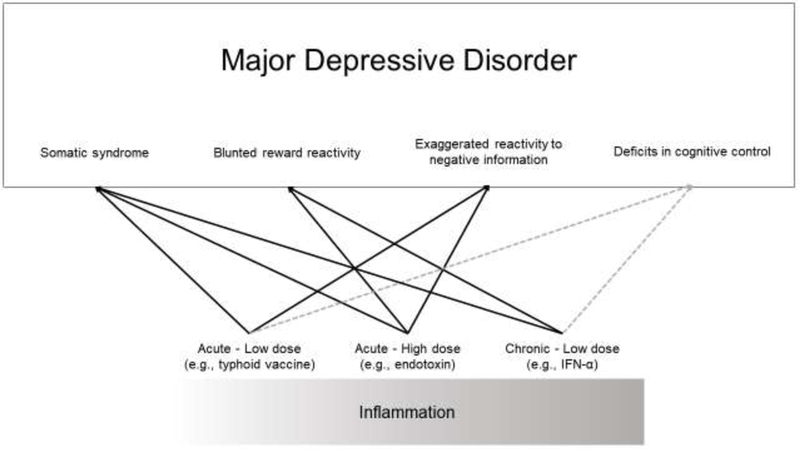
Core processes of depression that are influenced by inflammation.
Overall, there is strong and consistent evidence that inflammation leads to changes in somatic symptoms of depression across paradigms. Mild, short-term increases in inflammation are sufficient to cause somatic symptoms, particularly fatigue. This is exemplified by typhoid vaccine studies which can lead to increases in fatigue within 2–3 hours despite eliciting only mild, acute increases in inflammation (Brydon et al., 2008). Similar results have been observed in endotoxin studies, which document elevations in fatigue and other sickness symptoms that peak 2 hours post injection (Eisenberger et al., 2010; Moieni et al., 2015). Further, patients receiving (low-dose) IFN-α treatment for Hepatitis C exhibit increases in fatigue and decreases in vigor within four hours of initial injection that continue to rise across the 8 weeks of treatment (Dowell et al., 2016). Together, these findings indicate that fatigue is rapidly elicited by even modest perturbations in cytokine levels and there may be a dose response association between inflammation and the severity of somatic symptoms like fatigue (Dowell et al., 2016).
In general, research also supports an effect of inflammation on negative reactivity and reward processing, with most studies focusing on neural processes. Effects here may be more specific to the experimental paradigm. For example, only acute inflammatory challenges appear to induce increases in reactivity to the environment, whereas altered reward processing appears to only be observed in studies with high doses of inflammation regardless of their acute or chronic nature. That being said, there is no published evidence for or against the role of INF-α in reactivity to negative information, and little behavioral evidence on the role of mild inflammatory challenges in reward processing. Thus, our conclusions on this topic are limited by the studies published to date, and should be considered preliminary until more studies have been published.
Finally, we found very little evidence that cognitive control, as assessed by validated neuropsychological measures of executive function, was affected by exogenously-induced inflammation in either the endotoxin or the IFN-α model. In some ways, this is puzzling given that inflammatory processes have been shown to regulate learning and memory (e.g., Yirmiya and Goshen, 2011) and that correlational studies have identified associations between mild elevations in inflammatory markers and both self-reported (Davis et al., 2018; Kuhlman et al., 2018) and objective (Marsland et al., 2015) measures of cognitive function. Further, IFN-α and typhoid studies have documented links between increases in inflammation and subjective cognitive complaints (Brydon et al., 2009; Capuron et al., 2002; Harrison et al., 2009). However, results from both endotoxin and IFN-α studies were fairly consistent in showing no effects across a range of tasks assessing different aspects of executive function. It is possible that these tasks may not be sensitive to acute changes in inflammation, even at the high levels elicited by endotoxin, or to more “chronic” exposures elicited by IFN-α (that are still circumscribed compared to exposures seen in midlife and older adults). Alternatively, self-reported problems with basic daily cognitive processes may reflect another domain of functioning in depression that is not captured with executive functioning tasks. For example, cognitive control in these tasks are typically measured under ideal testing conditions that remove social and other contexts from the assessment. It is possible that inflammation may interfere with an individual’s sensitivity to context that makes daily cognitive tasks more effortful and distressing.
Altogether, it is important to examine different doses of inflammatory stimuli on the endophenotypes using stimuli that probe different aspects of the dimension in question (e.g., social vs. non-social reward) and that examine outcomes at several levels of analysis: neural, subjective, and behavioral. In addition, given that there are pronounced individual differences in response to a uniform inflammatory stimulus, it is important to examine how these differences influence outcomes. For this reason, studies that directly examine the effects of different doses of inflammatory stimuli (e.g., Grigoleit et al., 2011; Lasselin et al., 2016a) as well as individual differences in inflammatory responses to challenge (e.g., Kuhlman et al., 2018) on behavioral and neural outcomes are increasingly important to our understanding.
3.2. A Network Perspective
A network perspective of psychopathology assumes that symptoms causally influence one another over time, rather than emerge from a singular, common cause (Borkulo et al., 2015; Borsboom and Cramer, 2013). Taking a network perspective may enhance our understanding of the links between core features of depression. For example, it is possible that earlier appearing somatic symptoms may give rise to later-appearing affective symptoms (e.g., fatigue → mood symptoms), thus clarifying a biobehavioral pathway that informs interventions and their evaluation. Support for this idea derives from IFN-α studies, which show that although the large majority of patients rapidly develop somatic symptoms (e.g., fatigue), only a subgroup go on to develop other symptoms (e.g., feelings of guilt, suicidality) severe enough to qualify for a major depression diagnosis (Capuron et al., 2002; Capuron and Miller, 2004; Musselman et al., 2001). Indeed, these studies found that major depression was more likely to develop in individuals with pre-existing, subclinical cognitive disturbance (pessimistic thoughts) prior to the initiation of treatment (Capuron et al., 2004). Other IFN-α studies have begun to examine predictive relationships between symptoms (Dowell et al., 2016; Robaeys et al., 2007; Wichers et al., 2005).
This network model is also more synergistic with evolutionary conceptualizations of the role that inflammation can play in human behavior. When illness or injury befalls an organism, proinflammatory cytokines signal the central nervous system, triggering a constellation of behavioral changes (“sickness behaviors”) that represent an adaptive motivational response aimed at facilitating healing and recovery (Dantzer et al., 2008). The classic example of these adaptive behavioral changes is fatigue, which can promote recovery by driving the organism to rest and thereby allow the body to devote its energy to healing. Blunted responsivity to rewards may be similarly adaptive, minimizing an organism’s pursuit of rewards in favor of rest; the exception to this blunted reward responsivity could be social rewards, due to the likely benefits of having close others nearby for caretaking purposes (Eisenberger et al., 2016). Increased attention and reactivity to negative stimuli could also confer benefits; a vulnerable organism (due to illness, infection) may benefit from heightened vigilance to potential threats in the environment in order to prevent further injury while in an already compromised state. Thus, the inflammation-associated changes may be in the service of shielding the organism and promoting healing in the context of acute illness. In the short-term, these adaptations serve a clear benefit to the individual, but if sustained (such as through ongoing stress or immune dysregulation) could be iatrogenic in the long-term. Importantly, this approach also highlights the importance of contextual factors in driving the behavioral effects of inflammation. For example, heightened reactivity to negative stimuli may be beneficial when the threat is present, but not when an organism perceives safety.
One important issue to be disentangled using a network approach is whether the effect of inflammation on each of our identified endophenotypes occurs independent of the effect of inflammation on mood. There is evidence that somatic symptoms follow a different temporal course than depressed mood following inflammatory challenge and may have distinct neurobiological underpinnings. In particular, IFN-α therapy resulted in the emergence of a cluster of somatic symptoms that appeared in the majority of patients within two weeks of treatment initiation (including symptoms of anorexia, fatigue, and pain), whereas a cluster of affective/cognitive symptoms (including symptoms of negative mood, anxiety, and cognitive dysfunction) emerged later and in a smaller proportion of patients (Capuron et al., 2002). Further, the cognitive/affective symptoms were more responsive to paroxetine than symptoms of fatigue (Capuron et al., 2002). Similar findings have been observed in cancer patients, where paroxetine led to reductions in depressed mood but not fatigue in patients undergoing chemotherapy (Morrow et al., 2003). In addition, Eisenberger et al. (2010b) found that feelings of social disconnection following endotoxin persisted for two hours after depressed mood had recovered. Based on the studies to date, we expect that the effect of inflammation on somatic symptoms, negative reactivity, and reward sensitivity are independent of the effects of inflammation on mood, although only a handful of studies have directly addressed this issue. For example, IFN-α therapy resulted in the emergence of a cluster of somatic symptoms that appeared in the majority of patients within two weeks of treatment initiation (including symptoms of anorexia, fatigue, and pain), as well as cluster of affective/cognitive symptoms that emerged in a smaller proportion of patients (including symptoms of negative mood, anxiety, and cognitive dysfunction) (Capuron et al., 2002). Indeed, the association between inflammation and both attentional bias and fatigue remain when accounting for concurrent depressed mood (Bower et al., 2009; Boyle et al., 2017). Further, see Figure 1 of Eisenberger et al., (2010b) which illustrates that feelings of social disconnection following endotoxin persist for two hours after depressed mood has recovered. However, the majority of studies reviewed here did not control for depressed mood in their models, and therefore more careful testing of these associations is warranted to understand the independence or interdependence of depressed mood on these putative endophenotypes.
Overall, our review signals a clear benefit of deconstructing depression to examine links between inflammation and its underlying features, and provides substantial evidence supporting a relationship between inflammation and some of these key depressive processes. Moving forward, research in this area may benefit further from an updated conceptualization and modeling of endophenotypes and their position within explanatory models (Miller and Rockstroh, 2013). Genetic factors interact with psychological, environmental, and other biological factors and exert their influences at multiple points throughout the course of disorders. Instead, we might envision a network or web of influences that includes a multitude of factors and processes across multiple levels of analysis (e.g., genetic, neural, behavioral, developmental), with the recognition that none of these levels is more fundamental than any other (Miller and Rockstroh, 2013; Moore and Depue, 2016; Schmittmann et al., 2011). Network models offer the potential to develop more sophisticated and detailed models of the pathogenesis of depression, allow for the modeling of more complex (e.g., bidirectional, feedforward) associations, and are consistent with the RDoC initiative to capture pathological processes across multiple levels of analysis (e.g., interactions between symptoms, endophenotypic processes, genetic influences, environmental stressors, and time).
3.3. Clinical implications
If inflammation contributes causally to symptoms of depression, then anti-inflammatory medications would mitigate depressive symptoms. Indeed, antidepressant medications have anti-inflammatory effects on the body (Kenis and Maes, 2002), and remediate one of the sleep problems influenced by inflammation (Palagini et al., 2013; Riemann et al., 2001) while behavioral interventions that reduce somatic symptoms like fatigue also appear to work by reducing inflammatory signaling (Bower et al., 2014). There is also meta-analytic evidence that anti-inflammatory treatments can be effective means for treating depression (Köhler et al., 2014). Based on our review of the causal role of inflammation on core features of depression, we would expect anti-inflammatory treatments to benefit patients presenting with exaggerated reactivity to negative information, altered reward processes, and somatic symptoms but not necessarily those presenting primarily with difficulties in cognitive control. However, there are some published studies showing that supplementing anti-depressant treatment with anti-inflammatory drugs only benefit individuals with high peripheral inflammation (Raison et al., 2013), that inhibiting inflammation entirely has negative implications for learning and memory (Yirmiya and Goshen, 2011), and targeting inflammatory pathways may only benefit a subset of depressed patients (Raison, 2017). We posit that the effects of inflammation on specific features of depression, rather than the entire syndrome, may explain some of the mixed findings with regard to the effectiveness of targeting inflammatory pathways as part of depression treatment. Inflammation may only be relevant for some individuals with depression, while also being relevant to specific aspects of depression and not others. For example, only 33–47% of depressed individuals demonstrate elevated peripheral markers of inflammation (Raison and Miller, 2011; Rethorst et al., 2014). Further, although anti-inflammatory pharmacological interventions appear to improve depressive symptoms (Köhler et al., 2014), they may only be helpful for patients with elevations in peripheral inflammation (e.g., Raison et al., 2013). However, the impact of inflammation on mood and behavior occurs through inflammatory signaling in the brain or neuroinflammation, for which peripheral markers are only a proxy. Thus, it could be the case that depressed individuals have elevated neuroinflammation, but not elevated peripheral markers. What remains to be understood is whether and how acute increases in inflammation exert effects on some neural substrates but not others. It is possible that some people may be either genetically or environmentally predisposed to the effects of inflammation on specific receptors or neural substrates. For example, early life exposure to immune activation and exposure to chronic stress alter the density and morphology of microglia, particularly in regions of the brain involved in emotion and emotion regulation (Bilbo and Schwarz, 2012; Tynan et al., 2010). Thus early experiences may shape the effect that later inflammatory activation may have on behavior. Indeed, preclinical models have demonstrated that vulnerability factors like early life stress can alter the structure and function of microglia, leading to more neuroinflammation following an adult immune stimulus that their non-exposed peers (Bilbo and Schwarz, 2012), while minocycline treatment can rescue the effects of early life stress induced neuroinflammation during adulthood (Majidi et al., 2016). Further, there are health behaviors such as sleep and diet that can alter the permeability of the blood-brain barrier (He et al., 2014; Kanoski et al., 2010), thus rendering some individuals more vulnerable to the effects of peripheral inflammation on the brain. Taken together, these factors provide insight into how some individuals with depression do not exhibit elevated peripheral markers of inflammation while some individuals without depression do.
3.4. Limitations and Directions for Future Research
Despite the conclusions that can be drawn based on the studies to date, there are several limitations to our understanding that warrant consideration and further investigation. First among these is the fact that studies of exogenously activated inflammation have been limited to either clinical samples undergoing treatment for a serious health condition (e.g., cancer, hepatitis C), or in extremely healthy, young adult samples. Thus, it remains to be seen whether populations at risk for depression or with current depression would respond to inflammatory challenges in the same way. However, paradigms that can be extended to higher risk and more vulnerable populations are emerging which will bridge our understanding of the putative role of inflammation in the development of depressive symptoms (e.g. Kuhlman et al., 2018).
Much of the work on reactivity to negative information has focused on the impact of endotoxin-induced inflammation on negative reactivity to social threats (social evaluation, social exclusion, images of fearful faces) and sensitivity to social rewards. Thus, it remains unclear whether inflammation specifically upregulates reactivity to social stressors, or if reactivity to other types of negative information is also exaggerated. Relatedly, reactivity to negative information may be driven by cognitive biases toward negative information. Depressed individuals attend selectively to negative information (Gotlib and Joormann, 2010; Joormann and Quinn, 2014), and also interpret ambiguous information more negatively (Butler and Mathews, 1983; Dearing and Gotlib, 2009; Lawson et al., 2002; Rude et al., 2002). The only known study to have examined whether inflammation contributes to the cognitive bias toward negative information showed a correlation between CRP and attentional bias towards negative faces in a dot-probe task administered to breast cancer survivors (Boyle et al., 2017); this is an important area for future research.
Further, the accumulating evidence that elevated inflammation leads to blunted behavioral and neural reward reactivity, key elements of dampened positive affect and motivation in depression, in most cases comes from studies using monetary reward tasks. However, given the pronounced social deficits observed in depressed patients (Kupferberg et al., 2016), and the recent findings indicating that inflammation can lead to heightened VS reactivity specifically to social rewards (Inagaki et al., 2015; Muscatell et al., 2016), future work should explicitly test whether the effect of inflammation on reward reactivity is more pronounced for social compared to monetary rewards. This could be tested by directly comparing responsivity to different types of reward (e.g., social vs. monetary vs. primary rewards) under inflammatory conditions. Additionally, future work would benefit from examining the influence of inflammation on distinct domains of reward processing, including effects on reward anticipation (i.e., wanting), the consummatory reward response (i.e., liking), reward motivation, and reward-related learning, in order to understand more precisely the reward mechanisms affected by inflammation. Finally, research that incorporates subjective reports of cognitive function, current methods for assessing executive function (e.g., Snyder, Miyake, Hankin, 2015), and neuroimaging approaches are important to clarify effects of inflammation on cognitive control processes.
Researchers conducting studies on inflammation and depression should also consider assessing demonstrated vulnerability factors. Given that depressive symptoms only arise in a subset of individuals even under chronic inflammatory conditions, inflammation must interact with pre-existing host factors to determine mood outcomes. Potential risk factors include early life stress (Danese et al., 2008; Kuhlman et al., 2017) and certain genetic factors, such as the BDNF Met allele, which has been shown to moderate the association between CRP and cognitive depressive symptoms (Dooley et al., 2016). Another potentially important vulnerability factor to consider is sex. Despite the fact that depression is more prevalent among women than men (Kuehner, 2003; Nolen-Hoeksema, 2001, 1987), a preponderance of studies examining the effects of inflammation on mood and behavior have been conducted with solely male participants (Brydon et al., 2009, 2008; Harrison et al., 2014, 2009; Krabbe et al., 2005; Reichenberg et al., 2001; Wright et al., 2005). Studies that have included both male and female participants have revealed prominent sex differences in the psychological response to an inflammatory stimulus (See Lasselin et al., 2018 for review). For example, females exhibit greater increases in depressed mood and feelings of social disconnection following endotoxin exposure than males (Eisenberger et al., 2010a, 2009; Moieni et al., 2015). Moreover, endotoxin-induced increases in TNF-α and IL-6 were correlated with increases in social disconnection for females but not for males (Moieni et al., 2015). Additionally, among females but not males, endotoxin-induced increases in IL-6 have been associated with increased activation of threat-related neural regions (dACC, anterior insula), which mediate the relationship between IL-6 levels and depressed mood (Eisenberger et al., 2009). Taken together, females may be more sensitive to the negative psychological effects of inflammation via increased activation of threat circuitry. Early exploration of these pathways in preclinical models suggests that males have more microglia in early life and females have more activated or primed microglia during adolescence and adulthood (Schwarz et al., 2012). Further, gene expression profiles for a large number of cytokines and chemokines are sex-dependent (Schwarz et al., 2012). These gender differences deserve further investigation, as they may help to elucidate the mechanisms underlying the greater burden of depression among women that emerges in adolescence.
3.5. Conclusions
Using endophenotypes as intervention targets in depression has been a beneficial approach so far. For example, targeting the neural circuit associated with cognitive control using transcranial magnetic stimulation reduces depressive symptoms by at least 55% and is particularly effective in depressed patients with comorbid substance use disorders (Dunlop et al., 2017). Further, the field is shifting toward neuroscience-driven treatments for transdiagnostic endophenotypes (Craske et al., 2016). This is fitting given that the endophenotypes reviewed here are not specific to depression alone, but relevant to a number of psychiatric disorders. Targeting the biological underpinnings of these complex cognitive and behavioral processes could have profound implications for the treatment of psychiatric diseases. Yet, there remain important gaps in our knowledge that impede the translation of these findings to practice.
The current body of evidence indicates that inflammation is linked, in many cases causally, to alterations in core affective and cognitive processes (e.g., anhedonia; negative reactivity bias) and their neural circuits that are strongly implicated in current models of the etiology and treatment of depression. As such, the role of inflammation may be best understood as affecting these complex neurobiological, cognitive, and affective processes, rather than affecting the broad depressive syndrome directly. Our understanding of the role of inflammation in depression can be advanced by research that (1) deconstructs depression to effectively account for the heterogeneity across individuals; (2) considers the potentially distinct effects of different magnitudes and durations of inflammation exposure; (3) examines potential moderators (e.g., female gender, early life stress exposure) of the effects of inflammation, and (4) utilizes a network perspective that models these interdependent psychophysiological processes at multiple levels of analysis, as they play out over time. Research studies that incorporate such advances may elucidate the etiology of depression in its various iterations, and thus speed our progress towards alleviating the substantial suffering and burden that depression has on individuals, families, and societies.
Highlights.
Exogenously-induced inflammation leads to expression of 3 features of depression
They are neural reactivity to negative stimuli, reward processing, somatic symptoms
Little evidence for the role of inflammation in cognitive control was observed
Effects of inflammation on behavior depend on the timing and dose of the stimulus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Addington AM, Gallo JJ, Ford DE, Eaton WW, 2001. Epidemiology of unexplained fatigue and major depression in the community: The Baltimore ECA follow-up, 1981–1994. Psychol. Med 31, 1037–44. 10.1017/S0033291701004214 [DOI] [PubMed] [Google Scholar]
- Alam MN, McGinty D, Bashir T, Kumar S, Imeri L, Opp MR, Szymusiak R, 2004. Interleukin-1b modulates state-dependent discharge activity of preoptic area and basal forebrain neurons: Role in sleep regulation. Eur. J. Neurosci 20, 207–216. 10.1111/j.1460-9568.2004.03469.x [DOI] [PubMed] [Google Scholar]
- Alvarez JA, Emory E, 2006. Executive function and the frontal lobes: A meta-analytic review. Neuropsychol. Rev 16, 17–42. 10.1007/s11065-006-9002-x [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association, 2013. Diagnostic and Statistical Manual of Mental Disorders 10.1176/appi.books.9780890425596.744053 [DOI] [Google Scholar]
- Amodio P, De Toni EN, Cavalletto L, apelli D, Bernardinello E, Del Piccolo F, Bergamelli C, Costanzo R, Bergamaschi F, Poma Z, Chemello L, Gatta A, Perini G, 2005. Mood, cognition and EEG changes during interferon-a treatment for chronic hepatitis C. J. Affect. Disord 84, 93–98. 10.1016/j.jad.2004.09.004 [DOI] [PubMed] [Google Scholar]
- Andreasen S, Krabbe KS, Krogh-Madsen R, Taudorf S, Pedersen BK, Moller K, 2008. Human endotoxemia as a model of systemic inflammation. Curr. Med. Chem 15, 1697–1705. 10.2174/092986708784872393 [DOI] [PubMed] [Google Scholar]
- Anisman H, Matheson K, 2005. Stress, depression, and anhedonia: Caveats concerning animal models. Neurosci. Biobehav. Rev 29, 525–546. 10.1016/j.neubiorev.2005.03.007 [DOI] [PubMed] [Google Scholar]
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P, 1995. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron 15, 961–973. [DOI] [PubMed] [Google Scholar]
- Arnold LM, 2008. Understanding fatigue in major depressive disorder and other medical disorders. Psychosomatics 49, 185–90. 10.1176/appi.psy.49.3.185 [DOI] [PubMed] [Google Scholar]
- Banich MT, 2009. Executive function: The search for an integrated account. Curr. Dir. Psychol. Sci. 18, 89–94. 10.1111/j.1467-8721.2009.01615.x [DOI] [Google Scholar]
- Banks WA, Erickson MA, 2010. The blood–brain barrier and immune function and dysfunction. Neurobiol. Dis, Special Issue: Blood Brain Barrier 37, 26–32. 10.1016/j.nbd.2009.07.031 [DOI] [PubMed] [Google Scholar]
- Beck AT, 2004. Cognition and Psychotherapy Springer Science & Business Media, New York. [Google Scholar]
- Beevers CG, Clasen P, Stice E, Schnyer D, 2010. Depression symptoms and cognitive control of emotion cues:A functional magnetic resonance imaging study. Neuroscience 167, 97–103. 10.1016/j.neuroscience.2010.01.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bench J, Friston KJ, Brown RG, Frackowiak RS, Dolan RJ, 1993. Regional cerebral blood flow in depression measured by positron emission tomography: The relationship with clinical dimensions. Psychol. Med 23, 579–90. 10.1017/S0033291700025368 [DOI] [PubMed] [Google Scholar]
- Berridge K ., Kringelbach ML, 2008. Affective neuroscience of pleasure: Reward in humans and animals. Psychopharmacology (Berl.) 199, 457–480. 10.1007/s00213-008-1099-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesheuvel-Leliefeld KEM, Kok GD, Bockting CLH, Cuijpers P, Hollon SD, Marwijk H.W.J. van, Smit F, 2015. Effectiveness of psychological interventions in preventing recurrence of depressive disorder: Meta-analysis and meta-regression. J. Affect. Disord 174, 400–410. 10.1016/j.jad.2014.12.016 [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Schwarz JM, 2012. The immune system and developmental programming of brain and behavior. Front. Neuroendocrinol 33, 267–286. 10.1016/j.yfrne.2012.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan R, Pizzagalli DA, 2009. The heritability of hedonic capacity and perceived stress: A twin study evaluation of candidate depressive phenotypes. Psychol. Med 39, 211–218. 10.1017/S0033291708003619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkulo C. van , Boschloo L, Borsboom D, Penninx BWJH, Waldorp LJ, Schoevers RA, 2015. Association of symptom network structure with the course of depression. JAMA Psychiatry 72, 1219–1226. 10.1001/jamapsychiatry.2015.2079 [DOI] [PubMed] [Google Scholar]
- Borsboom D,Cramer AOJ,2013Network analysis: An integrative approach to the structure of psychopathology. Annu. Rev. Clin. Psychol 9, 91–121. 10.1146/annurev-clinpsy-050212-185608 [DOI] [PubMed] [Google Scholar]
- Bower JE, Ganz A, Tao ML, Hu W, Belin TR, Sepah S, Cole S, Aziz N, 2009. Inflammatory biomarkers and fatigue during radiation therapy for breast and prostate cancer. Clin. Cancer Res 15, 5534–5540. 10.1158/1078-0432.CCR-08-2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bower J, Greendale G, Crosswell AD, Garet D, Sternlieb B, Ganz PA, Irwin MR, Olmstead R, Arevalo J, Cole SW, 2014. Yoga reduces inflammatory signaling in fatigued breast cancer survivors: A randomized controlled trial. Psychoneuroendocrinology 43, 20–29. 10.1016/j.psyneuen.2014.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle CC, Ganz PA, Van Dyk KM, Bower JE, 2017. Inflammation and attentional bias in breast cancer survivors. Brain. Behav. Immun 66, 85–88. 10.1016/j.bbi.2017.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromet E, Andrade LH, Hwang I, Sampson NA, Alonso J, de Girolamo G, de Graaf R, Demyttenaere K, Hu C, Iwata N, Karam AN, Kaur J, Kostyuchenko S, Lépine J-P, Levinson D, Matschinger H, Mora MEM, Browne MO, Posada-Villa J, Viana MC, Williams DR, Kessler C, 2011. Cross-national epidemiology of DSM-IV major depressive episode. BMC Med 9, 90 10.1186/1741-7015-9-90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brydon L, Harrison NA, Walker C, Steptoe A, Critchley HD, 2008. Peripheral inflammation is associated with altered substantia nigra activity and psychomotor slowing in humans. Biol. Psychiatry 63, 1022–1029. https://doi.org/doi:10.1016/j.biopsych.2007.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brydon L, Walker C, Wawrzyniak A, Whitehead D, Okamura H, Yajima J, Tsuda A, Steptoe A, 2009. Synergistic effects of psychological and immune stressors on inflammatory cytokine and sickness responses in humans. Brain. Behav. Immun 23, 217–24. 10.1016/j.bbi.2008.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunzeck N, Düzel, ., 2006. Absolute coding of stimulus novelty in the human substantia nigra/VTA. Neuron 51, 369–79. 10.1016/j.neuron.2006.06.021 [DOI] [PubMed] [Google Scholar]
- Butler G, Mathews A, 1983. Cognitive processes in anxiety. Adv. Behav. Res. Ther 5, 51–62. 10.1016/0146-6402(83)90015-2 [DOI] [Google Scholar]
- Calabrese F, Rossetti AC, Racagni G, Gass P, Riva MA, Molteni R, 2014. Brain-derived neurotrophic factor: A bridge between inflammation and neuroplasticity. Front. Cell. Neurosci 8 10.3389/fncel.2014.00430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuron L, Gumnick JF, Musselman DL, Lawson DH, Reemsnyder A, Nemeroff CB, Miller AH, 2002. Neurobehavioral effects of interferon-α in cancer patients: Phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology 26, 643–652. 10.1016/S0893-133X(01)00407-9 [DOI] [PubMed] [Google Scholar]
- Capuron L, Miller AH, 2004. Cytokines and psychopathology: Lessons from interferon-α. Biol. Psychiatry 56, 819–824. 10.1016/j.biopsych.2004.02.009 [DOI] [PubMed] [Google Scholar]
- Capuron L, Pagnoni G, Demetrashvili MF, Lawson DH, Fornwalt FB, Woolwine B, Berns GS, Nemeroff CB, Miller AH, 2007. Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol 32, 2384–2392. 10.1038/sj.npp.1301362 [DOI] [PubMed] [Google Scholar]
- Capuron L, Pagnoni G, Drake DF, Woolwine BJ, Spivey JR, Crowe RJ, Votaw JR, Goodman MM, Miller AH, 2012. Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch. Gen. Psychiatry 69, 1044–53. 10.1001/archgenpsychiatry.2011.2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuron L, Raison L, Musselman DL, Lawson DH, Nemeroff CB, Miller AH, 2003. Association of exaggerated HPA axis response to the initial injection of interferon-alpha with development of depression during interferon-alpha therapy. Am. J. Psychiatry 160, 1342–1345. 10.1176/appi.ajp.160.7.1342 [DOI] [PubMed] [Google Scholar]
- Capuron L, Ravaud A, Dantzer R, 2001. Timing and specificity of the cognitive changes induced by interleukin-2 and interferon-alpha treatments in cancer patients. Psychosom. Med 63, 376–86. [DOI] [PubMed] [Google Scholar]
- Capuron L, Ravaud A, Miller AH, Dantzer R, 2004. Baseline mood and psychosocial characteristics of patients developing depressive symptoms during interleukin-2and/or interferon-alpha cancer therapy. Brain. Behav. Immun 18, 205–213. 10.1016/j.bbi.2003.11.004 [DOI] [PubMed] [Google Scholar]
- Caraceni A, Gangeri L, Martini C, Belli F, Brunelli C, Baldini M, Mascheroni L, Lenisa L, Cascinelli N, 1998. Neurotoxicity of interferon-α in melanoma therapy: Results from a randomized controlled trial. Cancer 83, 482–489. 10.1002/(SICI)1097-0142(19980801)83:3<482::AID-CNCR17>3.0.CO;2-S [DOI] [PubMed] [Google Scholar]
- Carty CL, Heagerty P, Nakayama K, McClung EC, Lewis J, Lum D, Boespflug E, McCloud-Gehring C, Soleimani BR, Ranchalis J, Bacus TJ, Furlong CE, Jarvik GP, 2006. Inflammatory response after influenza vaccination in men with and without carotid artery disease. Arterioscler. Thromb. Vasc. Biol 26, 2738–2744. 10.1161/01.ATV.0000248534.30057.b5 [DOI] [PubMed] [Google Scholar]
- Casper R, Redmond DE, Katz MM, Schaffer CB, Davis JM, Koslow SH, 1985. Somatic symptoms in primary affective disorder: Presence and relationship to the classification of depression. Arch. Gen. Psychiatry 42, 1098–1104. 10.1001/archpsyc.1985.01790340082012 [DOI] [PubMed] [Google Scholar]
- Castaneda AE, Tuulio-Henriksson A, Marttunen M, Suvisaari J, Lonnqvist J, 2008. A review on cognitive impairments in depressive and anxiety disorders with a focus on young adults. J. Affect. Disord 106, 1–27. 10.1016/j.jad.2007.06.006 [DOI] [PubMed] [Google Scholar]
- Christian LM, Iams JD, Porter K, Glaser R, 2011. Inflammatory responses to trivalent influenza virus vaccine among pregnant women. Vaccine 29,8982–8987. 10.1016/j.vaccine.2011.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriani A, Furukawa TA, Salanti G, Chaimani A, Atkinson LZ, Ogawa Y, Leucht S, Ruhe HG, Turner EH, Higgins JPT, Egger M, Takeshima N, Hayasaka Y, Imai H, Shinohara K, Tajika A, Ioannidis JA, Geddes JR, 2018. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: A systematic review and network meta-analysis. The Lancet 391, 1357–1366. 10.1016/S0140-6736(17)32802-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craske MG, 2012. The R-Doc Initiative: Science and Practice. Depress. Anxiety 29, 253–256. 10.1002/da.21930 [DOI] [PubMed] [Google Scholar]
- Craske MG, Meuret AE, Ritz T, Treanor M, Dour HJ, 2016. Treatment for anhedonia: A neuroscience driven approach. Depress. Anxiety 33, 927–938. 10.1002/da.22490 [DOI] [PubMed] [Google Scholar]
- Cuthbert B, Insel T, 2010. Commentary on “The need for patient-subjective data in the The Data of Diagnosis : New approaches to psychiatric classification”. Psychiatry 73, 311–315. 10.1521/psyc.2010.73.4.311 [DOI] [PubMed] [Google Scholar]
- Danese A, Moffitt TE, Pariante CM, Ambler A, Poulton R, Caspi A, 2008. Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch. Gen. Psychiatry 65, 409–15. 10.1001/archpsyc.65.4.409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, Heijnen CJ, Kavelaars A, Laye S, Capuron L, 2014. The neuroimmune basis of fatigue. Trends Neurosci. 37, 39–46. 10.1016/j.tins.2013.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW, 2008. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56. 10.1038/nrn2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Lawson MA, Kelley KW, 2011. Inflammation-associated depression: From serotonin to kynurenine. Psychoneuroendocrinology 36, 426–436. 10.1016/j.psyneuen.2010.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, Walker AK, 2014. Is there a role for glutamate-mediated excitotoxicity in inflammation-induced depression? J. Neural Transm. Vienna Austria 1996 121, 925–932. 10.1007/s00702-014-1187-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MC, Lemery-Chalfant K, Yeung EW, Luecken LJ, Zautra AJ, Irwin MR, 2018. Interleukin-6 and depressive mood symptoms: Mediators of the association between childhood abuse and cognitive performance in middle-aged adults. Ann. Behav. Med 10.1093/abm/kay014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Garza R, 2005. Endotoxin- or proinflammatory cytokine-induced sickness behavior as an animal model of depression: Focus on anhedonia. Neurosci. Biobehav. Rev 29, 761– 770. 10.1016/j.neubiorev.2005.03.016 [DOI] [PubMed] [Google Scholar]
- de Rooij SR, Schene H, Phillips DI, Roseboom TJ, 2010. Depression and anxiety: Associations with biological and perceived stress reactivity to a psychological stress protocol in a middle-aged population. Psychoneuroendocrinology 35, 866–877. 10.1016/j.psyneuen.2009.11.011 [DOI] [PubMed] [Google Scholar]
- Dearing KF, Gotlib IH, 2009. Interpretation of ambiguous information in girls at risk for depression 4, 79–91. 10.1126/scisignal.2001449.Engineering [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Arco A, Mora F, 2008. Prefrontal cortex-nucleus accumbens interaction: In vivo modulation by dopamine and glutamate in the prefrontal cortex. Pharmacol. Biochem. Behav. 90, 226–235. 10.1016/j.pbb.2008.04.011 [DOI] [PubMed] [Google Scholar]
- DellaGioia N, Hannestad J, 2010. A critical review of human endotoxin administration as an experimental paradigm of depression. Neurosci. Biobehav. Rev 34, 130–143. 10.1016/j.neubiorev.2009.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca J, Genova HM, Capili EJ, Wylie GR, 2009. Functional neuroimaging of fatigue. Phys. Med. Rehabil. Clin. N. Am., Fatigue 20, 325–337. 10.1016/j.pmr.2008.12.007 [DOI] [PubMed] [Google Scholar]
- Demyttenaere K, De Fruyt J, Stahl SM, 2005. The many faces of fatigue in major depressive disorder. Int. J. Neuropsychopharmacol. Off. Sci. J. Coll. Int. Neuropsychopharmacol. CINP 8, 93–105. 10.1017/S1461145704004729 [DOI] [PubMed] [Google Scholar]
- Deveney CM, Deldin PJ, 2004. Memory of faces: A slow wave ERP study of major depression. Emotion 4, 295–304. 10.1037/1528-3542.4.3.295\r2004-18367-008 [DOI] [PubMed] [Google Scholar]
- Disner SG, Beevers CG, Haigh EA, Beck AT, 2011. Neural mechanisms of the cognitive model of depression. Nat. Rev. Neurosci. 12, 467–477. 10.1038/nrn3027 [DOI] [PubMed] [Google Scholar]
- Dobryakova, ., Genova HM, DeLuca J, Wylie GR, 2015. The dopamine imbalance hypothesis of fatigue in multiple sclerosis and other neurological disorders. Front. Neurol 6 10.3389/fneur.2015.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley LN, Ganz PA, Cole SW, Crespi CM, Bower JE, 2016. Val66Met BDNF polymorphism as a vulnerability factor for inflammation-associated depressive symptoms in women with breast cancer. J. Affect. Disord 197, 43–50. 10.1016/j.jad.2016.02.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell NG, Cooper EA, Tibble J, Voon V,Critchley HD, Cercignani M, Harrison NA, 2016. Acute changes in striatal microstructure predict the development of interferon-alpha induced fatigue. Biol. Psychiatry 79, 320–328. 10.1016/j.biopsych.2015.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, Lanctôt KL, 2010. A meta-analysis of cytokines in Major Depression. Biol. Psychiatry, Cortical Inhibitory Deficits in Depression 67, 446–457. 10.1016/j.biopsych.2009.09.033 [DOI] [PubMed] [Google Scholar]
- Drevets WC, 2001. Neuroimaging and neuropathological studies of depression: Implications for the cognitive-emotional features of mood disorders. Curr. Opin. Neurobiol 11, 240–249. 10.1016/S0959-4388(00)00203-8 [DOI] [PubMed] [Google Scholar]
- Drozdz W, Borkowska A, Halota W, Rybakowski JK, 2008. Depressive symptoms and cognitive dysfunction in patients with hepatitis C treated with interferon-α and ribavirine. Arch. Psychiatry 85–91.
- Dunlop K, Hanlon CA, Downar J, 2017. Noninvasive brain stimulation treatments for addiction and major depression. Ann. N. Y. Acad. Sci 1394, 31–54. 10.1111/nyas.12985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn AJ, Swiergiel AH, De Beaurepaire R, 2005. Cytokines as mediators of depression: What can we learn from animal studies? Neurosci. Biobehav. Rev 29, 891–909. 10.1016/j.neubiorev.2005.03.023 [DOI] [PubMed] [Google Scholar]
- Dunn BD, Dalgleish T, Lawrence AD, Cusack R, Ogilvie AD, 2004. Categorical and dimensional reports of experienced affect to emotion-inducing pictures in depression. J. Abnorm. Psychol. 113, 654–660. 10.1037/0021-843X.113.4.654 [DOI] [PubMed] [Google Scholar]
- Eisenberger NI, Berkman ET, Inagaki TK, Rameson LT, Mashal NM, Irwin MR, 2010a. Inflammation-induced anhedonia: Endotoxin reduces ventrals triatum responses to reward. Biol. Psychiatry 68, 748–754. 10.1016/j.biopsych.2010.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberger NI, Cole SW, 2012. Social neuroscience and health: Neurophysiological mechanisms linking social ties with physical health. Nat. Neurosci 15, 669–74. 10.1038/nn.3086 [DOI] [PubMed] [Google Scholar]
- Eisenberger NI, Inagaki TK, Mashal NM, Irwin MR, 2010b. Inflammation and social experience: An inflammatory challenge induces feelings of social disconnection in addition to depressed mood. Brain. Behav. Immun 24, 558–563. 10.1016/j.bbi.2009.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberger NI, Inagaki TK, Rameson L ., Mashal M, Irwin MR, 2009. An fMRI study of cytokine-induced depressed mood and social pain: The role of sex differences. NeuroImage 47, 881–890. 10.1016/j.neuroimage.2009.04.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberger NI, Moieni M, Inagaki TK, Muscatell KA, Irwin MR, 2016. In sickness and in health: The co-regulation of inflammation and social behavior. Neuropsychopharmacology 1–12. 10.1038/npp.2016.141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein J, Pan H, Kocsis JH, Yang Y, Butler T, Chusid J, Hochberg H, Murrough J, Strohmayer, ., Stern E, Silbersweig DA, 2006. Lack of ventral striatal response to positive stimuli in depressed versus normal subjects . Am. J. Psychiatry 163, 1784–1790. 10.1176/appi.ajp.163.10.1784 [DOI] [PubMed] [Google Scholar]
- Etkin A, Egner T, Kalisch R, 2011. Emotional processing in anterior cingulate and medial prefrontal. Trends Cogn. Sci 15, 85–93. 10.1016/j.tics.2010.11.004.Emotional [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felger JC, 2017. The role of dopamine in inflammation-associated depression: Mechanisms and therapeutic implications. Curr. Top.Behav.Neurosci 31,199–219. 10.1007/7854_2016_13 [DOI] [PubMed] [Google Scholar]
- Felger JC, Mun J, Kimmel HL, Nye JA, Drake DF, Hernandez CR, Freeman AA, Rye DB, Goodman MM, Howell LL, Miller H, 2013Chronic interferon- a decreases dopamine 2 receptor binding and striatal dopamine release in association with anhedonia-like behavior in nonhuman primates. Neuropsychopharmacology 38, 2179– 2187. 10.1038/npp.2013.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felger JC, Treadway MT, 2016. Inflammation effects on motivation and motor activity: Role of dopamine. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 42, 1–88. 10.1038/npp.2016.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana RJ, Bieliauskas LA, Lindsay KL, Back-Madruga C, Wright EC, Snow KK, Lok SF, Kronfol Z, Padmanabhan L, 2007. Cognitive function does not worsen during pegylated interferon and ribavirin retreatment of chronic hepatitis C. Hepatology 45, 1154–1163. 10.1002/hep.21633 [DOI] [PubMed] [Google Scholar]
- Frank MG, Watkins LR, Maier SF, 2013. Stress-induced glucocorticoids as a neuroendocrine alarm signal of danger. Brain. Behav. Immun 33, 1 10.1016/j.bbi.2013.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried EI, Nesse RM, 2015. Depression is not a consistent syndrome : An investigation of unique symptom patterns in the STAR * D study. J. Affect. Disord 172, 96–102. 10.1016/j.jad.2014.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman NP, Miyake A, Young SE, DeFries JC, Corley RP, Hewitt JK, 2008. Individual differences in executive functions are almost entirely geneticin origin. J. Exp Psychol. Gen. 137, 201–225. 10.1037/0096-3445.137.2.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu CH, Williams SC, Cleare AJ, Brammer MJ, Walsh ND, Kim J, Andrew CM, Pich EM, Williams PM, Reed LJ, Mitterschiffthaler MT, Suckling J, Bullmore ET, 2004. Attenuation of the neural response to sad faces in major depression by antidepressant treatment: A prospective, event-related functional magnetic resonance imaging study. Arch. Gen. Psychiatry 61, 877–889. 10.1001/archpsyc.61.9.877 [DOI] [PubMed] [Google Scholar]
- Garvey MJ, Mungas D, Tollefson GD, 1984. Hypersomnia in major depressive disorders. J. Affect. Disord 6, 283–6. [DOI] [PubMed] [Google Scholar]
- Gimeno D, Kivimäki M, Brunner EJ, Elovainio M, De Vogli R, Steptoe A, Kumari M, Lowe GDO, Rumley A, Marmot MG, Ferrie JE, 2009. Associations of C-reactive protein and interleukin-6 with cognitive symptoms of depression: 12-year follow-up of the Whitehall II study. Psychol. Med 39, 413–423. 10.1017/S0033291708003723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn DC, Curran JE, Winkler AM, Carless MA, Kent JW, Charlesworth JC, Johnson MP, Göring HHH, Cole SA, Dyer TD, Moses EK, Olvera RL, Kochunov P, Duggirala R, Fox PT, Almasy L, Blangero J, 2012. High dimensional endophenotype ranking in the search for major depression risk genes. Biol. Psychiatry 71, 6–14. 10.1016/j.biopsych.2011.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein BL, Klein DN, 2014. A review of selected candidate endophenotypes for depression. Clin. Psychol. Rev 34, 417–427. 10.1016/j.cpr.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshen I, Yirmiya R, 2009. Interleukin-1(IL-1): A central regulator of stress responses. Front. Neuroendocrinol. 30, 30–45. 10.1016/j.yfrne.2008.10.001 [DOI] [PubMed] [Google Scholar]
- Gotlib IH, Hamilton JP, 2008. Neuroimaging and depression: Current status and unresolved issues. Curr. Dir. Psychol. Sci 17, 159–163. 10.1111/j.1467-8721.2008.00567.x [DOI] [Google Scholar]
- Gotlib IH, Hamilton JP, Cooney RE, Singh K, Henry ML, Joormann J, 2010. Neural processing of reward and loss in girls at risk for major depression. Arch. Gen. Psychiatry 67, 380–7. 10.1001/archgenpsychiatry.2010.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotlib IH, Joormann J, 2010. Cognition and depression: Current status and future directions. Annu. Rev. Clin. Psychol 6, 285–312. 10.1146/annurev.clinpsy.121208.131305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman II, Gould TD, 2003. The endophenotype concept in psychiatry: Etymology and strategic intentions. Am. J. Psychiatry 160, 636–645. 10.1176/appi.ajp.160.4.636 [DOI] [PubMed] [Google Scholar]
- Gould TD, Gottesman II, 2006. Psychiatric endophenotypes and the development of valid animal models. Genes Brain Behav 5, 113–119. 10.1111/j.1601-183X.2005.00186.x [DOI] [PubMed] [Google Scholar]
- Grigoleit J-S, Kullmann JS, Wolf OT, Hammes F, Wegner A, Jablonowski S, Engler H, Gizewski E, Oberbeck R, Schedlowski M, 2011. Dose-dependent effects of endotoxin on neurobehavioral functions in humans. PloS One 6, e28330–e28330. 10.1371/journal.pone.0028330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoleit JS, Oberbeck JR, Lichte P, Kobbe P, Wolf OT, Montag T, Rey A. del, Gizewski ER, Engler H, Schedlowsk M, 2010. Lipopolysaccharide-induced experimental immune activation does not impair memory functions in humans. Neurobiol. Learn. Mem 94, 561–567. 10.1016/j.nlm.2010.09.011 [DOI] [PubMed] [Google Scholar]
- Haapakoski R, Mathieu J, Ebmeier KP, Alenius H, Kivimäki,. 2015. Cumulative meta-analysis of interleukins 6 and 1β, tumour necrosis factor α and C-reactive protein in patients with major depressive disorder. Brain. Behav. Immun 49, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammar A, Ardal G, 2009. Cognitive functioning in major depression--a summary. Front. Hum. Neurosci. 3, 26–26. 10.3389/neuro.09.026.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haroon E, Miller AH, 2017. Inflammation effects on brain glutamate in depression: Mechanistic considerations and treatment implications. Curr. Top. Behav. Neurosci 31, 173–198. 10.1007/7854_2016_40 [DOI] [PubMed] [Google Scholar]
- Haroon E, Raison CL, Miller AH, 2012. Psychoneuroimmunology meets neuropsychopharmacology: Translational implications of the impact of inflammation on behavior. Neuropsychopharmacology 37, 137–162. 10.1038/npp.2011.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison NA, Brydon L, Walker C, Gray MA, Steptoe A, Critchley HD, 2009. Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol. Psychiatry 66, 407–414. 10.1016/j.biopsych.2009.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison NA, Cercignani M, Voon V, Critchley HD, 2015. Effects of inflammation on hippocampus and substantia nigra responses to novelty in healthy human participants. Neuropsychopharmacology 40, 831–838. 10.1038/npp.2014.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison NA, Doeller CF, Voon V, Burgess N, Critchley HD, 2014. Peripheral inflammation acutely impairs human spatial memory via actions on medial temporal lobe glucose metabolism. Biol. Psychiatry 76, 585–93. 10.1016/j.biopsych.2014.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey P-O, Pruessner J, Czechowska Y, Lepage M, 2007. Individual differences in trait anhedonia: A structural and functional magnetic resonance imaging study in non-clinical subjects. Mol. Psychiatry 12, 767–775. 10.1038/sj.mp.4002021 [DOI] [PubMed] [Google Scholar]
- Hasler G, Drevets WC, Gould TD, Gottesman II, Manji HK, 2006. Toward constructing an endophenotype strategy for bipolar disorders. Biol. Psychiatry 60, 93–105. 10.1016/j.biopsych.2005.11.006 [DOI] [PubMed] [Google Scholar]
- Hasler G, Drevets WC, Manji HK, Charney D ., 2004. Discovering endophenotypes for Major Depression. Neuropsychopharmacology 29, 1765–1781. 10.1038/sj.npp.1300506 [DOI] [PubMed] [Google Scholar]
- He J, Hsuchou H, He Y, Kastin AJ, Wang Y, Pan W, 2014. Sleep restriction impairs blood-brain barrier function. J. Neurosci. 34, 14697–14706. 10.1523/JNEUROSCI.2111-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller A, Johnstone T, Shackman A, Light S, Peterson M, Kolden G, Kalin N, Davidson R, 2009. Reduced capacity to sustain positive emotion in major depression reflects diminished maintenance of fronto-striatal brain activation. Proc. Natl. Acad. Sci. U. S. A 106, 22445–50. 10.1073/pnas.0910651106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann DM, Mullington J, Hinze-Selch D, Schreiber W, Galanos C, Pollmächer T, 1998. Endotoxin-induced changes in sleep and sleepiness during the day. Psychoneuroendocrinology 23, 427–437. 10.1016/S0306-4530(98)00030-4 [DOI] [PubMed] [Google Scholar]
- Hetta J, Rimón R, Almqvist M, 1985. Mood alterations and sleep. Ann.Clin.Res 17,252– 256. [PubMed] [Google Scholar]
- Hilsabeck RC, Hassanein TI, Ziegler EA, Carlson MD, Perry W, 2005. Effect of interferon-alpha on cognitive functioning in patients with chronic hepatitis C. J. Int. Neuropsychol. Soc. JINS 11, 16–22. 10.1017/1355617705050022 [DOI] [PubMed] [Google Scholar]
- Howren MB, Lamkin DM, Suls J, 2009. Associations of depression with C-reactive protein, IL-1, and IL-6: A meta-analysis. Psychosom. Med 71, 171–186. 10.1097/PSY.0b013e3181907c1b [DOI] [PubMed] [Google Scholar]
- Huang M, Su S, Goldberg J, Miller AH, urrah NV, Shallenberger L, Pimple P, Bremner JD, Vaccarino V, 2017. Pathway linking depression and inflammation: A 5-year longitudinal twin difference study. Circulation 135, AMP074–AMP074. [Google Scholar]
- Imeri L, Opp MR, 2009. How (and why) the immune system makes us sleep. Nat. Rev. Neurosci 10, 199–210. 10.1038/nrn2576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki TK, Muscatell KA, Irwin MR, Cole SW, Eisenberger NI, 2012. Inflammation selectively enhances amygdala activity to socially threatening images. NeuroImage 59, 3222–3226. 10.1016/j.neuroimage.2011.10.090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki K, Muscatell KA, Irwin MR, Moieni M, Dutcher JM, Jevtic I, Breen EC, Eisenberger NI, 2015. The role of the ventral striatum in inflammatory-induced approach toward support figures. Brain. Behav. Immun. 44, 247–252. 10.1016/j.bbi.2014.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, Sanislow C, Wang P, 2010. Research Domain Criteria (RDoC): Toward a new classification framework for research on mental disorders. Am. J. Psychiatry 167,748–751. 10.1176/appi.ajp.2010.09091379 [DOI] [PubMed] [Google Scholar]
- Irwin MR, 2002. Psychoneuroimmunology of depression: Clinical implications. Brain. Behav. Immun 16, 1–16. 10.1006/brbi.2001.0654 [DOI] [PubMed] [Google Scholar]
- Janssen HLA, Brouwer JT, van der Mast RC, Schalm SW, 1994. Suicide associated with alfa-interferon therapy for chronic viral hepatitis. J. Hepatol . 21, 241–243. 10.1016/S0168-8278(05)80402-7 [DOI] [PubMed] [Google Scholar]
- Joormann J, Quinn ME, 2014. Cognitive processes and emotion regulation in depression. Depress. Anxiety 31, 308–315. 10.1002/da.22264 [DOI] [PubMed] [Google Scholar]
- Kakade S, Dayan P, 2002. Dopamine: Generalization and bonuses. Neural Netw . 15, 549–559. 10.1016/S0893-6080(02)00048-5 [DOI] [PubMed] [Google Scholar]
- Kanoski SE, Zhang Y, Zheng W, Davidson TL, 2010. The effects of a high-energy diet on hippocampal function and blood-brain barrier integrity in the rat. J. Alzheimers Dis 21, 207–219. 10.3233/JAD-2010-091414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karshikoff B, Sundelin, ., Lasselin J, 2017. Role of inflammation in human fatigue: Relevance of multidimensional assessments and potential neuronal mechanisms. Front. Immunol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Sullivan PF, Evengård B, Pedersen NL, 2009. A population-based twin study of functional somatic syndromes. Psychol. Med 39, 497–505. 10.1017/S0033291708003784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keedwell PA, Andrew C, Williams SCR, Brammer MJ, Phillips ML, 2005. The neural correlates of anhedonia in major depressive disorder.Biol.Psychiatry 58,843–853. 10.1016/j.biopsych.2005.05.019 [DOI] [PubMed] [Google Scholar]
- Kendler KS, Neale MC, Kessler RC, Heath AC, Eaves LJ, 1992. Familial influences on the clinical characteristics of major depression: a twin study. Acta Psychiatr. Scand 86, 371–378. 10.1111/j.1600-0447.1992.tb03283.x [DOI] [PubMed] [Google Scholar]
- Kenis G, Maes M, 2002. Effects of antidepressants on the production of cytokines. Int. J. Neuropsychopharmacol 5, 401–412. 10.1017/S1461145702003164 [DOI] [PubMed] [Google Scholar]
- Kessler RC, Bromet EJ, 2013. The epidemiology of depression across cultures. Annu. Rev. Public Health 34, 119–38. 10.1146/annurev-publhealth-031912-114409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler O, Benros ME, Nordentoft M, Farkouh ME, Iyengar RL, Mors O, Krogh J, 2014. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: A systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry 71, 1381 10.1001/jamapsychiatry.2014.1611 [DOI] [PubMed] [Google Scholar]
- Korth C, Mullington J, Schreiber W, Pollmächer T, 1996. Influence of endotoxin on daytime sleep in humans. Infect. Immun 64, 1110–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krabbe KS, Reichenberg S, Yirmiya R, Smed A, Pedersen BK, Bruunsgaard H, 2005. Low-dose endotoxemia and human neuropsychological functions. Brain. Behav. Immun., Physical Activity, Behavior, Immunity and Health 19, 453–460. 10.1016/j.bbi.2005.04.010 [DOI] [PubMed] [Google Scholar]
- Krause AJ, Simon EB, Mander BA, Greer SM, Saletin JM, Goldstein-Piekarski AN, Walker MP, 2017. The sleep-deprived human brain. Nat. Rev. Neurosci 18, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroenke K, Price RK, 1993. Symptoms in the community. Prevalence, classification, and psychiatric comorbidity. Arch. Intern. Med 153, 2474–80. [PubMed] [Google Scholar]
- Kuehner C, 2003. Gender differences in unipolar depression:An update of epidemiological findings and possible explanations. Acta Psychiatr. Scand 108, 163–174. 10.1034/j.1600-0447.2003.00204.x [DOI] [PubMed] [Google Scholar]
- Kuhlman KR, Chiang JJ, Horn S, Bower JE, 2017. Developmental psychoneuroendocrine and psychoneuroimmune pathways from childhood adversity to disease. Neurosci. Biobehav. Rev 80, 166–184. 10.1016/j.neubiorev.2017.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman KR, Robles TF, Dooley LN, Boyle C, Haydon MD, Bower JE, 2018. Within-subject associations between inflammation and features of depression: Using the flu vaccine as a mild inflammatory stimulus. Brain. Behav. Immun 69, 540–547. 10.1016/j.bbi.2018.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupfer D, Harrow M, Detre T, 1969. Sleep patterns and psychopathology. Acta Psychiatr. Scand 45, 75–89. [DOI] [PubMed] [Google Scholar]
- Kupferberg A, Bicks L, Hasler G, 2016. Social functioning in major depressive disorder. Neurosci. Biobehav. Rev 69, 313–332. 10.1016/j.neubiorev.2016.07.002j.neubiorev.2016.07.002 [DOI] [PubMed] [Google Scholar]
- Larson SJ, Dunn a J., 2001. Behavioral effects of cytokines. Brain. Behav. Immun. 15, 371– 387. 10.1006/brbi.2001.0643 [DOI] [PubMed] [Google Scholar]
- Lasselin J, Elsenbruch S, Lekander M, Axelsson J, Karshikoff B, Grigoleit J-S, Engler H, Schedlowski M, Benson S, 2016a. Mood disturbance during experimental endotoxemia: Predictors of state anxiety as a psychological component of sickness behavior. Brain. Behav. Immun. 57, 30–37. 10.1016/j.bbi.2016.01.003 [DOI] [PubMed] [Google Scholar]
- Lasselin J, Lekander M, Axelsson J, Karshikoff B, 2018. Sex differences in how inflammation affects behavior: What we can learn from experimental inflammatory models in humans. Front. Neuroendocrinol . 10.1016/j.yfrne.2018.06.005 [DOI] [PubMed] [Google Scholar]
- Lasselin J, Treadway MT, Lacourt TE, Soop A, Olsson MJ, Karshikoff B, Paues-Göranson S, Axelsson J, Dantzer R, Lekander M, 2016b. Lipopolysaccharide alters motivated behavior in a monetary reward task: A randomized trial. Neuropsychopharmacology 1–10. 10.1038/npp.2016.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson C, MacLeod C, Hammond G, 2002. Interpretation revealed in the blink of an eye: Depressive bias in the resolution of ambiguity. J. Abnorm. Psychol. 111, 321–328. 10.1037//0021-843X.111.2.321 [DOI] [PubMed] [Google Scholar]
- Lieb K, Engelbrecht MA, Gut O, Fiebich BL, Bauer J, Janssen G, Schaefer,., 2006. Cognitive impairment in patients with chronic hepatitis treated with interferon alpha (IFN-a): Results from a prospective study. Eur. Psychiatry 21, 204–210. 10.1016/j.eurpsy.2004.09.030 [DOI] [PubMed] [Google Scholar]
- Lieberman M ., Eisenberger NI, 2009. Pains and pleasures of social life. Sci. Transl. Med 323, 890–891. 10.1126/science.1170008 [DOI] [PubMed] [Google Scholar]
- Lisman JE, Grace AA, 2005. The hippocampal-VTA loop: Controlling the entry of information into long-term memory. Neuron 46, 703–13. 10.1016/j.neuron.2005.05.002 [DOI] [PubMed] [Google Scholar]
- Lopez-Duran NL, Kovacs M, George CJ, 2009. Hypothalamic-pituitary-adrenal axis dysregulation in depressed children and adolescents: A meta-analysis. Psychoneuroendocrinology 34, 1272–1283. 10.1016/j.psyneuen.2009.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotrich FE, Rabinovitz M, Gironda P, Pollock BG, 2007. Depression following pegylated interferon-alpha: Characteristics and vulnerability. J. Psychosom. Res 63, 131–135. 10.1016/j.jpsychores.2007.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majer M, Welberg LAM, Capuron L, Pagnoni G, Raison L, Miller AH, 2008. IFN-alpha-induced motor slowing is associated with increased depression and fatigue in patients with chronic hepatitis C. Brain. Behav. Immun 22, 870–880. 10.1016/j.bbi.2007.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majidi J, Kosari-Nasab M, Salari A-A, 2016. Developmental minocycline treatment reverses the effects of neonatal immune activation on anxiety- and depression-like behaviors, hippocampal inflammation, and HPA axis activity in adult mice. Brain Res. Bull 120, 1–13. 10.1016/j.brainresbull.2015.10.009 [DOI] [PubMed] [Google Scholar]
- Malarkey EB, Parpura V, 2008. Mechanisms of glutamate release from astrocytes. Neurochem. Int 52, 142–154. 10.1016/j.neuint.2007.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsland AL, Gianaros PJ, Kuan DC, Sheu LK, Krajina K, Manuck SB, 2015. Brain morphology links systemic inflammation to cognitive function in midlife adults. Brain. Behav. Immun 48, 195 10.1016/j.bbi.2015.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinot ML, Bragulat V, Artiges E, Dollé F, Hinnen F, Jouvent R, Martinot JL, 2001. Decreased presynaptic dopamine function in the left caudate of depressed patients with affective flattening and psychomotor retardation. Am. J. Psychiatry 158, 314–316. 10.1176/appi.ajp.158.2.314 [DOI] [PubMed] [Google Scholar]
- Matthews KA, Schott LL, Bromberger JT, Cyranowski JM, Everson-Rose SA, Sowers M, 2010. Are there bi-directional associations between depressive symptoms and C-reactive protein in mid-life women? Brain. Behav. Immun 24, 96–101. 10.1016/j.bbi.2009.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayberg HS, 2003. Positron emission tomography imaging indepression:Aneural systems perspective. Neuroimaging Clin. N. Am 13, 805–815. 10.1016/S1052-5149(03)00104-7 [DOI] [PubMed] [Google Scholar]
- Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA, Silva JA, Tekell JL, Martin CC, Lancaster JL, Fox PT, 1999. Reciprocal limbic-cortical function and negative mood: Converging PET findings in depression and normal sadness. Am. J. Psychiatry 156, 675–682. 10.1176/ajp.156.5.675 [DOI] [PubMed] [Google Scholar]
- Miller AH, Haroon E, Raison CL, Felger J ., 2013. Cytokine targets in the brain: Impact on neurotransmitters and neurocircuits. Depress. Anxiety 30, 297–306. 10.1002/da.22084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AH, Maletic V, Raison CL, 2009. Inflammation and its discontents: The role of cytokines in the pathophysiology of Major Depression. Biol. Psychiatry 65, 732–741. 10.1016/j.biopsych.2008.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AH, Raison CL, 2016. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol 16, 22–34. 10.1038/nri.2015.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GA ., Rockstroh B, 2013. Endophenotypes in psychopathology research: Where do we stand? Annu. Rev. Clin. Psychol 9, 177–213. 10.1146/annurev-clinpsy-050212-185540 [DOI] [PubMed] [Google Scholar]
- Moieni M, Irwin MR, Jevtic I, Olmstead R, Breen EC, Eisenberger NI, 2015. Sex differences in depressive and socioemotional responses to an inflammatory challenge: Implications for sex differences in depression. Neuropsychopharmacology 40, 1709–1716. 10.1038/npp.2015.17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore SR, Depue RA, 2016. Neurobehavioral foundation of environmental reactivity. Psychol. Bull 142, 107–164. 10.1037/bul0000028 [DOI] [PubMed] [Google Scholar]
- Morris BH, Bylsma LM, Rottenberg J, 2009. Does emotion predict the course of major depressive disorder? A review of prospective studies. Br. J. Clin. Psychol 48, 255–273. 10.1348/014466508X396549 [DOI] [PubMed] [Google Scholar]
- Morrow GR, Hickok JT, Roscoe JA, Raubertas F, Andrews PLR, Flynn PJ, Hynes HE, Banerjee TK, Kirshner JJ, King DK, 2003. Differential effects of paroxetine on fatigue and depression: A randomized, double-blind trial from the University of Rochester Cancer Center Community Clinical Oncology Program. J. Clin. Oncol 21,4635–4641. 10.1200/JCO.2003.04.070 [DOI] [PubMed] [Google Scholar]
- Mullington J, Korth C, Hermann DM, Orth A, Galanos C, Holsboer F, Pollmächer T, 2000. Dose-dependent effects of endotoxin on human sleep. Am J Physiol Regul. Integr. Comp Physiol 278, R947–55. [DOI] [PubMed] [Google Scholar]
- Muscatell KA, Eisenberger NI, 2012. A social neuroscience perspective on stress and health. Soc. Personal. Psychol. Compass 6, 890–904. 10.1111/j.1751-9004.2012.00467.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscatell K, Moieni M, Inagaki TK, Dutcher JM, Jevtic I, Breen EC, Irwin MR, Eisenberger NI, 2016. Exposure to an inflammatory challenge enhances neural sensitivity to negative and positive social feedback. Brain. Behav. Immun 57, 21–29. 10.1016/j.bbi.2016.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman DL, Lawson DH, Gumnick JF, Manatunga AK, Penna S, Goodkin RS,Greiner K, Nemeroff CB, Miller AH, 2001. Paroxetine for the prevention of depression induced by high-dose interferon alpha. Engl. J. Med. 344,961–966. [DOI] [PubMed] [Google Scholar]
- Myin-Germeys I, Peeters F, Havermans R, Nicolson NA, deVries MW, Delespaul P,van Os J, 2003. Emotional reactivity to daily life stress in psychosis and affective disorder: An experience sampling study. Acta Psychiatr. Scand. 107, 124–131. 10.1034/j.1600-0447.2003.02025.x [DOI] [PubMed] [Google Scholar]
- Nair A, Bonneau RH, 2006. Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation. J. euroimmunol 171, 72 10.1016/j.jneuroim.2005.09.012 [DOI] [PubMed] [Google Scholar]
- Nemeroff CB, 2007. Prevalence and management of treatment-resistant depression. J. Clin. Psychiatry 68, 17–25. [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WA, 2006. The mesolimbic dopamine reward circuit in depression. Biol. Psychiatry 59, 1151–1159. 10.1016/j.biopsych.2005.09.018 [DOI] [PubMed] [Google Scholar]
- Nikkheslat N, Pariante CM, Zunszain PA, 2018. Chapter 3 - Neuroendocrine abnormalities in Major Depression: An insight into glucocorticoids, cytokines, and the kynurenine pathway, in: Baune B . (Ed.), Inflammation and Immunity in Depression Academic Press, pp. 45–60. 10.1016/B978-0-12-811073-7.00003-9 [DOI] [Google Scholar]
- Nolen-Hoeksema S, 2001. Gender differences in depression. Curr. Dir. Psychol. Sci 10, 173– 176. 10.1111/1467-8721.00142 [DOI] [Google Scholar]
- Nolen-Hoeksema S, 1987. Sex differences in unipolar depression: Evidence and theory. Psychol. Bull 101, 259–282. 10.1037/0033-2909.101.2.259 [DOI] [PubMed] [Google Scholar]
- Nutt D, Demyttenaere K, Janka Z, Aarre T, Bourin M, Canonico PL, Carrasco JL, Stahl S, 2007. The other face of depression, reduced positive affect: the role ofcatecholamines in causation and cure . J. Psychopharmacol. Oxf. Engl 21,461–471. 10.1177/0269881106069938 [DOI] [PubMed] [Google Scholar]
- O’Connor JC, Lawson MA, André C, Moreau, ., Lestage J, Castanon N, Kelley KW, Dantzer R, 2009. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol. Psychiatry 14, 511–522. 10.1038/sj.mp.4002148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Farrell K, Harkin A, 2017. Stress-related regulation of the kynurenine pathway: Relevance to neuropsychiatric and degenerative disorders. Neuropharmacology, The Kynurenine Pathway in Health and Disease 112, 307–323. 10.1016/j.neuropharm.2015.12.004 [DOI] [PubMed] [Google Scholar]
- Paine NJ, Ring C, Bosch JA, Drayson MT, Veldhuijzen van Zanten JJCS, 2013. The time course of the inflammatory response to the Salmonella typhi vaccination. Brain. Behav. Immun 30, 73–79. 10.1016/j.bbi.2013.01.004 [DOI] [PubMed] [Google Scholar]
- Palagini L, Baglioni C, Ciapparelli A, Gemignani A, Riemann D, 2013. REM sleep dysregulation in depression: State of the art. Sleep Med. Rev 17, 377–390. 10.1016/j.smrv.2012.11.001 [DOI] [PubMed] [Google Scholar]
- Pariante M, Lightman SL, 2008. The HPA axis in major depression: Classical theories and new developments. Trends Neurosci 31, 464–468. 10.1016/j.tins.2008.06.006 [DOI] [PubMed] [Google Scholar]
- Pavol MA, Meyers CA, Rexer JL, Valentine AD, Mattis PJ, Talpaz M, 1995. Pattern of neurobehavioral deficits associated with interferon alfa therapy for leukemia. Neurology 45, 947–950. 10.1212/WNL.45.5.947 [DOI] [PubMed] [Google Scholar]
- Pecchi E, Dallaporta M, Jean A, Thirion S, Troadec JD, 2009. Prostaglandins and sickness 10.1016/j.physbeh.2009.02.040 [DOI] [PubMed] [Google Scholar]
- Piet J, Hougaard E, 2011. The effect of mindfulness-based cognitive therapy for prevention of relapse in recurrent major depressive disorder: a systematic review and meta-analysis. Clin. Psychol. Rev 31, 1032–1040. 10.1016/j.cpr.2011.05.002 [DOI] [PubMed] [Google Scholar]
- Pizzagalli DA, Holmes AJ, Dillon DG, Goetz EL, Birk JL, Bogdan R, Dougherty DD, Iosifescu DV, Rauch SL, Fava, ., 2009. Reduced caudate and nucleus accumbens response to rewards in unmedicated subjects with major depressive disorder. Am. J. Psychiatry 166, 702–710. 10.1176/appi.ajp.2008.08081201.Reduced [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzagalli DA, Iosifescu D, Hallett LA, Ratner KG, Fava M, 2008. Reduced hedonic capacity in major depressive disorder: Evidence from a probabilistic reward task. J. Psychiatr. Res 43, 76–87. 10.1016/j.jpsychires.2008.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong J-S, Knapp DJ, Crews FT, 2007. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55, 453–462. 10.1002/glia.20467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raison CL, 2017. The promise and limitations of anti-inflammatory agents for the treatment of Major Depressive Disorder. Curr. Top. Behav. Neurosci 31, 287–302. 10.1007/7854_2016_26 [DOI] [PubMed] [Google Scholar]
- Raison CL, Capuron L, Miller AH, 2006. Cytokines sing the blues: Inflammation and the pathogenesis of depression. Trends Immunol 27, 24–31. 10.1016/j.it.2005.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raison CL, Demetrashvili M, Capuron L, Miller AH, 2005. Neuropsychiatric adverse effects of interferon-alpha. CNS Drugs 19,105–123. 10.2165/00023210-200519020-00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raison CL, Miller AH, 2011. Is depression an inflammatory disorder? Curr. Psychiatry Rep 13, 467–475. 10.1007/s11920-011-0232-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raison CL, Rutherford RE, Woolwine BJ, Shuo C, Schettler P, Drake DF, Haroon E, Miller AH, 2013. A randomized controlled trial of the tumor necrosis factor-alpha antagonist infliximab in treatment resistant depression: Role of baseline inflammatory biomarkers. JAMA Psychiatry 70, 31–41. 10.1001/2013.jamapsychiatry.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raison CL, Rye DB, Woolwine BJ, Vogt GJ, Bautista BM, Spivey JR, Miller AH,, 2010Chronic interferon-alpha administration disrupts sleep continuity and depth in patients with hepatitis C: Association with fatigue, motor slowing, and increased evening cortisol. Biol. Psychiatry 68, 942–949. 10.1016/j.biopsych.2010.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichenberg A, Yirmiya R, Schuld A, Kraus T, Haack M, Morag A, Pollmächer T, 2001. Cytokine-associated emotional and cognitive disturbances in humans. Arch. Gen. Psychiatry 58, 445–452. 10.1001/archpsyc.58.5.445 [DOI] [PubMed] [Google Scholar]
- Reinert KRS, Umphlet D, Quattlebaum A, Boger HA, 2014. Short-term effects of an endotoxin on substantia nigra dopamine neurons. Brain Res 1557, 164–170. 10.1016/j.brainres.2014.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rethorst CD, Bernstein I, Trivedi MH, 2014. Inflammation, obesity, and metabolic syndrome in depression: analysis of the 2009–2010 National Health and Nutrition Examination Survey (NHANES). J. Clin. Psychiatry 75, e1428–32. 10.4088/JCP.14m09009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemann D, Berger M, Voderholzer U, 2001. Sleep and depression--resultsfrom psychobiological studies: An overview. Biol. Psychol 57, 67–103. 10.1016/S0301-0511(01)00090-4 [DOI] [PubMed] [Google Scholar]
- Robaeys G, De Bie L, Wichers MC, Bruckers L, Nevens F, Michielsen P, Van Ranst M, Buntinx F, 2007. Early prediction of major depression in chronic hepatitis C patients during peg-interferon alpha-2b treatment by assessment of vegetative-depressive symptoms after four weeks. World J. Gastroenterol . 13, 5736–5740. 10.3748/wjg.v13.i43.5736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers D, Lees AJ, Smith E, Trimble M, Stern GM, 1987. Bradyphrenia in Parkinson’s Disease and psychomotor retardation in depressive illness. An experimental study. Brain J. Neurol 110 ( Pt 3, 761–76. [DOI] [PubMed] [Google Scholar]
- Rude SS, Wenzlaff RM, Gibbs B, Vane J, Whitney T, 2002. Negative processing biases predict subsequent depressive symptoms. Cogn. Emot 16, 423–440. 10.1080/02699930143000554 [DOI] [Google Scholar]
- Sacher J, Neumann J, Fünfstück, ., Soliman A, Villringer A, Schroeter ML, 2012. Mapping the depressed brain: A meta-analysis of structural and functional alterations in major depressive disorder. J. Affect. Disord 140, 142–148. 10.1016/j.jad.2011.08.001 [DOI] [PubMed] [Google Scholar]
- Sanislow CA, Pine DS, Quinn KJ, Kozak MJ, Garvey MA, Heinssen RK, Wang PS-E, Cuthbert BN, 2010. Developing constructs for psychopathology research: Research domain criteria. J. Abnorm. Psychol 119, 631–639. 10.1037/a0020909 [DOI] [PubMed] [Google Scholar]
- Schmittmann VD, Cramer AOJ, Waldorp LJ, Epskamp S, Kievit RA, Borsboom D, 2011. Deconstructing the construct: A network perspective on psychological phenomena. New Ideas Psychol 31, 43–53. 10.1016/j.newideapsych.2011.02.007 [DOI] [Google Scholar]
- Schwarz JM, Sholar PW, Bilbo SD, 2012. Sex differences in microglial colonization of the developing rat brain. J. Neurochem 120, 948–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PJ, Ogilvie AD, Goodwin GM, Ebmeier KP, 1997. Clinical and psychometric correlates of dopamine D2 binding in depression. Psychol. Med 27, 1247–56. 10.1017/S0033291797005382 [DOI] [PubMed] [Google Scholar]
- Shea MT, Elkin I, Imber SD, Sotsky SM, Watkins JJ, Collins JF, Pilkonis PA, Beckham E,Glass DR,Dolan RT, Parloff MB, 1992. Course of depressive symptoms over follow-up: Findings from the National Institute of Mental Health Treatment of Depression Collaborative Research Program. Arch. Gen. Psychiatry 49, 782–787. 10.1001/archpsyc.1992.01820100026006 [DOI] [PubMed] [Google Scholar]
- Siegle GJ, Steinhauer SR, Thase M ., Stenger VA, Carter CS, 2002. Can’t shake that feeling: Assessment of sustained event-related fMRI amygdala activity in response to emotional information in depressed individuals. Biol. Psychiatry 51, 693–707. 10.1016/S0006-3223(02)01314-8 [DOI] [PubMed] [Google Scholar]
- Simons CJP, Jacobs N, Derom C, Thiery E, Jolles J, Van Os J, Krabbendam L, 2009. Cognition as predictor of current and follow-up depressive symptoms in the general population. Acta Psychiatr. Scand. 120, 45–52. 10.1111/j.1600-0447.2008.01339.x [DOI] [PubMed] [Google Scholar]
- Slavich GM, Irwin MR, 2014. From stress to inflammation and major depressive disorder: A social signal transduction theory of depression. Psychol. Bull 140, 774–815. 10.1037/a0035302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DM, Strauss ME, Quirk SW, Sajatovic M, 1997. Subjective and expressive emotional responses in depression. J. Affect. Disord 46, 135–141. 10.1016/S0165-0327(97)00097-9 [DOI] [PubMed] [Google Scholar]
- Sloan DM, Strauss ME, Wisner KL, 2001. Diminished response to pleasant stimuli by depressed women. J. Abnorm. Psychol 110, 488–493. 10.1037/0021-843X.110.3.488 [DOI] [PubMed] [Google Scholar]
- Snyder HR, 2014. Major depressive disorder is associated with broad impairments on neuropsychological measures of executive function: meta-analysis and review. Psychol. Bull 139,81–132. 10.1037/a0028727.Major [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens FL, Hurley RA, Taber KH, 2011. Anterior cingulate cortex: Unique role in cognition and emotion. J. Neuropsychiatry Clin. Neurosci 23, 121–125. 10.1176/jnp.23.2.jnp121 [DOI] [PubMed] [Google Scholar]
- Stewart JC, Rand KL, Muldoon MF, Kamarck TW, 2009. A prospective evaluation of the directionality of the depression–inflammation relationship. Brain. Behav. Immun 23, 936–944. 10.1016/j.bbi.2009.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhrmann S, Dohm K, Kugel H, Zwanzger P, Redlich R, Grotegerd D, Rauch AV, Arolt V, Heindel W, Suslow T, Zwitserlood P, Dannlowski U, 2013. Mood-congruent amygdala responses to subliminally presented facial expressions in major depression: Associations with anhedonia. J. Psychiatry Neurosci. JPN 38, 249–58. 10.1503/jpn.120060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swardfager W, Rosenblat JD, Benlamri M, McIntyre RS, 2016. Mapping inflammation onto mood: Inflammatory mediators of anhedonia. Neurosci. Biobehav. Rev 64, 148– 166. 10.1016/j.neubiorev.2016.02.017 [DOI] [PubMed] [Google Scholar]
- Targum SD, Fava M, 2011. Fatigue as a residual symptom of depression. Innov. Clin. Neurosci 8, 40–3. [PMC free article] [PubMed] [Google Scholar]
- Taylor BP, Bruder GE, Stewart JW, McGrath PJ, Halperin J, Ehrlichman H, Quitkin FM, 2006. Psychomotor slowing as a predictor of fluoxetine nonresponse in depressed outpatients. Am. J. Psychiatry 163, 73–78. 10.1176/appi.ajp.163.1.73 [DOI] [PubMed] [Google Scholar]
- Taylor JL, Grossberg SE, 1998. The effects of interferon-alpha on the production and action of other cytokines. Semin. Oncol 25, 23–29. [PubMed] [Google Scholar]
- Treadway MT, Buckholtz JW, Schwartzman N, Lambert WE, Zald DH, 2009. Worth the”EEfRT”?Theeffortexpenditure for rewards task as an objective measure of motivation and anhedonia. PLoS ONE 4, 1–9. 10.1371/journal.pone.0006598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treadway MT, Zald DH, 2011. Reconsidering anhedonia in depression: Lessons from translational neuroscience. Neurosci. Biobehav. Rev 35, 537–555. 10.1016/j.neubiorev.2010.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay LK, Naranjo A, Graham SJ, Herrmann N, Mayberg HS, Hevenor SJ, Busto UE, 2005. Functional neuroanatomical substrates of altered reward processing in major depressive disorder revealed by a dopaminergic probe. Arch. Gen. Psychiatry 62, 1228–36. 10.1001/archpsyc.62.11.1228 [DOI] [PubMed] [Google Scholar]
- Tsai MY, Hanson NQ, Straka RJ, Hoke TR, Ordovas JM, Peacock JM, Arends VL, Arnett DK, 2005. Effect of influenza vaccine on markers of inflammation and lipid profile. J. Lab. Clin. Med 145, 323–327. 10.1016/j.lab.2005.03.009 [DOI] [PubMed] [Google Scholar]
- Tsuno N, Besset A, Ritchie K, 2005. Sleep and depression. J. Clin. Psychiatry 66, 1254–1269. 10.4088/JCP.v66n1008 [DOI] [PubMed] [Google Scholar]
- Tylee A, Gastpar M, Lépine JP, Mendlewicz J, 1999. Identification of depressed patient types in the community and their treatment needs: findings from the DEP ES II (Depression Research in European Society II) survey. DEPRES Steering Committee. Int. Clin. Psychopharmacol 14, 153–65. [PubMed] [Google Scholar]
- Tynan RJ, Naicker S, Hinwood M, Nalivaiko E, Buller KM, Pow DV, Day TA, Walker FR, 2010. Chronic stress alters the density and morphology of microglia in a subset of stress-responsive brain regions. Brain. Behav. Immun 24, 1058 10.1016/j.bbi.2010.02.001 [DOI] [PubMed] [Google Scholar]
- Valkanova V,Ebmeier KP,Allan CL, 2013. CRP, IL-6 and depression: systematic review and meta-analysis of longitudinal studies. J. Affect. Disord 150, 736–44. 10.1016/j.jad.2013.06.004 [DOI] [PubMed] [Google Scholar]
- van den Biggelaar AHJ, Gussekloo J, de Craen AJM, Frölich M, Stek ML, van der Mast RC, Westendorp RGJ, 2007. Inflammation and interleukin-1 signaling network contribute to depressive symptoms but not cognitive decline in old age. Exp. Gerontol 42, 693–701. 10.1016/j.exger.2007.01.011 [DOI] [PubMed] [Google Scholar]
- van den Boogaard M, Ramakers B, van Alfen N, van der Werf SP, Fick WF, Hoedemaekers CW, Verbeek MM, Schoonhoven L, van der Hoeven JG, Pickkers P, 2010. Endotoxemia-induced inflammation and the effect on the human brain. Crit. Care Lond. Engl 14, R81–R81. 10.1186/cc9001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praag HM, Korf J, 1971. Retarded depression and the dopamine metabolism. Psychopharmacologia 19, 199–203. 10.1007/BF00402643 [DOI] [PubMed] [Google Scholar]
- Vgontzas AN, Bixler EO, Lin H-M, Prolo P, Trakada G, Chrousos GP, 2005. IL-6 and its circadian secretion in humans. Neuroimmunomodulation 12, 131–140. 10.1159/000084844 [DOI] [PubMed] [Google Scholar]
- Videbech P, Ravnkilde B, Pedersen TH, Hartvig H, Egander A, lemmensen K, Rasmussen NA, Andersen F, Gjedde A, Rosenberg R, 2002. The Danish PET/depression project: Clinical symptoms and cerebral blood flow. A regions-of-interest analysis. Acta Psychiatr. Scand 106, 35–44. [DOI] [PubMed] [Google Scholar]
- Vittengl JR, Clark LA, Dunn TW, Jarrett B, 2007. Reducing relapse and recurrence in unipolar depression: A comparative meta-analysis of cognitive–behavioral therapy’s effects. J. Consult. Clin. Psychol 75, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker J, Dillon DG, Pizzagalli DA, 2009. The role of the nucleus accumbens and rostral anterior cingulate cortex in anhedonia: Integration of resting EEG, fMRI, and volumetric techniques. NeuroImage 46, 327–337. 10.1016/j.neuroimage.2009.01.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker EA, Katon WJ, Jemelka RP, 1993. Psychiatric disorders and medical care utilization among people in the general population who report fatigue. J. Gen. Intern. Med. 8, 436–40. 10.1007/BF02599621 [DOI] [PubMed] [Google Scholar]
- Webb A, Dillon DG, Pechtel P, Goer FK, Murray L, Huys QJ, Fava M, McGrath PJ, Weissman M, Parsey R, Kurian BT, Adams P, Weyandt S, Trombello JM, Grannemann B, Cooper CM, Deldin P, Tenke C, Trivedi M, Bruder G, Pizzagalli DA, 2016. Neural Correlates of three promising endophenotypes of depression: Evidence from the EMBARC study. Neuropsychopharmacology 41, 454– 463. 10.1038/npp.2015.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein AA, Ph D, Deuster PA, Francis JL, Robert W, Tracy RP, Kop WJ, 2010. Acute mental stress in depression. Russell J. Bertr and Russell Arch 84, 228–234. 10.1016/j.biopsycho.2010.01.016.Neurohormonal [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO | Depression [WWW Document], 2017. . WHO; URL http://www.who.int/mediacentre/factsheets/fs369/en/ (accessed 11.2.17). [Google Scholar]
- Wichers MC, Koek GH, Robaeys G, Praamstra J, Maes M, 2005. Early increase in vegetative symptoms predicts IFN-alpha-induced cognitive-depressive changes. Psychol. Med 35, 433–41. 10.1017/S0033291704003526 [DOI] [PubMed] [Google Scholar]
- Winer ES, Salem T, 2015. Reward devaluation: Dot-probe meta-analytic evidence of avoidance of positive information in depressed persons. Psychol. Bull 142, 18–78. 10.1037/bul0000022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wium-Andersen MK, Ørsted DD, Nielsen SF, Nordestgaard BG, 2013. Elevated C-reactive protein levels, psychological distress, and depression in 73,131 individuals . JAMA Psychiatry 70, 176–176. 10.1001/2013.jamapsychiatry.102 [DOI] [PubMed] [Google Scholar]
- Wright E, Strike PC, Brydon L, Steptoe A, 2005. Acute inflammation and negative mood: Mediation by cytokine activation. Brain. Behav. Immun 19, 345–350. 10.1016/j.bbi.2004.10.003 [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I, 2011. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain. Behav. Immun 25, 181–213. 10.1016/j.bbi.2010.10.015 [DOI] [PubMed] [Google Scholar]
- Young JJ, Bruno D, Pomara N, 2014. A review of the relationship between proinflammatory cytokines and major depressive disorder. J. Affect. Disord 169, 15–20. 10.1016/j.jad.2014.07.032 [DOI] [PubMed] [Google Scholar]
- Zunszain PA, Anacker C, Cattaneo A, Choudhury S, Musaelyan K, Myint AM, Thuret S, Price J, Pariante CM, 2012. Interleukin-1β:new regulator of the kynurenine pathway affecting human hippocampal neurogenesis. Neuropsychopharmacology 37, 939–949. 10.1038/npp.2011.277 [DOI] [PMC free article] [PubMed] [Google Scholar]