

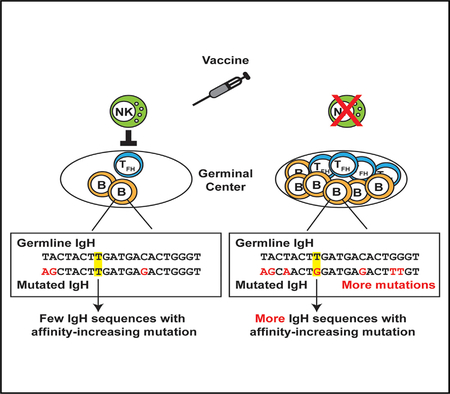
In Brief
Natural killer (NK) cells limit immunization-elicited follicular helper T cell and germinal center B cell responses. Rydyznski et al. link perforin-dependent functions of NK cells to a reduced frequency and quality of somatic hypermutation within antigen-specific B cells. Strategies targeting this NK cell activity may enhance vaccination-induced generation of high-affinity protective antibodies.
SUMMARY
Somatic hypermutation of immunoglobulin sequences in germinal center (GC) reactions must be optimized to elicit high-affinity, protective antibodies after vaccination. We expose natural killer (NK) cells as robust negative regulators of somatic hypermutation in antigen-reactive B cells. NK cells restrict follicular helper T cell (TFH) and GC B cell frequencies and titers of antigen-specific immunoglobulin after administration of alum-adjuvanted hapten-protein conjugate vaccines. This inhibition is perforin dependent, suggesting that NK cells kill one or more cells critical for GC development. In the presence of perforin-competent NK cells, antigen-specific GC B cells acquire fewer mutations, including less frequent generation of non-synonymous substitu-tions and mutations associated with increased antibody affinity. Thus, NK cells limit the magnitude of GC reactions and thereby restrain vaccine elicitation of high-affinity antibodies. Circumventing this activity of NK cells during vaccination has strong potential to enhance humoral immunity and facilitate vaccine-elicited prevention of disease.
Graphical Abstract
INTRODUCTION
Infection and immunization induce formation of germinal centers (GCs), which facilitate follicular helper T cell (TFH) interaction with B cells to promote protective humoral immunity (Mesin et al., 2016). The GC crucially promotes affinity maturation of immuno-globulin responses through iterative rounds of somatic hypermutation (SHM) and Darwinian selection of mutant B cells with higher affinity immunoglobulin sequences. Thus, the GC aids generation of long-lived B cells, producing antibodies of greateraffinity than would be possible in the germline immunoglobulin repertoire.
Multiple mechanisms contribute to regulating the formation and dissolution of GCs. This regulation is vital to optimize the output of long-lived protective B cells while preventing aberrant responses that can lead to autoimmunity. Several different cell types play either supportive or inhibitory roles in determining the development, maintenance, and resolution of GCs. Recently, natural killer (NK) cells were discovered to be an additional inhibitor of TFH and GC B cell responses during virus infection of mice (Cook et al., 2015; Rydyznski et al., 2015).
NK cells are classically valued for their ability to kill virus-infected and transformed cells, but these innate cells can also suppress antiviral T cells to limit disease associated with chronic inflammation (Andrews et al., 2010; Crouse et al., 2015; Welsh and Waggoner, 2013). NK cell immunosuppressive function is contextually linked to secretion of the anti-inflammatory cytokine interleukin-10 (De Maria et al., 2007; Deniz et al., 2008; Lee et al., 2009; Perona-Wright et al., 2009), immune editing of dendritic cells (Ferlazzo et al., 2002; Piccioli et al., 2002; Wilson et al., 1999), and direct lysis of activated T cells (Crouse et al., 2014; Lang et al., 2012; Rabinovich et al., 2003; Waggoner et al., 2011; Xu et al., 2014). In the context of lymphocytic choriomenin-gitis (LCMV) virus infection, NK cells eliminate activated CD4 T cells (Waggoner et al., 2011), resulting in a diminished magnitude of GC responses (Cook et al., 2015; Rydyznski et al., 2015) as well as weak induction of both long-lived LCMV-specific B cells and virus-specific neutralizing antibodies (Rydyznski et al., 2015). Whether NK-cell-mediated decreases in GC magnitude translate to reduced SHM of immunoglobulin in antigen-specific B cells and whether this immunoregulatory function is generalizable to non-viral vaccine regimens remains unclear.
To determine whether NK-cell-regulatory activity inhibits SHM during immunization, we used the well-established mouse model of 4-hydroxy-3-nitrophenylacetyl (NP) conjugated to keyhole limpet hemocyanin (KLH) hapten-carrier conjugate (NP-KLH) immunization (Jack et al., 1977; Mäkelä and Karjalainen, 1977; Reth et al., 1978). Because previous analyses of immunoregulatory NK cells were performed in the context of highly inflammatory live-virus infection (Cook et al., 2015; Rydyznski et al., 2015; Waggoner et al., 2011; Xu et al., 2014), we adopted a regimen of repeat injections of NP-KLH (adapted from Schwickert et al., 2009) to ensure an adequate response by NK cells. The regulatory activity of NK cells was ablated using regimens of mono-clonal antibodies shown to selectively deplete NK cells (Waggoner et al., 2011) or via analysis of perforin-deficient (Prf1−/−) mice in which the granule pathway of NK cell cytolysis is inactivated (Kägi et al., 1994). We previously found that perforin is a crucial mediator of NK cell suppression of T and B cells (Rydyznski et al., 2015; Waggoner et al., 2011). The results of these experiments reveal that NK cells are important regulators of SHM in GC reactions.
RESULTS
NK Cells Restrain GC Responses following Immunization
To test whether NK cells suppress humoral immune responses after adjuvanted protein-conjugate immunization, we intraperitoneally inoculated mice with 100 µg NP-KLH in alum on day 0 and day 7. One day prior to immunization, groups of C57BL/6 mice were selectively and efficiently depleted of NK cells (Rydyznski et al., 2015; Waggoner et al., 2011) via intra-peritoneal (i.p.) administration of 25 µg of anti-NK1.1-depleting antibody (α-NK1.1) or mouse IgG2a isotype (control). The frequency of splenic CD4 NKp46+ NK cells remained significantly reduced for at least the duration of our immunization protocol (control = 1.78% ± 0.094% cells; α-NK1.1 = 0.52% ± 0.012%; n = 4/group; p < 0.0001; Student’s t test; day 8 post-depletion). NP-KLH-immunized, NK-cell-depleted animals harbored an increased frequency of splenic GC TFH (CD4+CD44hi and CXCR5+ PD-1hi) cells (Figures 1A and 1B) compared to immunized control mice. The total number of spleen CD4 cells (control = 1.1 ± 0.68 × 107; α-NK1.1 = 1.1 ± 0.11 × 107; n = 9 or 10/group; p = 0.85; Student’s t test) was similar between immunized α-NK1.1 and control mice. Consistent with enhanced frequencies of TFH after immunization in the absence of NK cells, there was a greater proportion (Figure 1C) and total number (Figure 1D) of B220+ CD19+ GL-7+ Fas+ GC B cell responses in the spleen of NK-cell-depleted mice relative to their NK-cell-sufficient counterparts after immunization. Thus, NK cells constrain GC responses following administration of an alum-adjuvanted non-replicating immunogen.
Figure 1. NK-Cell-Depleted Mice Have Larger TFH and GC B Cell Populations Relative to NK-Cell-Replete Controls.
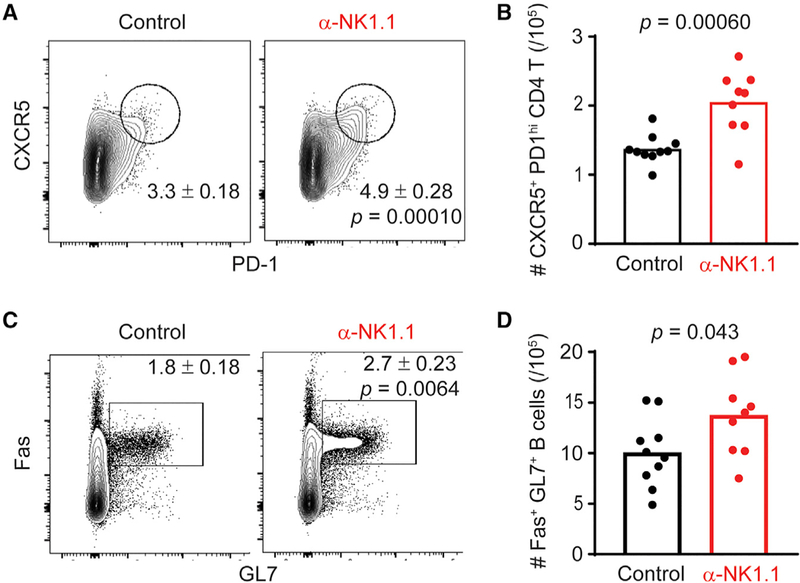
(A) Frequency of GC TFH (CD4+ CD44hi CXCR5+ PD-1hi) cells in control or α-NK1.1 mice twelve days post-immunization with NP-KLH in alum.
(B) Total number of GC TFH day 12 post-immunization in spleen.
(C and D) Frequency of germinal center B (CD19+B220+Fas+GL-7+) cells in the spleens of control or α-NK1.1 mice day 12 post-immunization (C) and total number of GC B cells in spleen day 12 post-immunization (D). Data were analyzed via Student’s t test; n = 9–10 mice/group. Experiments were repeated 3 times.
Reduced NP-Specific B Cell Frequency and Immunoglobulin Titers in Presence of NK Cells
Antigen-specific B cells generated in response to alum-adjuvanted immunogens predominately class switch to IgG1 (Lindblad et al., 1997; McHeyzer-Williams et al., 1993). Staining with fluorescently tagged NP reveals a two-fold increase in the pro-portion (Figure 2A) and total number (Figure 2B) of NP-specific IgG1+ GC B cells in NK-cell-depleted mice relative to control mice after immunization. In contrast, the proportion and total number of NP-specific IgG1neg GC B cells was similar in NK-cell-depleted and control mice (Figures 2A and 2C). Moreover, the proportions of NP-specific GC B cells expressing immuno-globulin M (IgM) (control = 3.3 ± 0.17; α-NK1.1 = 3.8% ± 0.40%; n = 4/group; p = 0.34; Student’s t test) or IgG3 (control = 2.0 ± 0.36; α-NK1.1 = 1.9 ± 0.33; n = 4/group; p = 0.82; Student’s t test) in the spleen at day 12 post-immunization were not enhanced with NK cell depletion. In accordance with increased frequencies of NP-specific IgG1+ GC B cells in the absence of NK cells, NK-cell-depleted animals had higher titers of sera NP-specific IgG1 captured by both high (NP4)- and low (NP20)-affinity conjugates of NP in ELISA (Figures 2D and 2E). Together, these data demonstrate that NK cell suppression predominately impacts IgG1-switched NP-specific B cells in the GC and subsequent elaboration of NP-specific IgG1.
Figure 2. NK-Cell-Depleted Mice Have Higher Levels of NP-Specific GC B Cells and NP-Specific Immunoglobulin.
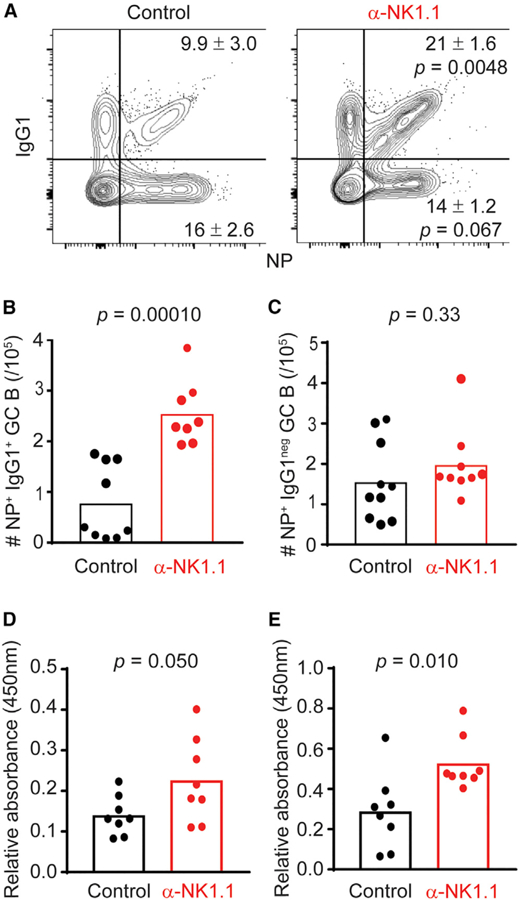
(A) Frequency of NP+IgG1+ GC B cells and NP+ IgG1 GC B cells in the spleens of control or α-NK1.1 mice day twelve post-immunization with NP-KLH in alum.
(B and C) Total number of NP+IgG1+ (B) and NP+IgG1 GC B cells day 12 post-immunization (C).
(D and E) Relative levels of high (NP4; D) and low (NP20; E) NP-specific IgG1 from sera of control or α-NK1.1 mice 12 days post-NP-KLH immunization. Data were analyzed via Student’s t test; n = 8–10 mice/group. Experiments were repeated 3 times.
NK Cell Suppression Limits Quantity and Quality of Immunoglobulin Somatic Hypermutation
We hypothesize that larger GC reactions following immunization in the absence of NK cells (Figures 1 and 2) will support a greater quantity and quality of SHM in GC B cells. The NP-KLH model is highly amenable to analysis of SHM in NP-specific B cells (Smith et al., 1997), in part because the majority of responding GC B cells carry the VH186.2 heavy chain (Cumano and Rajewsky, 1986). Moreover, mutations engendering a tryptophan to leucine switch at position 33 of VH186.2 confer an approximately ten-fold increase in antibody affinity for NP (Xu et al., 2015).
Sequencing of the VH186.2 heavy chain in sorted splenic NP-specific GC B cells 12 days after immunization reveals an increased average number of somatic heavy-chain mutations in B cells from NK-cell-depleted mice relative to control animals (Figure 3A). In addition, NP-KLH immunization in the absence of NK cells resulted in an enhanced proportion of sequences bearing the affinity-conferring W33L mutation compared to non-depleted control mice (Figures 3B and 3C). NP-specific GC B cells from NK-cell-depleted animals also exhibited more replacement mutations (p = 1.38 × 10–4) across the entire length of VH186.2 BCR sequences compared to controls when compared on a codon-by-codon basis (Figure 3C). Overall, these data suggest that NK cell suppression of GC responses constrains the frequency and quality of somatic mutations.
Figure 3. NK Cell Suppression of GC Responses Has Consequences for Somatic Hypermutation.

(A) Total number of mutations in unique VH186.2 BCR sequences isolated from spleens of control or α-NK1.1 mice day 12 post-immunization with NP-KLH in alum. Data were analyzed via Mann-Whit-ney with median displayed; n = 5–6 mice/group.
(B) The proportion of unique VH186.2 sequences from (A) containing the W33L mutation in control and α-NK1.1 mice.
(C) The frequency of either replacement or silent mutations at all positions within the VH186.2 sequence between control or α-NK1.1 mice. Experiments were repeated 2 or 3 times.
Diminished GCs in Presence of NK Cells Is Driven by a Perforin-Dependent Mechanism
Perforin-containing cytolytic granules represent a major mechanism by which NK cells trigger target cell death (Kägi et al., 1994) that is implicated in NK cell killing of activated CD4 T cells (Waggoner et al., 2011). Thus, we use perforin-deficient (Prf1−/−) mice to evaluate the role of perforin in NK cell suppression of GC-associated SHM. Similar to NK cell depletion in C57BL/6 mice (Figure 1), NP-KLH in alum immunization of non-depleted Prf1−/−mice results in increased frequencies of splenic GC TFH (Figure 4A) and NP-specific IgG1+ GC B cells (Figure 4B) relative to C57BL/6 controls. Additionally, there was no enhancement of IgM+ (wild-type [WT] control = 12.0 ± 1.47 × 104; WT α-NK1.1 = 9.95 ± 1.34 × 104; Prf1−/−control = 9.73 ± 1.30 × 104; Prf1−/− α-NK1.1 = 13.6 ± 1.42 × 104; n = 4/group; p = 0.67; one-way ANOVA) or IgG3+ (WT control = 9.35 ± 2.50 × 104; WT α-NK1.1 = 7.90 ± 0.910 × 104; Prf1−/−control = 8.25 ± 1.51 × 104; Prf1−/− α-NK1.1 = 11.2 ± 0.810 × 104; n = 4/group; p = 0.65; one-way ANOVA) NP-specific GC B cells in Prf1−/−or anti-NK1.1-treated WT mice relative to non-depleted C57BL/6 WT controls. The elevated magnitudes of TFH (control = 11.6 ± 0.97; α-NK1.1 = 10.6 ± 0.54; n = 3 or 4/group; p = 0.38; Student’s t test) and NP-specific IgG1+GC B cells (control = 29.5 ± 1.9; α-NK1.1 = 29.2 ± 1.5; n = 3 or 4/group; p = 0.91; Student’s t test) in Prf1−/−mice were not further enhanced by NK cell depletion.
Figure 4. Perforin-Deficient Animals Have Enhanced GC Expansion and Somatic Mutation Rates Comparable to NK-Cell-Depleted Animals.
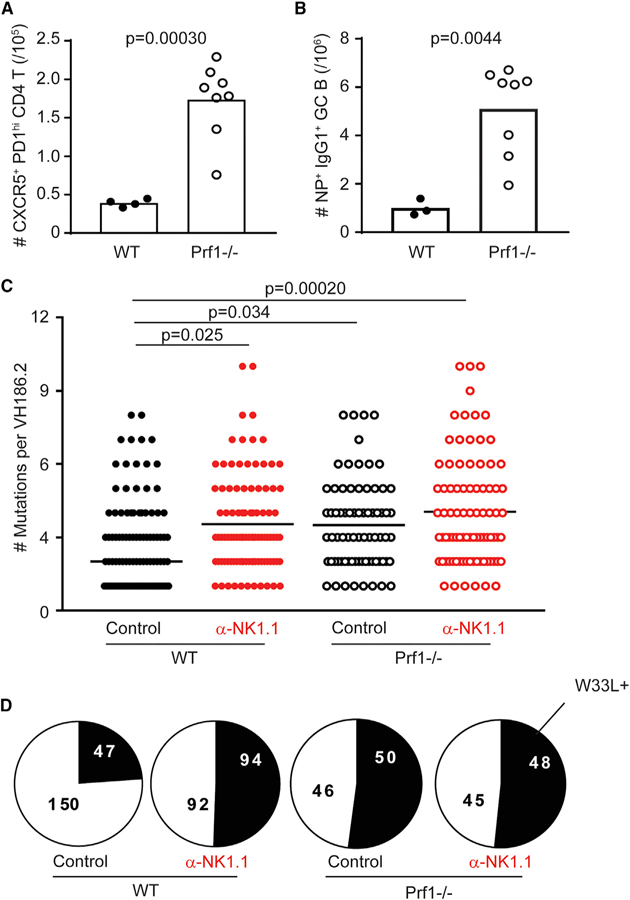
(A) Total number of GC TFH (CD4+CD44hiCXCR5+ PD-1hi) cells in the spleen of C57BL/6 (WT) or perforin-deficient (Prf1−/−) control-treated (mIgG2a) animals day 12 post-NP-KLH immunization.
(B) Total number of NP+IgG1+ GC B (CD19+B220+ Fas+GL-7+) cells. Data were analyzed via Stu-dent’s t test; n = 3–8 mice/group.
(C) Total number of mutations in control or α-NK1.1-treated WT or Prf1−/−animals day 12 post-immunization. Data were analyzed via Kruskal-Wallis with multiple testing correction; n = 5–6 mice/group.
(D) Proportion of unique VH186.2 sequences from (C) bearing W33L mutation. Experiments were repeated 2 or 3 times.
Importantly, analysis of VH186.2 heavy-chain sequences in sorted splenic NP-specific GC B cells reveals a similar (p = 0.11) average number of VH186.2 mutations among mice lacking NK cells, perforin, or both, where all three groups of mice demonstrate enhanced mutation frequencies (p < 0.01) compared to control wild-type (C57BL/6) mice (Figure 4C). VH186.2 sequences in non-depleted Prf1−/−mice and NK-cell-depleted mice, regardless of strain, harbor mutations conferring the W33L change roughly two-fold more often than sequences from non-depleted control mice (Figure 4D). Together, these data suggest that NK-cell-derived perforin is important for suppression of the magnitude and quality of GC responses after immunization.
DISCUSSION
A major goal of vaccine development, particularly in the HIV field, is identification of mechanisms that limit affinity maturation of antibodies in GCs after immunization. We demonstrate that NK cells impose stringent limitations on GC B cell responses in the context of immunization with either live virus (Rydyznski et al., 2015) or alum-adjuvanted protein-conjugate immunogens. This NK-cell-mediated inhibition limits the efficiency of SHM by reducing the rate of mutation and by decreasing the frequency of replacement mutations, including those conferring large improvements in immunoglobulin affinity. Mechanistically, perforin-dependent functions of NK cells are critical in suppression of the GC. Our discovery of a crucial contribution of NK cells that limits vaccine-elicited SHM in antigen-specific GC B cells has major implications for vaccine design.
One goal of our study was to determine whether NK cells could suppress B cell responses against a non-replicating antigen coupled with a vaccine-relevant adjuvant (alum). In the context of LCMV infection, potent and sustained induction of type I interferon (IFN) and interleukin-15 (IL-15) activates NK cells (Nguyen et al., 2002) and likely provokes NK cell suppression of GC reactions (Cook et al., 2015; Rydyznski et al., 2015). In contrast, aluminum compounds (alum) used as adjuvants in human vaccines (Oleszycka and Lavelle, 2014) trigger markedly less inflammation than virus infection. Nevertheless, alum stimulates inflammasome-dependent release of IL-1 and IL-18 (Eisenbarth et al., 2008; Pollock et al., 2003), where the latter is a known stim-ulus for NK cell activity. This likely explained the capacity of alum to induce NK-cell-suppressive functions after NP-KLH immunization. Overall, our results demonstrate that NK cells are sufficiently activated and GCs effectively receptive to NK cell suppression in the context of NP-KLH/alum immunization to provoke suboptimal affinity maturation of immunoglobulin in GCs. Suppression was most evident among IgG1-switched GC B cells that dominate the alum-adjuvanted NP-specific response. The focused effect of NK cells on IgG1 may simply reflect the IgG1-dominant nature of our schema or may be a consequence of the greater reliance of IgG1 responses on CD4 T cell help than has been noted for IgG3 responses (Mongini et al., 1981; Snapper et al., 1992). Of note, our sequencing experiments excluded analysis of non-IgG1 B cells, thereby precluding determination of the effect of NK cells on SHM in B cells using different isotypes. Consistent with prior studies (Deauvieau et al., 2016), NK cells did not appreciably alter overall isotype usage in the NP-specific B cell response, suggesting that specific impacts of NK cells on B cells using discrete isotypes likely depends on adjuvant-stimulated class switching and the requirement for T cell help. Thus, NK cell suppression represents an innovative target for interventions to enhance SHM after immunization.
Importantly, depletion of NK cells or genetic ablation of perforin has similar, non-additive effects on GC responses. Therefore, GC suppression by NK cells is likely a product of NK cell killing of cells critical for GC formation, including TFH cells. In fact, the earliest effect we observe in the absence of perforin or NK cells is an increased magnitude of activated CD4 T cells as well as a bias for TFH versus TH1 differentiation of these cells (Rydyznski et al., 2015). Therefore, NK cells may shape the pool of TFH cells available to support B cell expansion and affinity maturation in GCs. We did not observe any changes in the frequency of follicular regulatory CD4 T cells, suggesting that alterations in regulatory T cell development are not involved in GC suppression by NK cells. Nevertheless, we cannot rule out perforin-dependent lysis of antigen-presenting cells or even B cells themselves as phenomenon occurring in parallel to lysis of CD4 T cells, potentially creating a complex regulatory environment enforced by NK cells. The observation of perforin-dependent, NK-cell-mediated sup-pression of TFH and GC B cell responses provides important mechanistic insights into how NK cells suppress humoral immune responses during immunization.
A deeper understanding of the impact of NK cell suppression of GC magnitude on the quality of resulting antigen-specific B cell re-sponses was a major goal of this study. We adopted a two-dose immunization scheme in an effort to maintain a robust GC response from which we could measure and compare SHM rates. Importantly, we observed an increased number of mutations in the heavy-chain gene of NP-specific GC B cells following NP-KLH immunization of NK-cell-deficient mice. NK cells not only reduced mutation rates, but their suppressive activity was associated with less efficient induction of affinity-conferring W33L mutations and a reduced ratio of replacement to silent mutations across all nucleotide positions within the complementarity determining regions of immunoglobulin heavy chains. Concurrent ELISA analysis revealed higher sera titers of NP-specific immuno-globulin in the absence of NK cells. However, measurements of both high- and low-affinity antibodies were elevated, with no significant skewing of the affinity ratio at the two-week time point. This result likely reflects a need for the more mutated GC B cells in NK-cell-depleted mice to egress from the GC before contributing to the peripheral pool of immunoglobulins.
Our results reveal that NK cells quantitatively and qualitatively restrain GC B cell responses following immunization. Of note, NP-KLH presents a relatively simple antigen necessitating a small number of specific mutations in a relatively large pool of naive NP-specific B cells to confer large increases in immunoglobulin affinity for antigen. By contrast, GC responses against HIV envelope originate from very small numbers of precursor cells and require both years of sustained inflammation and exposure to antigen to elicit very large numbers of mutations to enable development of broadly neutralizing antibodies. Despite these distinctions, our results highlight the potential for interventions that limit NK cell suppression of GC reactions as a strategy to potentiate vaccine-induced, GC-mediated generation of heavily mutated high-affinity antibodies capable of broad neutralization of HIV infection.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-NK1.1 (clone pk136) | Bio-X-Cell | BE0036; RRID: AB_1107737 |
| Mouse anti-IgG2a isotype control (clone C1.18.4) | Bio-X-Cell | BE0085; RRID: AB_1107771 |
| Mouse anti-CD4 AF700 | Biolegend | 100430; RRID: AB_493699 |
| Mouse anti-CD44 BV510 | Biolegend | 103044; RRID: AB_2650923 |
| Mouse anti-PD-1 ef450 | eBiosciences | 48-9981-82; RRID: AB_2574139 |
| Mouse anti-CXCR5 PE-Cy7 | Biolegend | 145516; RRID: AB_2562209 |
| Mouse anti-CD49d FITC | Biolegend | 103606; RRID: AB_313037 |
| Mouse anti-IgM FITC | Biolegend | 406506; RRID: AB_315056 |
| Mouse anti-IgD FITC | Biolegend | 405704; RRID: AB_315025 |
| Mouse anti-CD95 PE-Cy7 | BD Biosciences | 557653; RRID: AB_396768 |
| Mouse/human anti-B220 AF700 | Biolegend | 103232; RRID: AB_493717 |
| Mouse anti-GL-7 ef450 | eBiosiences | 48-5902-82; RRID: AB_10870775 |
| Mouse anti-IgG1 APC | Biolegend | 406610; RRID:AB_10696420 |
| Mouse anti-IgG3 BV711 | BD Biosciences | 565809 |
| Mouse anti-CD19 BV510 | Biolegend | 115546; RRID: AB_2562136 |
| Bacterial and Virus Strains | ||
| XL-10 Gold Ultracompetent e.coli | Agilent | 200314 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NP-KLH | Biosearch Technologies | N-5060-25 |
| TRIzol | ThermoFisher Scientific | 15596026 |
| NP-PE | Biosearch Technologies | N-5070-1 |
| NP-4-BSA | Biosearch Technologies | N-5050L-10 |
| NP-20-BSA | Biosearch Technologies | N-5050H-100 |
| LIVE/DEAD Fixable Aqua dead cell stain kit | Life Technologies | L34957 |
| GoTaq Flexi DNA polymerase | Promega | M8291 |
| Critical Commercial Assays | ||
| Superscript IV reverse transcription kit | Invitrogen | 18090010 |
| GeneJet gel extraction and cleanup kit | QIAGEN | K0691 |
| pGEM-T vector system | Promega | A3600 |
| Qiaprep spin miniprep kit | QIAGEN | 27104 |
| Deposited Data | ||
| Vh186.2 DNA sequences | This paper | https://tf.cchmc.org/external/rydlb5/Rydyznski_SHM_NK_data.zip |
| Experimental Models: Organisms/Strains | ||
| C57BL/6 mice | Jackson Labs | JAX:000664 |
| Oligonucleotides | ||
| CTCTTCTTGGCAGCAACAGC-Forward Primer 1 | This paper | N/A |
| GCTGCTCAGAGTGTAGAGGTC-Reverse Primer 1 | This paper | N/A |
| GTGTCCACTCCCAGGTCCAAC-Forward Primer 2 | This paper | N/A |
| GTTCCAGGTCACTGTCACTG-Reverse Primer 2 | This paper | N/A |
| Software and Algorithms | ||
| pRESTO | Vander Heiden et al., 2014 | https://prestodb.io/ |
| Change-O | Gupta et al., 2015 | https://changeo.readthedocs.io/en/version-0.4.1—airr-standards/ |
| R | R Development Core Team, 2014 | https://www.r-project.org/ |
| NCBI IgBlast package | Ye et al., 2013 | https://www.ncbi.nlm.nih.gov/igbalst/ |
| IMGT/GENE-DB | Giudicelli et al., 2005 | https://www.imgt.org/genedb/ |
| Other | ||
| Xgal-IPTG-Ampicillin agar plates | KD Medical | BPL-2310 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephen Waggoner (stephen.waggoner@cchmc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6 and Prf1−/−mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Male mice 6–12 weeks of age (at onset of immunizations) were routinely utilized in experiments. Mice were housed under pathogen-free conditions, and experiments were per-formed using the ethical guidelines approved by the Institutional Animal Use and Care Committees of Cincinnati Children’s Hospital Medical Center. In many experiments, randomization was achieved by randomly assigning mice within a cage to different experi-mental groups or by modulating the processing order of the mice at the time of harvest.
METHOD DETAILS
In vivo NK-cell depletion
One day before infection, selective depletion of NK cells was attained though a single i.p. injection of 25 µg per mouse anti-NK1.1 monoclonal antibody (PK136) or 25 µg per mouse of a control mouse IgG2a (C1.18.4) produced by Bio-X-Cell (West Lebanon, NH).
Immunizations
4-hydroxy-3-nitrophenylacetyl conjugated to keyhole limpet hemocyanin (NP-KLH) was purchased from Biosearch Technologies (Petaluma, CA). Prior to immunization, NP-KLH was adsorbed to alum (Imject alum from Thermo Fishe) at a 1:1 volumetric ratio (100 µL of a 1 mg/ml NP-KLH stock with 100 µL alum) on a rotating mixer for one hour at room temperature. Mice were administered 200 µL of NP-KLH/alum intraperitoneally.
Enzyme-linked immunosorbent assays (ELISAs)
NP-specific IgG1, ELISA plates were coated overnight with 5 µg/ml NP(4) or NP(20) conjugated to bovine serum albumin (BSA) in PBS. The next day, plates were washed and blocked with 2x PBS coating buffer at room temp for 2 hours. Following blocking, plates were washed and serum samples were serially diluted and plated in duplicate, and incubated for 2 hours. Plates were then washed 4x with PBS and biotinylated IgG or IgG1-HRP was added to the plates and incubated for 1 hour. Streptavidin-HRP was added to biotin-IgG plates and incubated for 30 minutes. All plates were washed and developed with the addition of TMB substrate for 15 minutes before addition of stop solution (2N H2SO4), and read in a plate reader at 450 nm. Values were reported as relative absorbance.
Flow cytometry
Single-cell leukocyte suspensions were prepared from spleens and LNs by mechanical homogenization of tissues between glass slides and filtered through nylon mesh. Red blood cells were then lysed via the addition of ACK lysis buffer (Thermo Fisher) for 5 minutes at 37 C. Cells were then washed, pelleted and resuspended in flow cytometry buffer (HBSS + 5% fetal bovine serum). Single-cell suspensions were plated at 2 × 106 cells per well in 96-well plates and then incubated with a 1:200 dilution of anti-CD16/32 (clone 2.4G2 Tonbo Biosciences) in flow cytometry buffer. Subsequently, cells were stained for 20 minutes, or 90 minutes for NP-PE staining, at 4°C with various combinations of fluorescently-tagged antibodies. The following antibodies/reagents were used for flow cytometry: anti-CD4 AF700 (clone GK1.5 Biolegend), anti-CD44 BV510 (clone IM-7 Biolegend), anti-PD-1 ef450 (clone J43 eBiosciences), anti-CXCR5 PE-Cy7 (clone L138D7 Biolegend), anti-NK1.1 PE (clone PK136 Biolegend), anti-NKp46 APC (clone 29A1.4 Biolegend), anti-CD49d FITC (clone R1–2 Biolegend), anti-IgM FITC (clone RMM-1 Biolegend), anti-IgD FITC (clone 11–26c.2a Biolegend), NP-PE (Biosearch Technologies), anti-CD95 PE-Cy7 (clone Jo.2 BD Biosciences), anti-B220 AF700 (clone RA3–6B2 Biolegend), anti-CD19 BV510 (clone 6D5 Biolegend), anti-GL-7 ef450 (clone GL7 eBiosciences), anti-IgG1 APC (clone RMG1–1 Biolegend), anti-IgG3 BV711 (clone R40–82 BD Biosciences). Following staining, cells were washed and fixed with BD fixation buffer (BD Biosciences) for 3 minutes at RT. Cells were washed twice and resuspended in 200µl flow cytometry buffer and analyzed on a BD Fortessa or LSRII flow cytometer.
Cell sorting
Single cell suspensions were prepared from spleens by mechanical homogenization between glass slides, filtered through nylon mesh, and RBC lysed with ACK lysis buffer (Thermo Fisher) for 5 minutes at 37°C. Cells were then washed and resuspended in cell sorting buffer (HBSS + 5% FBS + 5 mM EDTA). Cells were stained for 20–45 minutes at 4°C. The following antibodies/reagents were used for cell sorting experiments: anti-CD19 FITC (clone 6D5 Biolegend), anti-B220 AF700 (clone RA3–6B2 Biolegend), anti-CD95 PE-Cy7 (clone Jo.2 BD Biosciences), anti-GL7 ef450 (clone GL7 eBiosciences), NP-PE (Biosearch Technologies), anti-IgG1 APC (clone RMG1–1 Biolegend), LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Life Technologies). Cells were washed twice and resuspended in 500µl cell sorting buffer. Bulk GC B cells (300,000 – 500,000 cells) from pooled (n = 4/group) spleens of NK cell depleted or control treated animals or from spleens of individual mice (n = 4–8/group) receiving either NK-depleting antibody or isotype control were sorted on a BD FacsAria cell sorter into 1.5 mL Eppendorf tubes containing 200 µL of cell sorting buffer.
VH186.2 BCR sequencing
Following sorting, cells were pelleted and bulk RNA was isolated via Trizol/chloroform extraction. Extracted RNA was resuspended in 20 µL nuclease free water. Total cDNA was transcribed using the SuperScript IV reverse transcription kit. cDNA was then subjected to two rounds of nested PCR to amplify the Vh186.2 NP-specific BCR heavy chain (Forward primer 1: CTCTTCTTGGCAG CAACAGC, Reverse primer 1: GCTGCTCAGAGTGTAGAGGTC, Forward primer 2: GTGTCCACTCCCAGGTCCAAC, Reverse primer GTTCCAGGTCACTGTCACTG). For the first PCR reaction, 2µl of cDNA was used for an amplification reaction in a total volume of 20µl containing 13.7µl nuclease-free H2O, 2µl 5x PCR buffer, 0.4µl each of 25mM MgCl2 and dNTPs, 0.5µl each of the first forward and first reverse primers, 0.5µl Taq polymerase and amplified with the following program: 94°C for 10 min, then 40 cycles of 94°C for 30 s, 56°C for 30 s, 72°C for 1 min, then 72°C for 10 min. For the second PCR reaction, 4µl of product from the first reaction was used for an amplification reaction in a total volume of 40ml containing 27.4µl nuclease-free H2O, 4µl 5x PCR buffer, 0.8µl each of 25mM MgCl2 and dNTPs, 1µl each of the first forward and first reverse primers, 1µl Taq polymerase and amplified with the following pro-gram: 94 C for 10 min, then 40 cycles of 94°C for 30 s, 56°C for 30 s, 72° C for 1 min, then 72° C for 10 min. Amplified sequences were then purified on a 1% agarose gel (product is 500 base pairs), and gel extracted using the GeneJet gel extraction (ThermoFisher Scientific) and cleanup kit. Purified products were then ligated into pGEM-T vectors (Promega) overnight at 4°C, transformed into XL-10 Gold ultracompetent E. coli (Agilent), plated on Xgal-IPTG-Amp agar plates (KD Medical) in triplicate, and incubated overnight at 37°C. The next day, white colonies were selected on plates and sent for direct colony sequencing at Genewiz, or white colonies were selected in the lab and grown overnight in 3ml LB with shaking at 37°C. When colonies were grown ON in house, the next morning, bacteria were pelleted and DNA extracted with the QIAprep spin miniprep kit (QIAGEN). Purified DNA was sent to the CCHMC sequencing core for Sanger sequencing.
VH186.2 sequence alignment
Sequences were aligned to the Vh186.2 germline sequence using the NCBI IgBlast browser. Mutation data was only recorded if the entire (98aa) sequence was intact. We recorded the total number of mutations along the sequence as well as the type of mutation as either silent or amino acid changing. We also recorded whether the mutation occurred in framework regions or complementarity determining regions (CDRs) and whether the tryptophan to leucine (W33L) mutation was present. Due to an inability to determine whether multiple identical sequences came from truly separate cells representing a clonally dominant population or from potential biases introduced during the post-sorting processing steps, only unique sequences were plotted and used for statistical analysis.
Mutation pattern analysis
Raw sequencing data was processed using pRESTO (v0.3.5) (Vander Heiden et al., 2014), during which duplicate sequences were removed using ‘‘collapseSeq.py.’’ VDJ assignment was performed using ‘‘igblastn’’ from the standalone NCBI IgBlast package (v1.6.1) (Ye et al., 2013) against germline references from IMGT/GENE-DB (v.3.1.14) (Giudicelli et al., 2005). Default alignment parameters were used. For V, the mouse germline IGHV1–72*01 allele (UniProt: AC163348), which is identical to NCBI V186.2, was used. For D and J, the full IMGT mouse D- and J-references were used. Final processing was performed in Change-O (v0.3.5) (Gupta et al., 2015), during which non-productively rearranged sequences and sequences with V regions shorter than 294nt (98aa) were filtered. Mutation pattern analysis was performed for the V region using customized scripts in R (v3.3.3) (R Development Core Team, 2014). The IMGT unique numbering scheme (Lefranc et al., 2003) was used. Under this scheme, the V region of IGHV1-72*01 is numbered as follows: FWR1 = aa 1..26 (aa 10 unused); CDR1 = aa 27..38 (aa 31..34 unused); FWR2 = aa 39..55; CDR2 = aa 56..65 (aa 60–61 un-used); FWR3 = aa 66..104 (aa 73 unused); and CDR3 = aa 105..106 (Giudicelli et al., 2005). The codon known for Trp to Leu (W33L) mutation in NP mice is numbered aa 38. The amino acids encoded by the observed and germline sequences were compared codon-by-codon to determine the mutation type (silent or replacement). At each codon, the proportions of replacement and silent mutations across sequences were calculated. Two-sample, paired, and one-sided Wilcoxon signed rank tests were performed to compare the proportions across codons of silent mutations in Control and in ΔNK, and of replacement mutations in Control and in α-NK1.1 mice.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical details are reported in the figure legends. For Figures 1 and 2, data were analyzed via two-tailed unpaired Student’s t test with biological replicates, n = 8–10 mice/group. Due to non-normal distributions in the data depicted in Figures 3 and 4, we performed non-parametric analyses. In Figure 3, differences in the total number of somatic mutations were analyzed via Mann-Whitney with median displayed with n = 67–97 unique VH186.2 sequences isolated from a total of 5–6 mice/group. In Figure 4, to compare non-normal data from more than two groups we used a Kruskal-Wallis test and Dunn’s multiple testing correction with n = 74–99 unique VH186.2 sequences isolated from a total of 5–6 mice/group. In Figure 4, differences in TFH and GC B cell numbers between WT and Prf−/−mice were analyzed by two-tailed unpaired Student’s t test, n = 3–8 mice/group. Significance was accepted when p < 0.05.
Highlights.
NK cells limit germinal center responses following protein immunization
NK cells repress antigen-specific B cell and immunoglobulin production
Quantity and quality of immunoglobulin somatic mutations are inhibited by NK cells
NK cell suppression of affinity maturation is perforin dependent
ACKNOWLEDGMENTS
Cell sorting and flow cytometric data were acquired using equipment maintained by CCHMC Research Flow Cytometry Core with support from NIH grants AR047363, AR070549, DK078392, and DK090971. Work was supported by NIH grants AI104739 (S.H.K.), AI118179 (C.E.R.), and DA038017 (S.N.W.); New Scholar Award from The Lawrence Ellison Foundation (S.N.W.); start-up funds from the Cincinnati Children’s Research Foundation (S.N.W.); Albert J. Ryan Fellowship Program (C.E.R.); and Gruber Science Fellowship (J.Q.Z.). We thank Drs. S.S. Way and F.D. Finkelman for discussion of manuscript and Dr. L. Kottyan and Kevin Ernst for help with data management.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
The raw Vh186.2 DNA sequences can be accessed at https://tf.cchmc.org/external/rydlb5/Rydyznski_SHM_NK_data.zip.
REFERENCES
- Andrews DM, Estcourt MJ, Andoniou CE, Wikstrom ME, Khong A, Voigt V, Fleming P, Tabarias H, Hill GR, van der Most RG, et al. (2010). Innate immunity defines the capacity of antiviral T cells to limit persistent infection. J. Exp. Med 207, 1333–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook KD, Kline HC, and Whitmire JK (2015). NK cells inhibit humoral immunity by reducing the abundance of CD4+ T follicular helper cells during a chronic virus infection. J. Leukoc. Biol 98, 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouse J, Bedenikovic G, Wiesel M, Ibberson M, Xenarios I, Von Laer D, Kalinke U, Vivier E, Jonjic S, and Oxenius A (2014). Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity 40, 961–973. [DOI] [PubMed] [Google Scholar]
- Crouse J, Xu HC, Lang PA, and Oxenius A (2015). NK cells regulating T cell responses: mechanisms and outcome. Trends Immunol 36, 49–58. [DOI] [PubMed] [Google Scholar]
- Cumano A, and Rajewsky K (1986). Clonal recruitment and somatic mutation in the generation of immunological memory to the hapten NP. EMBO J 5, 2459–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Maria A, Fogli M, Mazza S, Basso M, Picciotto A, Costa P, Congia S, Mingari MC, and Moretta L (2007). Increased natural cytotoxicity receptor expression and relevant IL-10 production in NK cells from chronically infected viremic HCV patients. Eur. J. Immunol 37, 445–455. [DOI] [PubMed] [Google Scholar]
- Deauvieau F, Fenis A, Dalençon F, Burdin N, Vivier E, and Kerdiles Y. (2016). Lessons from NK cell deficiencies in the mouse. Curr. Top. Microbiol. Immunol 395, 173–190. [DOI] [PubMed] [Google Scholar]
- Deniz G, Erten G, Kücüuksezer UC, Kocacik D, Karagiannidis C, Aktas E, Akdis CA, and Akdis M. (2008). Regulatory NK cells suppress antigen-specific T cell responses. J. Immunol 180, 850–857. [DOI] [PubMed] [Google Scholar]
- Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, and Flavell RA (2008). Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 453, 1122–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlazzo G, Tsang ML, Moretta L, Melioli G, Steinman RM, and Münz C (2002). Human dendritic cells activate resting natural killer (NK) cells and are recognized via the NKp30 receptor by activated NK cells. J. Exp. Med 195, 343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudicelli V, Chaume D, and Lefranc MP (2005). IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell re-ceptor genes. Nucleic Acids Res 33, D256–D261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, and Kleinstein SH (2015). Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31, 3356–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack RS, Imanishi-Kari T, and Rajewsky K (1977). Idiotypic analysis of the response of C57BL/6 mice to the (4-hydroxy-3-nitrophenyl)acetyl group. Eur. J. Immunol 7, 559–565. [DOI] [PubMed] [Google Scholar]
- Kägi D, Ledermann B, Bürki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, and Hengartner H (1994). Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 369, 31–37. [DOI] [PubMed] [Google Scholar]
- Lang PA, Lang KS, Xu HC, Grusdat M, Parish IA, Recher M, Elford AR, Dhanji S, Shaabani N, Tran CW, et al. (2012). Natural killer cell activation enhances immune pathology and promotes chronic infection by limiting CD8+ T-cell immunity. Proc. Natl. Acad. Sci. USA 109, 1210–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Kim KS, Fodil-Cornu N, Vidal SM, and Biron CA (2009). Activating receptors promote NK cell expansion for maintenance, IL-10 production, and CD8 T cell regulation during viral infection. J. Exp. Med 206, 2235–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc MP, Pommié C, Ruiz M, Giudicelli V, Foulquier E, Truong L, Thouvenin-Contet V, and Lefranc G (2003). IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev. Comp. Immunol 27, 55–77. [DOI] [PubMed] [Google Scholar]
- Lindblad EB, Elhay MJ, Silva R, Appelberg R, and Andersen P (1997). Adjuvant modulation of immune responses to tuberculosis subunit vaccines. Infect. Immun 65, 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkelä O, and Karjalainen K (1977). Inherited immunoglobulin idiotypes of the mouse. Immunol. Rev 34, 119–138. [DOI] [PubMed] [Google Scholar]
- McHeyzer-Williams MG, McLean MJ, Lalor PA, and Nossal GJ (1993). Antigen-driven B cell differentiation in vivo. J. Exp. Med 178, 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesin L, Ersching J, and Victora GD (2016). Germinal center B cell dynamics. Immunity 45, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongini PK, Stein KE, and Paul WE (1981). T cell regulation of IgG sub-class antibody production in response to T-independent antigens. J. Exp. Med 153, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen KB, Salazar-Mather TP, Dalod MY, Van Deusen JB, Wei XQ, Liew FY, Caligiuri MA, Durbin JE, and Biron CA (2002). Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J. Immunol 169, 4279–4287. [DOI] [PubMed] [Google Scholar]
- Oleszycka E, and Lavelle EC (2014). Immunomodulatory properties of the vaccine adjuvant alum. Curr. Opin. Immunol 28, 1–5. [DOI] [PubMed] [Google Scholar]
- Perona-Wright G, Mohrs K, Szaba FM, Kummer LW, Madan R, Karp CL, Johnson LL, Smiley ST, and Mohrs M (2009). Systemic but not local infections elicit immunosuppressive IL-10 production by natural killer cells. Cell Host Microbe 6, 503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccioli D, Sbrana S, Melandri E, and Valiante NM (2002). Contact-dependent stimulation and inhibition of dendritic cells by natural killer cells. J. Exp. Med 195, 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock KG, Conacher M, Wei XQ, Alexander J, and Brewer JM (2003). Interleukin-18 plays a role in both the alum-induced T helper 2 response and the T helper 1 response induced by alum-adsorbed interleukin-12. Immunology 108, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich BA, Li J, Shannon J, Hurren R, Chalupny J, Cosman D, and Miller RG (2003). Activated, but not resting, T cells can be recognized and killed by syngeneic NK cells. J. Immunol 170, 3572–3576. [DOI] [PubMed] [Google Scholar]
- Reth M, Hämmerling GJ, and Rajewsky K (1978). Analysis of the repertoire of anti-NP antibodies in C57BL/6 mice by cell fusion. I. Characterization of antibody families in the primary and hyperimmune response. Eur. J. Immunol 8, 393–400. [DOI] [PubMed] [Google Scholar]
- Rydyznski C, Daniels KA, Karmele EP, Brooks TR, Mahl SE, Moran MT, Li C, Sutiwisesak R, Welsh RM, and Waggoner SN (2015). Gener-ation of cellular immune memory and B-cell immunity is impaired by natural killer cells. Nat. Commun 6, 6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwickert TA, Alabyev B, Manser T, and Nussenzweig MC (2009). Germinal center reutilization by newly activated B cells. J. Exp. Med 206, 2907–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KG, Light A, Nossal GJ, and Tarlinton DM (1997). The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J 16, 2996–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapper CM, McIntyre TM, Mandler R, Pecanha LM, Finkelman FD, Lees A, and Mond JJ (1992). Induction of IgG3 secretion by interferon gamma: a model for T cell-independent class switching in response to T cell-independent type 2 antigens. J. Exp. Med 175, 1367–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2014). R: A language and environment for statistical computing (R Foundation for Statistical Computing)
- Vander Heiden JA, Yaari G, Uduman M, Stern JN, O’Connor KC, Hafler DA, Vigneault F, and Kleinstein SH (2014). pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics 30, 1930–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggoner SN, Cornberg M, Selin LK, and Welsh RM (2011). Natural killer cells act as rheostats modulating antiviral T cells. Nature 481, 394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh RM, and Waggoner SN (2013). NK cells controlling virus-specific T cells: Rheostats for acute vs. persistent infections. Virology 435, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JL, Heffler LC, Charo J, Scheynius A, Bejarano MT, and Ljunggren HG (1999). Targeting of human dendritic cells by autologous NK cells. J. Immunol 163, 6365–6370. [PubMed] [Google Scholar]
- Xu HC, Grusdat M, Pandyra AA, Polz R, Huang J, Sharma P, Deenen R, Köhrer K, Rahbar R, Diefenbach A, et al. (2014). Type I interferon pro-tects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity 40, 949–960. [DOI] [PubMed] [Google Scholar]
- Xu H, Chaudhri VK, Wu Z, Biliouris K, Dienger-Stambaugh K, Rochman Y, and Singh H (2015). Regulation of bifurcating B cell trajectories by mutual antagonism between transcription factors IRF4 and IRF8. Nat. Immunol 16 , 1274–1281. [DOI] [PubMed] [Google Scholar]
- Ye J, Ma N, Madden TL, and Ostell JM (2013). IgBLAST: an immuno-globulin variable domain sequence analysis tool. Nucleic Acids Res 41, W34–W40. [DOI] [PMC free article] [PubMed] [Google Scholar]