

This article reviews the management of peripheral T cell lymphoma, focusing on new agents and therapeutic combinations to better understand the biology and pathogenesis of the disease.
Keywords: Lymphoma, New agents, T cell, Therapeutic, Signaling pathway, Drug targeting
Abstract
Peripheral T‐cell lymphoma (PTCL) is a heterogeneous group of clinically aggressive diseases associated with poor outcome. Despite progress in the last several years, resulting in a deeper understanding of the natural history and biology of PTCL based on molecular profiling and next‐generation sequencing, there is a need for improvement in efficacy of chemotherapeutic regimens for newly diagnosed patients. Treatment in the front‐line setting is most often cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or CHOP‐like regimens, which are associated with a high failure rate and frequent relapses. Trials evaluating intensive chemotherapy have resulted in variable success in prolonging event‐free survival, but overall survival has remained unchanged. Furthermore, this strategy is limited to patients who are in complete remission after initial anthracycline‐based chemotherapy. Many patients are ineligible for hematopoietic stem cell transplantation because of age or failure to achieve remission. For relapsed disease, advances have been made in the therapeutic arsenal for PTCL. New drugs investigated in phase II studies have achieved response rates between 10% and 30%. However, to date the identification of new therapies has been largely empiric, and long‐term remissions are the exception to the rule. Current patient outcomes suggest the need for the identification and development of active and biologically rational therapies to improve disease management and to extend the duration of response with iterative biomarker evaluation. This review covers the management of PTCL and focuses on new agents and therapeutic combinations, based on a better understanding of biology and pathogenesis of the disease.
Implications for Practice.
Recent progress in understanding of the biology and pathogenesis of peripheral T‐cell lymphoma has led to the emergence of new drugs. Unfortunately, this has not been met with similar advances in outcome improvement. Anthracycline‐containing regimens, mostly cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP), are considered the standard of care, although the best first‐line approach remains to be defined. In the relapsed and refractory settings, several new agents achieved response rates between 10% and 30%, although these drugs do not significantly affect survival rates. Therapeutic options based on better molecular characterization of various histological types and combinations with the CHOP regimen or synergic combinations of new drugs may lead to better outcomes.
Introduction
Peripheral T‐cell lymphoma (PTCL) is a rare and heterogeneous group of clinically aggressive diseases associated with poor prognosis, representing 10%–15% of non‐Hodgkin lymphomas (NHLs) in Western countries (Table 1) [1], [2]. Many advances have occurred in the classification of mature T‐cell and natural killer‐cell (NK‐cell) neoplasms with the introduction of new provisional entities in the World Health Organization classification system of 2016 (Table 2). These modifications are the result of genomic studies using approaches to analyze Gene expression profiling and the genetic landscape of T‐cell and NK‐cell neoplasms.
Table 1. Incidence and survival by histological type.
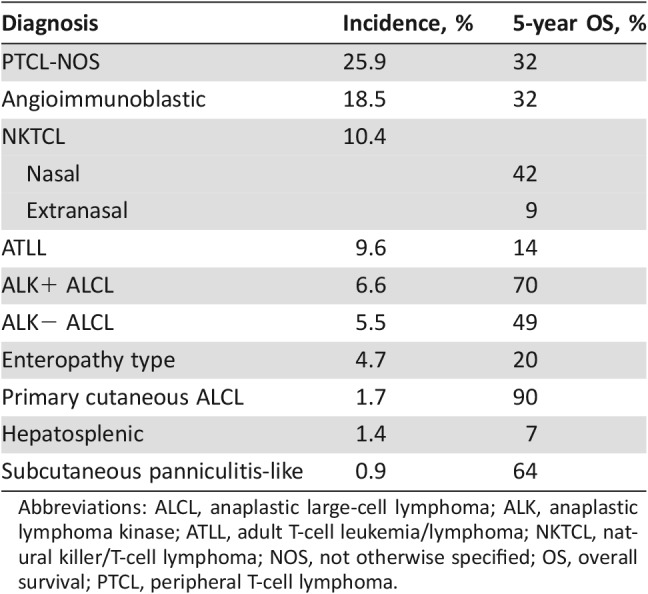
Abbreviations: ALCL, anaplastic large‐cell lymphoma; ALK, anaplastic lymphoma kinase; ATLL, adult T‐cell leukemia/lymphoma; NKTCL, natural killer/T‐cell lymphoma; NOS, not otherwise specified; OS, overall survival; PTCL, peripheral T‐cell lymphoma.
Table 2. Mature T‐cell and NK‐cell neoplasms: WHO classification 2016.
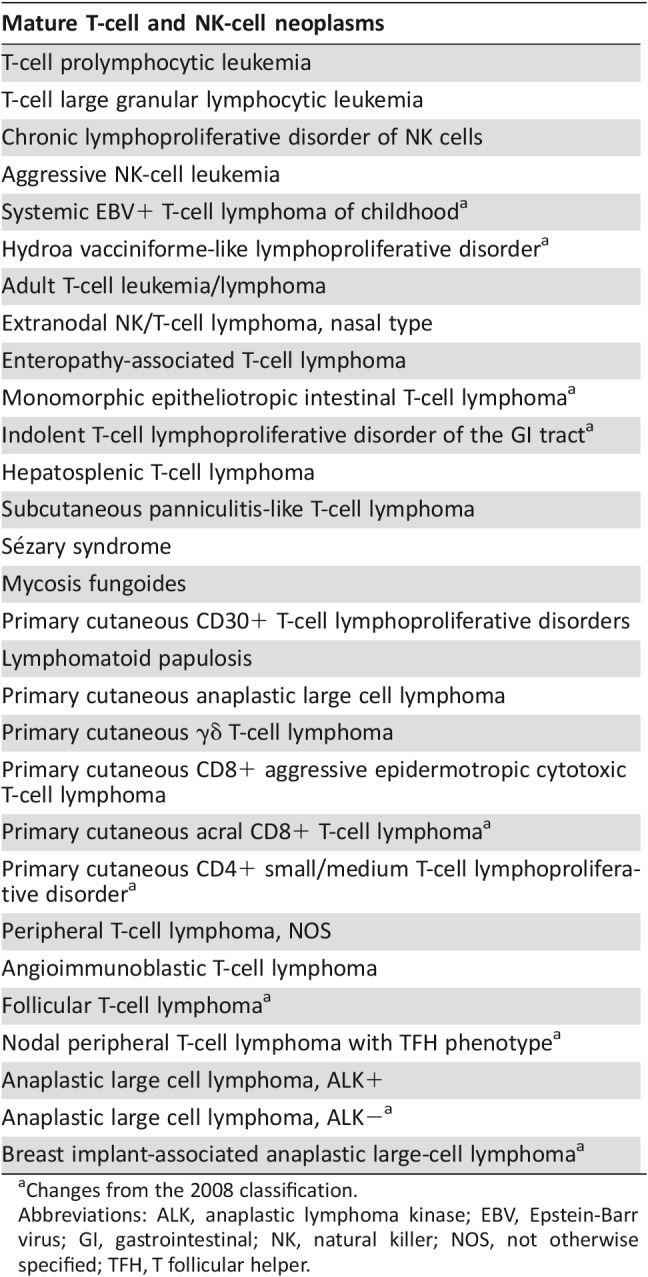
Changes from the 2008 classification.
Abbreviations: ALK, anaplastic lymphoma kinase; EBV, Epstein‐Barr virus; GI, gastrointestinal; NK, natural killer; NOS, not otherwise specified; TFH, T follicular helper.
Patients with PTCLs demonstrate more aggressive clinical features than those with B‐cell lymphoma, including B‐symptoms, diffuse disease, extranodal disease, increased serum lactate dehydrogenase (LDH) levels, increased serum beta‐2‐microglobulin levels, bulky disease, elevated Ki‐67, and overexpression of p53 [3], [4], [5].
The approach to treat PTCL has traditionally been similar to that for diffuse large B‐cell lymphoma. However, outcomes are poor when PTCL is treated according to strategies established for aggressive B‐cell lymphomas, with early relapse, progression‐free survival (PFS) of less than 1 year, and overall survival (OS) of less than 2 years [6], [7], [8], [9], [10], [11].
The exact roles of high‐dose therapy (HDT) and autologous stem cell transplantation (ASCT) support remain undefined. Several retrospective studies suggest that there are populations of patients with PTCL who will benefit from transplantation for whom disease status at transplantation is a major predictor of success. However, the interpretation of these studies is complicated by the heterogeneous histologic subtype frequencies and the enrollment of patients with anaplastic lymphoma kinase‐positive (ALK+) anaplastic large‐cell lymphoma (ALCL) in some series, who had a more favorable outcome. [12], [13], [14], [15], [16], [17].
The role of allogeneic stem cell transplantation (allo‐SCT) has been specifically investigated in relapsed and/or refractory PTCL in retrospective series with limited data. Allo‐SCT is a viable option, particularly in refractory patients. The advantage comes from lower relapse rate in patients developing chronic graft‐versus‐host disease, as well as some responses obtained in patients who received donor lymphocyte infusions, supporting the existence of a graft‐versus‐lymphoma (GVL) effect in PTCL [18], [19], [20]. A better understanding of PTCL biology, based on molecular profiling and next‐generation sequencing, has led to advances in the treatment of PTCL. Four novel agents have recently been approved by the U.S. Food and Drug Administration (FDA) for the treatment of relapsed and refractory PTCL and are being investigated in the front‐line setting: pralatrexate, brentuximab vedotin, romidepsin, and belinostat. Ongoing studies are testing these new agents in combination with chemotherapy in the front‐line setting. Other new agents are also being tested in the relapsed/refractory (R/R) setting. They are also being tested in combination (chemo‐free) in other less‐advanced studies and are likely to be used in the future.
Methods
We highlighted all pertinent published and unpublished studies that analyzed treatment of peripheral T‐cell lymphoma through 2017. Selection criteria were established before data collection. An exhaustive literature search was performed on the MEDLINE and ScienceDirect databases. The search criteria were peripheral T‐cell lymphoma and treatment. We manually checked the references of all identified articles and also searched Google Scholar and conference proceedings in the fields of hematology and oncology to find additional articles.
Diagnosis
Clinical presentations of PTCL are heterogeneous with more disseminated disease (stage III or IV disease) compared with aggressive B‐cell lymphomas, a poorer performance status, an increased incidence of B‐symptoms, and a common extranodal localization [21], [22]. Patients occasionally present with eosinophilia, pruritis, hemophagocytic syndromes, or autoimmune manifestations [23]. Bone marrow involvement is more frequent than that observed in diffuse large B‐cell lymphoma [2].
Diagnosis requires examination of peripheral blood or tissue biopsy for histological characterization, completed by detailed immunohistochemistry, flow cytometry, cytogenetics, and molecular genetics. Clonality should be assessed by polymerase chain reaction (PCR) for T‐cell receptor gene rearrangements.
The staging system for PTCL is the same for all non‐Hodgkin lymphomas, including tests to perform an extension assessment and to assign a prognostic score. Investigations include performance status, physical examination, a complete blood count, and routine blood chemistry, including LDH, uric acid, serum calcium, and beta‐2‐microglobulin, and screening tests for HIV, human T‐cell lymphotropic virus type 1 (HTLV‐1), and hepatitis B and C are also required.
At diagnosis, patients should have at least a computed tomography (CT) scan of chest, abdomen, and pelvis, as well as a bone marrow aspirate and biopsy. Most PTCLs are FDG‐avid; for this reason, 18‐FDG positron emission tomography combined with CT is now recommended by the International Conference on Malignant Lymphoma group for PTCL staging [24].
Some extranodal lymphomas displaying particular biological behavior need specific endoscopic investigations, barium study, or CT enterography for gastrointestinal lymphoma. Similarly, lumbar puncture and magnetic resonance imaging of the brain should be performed if neurological symptoms are suspected.
For clinical practice purposes, the International Prognostic Index (IPI) is the recommended prognostic tool in nodal PTCL [25].
Conventional Chemotherapy of Up‐Front Treatment in PTCL
Anthracycline‐Based Regimens
Standard first‐line therapy still consists of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) or a CHOP‐like regimen. It is based on treatment paradigms for diffuse large B‐cell lymphoma, despite the lack of high‐level evidence, a disease rationale, or a biological basis. Therapeutic responses to this approach have been neither sufficient nor durable [2], [9], [10].
The International Peripheral T‐Cell Lymphoma Project reported a 5‐year OS of 32% and a 5‐year failure‐free survival of only 20% in a cohort of 1,314 cases of previously untreated PTCL and natural killer/T‐cell lymphoma, of which there were 340 cases of PTCL not otherwise specified (PTCL‐NOS) treated with an anthracycline‐based regimen. The use of an anthracycline‐containing regimen was not associated with an improved outcome in PTCL‐NOS or angioimmunoblastic T‐cell lymphoma (AITL), but was associated with an improved outcome in ALK+ ALCL, with an overall response (OR) rate of 75% and an OS rate of 60% after 5 years [2]. In contrast, the OR and OS rates of the anaplastic lymphoma kinase negative (ALK−) ALCL were identical to those of other PTCL subtypes [2], [10].
The German High‐Grade Non‐Hodgkin Lymphoma Study Group evaluated the addition of etoposide to the CHOP regimen (CHOEP) administered every 3 weeks or reducing the interval between courses of CHOEP to 2 weeks in 343 patients with T‐cell lymphoma. The addition of etoposide to CHOP significantly improved event‐free survival (EFS; 75.4% vs. 51%; p = .003), but not OS of patients under 60 years of age with normal LDH levels. However, the addition of etoposide or reducing the interval between chemotherapy cycles did not affect OS or PFS of patients aged over 60 years [9].
A study from the Swedish Lymphoma Registry analyzed prognostic factors and outcome of 755 patients with PTCL diagnosed during a 10‐year period. The 5‐year OS and 5‐year PFS were 34.1% and 25.7%, respectively. Patients with ALK+ ALCL had superior survival compared with all other groups (p < .001) except for subcutaneous panniculitis‐like T‐cell lymphoma. The 5‐year OS and 5‐year PFS for the ALK+ ALCL subtype were 79.4% and 63.2%, respectively. International Prognostic Index factors and male gender were associated with an adverse OS and PFS. In an intention‐to‐treat analysis in 252 patients with nodal PTCL and enteropathy‐associated T‐cell lymphoma (excluding ALK+ ALCL), up‐front ASCT was associated with a superior OS and PFS compared with patients treated without ASCT. The 5‐year OS and PFS in the up‐front ASCT group were 48% and 41%, respectively, whereas they were 26% and 21%, respectively, in the group without ASCT. The addition of etoposide to CHOP resulted in superior PFS in patients aged ≤60 years. The median OS after relapse or progression in 211 patients initially responding (partial remission [PR] or complete remission [CR]) to primary treatment was 6.0 months [26].
More recently, a prospective multicenter cohort study of patients with a new diagnosis of nodal PTCL in the U.S. (COMPLETE registry) analyzed treatment patterns and outcomes of 499 patients. Among them, 256 (51.3%) had nodal PTCL and completed their planned treatment. The estimated median survival was 43.0 months, with 12‐month and 24‐month survival rates of 71.7% and 58.7%, respectively. Patients who received doxorubicin had more favorable survival (log‐rank p = .03).
PTCL subtype and IPI score were found to have a significant effect on overall survival, and patients with ALCL had better overall survival compared with those with AITL and PTCL‐NOS.
After controlling for disease histology and IPI score, there was a trend toward statistical significance in mortality reduction among those patients treated with doxorubicin (p = .09), whereas the addition of etoposide as a component of initial therapy was not observed to be significantly associated with survival [27].
Non‐Anthracycline‐Based Regimens
The Southwest Oncology Group tested a combination of platinum, etoposide, gemcitabine, and methylprednisolone in a phase II trial in 33 patients with PTCL, among whom 79% were newly diagnosed. They received 25 mg/m2 cisplatin, 40 mg/m2 etoposide, and 250 mg methylprednisolone on days 1 through 4 and 1000 mg/m2 gemcitabine on day 1 of a 21‐day cycle for six cycles. The overall response rate (ORR) and the PFS rate after 2 years were 39% and 12%, respectively. The median PFS and OS were 7 and 17 months, respectively. The OS was not considered to be promising. Adverse events included one grade 5 infection with grade 3–4 neutropenia and nine grade 4 hematological toxicities [28].
Conventional Chemotherapy in Relapsed/Refractory PTCL
Salvage treatment by conventional chemotherapy without intensification is still lacking efficacy. The British Columbia Cancer Agency studied the outcome of 153 patients with PTCL after relapse or progression in the absence of hematopoietic stem‐cell transplantation and explored factors influencing survival. The median time from diagnosis to first relapse or progression after primary therapy was 6.7 months. Median OS and PFS after second relapse or progression were 5.5 and 3.1 months, respectively, and were only marginally better than in patients who received chemotherapy at relapse: 6.5 and 3.7 months, respectively [29].
Gemcitabine Containing Regimens
The gemcitabine (1 g/m2 on days 1, 8, and 15), cisplatin (100 mg/m2 on day 15), and methylprednisolone (1 g on days 1–5) regimen repeated every 28 days was evaluated in a cohort of 16 patients with R/R PTCL and showed an ORR and CR rate of 69% and 19%, respectively. The median time to disease progression was 4.1 months, and the median OS was not reached after a median follow‐up of 17.4 months. The probability of survival after 1 year was 68.2%. The main grade 3–4 toxicities were neutropenia (62%) and anemia (12%) [30].
Hematopoietic Stem Cell Transplantation
Autologous Stem Cell Transplantation
Several retrospective and prospective ASCT studies have shown an improved outcome in patients with PTCL with an OS rate of 60%–70% [12], [31], [32]. The results between different studies are relatively discordant and are associated with differences in the number of patients included and the regimen used before ASCT, as well as when the autograft was performed (first‐line or salvage therapy), the heterogeneity of PTCLs included, and the enrollment of patients with ALK+ ALCL in some series. It is likely that retrospective studies show a better survival outcome compared with prospective studies because only the patients who finally underwent ASCT were enrolled.
Autologous Stem Cell Transplantation in Relapsed PTCL
There have been no prospective trials evaluating high‐dose regimens of chemotherapy followed by ASCT in patients with relapsed PTCL.
The GELTAMO group reviewed 123 patients with relapsed PTCL treated with high‐dose chemotherapy followed by ASCT (median age, 43.5 years). After a median follow‐up of 61 months, the 5‐year OS and PFS rates were 45% and 34%, respectively. The a‐Index Pronostic International (a‐IPI) and beta‐2‐microglobulin level at transplantation were identified as adverse prognostic factors for both overall and progression‐free survival [13].
The Stanford group reported a retrospective series of 53 patients who underwent ASCT for PTCL. The 5‐year PFS for patients in first CR or PR, in second or greater CR or PR, or with refractory disease was 51%, 12%, and 0%, respectively, and the corresponding OS values were 76%, 40%, and 30%, respectively. The pretransplant factors that influenced survival were the number of prior regimens and disease status [31].
Autologous Stem Cell Transplantation in the Front‐Line Setting
Results from prospective studies suggest a substantial effect of up‐front ASCT on the outcome of patients with PTCL, which should be further evaluated in randomized trials. The global conclusion of reported trials is that pretransplantation treatment must be improved to increase the transplantation success and that one of the major challenges is knowing which patients with PTCL in first remission to select for consolidative ASCT, as patients with low IPI, ALK+ ALCL disease in remission do not need consolidation transplant. For patients with ALK+ ALCL with high IPI score and poor outcomes, alternate strategies, including ASCT, should be considered [8], [9].
The first prospective multicenter study on up‐front ASCT in PTCL reported the data of 83 patients (excluding ALK+ ALCL) treated with four to six cycles of CHOP followed by ASCT. Fifty‐five patients (66%) who achieved PR or CR underwent fractionated total‐body irradiation and high‐dose cyclophosphamide followed by ASCT. In an intent‐to‐treat analysis, the ORR after myeloablative therapy was 66% (56% CR and 8% PR). With a median follow‐up time of 33 months, the estimated 3‐year OS, disease‐free survival for patients in CR (calculated from CR to the date of relapse), and PFS rates were 48%, 53%, and 36%, respectively. The main reason for not receiving ASCT was progressive disease [14].
The Nordic group reported the results of a prospective phase II trial (NLG‐T‐01) for untreated systemic PTCL (excluding ALK+ ALCL) to evaluate the efficacy of a dose‐dense approach consolidated by up‐front HDT and ASCT. A total of 160 patients received an induction regimen of six cycles of biweekly CHOEP. Etoposide was excluded for patients aged over 60 years. Patients in CR or PR received consolidated treatment with HDT and ASCT. A total of 115 patients underwent HDT/ASCT, with 90 in CR 3 months after transplantation. Early failures occurred in 26% of patients. Treatment‐related mortality was 4%. With a median follow‐up of 60.5 months, the 5‐year OS and PFS for the entire cohort were 51% and 44%, respectively. The best results were shown in ALK− ALCL [32].
High‐dose chemotherapy followed by ASCT may improve the outcome in PTCL, but the available data come from nonrandomized studies, meaning definitive recommendations cannot be made. The achievement of a first complete remission before ASCT has proven to be a strong predictor of improved outcome. However, this conclusion reflects selection bias because patients who achieve first complete remission have an intrinsically more chemosensitive disease and a higher probability of undergoing HDC/ASCT. This is supported by a large U.S. multicenter cohort of 341 patients with newly diagnosed PTCL, of whom only 8% underwent first‐line high‐dose chemotherapy followed by ASCT. There was no difference in OS based on the choice of up‐front regimen or SCT in first remission by multivariate analyses after adjusting for the initial treatment response [17].
Allogenic Stem Cell Transplantation in PTCL
The role of allo‐SCT has been especially investigated in relapsed and/or refractory PTCL in retrospective series with limited data.
In a phase II trial with a limited number of patients, the outcome of 17 patients with relapsed or refractory PTCL (2 median previous lines) was evaluated, including 8 patients who had relapsed after ASCT. Patients received reduced‐intensity allo‐SCT combining thiotepa, fludarabine, and cyclophosphamide after salvage therapy by four to six sequences of dexaméthasone, cytarabine and cisplatine (DHAP). After a median follow‐up time of 28 months, the estimated 3‐year OS and PFS were 81% and 64%, respectively. The transplant‐related mortality rate was 12%. Donor lymphocyte infusions induced a response in two patients with progressive disease, suggesting the existence of a GVL effect [18].
The British Society of Bone Marrow Transplantation and the Australian Bone Marrow Transplant Recipient Registry reported a series of 82 patients with relapsed and/or refractory PTCL who received high‐dose therapy with ASCT (n = 64) or allo‐SCT (n = 18). In the group of patients who received allo‐SCT, the median number of pretransplant treatment lines was two, and two patients had received a prior ASCT. Thirty‐three percent received total body irradiation, and 67% received chemotherapy‐only conditioning regimens. After a median follow‐up time of 57 months, five patients were alive, five died of progressive disease, and eight died from nonrelapse mortality. The estimated 3‐year OS, PFS, and relapse rates were 39%, 33%, and 28%, respectively. However, the use of full‐intensity allogeneic transplantation was limited by high transplant‐related mortality [33].
The outcomes of 44 allogeneic blood or bone marrow transplants (BMTs) from related donors for PTCL were reviewed, with 24 patients receiving reduced‐intensity conditioning (RIC) and 20 with myeloablative conditioning (MAC). The estimated 2‐year PFS and OS rates were 40% and 43%, respectively. The 1‐year cumulative incidence of relapse was 38% for MAC or human leukocyte antigen‐identical allogeneic BMTs and 34% for RIC or haploidentical allogeneic BMTs by competing risk analysis. On unadjusted analysis, patients with acute grade 2–4 or chronic graft‐versus‐host disease (GVHD) had a 17% probability of relapse compared with 66% in patients without GVHD by unadjusted analysis (p = .04) [19].
The European Group for Blood and Marrow Transplantation reviewed the outcome of 45 patients with AITL who underwent allo‐SCT in a retrospective study. Thirty‐four patients had received two or more lines of chemotherapy before allo‐SCT, and 11 had experienced treatment failure with a prior ASCT. Twenty‐five patients underwent a myeloablative allo‐SCT, and 20 a reduced‐intensity allo‐SCT. The estimated 3‐year OS, PFS, and relapse rates were 64%, 53%, and 20% (lower in patients who developed GVHD), respectively. The lower relapse rate after transplantation and in patients who develop chronic GVHD (cGVHD) after the allo‐SCT suggests the existence of a clinically relevant GVL effect [20].
Patients with a related or unrelated matched donor and a good performance status may be eligible for this procedure. Utilization of RIC and alternative donors expands treatment options in PTCL to older patients, who are unable to tolerate high‐dose conditioning, with outcomes comparable to approaches using myeloablative regimens and HLA‐matched donors. Allo‐SCT may be appropriate for selected high‐risk cases in first remission and may offer a benefit for patients with PTCL who relapse after ASCT.
Novel Agents Alone or in Combination with Conventional Chemotherapy
To improve the poor outcome of PTCL, novel agents that target various pathways are required. The most significant progress in recent years has been made by evaluating a large number of novel agents alone, or in combination with other therapies, including histone deacetylase (HDAC) inhibitors, monoclonal antibodies, immunoconjugates, antifolates, immunomodulatory agents, nucleoside analogs, proteasome inhibitors, kinase inhibitors, and other targeted agents.
Licensed Drugs in Relapsed and Refractory PTCL
Four next‐generation drugs (pralatrexate, romidepsin, brentuximab vedotin, and belinostat) have recently been approved by the FDA for the treatment of relapsed and refractory PTCL based on response rates (Table 3). However, none of these agents led to survival improvement. The European Medicines Agency (EMA) has rejected approval of both pralatrexate and romidepsin because of a lack of evident clinical benefit; belinostat is not licensed in Europe, although it has been granted orphan designation status by the EMA. Brentuximab vedotin is the only drug approved by the EMA for the treatment of relapsed or refractory systemic ALCL based on a better benefits‐risk ratio.
The European Medicines Agency (EMA) has rejected approval of both pralatrexate and romidepsin because of a lack of evident clinical benefit; belinostat is not licensed in Europe, although it has been granted orphan designation status by the EMA.
Table 3. Clinical trial efficacy data of licensed and available drug monotherapy.

Only the data of patients who had relapsed/refractory PTCL were reported.
Abbreviations: —, not available; ALCL, anaplastic large‐cell lymphoma; CR, complete remission; CRu, unconfirmed complete remission; DoR, duration of response; HDAC, histone deacetylase; MoAb, monoclonal antibodies; NR, not reached; OS, overall survival; PFS, progression‐free survival; PTCL, peripheral T‐cell lymphoma; Ref., reference; TCL, T‐cell lymphoma.
Pralatrexate
Pralatrexate is an antifolate (a folate analogue metabolic inhibitor) designed to preferentially accumulate in cancer cells. Pralatrexate was studied in a large prospective phase II trial (the pivotal PROPEL trial) in patients with relapsed or refractory PTCL. Of the 115 patients enrolled in the trial, 109 were evaluable. Patients received pralatrexate i.v. at a dose of 30 mg/m2 per week for 6 weeks in 7‐week cycles until disease progression or the development of unacceptable toxicity. The ORR was 29%, including 11% CR or unconfirmed CR (CRu). The median duration of response was 10.1 months. The median PFS and OS were 3.5 and 14.5 months, respectively. The most common grade 3–4 adverse events (AEs) were thrombocytopenia (33%), mucositis (22%), neutropenia (22%), and anemia (18%). These results formed the basis for the FDA approval of pralatrexate in R/R PTCL [34].
Shustov et al. performed a subanalysis in the PROPEL data set and analyzed the impact of prior therapy on response in 15 patients who had previously received a CHOP‐based regimen. Seven patients achieved CR and 4 achieved PR. The ORR and CR were 47% and 20%, respectively. Two patients remained on treatment at the time of data cutoff (12.9 and 18.5 months). Median duration of response and PFS were not reached and 8.4 months, respectively, based on the independent central review [35].
Romidepsin
The rationale for using epigenetic therapies is supported by several studies that have shown mutations in epigenetic genes in different PTCL subtypes. Mutations affecting DNA methylation have been reported in AITL and PTCL‐NOS. Recent studies have identified mutations in TET2, IDH2, RHOA, DNMT3A, and FYN in PTCL [36], [37], [38], [39], [40].
The rationale for using epigenetic therapies is supported by several studies that have shown mutations in epigenetic genes in different PTCL subtypes. Mutations affecting DNA methylation have been reported in AITL and PTCL‐NOS. Recent studies have identified mutations in TET2, IDH2, RHOA, DNMT3A, and FYN in PTCL
A pivotal, open‐label, phase II study of a selective HDAC inhibitor, romidepsin, in patients with relapsed or refractory PTCL who were refractory to at least one prior systemic therapy or for whom at least one prior systemic therapy failed, enrolled 130 patients with a median of two previous lines of treatment. Patients received 14 mg/m2 romidepsin as a 4‐hour i.v. infusion on days 1, 8, and 15 every 28 days. The ORR was 25%, including 15% CR/CRu. An update of these results showed a median duration of response for all responders of 28 months and was not reached for those who achieved CR/CRu. Of the 19 patients who achieved CR/CRu, 10 had long‐term (≥12 months) responses. Patients who achieved a CR/CRu of ≥12 months had a median PFS of 29 months and a significantly longer OR than those with a CR/CRu of <12 months or a PR. The most common grade 3–4 AEs were thrombocytopenia (24%), neutropenia (20%), and infections (19%). This study also demonstrated that prolonged romidepsin treatment provided clinical benefits for R/R patients who had achieved at least stable disease [41], [42]. Based on this, romidepsin was approved by the FDA for this indication. Romidepsin has been recommended by the National Comprehensive Cancer Network (NCCN) as a second‐line and subsequent therapy in patients, regardless of intention to proceed to high‐dose therapy or SCT [43].
Belinostat
Belinostat is a hydroxamic acid‐derived pan‐HDAC inhibitor that broadly inhibits all zinc‐dependent HDAC enzymes. The BELIEF trial reported the results of 120 patients with PTCL who experienced progression after one or more prior therapies (median, 2), who received daily 30‐minute infusions of 1,000 mg/m2 belinostat on days 1–5 in 21‐day cycles. The ORR was 25.8%, including 10.8% CR. For the three most common subtypes, the ORR was 23% in patients with PTCL‐NOS, 46% in patients with AITL, and 15% in patients with ALK− ALCL. The median duration of response was 13.6 months. Median PFS and OR were 1.6 and 7.9 months, respectively. Twelve of the enrolled patients underwent SCT after belinostat monotherapy. The most common grade 3–4 AEs were anemia (10.8%), thrombocytopenia (7%), dyspnea (6.2%), and neutropenia (6.2%). These results demonstrated the antitumor activity of belinostat, resulting in its FDA approval for patients with R/R PTCL [44].
Brentuximab Vedotin
Brentuximab vedotin (SGN‐35; BV) is a novel anti‐CD30 antibody linked to monomethyl auristatin E (MMAE), a potent antimicrotubule agent, via a protease‐cleavable linker. MMAE is a mitotic spindle poison that induces G2‐M cell cycle arrest and apoptosis [45]. BV is FDA and EMA approved for use in patients with systemic ALCL after failure of one or more multiagent chemotherapy regimens [46].
Pro et al. reported recently the 5‐year results of their pivotal, multicenter, phase II study that evaluated the efficacy and safety of brentuximab vedotin as a single agent in patients with relapsed or refractory systemic ALCL. Fifty‐eight patients with systemic ALCL and recurrent disease after at least one prior therapy received BV 1.8 mg/kg intravenously every 3 weeks for up to 16 cycles. Sixteen patients (28%) had ALK+ disease, and 42 patients (72%) had ALK− disease. After a median observation of 71.4 months and a median follow‐up time from end of treatment of 58.4 months, the estimated 5‐year OS and PFS were 60% and 39%, respectively. The 5‐year OS and PFS for patients who achieved a best response of CR (38/58; 66%) were 79% and 57%, respectively, whereas the 5‐year OS for patients who did not achieve CR was 25% [47].
BV was investigated in another multicenter, open‐label, phase II trial of 35 patients with R/R PTCL, specifically AITL (n = 13) and PTCL‐NOS (n = 22). The median number of prior therapies was two, and 63% of patients were refractory to their most recent therapy. BV (1.8 mg/kg) was administered every 3 weeks until progression or unacceptable toxicity. The ORR for all evaluable patients was 41%, and it was 54% in patients with AITL. The median PFS and median duration of response were 2.6 and 7.6 months, respectively. The most common grade 3–4 AEs were neutropenia (14%), peripheral sensory neuropathy, and hyperkalemia (9% each). No correlation between CD30 expression (per central review) and response was observed [48].
Relevant Combinations for Licensed Drugs
To improve the response to treatment in PTCL, romidepsin was combined with either chemotherapy or new agents. Romidepsin was combined with a standard CHOP regimen in the front‐line setting. The LYSA group reported the data of 37 patients with previously untreated PTCL treated with eight 3‐week cycles of CHOP combined with various doses of romidepsin (8, 10, or 12 mg/m2 on days 1 and 8) in a single‐arm, phase Ib/II study. The recommended dose of romidepsin for the phase II study was 12 mg/m2 based on this protocol. Among 35 evaluable patients, the ORR was 69%, including 51% with a CR. With a median follow‐up of 30 months, the estimated PFS and OS were 41% and 71%, respectively. No deaths were attributable to toxicity in the reported data. Three patients had early cardiac events, and 25 had at least one serious AE; the most common AEs were febrile neutropenia (14%), deterioration of physical health (14%), lung infection (11%), and vomiting (8%; Table 4) [49]. Based on these promising results, a phase III, randomized trial of romidepsin + CHOP versus CHOP in patients with untreated PTCL is ongoing (NCT01796002).
Table 4. Licensed drugs in combination with CHOP or CHOP‐like chemotherapies in the front‐line setting.

82% ALCL.
Abbreviations: —, not available; ALCL, anaplastic large‐cell lymphoma; CEOP, cyclophosphamide, etoposide, vincristine, and prednisone; CHOP, cyclophosphamide, doxorubicin, vincristine, and prednisone; CHP, cyclophosphamide, doxorubicin, and prednisone; CR, complete remission; ORR, overall response rate; OS, overall survival; PFS, progression‐free survival; PTCL, peripheral T‐cell lymphoma; Ref., reference; T/NK cell, T‐cell/natural killer cell.
Synergistic activity has been seen for the combination of pralatrexate with romidepsin in preclinical models of PTCL. A phase I–II trial was initiated to evaluate the safety and clinical activity of this combination in 26 patients with R/R lymphomas, including 13 with PTCL. Pralatrexate was administered at a dose of 10 mg/m2 and romidepsin at a dose of 12 mg/m2, with escalation to 25 mg/m2 for pralatrexate and 14 mg/m2 for romidepsin. Patients were treated using one of three dosing schedules (three times weekly every 28 days; twice weekly every 21 days, and QOW Q28DT). The median number of prior therapies was three. The grade 3–4 toxicities reported in >5% of patients were neutropenia (31%), thrombocytopenia (31%), anemia (23%), oral mucositis (15%), hyponatremia (8%), pneumonia (8%), and sepsis (8%). Twenty‐two patients were evaluable. The ORR in patients with PTCL was 77%, with CR for 31% [50].
These data support the therapeutic potential of this combination in this setting, which is now being expanded to a multicenter phase II trial for PTCL (NCT01947140).
A phase I–II study was initiated to evaluate the safety and efficacy of romidepsin combined with lenalidomide in patients with T‐cell lymphoma (10 patients with cutaneous T‐cell lymphoma [CTCL] and 11 patients with PTCL), B‐cell lymphoma, and multiple myeloma. The maximal tolerated dose (MTD) defined in cycle 1 was romidepsin 14 mg/m2 i.v. on days 1, 8, and 15 and lenalidomide 25 mg orally on days 1–21, every 28 days. Nineteen patients with T‐cell lymphoma (TCL) were evaluable for efficacy, with an ORR of 53%. The ORR in PTCL was 50% (5/10, 5 PR). Median EFS was 15.5 weeks (CTCL, 30.0 weeks; PTCL, 13.5 weeks). Median OS was not reached. The grade 3–4 toxicities reported in ≥10% of patients were neutropenia (48%), thrombocytopenia (38%), anemia (33%), and electrolyte abnormalities (43%) [51].
Romidepsin was also evaluated in combination with bortezomib in a phase I study in 18 patients with R/R lymphoma. The MTD was 1.3 mg/m2 bortezomib and 10 mg/m2 romidepsin. The dose‐limiting toxicities were grade 3 fatigue, vomiting, and chills. Two patients with PTCL were evaluated and neither responded to treatment [52].
Considering the potential synergy of proteasome inhibitors with histone deacetylase inhibitors and lenalidomide, a phase Ib/IIa trial was initiated to evaluate the safety and clinical activity of romidepsin and lenalidomide in combination with carfilzomib in 27 patients with R/R lymphomas, including 16 with TCL. Romidepsin and carfilzomib were given i.v. on days 1 and 8, and lenalidomide was given orally on days 1–15 every 21 days, in a standard 3 + 3 dose escalation design. The starting dose was romidepsin 8 mg/m2, lenalidomide 15 mg, and carfilzomib 36 mg/m2. Grade 3–4 toxicities reported in >10% of patients were neutropenia and thrombocytopenia. Of the 16 evaluable patients with T‐cell lymphoma, the OR and CR rates were 50% and 31%, respectively. Four of 5 patients with AITL achieved CR. The median duration of response and EFS for patients with TCL were 38.7 weeks and 13.57 weeks, respectively. The preliminary OR and CR rates of this combination are promising in PTCL, particularly in the AITL subtype, and warrant further study [53].
Pralatrexate was investigated in the front‐line setting in combination with a cyclophosphamide, etoposide, vincristine, and prednisone (CEOP) regimen in a phase II trial in 33 patients with PTCL. Patients achieving CR or PR underwent consolidation by ASCT after four cycles. The ORR was 70%, with 17 patients (52%) achieving a CR. The 2‐year PFS and OS were 39% and 60%, respectively. The most common grade 3–4 AEs were anemia (27%), thrombocytopenia (12%), febrile neutropenia (18%), mucositis (18%), sepsis (15%), increased creatinine (12%), and liver transaminases (12%). This combination did not improve outcomes relative to those historically achieved using CHOP [54].
More recently, pralatrexate was evaluated in combination with the CHOP regimen in previously untreated patients with PTCL in a 3 + 3 phase I dose‐escalation study. The MTD was not reached at protocol‐defined highest administration dose. In the second part, a pralatrexate dose of 30 mg/m2 was administered, on days 1 and 8 of a standard 21‐day CHOP regimen for six cycles or until toxicity or disease progression, as the protocol‐defined maximum dose. The treatment association was generally well tolerated, with median relative dose intensity of 98%. The only grade >3 treatment‐related AE (≥10%) was neutropenia (n = 4). In the 29 patients evaluable for response, the investigator found that objective response and complete response rates were 90% and 66%, respectively [55].
Belinostat (1,000 mg/m2) on various schedules repeated every 21 days for up to six cycles in combination with the CHOP regimen was tested in 23 patients with PTCL. The maximum tolerated dose was determined to be 1,000 mg/m2 on days 1–5. The most frequent grade 3–4 AEs were hematological in nature: decreased neutrophil counts (26%), anemia (22%), neutropenia (17%), and decreased white blood cell counts (17%). The ORR for the first 18 evaluable patients was 89% (16/18), with 72% CR. These data demonstrate that the combination of belinostat with CHOP was well tolerated, with strong clinical efficacy. This combination is a promising new regimen for the treatment of PTCL that will be further tested in a phase III randomized clinical trial [56].
Beyond the relapse setting, BV has been combined in a phase I open‐label trial at full dose with standard CHOP or CHP (CHOP without vincristine) chemotherapy for the front‐line treatment of systemic ALCL and other mature CD30+ T/NK‐cell lymphomas. Patients received sequential treatment (once every 3 weeks) with 1.8 mg/kg BV (two cycles) followed by CHOP (six cycles) or 1.8 mg/kg BV plus CHP (BV + CHP) for six cycles (once every 3 weeks). Responders received single‐agent BV for 8–10 additional cycles (for a total of 16 cycles). Thirty‐nine patients were enrolled in the study (13 and 26 patients in the sequential‐ and combination‐treatment groups, respectively). After sequential treatment, 11 (85%) of 13 patients achieved an OR, including 62% CR; the estimated 1‐year PFS rate was 77%. At the end of the combination treatment, all patients (n = 26) achieved an OR, including 88% CR; the estimated 1‐year PFS rate was 71%. All seven patients without ALCL achieved CR. The most frequent grade 3–4 AEs in the combination‐treatment group were febrile neutropenia (31%), neutropenia (23%), anemia (15%), and pulmonary embolism (12%) [57].
Recently, 4‐year updated data from the BV + CHP combination treatment arm were reported. Twenty‐one of the 26 patients who achieved remission with the BV + CHP combination continued to receive single‐agent BV. The median number of cycles of BV was 13.
After a median follow‐up of 52 months, 18 patients remained on study. The estimated 4‐year PFS and OS rates were 52% and 80%, respectively. The median PFS had not been reached. Of 26 patients treated with the BV + CHP combination, 19 (73%) developed peripheral neuropathy. Eighteen patients (95%) experienced complete resolution (42%) or partial resolution, defined as a decrease by at least one grade from worst grade (58%). The median time to resolution of peripheral neuropathy symptoms was 5.7 months [58]. The results from the current phase II trial in PTCL, coupled with the ability to combine BV with standard‐induction chemotherapy, updated above, resulted in the initiation of a phase III, double‐blind, randomized study of BV plus CHP versus CHOP for untreated patients with CD30‐expressing PTCL (NCT01777152).
In relapsed or refractory settings, it would be interesting to incorporate these new drugs in earlier lines of therapy with responses that could be sustained in patients treated at first relapse by pralatrexate and romidepsin.
The high response rates shown in patients with AITL treated with belinostat should be confirmed by specific dedicated studies to explore the effectiveness of belinostat in this specific subpopulation of patients. The results of the 5‐year follow‐up of brentuximab vedotin therapy in patients with relapsed or refractory systemic ALCL are very promising.
In the first‐line setting, results of the romidepsin + CHOP versus CHOP phase III trial and the BV + CHP versus CHOP echelon 2, phase III trial are pending. However, initial trials incorporating new agents into conventional treatment backbones (alemtuzumab‐CHOP, denileukin diftitox‐CHOP, pralatrexate + CEOP, etc.) have not yielded improvement. Based on a better understanding of biology and pathogenesis of the disease, several early‐phase trials tested chemo‐free combinations with encouraging results and are expected to improve disease management and to extend the duration of response with iterative biomarker evaluation.
Additional Drugs Listed in NCCN Guidelines for Relapsed PTCL
Bendamustine
Bendamustine is an alkylating agent with properties of the nitrogen mustard, mechlorethamine, and the purine analogue, fludarabine. In the Bently trial for the LYSA group, the bendamustine regimen was administered to 60 patients with CTCL and PTCL (median number of previous treatments, 1) at a dose of 120 mg/m2 on days 1 and 2, every 3 weeks, for six cycles. The ORR and CR rate were 50% and 28%, respectively. The median duration of response was 6.6 months. The median PFS and OS were 3.6 and 6.2 months, respectively [59]. Because of its encouraging response rate and acceptable toxicity, the National Comprehensive Cancer Network (NCCN) has recommended bendamustine as a second‐line and subsequent therapy, regardless of high‐dose therapy and SCT [43].
Lenalidomide
Lenalidomide is an immunomodulatory drug initially intended as a treatment for multiple myeloma with three main mechanisms of action: direct antitumor effect, inhibition of angiogenesis, and immunomodulation activity.
The outcome of 40 patients with R/R PTCL or untreated PTCL who were not candidates for combination chemotherapy was studied in a phase II clinical trial. Patients received oral lenalidomide at a dose of 25 mg, daily, on days 1–21 of each 28‐day cycle. The ORR was 26%; 8% of patients had a CR. The median PFS and OS were 4 months and 12 months, respectively. The median duration of response was 13 months. Among the patients who had R/R PTCL (29 patients), the ORR was 24%, and the median OS, PFS, and duration of response were 12 months, 4 months, and 5 months, respectively. The most common grade 4 AEs were pain, not otherwise specified (21%); neutropenia (13%); muscle weakness (10%); and dyspnea (10%) [60].
A multicenter, open‐label phase II trial (EXPECT) tested the efficacy and safety of 25 mg lenalidomide, taken orally once daily on days 1–21 of each 28‐day cycle, for a maximum of 24 months, until disease progression or development of unacceptable toxicity, in 54 patients with R/R PTCL, mostly with AITL (n = 26; 48%) and PTCL‐NOS (n = 20; 37%). The ORR was 22%, including CR or unconfirmed CR/CRu in 11% of patients. The median PFS and duration of response were 2.5 and 3.6 months, respectively. The most common grade 3–4 AEs included thrombocytopenia (20%), gastrointestinal disorders (17%), neutropenia (15%), and infections (15%) [61].
Bortezomib
Bortezomib is a proteasome inhibitor with clinical activity in multiple myeloma and mantle cell lymphoma. A phase II trial evaluated the efficacy and tolerability of bortezomib (1.3 mg/m2 i.v. on days 1, 4, 8, and 11, every 21 days, for a total of six cycles) in a series of 15 patients with relapsed or refractory CTCL or PTCL with isolated skin involvement. Twelve patients were evaluable. The ORR was 67%, with 17% CR. The most common grade 3 AEs were neutropenia (n = 2), thrombocytopenia (n = 2), and sensory neuropathy (n = 2). There was no reported grade 4 toxicity [62].
Bortezomib was evaluated in combination with a standard CHOP regimen in a phase II study as first‐line treatment for patients with PTCL (n = 41) and CTCL (n = 5). Bortezomib was administered on days 1 and 8 at a dose of 1.6 mg/m2 in addition to CHOP every 3 weeks for a total of six cycles. The ORR was 76%, with 65% of patients achieving CR. The 3‐year OS and PFS were 47% and 35%, respectively, because of frequent relapse after remission. Grade 3–4 leucopenia was the most frequent toxicity, whereas neurotoxicity was tolerable—grade 1 or 2 of peripheral neuropathy. The outcome was similar to CHOP alone [63].
Alemtuzumab
Alemtuzumab is a monoclonal antibody that binds to CD52, a protein that is present on the surface of mature lymphocytes but not on the stem cells from which they are derived. CD52‐bearing lymphocytes are targeted for destruction after treatment with alemtuzumab. Alemtuzumab was evaluated in 14 patients with R/R PTCL. Patients received a rapidly escalating dosage of alemtuzumab during the first week and then 30 mg i.v. three times per week for a maximum of 12 weeks. The ORR rate was 35.7%, with three patients achieving CR (21.4%) and two achieving PR (14.3%). However, significant hematologic toxicity and infectious complications were reported (cytomegalovirus reactivation in six patients, pulmonary aspergillosis in two patients, pancytopenia in four patients, and Epstein‐Barr virus‐related hemophagocytosis in two patients). Five patients died of causes related to the treatment in combination with advanced disease. The toxicity revealed by this study suggests that lower doses and/or different schedules should be explored [64].
A subcutaneous form of alemtuzumab was tested in combination with the CHOP regimen to reduce treatment‐related toxicity in a prospective multicenter trial in untreated patients with PTCL. Twenty‐four patients were successively included and received eight cycles of CHOP plus 30 mg alemtuzumab subcutaneously on day −1 initially for the first four courses (four patients), and then for all eight courses (20 patients). CR was achieved in 17 patients (71%), and one had a PR. After a median follow‐up of 16 months, 58% of patients were alive, 38% died from disease progression, and one died from pneumonia after a CR. The most frequent side effects were grade 4 neutropenia and cytomegalovirus (CMV) reactivation. Major infections were Jakob‐Creutzfeldt virus reactivation, pulmonary invasive aspergillosis, Staphylococcus sepsis, and pneumonia [65].
Emerging Drugs in Late‐Phase Trials
Mogamulizumab
Mogamulizumab (KW‐0761) is a defucosylated humanized anti‐CCR4 antibody engineered to exert potent antibody‐dependent cellular cytotoxicity.
The efficacy and safety of mogamulizumab were studied in a multicenter phase II trial in patients with relapsed CCR4‐positive PTCL or CTCL. Mogamulizumab (1.0 mg/kg) was administered i.v. once per week for 8 weeks to 37 patients. The ORR was 35%, including 14% CR; the median PFS was 3 months. The most common grade 3–4 AEs were lymphocytopenia (73%) and neutropenia (19%) [66].
More recently, a prospective, multicenter, randomized trial of mogamulizumab (1.0 mg/kg), administered weekly for the first 4‐week cycle and then biweekly versus one of three investigator‐chosen regimens (gemcitabine and oxaliplatin, DHAP, or pralatrexate) to patients with R/R adult T‐cell leukemia/lymphoma (ATLL), was performed. Seventy‐one patients were randomized (47 to mogamulizumab, 24 to investigator's choice). In the mogamulizumab‐treated group, the ORR was 23.4% (11/47); in the investigator's choice group, the ORR was 8.3% (2/24). Among the investigator's choice patients, 18 crossed over to mogamulizumab, and 3 of these were responders. The median duration of response for mogamulizumab was 5.0 months, and one patient had a CR lasting >9 months. The most common drug‐related AEs in the mogamulizumab group were infusion reactions (46.8%), rash/drug eruption (25.5%), and infections (14.9%). These data are promising for the treatment of aggressive R/R ATLL, which usually responds poorly to cytotoxic regimens [67].
Alisertib
Aurora A kinase (AAK) is overexpressed in aggressive lymphomas and can correlate with more histologically aggressive forms of the disease. AAK has been found to be upregulated in PTCL, most strongly in ALK+ ALCL, followed by ALK− ALCL and PTCL‐NOS [68].
Alisertib is a novel oral AAK inhibitor without adverse safety signals in early‐phase studies that demonstrated preliminary activity in TCL. The SWOG group reported the data of 37 patients with R/R PTCL or transformed mycosis fungoides (tMF) treated with 50 mg alisertib, twice a day, for 7 days on 21‐day cycles in a phase II trial. The ORR was 30% among the PTCL subtypes, whereas no responses were observed in patients with tMF. The median duration of response was 3 months. The median PFS and OS were 3 and 8 months, respectively. The most common grade 3–4 AEs were neutropenia (32%), anemia (30%), thrombocytopenia (24%), febrile neutropenia (14%), mucositis (11%), and rash (5%). Treatment was most frequently discontinued because of disease progression [69].
Preclinical studies support the combination of alisertib with an HDAC inhibitor. Alisertib and romidepsin showed synergy in vivo on a TCL cell line inoculated into severe combined immunodeficiency mice [70].
Based on these data, a phase I clinical trial was performed to evaluate the combination of alisertib and romidepsin in R/R PTCL and B‐cell lymphoma. Patients were treated with alisertib orally on days 1–7 and romidepsin i.v. on days 1 and 8, with five planned dose escalations. The median number of cycles was 1.5 (range, 1–8) with a median time for retreatment of 28.5 days (range, 22–40).
Eight of nine enrolled patients were evaluable for response, including three with PTCL, with a median number of four prior therapies. Grade 3–4 toxicities were neutropenia, thrombocytopenia, and anemia in 45%, 45%, and 20% of the cycles, respectively. One patient with PTCL achieved a CR, one had stable disease, and one had disease progression. Further enrollment in this trial is ongoing [71].
Crizotinib
Patients with ALK+ lymphomas have a relatively favorable outcome to anthracycline‐based chemotherapy, but those who relapse have a poor prognosis [72]. Crizotinib is an oral, small‐molecule tyrosine kinase inhibitor, with specificity for ALK and ROS1 (c‐ros oncogene 1), which has been approved in the U.S. and several other countries for the treatment of some non‐small cell lung carcinomas harboring a translocation of the ALK gene. It was tested in a small series of 11 patients with refractory ALK+ lymphoma, 9 with ALCL histology. It was administered at a dose of 250 mg twice daily until disease progression. All nine patients with ALK+ ALCL achieved a CR. OS and PFS rates after 2 years were 72.7% and 63.7%, respectively. Three patients had a CR lasting >30 months under continuous crizotinib administration. All toxicities were grade 1–2, including ocular flashes, peripheral edema, and neutropenia [73].
A pilot phase II study of crizotinib in patients with ALK+ lymphomas resistant or refractory to standard cytotoxic treatment is ongoing (NCT02419287).
Duvelisib
Duvelisib, previously known as IPI‐145, is an oral inhibitor of phosphoinositide‐3 kinase (PI3K) ‐δ and ‐γ being tested as a treatment for hematological malignancies, as well as a broad range of inflammatory conditions. The safety and efficacy of duvelisib has been evaluated in a phase I trial, in 210 patients with advanced hematologic malignancies; 63% had received ≥3 prior systemic anticancer regimens. Thirty‐one patients received duvelisib 8–100 mg twice daily in the dose escalation phase, and 179 in the expansion phase were treated with 25 or 75 mg duvelisib twice daily continuously. The ORR rate of patients with PTCL (n = 16) was 50%, including three CRs. Neutropenia was the most frequent hematologic AE, occurring in 39% of patients of the entire cohort, with 32% of patients experiencing grade ≥3 events. The most common nonhematological AEs overall and the most common nonhematologic events of grade ≥3 were transaminase (ALT/AST) elevations (38%) and diarrhea (42%). The grade 3–4 toxicity, reported in >5% of patients, was pneumonia in 28 patients (13%). Pneumocystis jirovecii pneumonia was reported in three cases, and CMV infection was reported in two cases. Among the 11 fatal AEs other than disease progression in the entire cohort, two patients with TCL and R/R disease experienced fatal AEs [74]. These data support further investigation of duvelisib in a multicenter, phase II, open‐label, parallel cohort study in patients with R/R PTCL (NCT03372057).
Parallel phase I studies of duvelisib plus romidepsin or bortezomib were recently reported in a 3 + 3 design study with dose expansion at maximum tolerated dose in patients with R/R TCL. Patients received romidepsin at 10 mg/m2 i.v. at days 1, 8, and 15 and duvelisib at 75 mg, PO, twice daily, days 1–28, in the MTD arm A, dose level 3. Eleven patients with PTCL were evaluable for responses. The ORR and CR rate were 64% and 36%, respectively. In Arm B, patients received duvelisib at 25 mg, PO, twice daily, days 1–28, and bortezomib at 1 mg/m2 subcutaneously on days 1,4, 8, and 11, at the MTD dose level 1. Ten patients with PTCL were evaluable for responses. The ORR and CR rate were 50% and 30%, respectively. The duvelisib plus romidepsin combination was well tolerated, unlike the duvelisib plus bortezomib combination, in which AST/ALT elevation limited tolerability upon dose escalation. Further expansion of the duvelisib + romidepsin cohort is planned to further evaluate the efficacy of this combination (Table 5) [75].
Table 5. Clinical trial efficacy data of licensed or emergent combinations.

TCL.
Angioimmunoblastic T‐cell lymphoma.
Abbreviations: BCL, B‐cell lymphoma; CLL, chronic lymphocytic leukemia; CR, complete remission; DoR, duration of response; ORR, overall response rate; PTCL, peripheral T‐cell lymphoma; Ref., reference; TCL, T‐cell lymphoma.
Other Agents in Early‐Phase Trials
Frequent mutations affecting the IDH2 and TET2 genes have been identified in AITL [36], [38], [39], [40]. TET2 mutations have been reported in 47%–81% of all AITL analyzed [76], [77].
The LYSA group reported the results of 5‐azacytidine (5‐AZA) treatment in 19 patients with R/R PTCL (12 with AITL). TET2 was sequenced in 14 patients. It was mutated in 8/10 (80%) with AITL and 1/4 (25%) with other PTCL. 5‐AZA was administered by subcutaneous injection of 75 mg/m2 during 7 consecutive days every 28 days, until progression or unacceptable tolerability. Ten patients had a previous or concomitant diagnosis of myelodysplastic syndrome (MDS). The ORR was 53% (10/19) and was significantly higher in patients with AITL than in patients with other PTCL subtypes (9/12, 75% vs. 1/7, 15%). Five patients with AITL achieved CR, and all of them were TET2 mutated. Among the nine patients with AITL who responded, only two patients experienced progression, after 86 and 499 days, respectively [78].
Furthermore, a phase I–II, multicenter study is ongoing to evaluate the safety and clinical activity of AG‐221, a first‐in‐class, oral, selective, potent inhibitor of mutant IDH2 in patients with advanced solid tumors and AITL with an IDH2 mutation (NCT02273739).
Tenalisib, also known as RP6530, is a highly specific inhibitor of PI3K δ/γ isoforms and an anticellular proliferation agent for treatment of hematologic malignancies. Tenalisib was investigated to evaluate safety and efficacy in a phase I/Ib study using a standard 3 + 3 dose escalation schema in 11 patients with R/R mature T‐cell neoplasms (median number of previous treatments, 3). To date, 6 patients with PTCL and 5 patients with CTCL have been enrolled at three dose levels: 200 mg twice daily, 400 mg twice daily, and 800 mg twice daily. No grade 3–4 drug‐related AEs have been observed, except for ALT/AST elevation in one patient. Five patients have been evaluated for responses at cycle 3, day,1. Two patients (one with PTCL and one with CTCL) experienced PRs (40%) that have been ongoing for >5 months [79].
Nivolumab is a humanized IgG4 anti‐programmed death 1 (PD‐1) monoclonal antibody that potentiates T‐cell activity. It has demonstrated clinical efficacy in various solid tumors and has been tested in an open‐label trial enrolling patients with R/R lymphoid malignancies, including B‐cell NHL, T‐cell NHL, multiple myeloma, and classical Hodgkin lymphoma.
Eighty‐two heavily pretreated patients (≥3 prior treatment regimens, 78%), including 23 with T‐NHL (13 with mycosis fungoides, 5 with PTCL, and 5 with other T‐cell lymphomas), received nivolumab in a dose‐escalation study (1 mg/kg and 3 mg/kg) every 2 weeks for up to 2 years. Drug‐related AEs occurred in 65% of patients with T‐NHL. Serious AEs were pneumonitis, rash, and sepsis, each occurring in 4% of patients. The ORR was 17% (no CR), including an ORR of 40% in the five patients with PTCL. With median follow‐up times of 44 weeks, response durations for the two patients with PTCL were 10.6 and 78.6 + weeks, respectively. The median duration of stable disease (SD) was 11.0 weeks (range, 7·1–42·9+ weeks) for 10 patients with T‐cell lymphomas (Table 6) [80].
Table 6. Clinical trial efficacy data of new drug monotherapy.

Abbreviations: —, not available; ALK, anaplastic lymphoma kinase; CTCL, cutaneous T‐cell lymphoma; CR, complete remission; Cru, unconfirmed complete remission; DoR, duration of response; ORR, overall response rate; OS, overall survival; MoAb, monoclonal antibodies; NK, natural killer; PFS, progression‐free survival; PI3K, phosphoinositide‐3 kinase; PTCL, peripheral T‐cell lymphoma; Ref., reference.
Several cancers, including lymphoma, have been associated with activation of the JAK/STAT pathway by multiple mechanisms, including inappropriate autocrine and paracrine cytokine stimulation [81] as well as activating mutations [82].
Ruxolitinib, a selective JAK1/2 inhibitor approved by the FDA for myeloproliferative neoplasms, is being studied in relapsed B‐cell NHL and PTCL (NCT01431209).
NK‐/T‐Cell Lymphoma (Nasal and Extranasal)
Although rare in Europe and the U.S., extranodal NK/T‐cell lymphoma (ENKTL) is diagnosed comparatively frequently in Asia and Latin America and represents 5%–10% of all malignant lymphomas in China [83], [84].
ENKTL has a predilection for the nasal cavity and upper aerodigestive tract, and skin and soft tissue infiltration have also been reported [85]. ENKTL is characterized by the expression of P‐glycoprotein, which is related to multidrug resistance (MDR), for which reason ENKTL is resistant to anthracycline‐based chemotherapies, and it is associated with Epstein‐Barr virus (EBV). In the first setting, guidelines recommend treatments, including local radiotherapy for patients with nasal type ENKTL, stage I, and contiguous stage II disease with cervical lymph node involvement and chemotherapy with L‐asparaginase and/or nonrelated MDR compounds for patients with advanced extranodal NK/T‐cell lymphoma (ENKTL) [25], [43].
Conventional Chemotherapy and Radiotherapy
Recently, a cooperative study in Japan retrospectively analyzed the management and outcomes of 358 patients diagnosed between 2000 and 2003 with nasal type ENKTL; 82% of them received a regimen of concurrent radiotherapy + dexamethasone, etoposide, ifosfamide, and carboplatin (RT‐DeVIC). Of 257 patients with localized disease, 66% received RT‐DeVIC as first‐line therapy. The 5‐year OS for localized and advanced ENKTL were 68% and 24%, respectively. The 5‐year OS and PFS in 150 patients treated with RT‐DeVIC in clinical practice were 72% and 61%, respectively [86].
Zhang et al. reported the 5‐year analysis of a phase II trial of “sandwich” chemo‐radiotherapy, comprising L‐asparaginase, vincristine, and prednisone chemotherapy + radiotherapy, in patients with newly diagnosed, stage IE–IIE, nasal type ENKTL. The 5‐year OS and PFS rates were both 64%. The initial therapeutic response (odds ratio, 5.83; p = .001) and B symptoms (odds ratio, 6.13; p = .043) were significant prognostic factors for OS, whereas the IPI score was not significant for PFS or OS [87].
Suzuki et al. reported the 5‐year follow‐up of the phase II study of steroid (dexamethasone), methotrexate, ifosfamide, L‐asparaginase, and etoposide (SMILE) in 38 patients with newly diagnosed, stage IV, relapsed or refractory nasal‐type ENKTL. Twenty patients had newly diagnosed lymphoma, 14 had first relapse, and 4 had refractory disease. The ORR and complete response rate after two cycles of the SMILE regimen were 79% and 45%, respectively, and the 5‐year OS rate was 47%. Grade 4 neutropenia occurred in 92% of the patients. The most common grade 3 or 4 nonhematologic complication was infection (61%) [88].
Li et al. reported the results of the randomized, controlled, open‐label, multicenter study that compared the efficacy and safety of six cycles of a regimen with pegaspargase, gemcitabine, and cisplatin‐dexamethasone (DDGP) with 6 cycles of the SMILE regimen in 42 patients (21 vs. 21) with newly diagnosed ENKTL in stages III–IV. The 1‐year PFS (86% vs. 38%, p = .006) and 2‐year OS rates (74% vs. 45%, p = .027), as well as the ORR rates (95% vs. 67%, p = .018) and CR rates (71% vs. 29%, p = .005), were better in the DDGP group than in the SMILE group. Furthermore, patients in the SMILE group developed more serious leucopenia (p = .030) and severe allergic reaction (p = .015) than in the DDGP group [89].
Wang et al. retrospectively studied a cohort of 117 patients with newly diagnosed or relapsed/refractory ENKTL treated with a regimen of gemcitabine, oxaliplatin, and pegaspargase. The OR rate on completion of treatment was 88.8%, and responses were similar for newly diagnosed and relapsed/refractory patients. The 3‐year OS and PFS rates were 72.7% and 57.8%, respectively [90].
The Korean Consortium for Improving Survival of Lymphoma analyzed 62 patients with newly diagnosed ENKTL who underwent up‐front ASCT after primary therapy. Thirty‐one had advanced stage (50%), 16 (25.8%), had high‐intermediate‐ to high‐risk IPI, and 42 (67.7%) were in group 3–4 of the NK/T Cell Lymphoma Prognostic Index. The pretransplant CR rate and PR were 61.3% and 38.7%, respectively, whereas the post‐transplantation final CR rate was 78.3%. The 3‐year PFS and OS were 52.4% and 60.0%, respectively. Patients with limited disease had significantly better 3‐year PFS (64.5% vs. 40.1%, p = .017) and OS (67.6% vs. 52.3%, p = .048) than those with advanced disease [91].
New Agents
EBV‐infected ENKTL upregulates programmed death ligand 1 (PD‐L1), the ligand of the inhibitory receptor PD‐1 on T‐cells. Thus, the PD‐L1/PD‐1 axis is a potential mechanism for NK/T‐cell lymphomas to avoid effector T‐cell targeting. Kwong et al. treated seven patients with relapsed/refractory NK/T‐cell lymphoma with the anti‐PD‐1 antibody pembrolizumab; the patients had failed a median of two previous regimens, including L‐asparaginase regimens and allogeneic hematopoietic stem‐cell transplantation (HSCT) in two cases. The patients received a median of 7 (range, 2–13) cycles of pembrolizumab. All patients were responders, two achieved CR in all tested parameters (clinical, radiologic [positron emission tomography], morphologic, and molecular [circulating EBV DNA]), three patients achieved clinical and radiologic CRs, and two achieved PR. After a median of 6 months, all five CR patients were still in remission. One patient with previous allogeneic HSCT developed grade 2 skin GVH‐disease. Strong PD‐L1 expression was correlated with excellent responses: Four patients had strong expression of the PD‐L1 ligand; among them, three achieved CR. One patient with a weak expression of the PD‐L1 ligand achieved a PR. Data were not available for two patients [92].
Wang et al. measured pre‐ and post‐treatment serum soluble PD‐L1 (sPD‐L1) levels in 97 patients with newly diagnosed, early‐stage ENKTL treated with asparaginase‐based chemotherapy followed by radiotherapy. Patients with high pretreatment (>3.23 ng/mL) serum sPD‐L1 levels had shorter PFS and OS. In a multivariate survival analysis, post‐treatment sPD‐L1 >1.12 ng/mL, treatment response (CR vs. incomplete response), and stage II disease were independent prognostic factors for shorter PFS and OS. Furthermore, post‐treatment sPD‐L1 >1.12 ng/mL was associated with shorter PFS and OS in patients who achieved CR, suggesting the use of sPD‐L1 as a marker of minimal residual disease [93].
Hari et al. reported a case of relapsed ENKTL‐nasal type with circulating aberrant CD38+ NK cells, previously treated with pegylated asparaginase‐based combination chemotherapy followed by allogeneic hematopoietic‐cell transplantation. The patient then received off‐label therapy with daratumumab, an IgG1k monoclonal antibody directed against CD38, inducing potent antibody‐dependent cellular cytotoxicity and complement‐dependent cytotoxicity. Daratumumab was given at a dose of 16 mg/kg per week. The patient achieved complete remission with undetectable EBV by PCR after 6 weeks and was still in CR at 21 weeks of follow‐up [94].
Kim et al. reported a case of refractory CD30‐positive ENKTL, heavily pretreated. The patient received four cycles of the single‐agent BV and achieved CR, but the treatment was stopped for 3 months because of dyspnea, and relapse was observed [95].
Conclusion
Recent advances in our understanding of the biology of PTCLs and their improved molecular characterization have allowed the emergence of promising new drugs, some of which have been recently approved by the FDA for the treatment of R/R PTCL. However, a large proportion of new drugs being tested display only moderate efficacy when used alone. A better understanding of their modes of action, targeted pathways, and synergistic actions when combined with other agents will allow the development of more effective drug combinations. These novel drugs are being evaluated in combination with CHOP, CHOP‐like, or non‐anthracycline‐based chemotherapy in the front‐line setting and are also being tested together in combination in the R/R setting.
The development of novel therapies requires the development of new treatment paradigms that combine these agents to improve response rates and durability of responses. There are still many questions that can only be addressed by prospective, randomized, phase III trials with the least possible bias. They must take into account the various elements of therapeutic management, including response rates; EFS; OS; toxicity and its short, medium, and long‐term consequences, as well as the costs of these new drugs, often used continuously. The therapeutic management must be well adapted to the patient and take into account the different epidemiological, clinical, histological, and molecular aspects of the tumor.
The recent progress made with the four recently FDA‐approved drugs, pralatrexate, brentuximab vedotin, romidepsin and belinostat, must be confirmed in the front‐line setting either by substituting older, less effective drugs (brentuximab vedotin plus CHP vs. CHOP), or in combination with chemotherapy (CHOP + romidepsine vs. CHOP). They should then be tested in combinations without chemotherapy with the goal to eventually replace chemotherapy.
Author Contributions
Conception/design: Kamel Laribi
Collection and/or assembly of data: Kamel Laribi, Mustapha Alani, Catherine Truong, Alix Baugier de Materre
Data analysis and interpretation: Kamel Laribi, Mustapha Alani, Catherine Truong, Alix Baugier de Materre
Manuscript writing: Kamel Laribi, Mustapha Alani, Alix Baugier de Materre
Final approval of manuscript: Kamel Laribi, Mustapha Alani, Catherine Truong, Alix Baugier de Materre
Disclosures
Kamel Laribi: Hospira, Roche, Teva, Janssen‐Cilag, Mundipharma (RF), Amgen, Takeda, Novartis (C/A). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1. Swerdlow SH, Campo E, Pileri SA et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016;127:2375–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Vose J, Armitage J, Weisenburger D; International T‐Cell Lymphoma Project . International peripheral T‐cell and natural killer/T‐cell lymphoma study: Pathology findings and clinical outcomes. J Clin Oncol 2008;26:4124–4130. [DOI] [PubMed] [Google Scholar]
- 3. Rodríguez J, Conde E, Gutiérrez A et al. The adjusted International Prognostic Index and beta‐2‐microglobulin predict the outcome after autologous stem cell transplantation in relapsing/refractory peripheral T‐cell lymphoma. Haematologica 2007;92:1067–1074. [DOI] [PubMed] [Google Scholar]
- 4. Went P, Agostinelli C, Gallamini A et al. Marker expression in peripheral T‐cell lymphoma: A proposed clinical‐pathologic prognostic score. J Clin Oncol 2006;24:2472–2479. [DOI] [PubMed] [Google Scholar]
- 5. Pescarmona E, Pignoloni P, Puopolo M et al. p53 over‐expression identifies a subset of nodal peripheral T‐cell lymphomas with a distinctive biological profile and poor clinical outcome. J Pathol 2001;195:361–366. [DOI] [PubMed] [Google Scholar]
- 6. Foss F. Hematology: Relapsed and refractory PTCL–into the therapeutic abyss. Nat Rev Clin Oncol 2011;8:321–322. [DOI] [PubMed] [Google Scholar]
- 7. Abouyabis AN, Shenoy PJ, Sinha R et al. A systematic review and meta‐analysis of front‐line anthracycline‐based chemotherapy regimens for peripheral T‐cell lymphoma. ISRN Hematol 2011;2011:623924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Savage KJ, Harris NL, Vose JM et al. ALK− anaplastic large‐cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T‐cell lymphoma, not otherwise specified: Report from the International Peripheral T‐Cell Lymphoma Project. Blood 2008;111:5496–5504. [DOI] [PubMed] [Google Scholar]
- 9. Schmitz N, Trümper L, Ziepert M et al. Treatment and prognosis of mature T‐cell and NK‐cell lymphoma: An analysis of patients with T‐cell lymphoma treated in studies of the German High‐Grade Non‐Hodgkin Lymphoma Study Group. Blood 2010;116:3418–3425. [DOI] [PubMed] [Google Scholar]
- 10. Weisenburger DD, Savage KJ, Harris NL et al. Peripheral T‐cell lymphoma, not otherwise specified: A report of 340 cases from the International Peripheral T‐cell Lymphoma Project. Blood 2011;117:3402–3408. [DOI] [PubMed] [Google Scholar]
- 11. Pfreundschuh M, Trümper L, Kloess M et al.; German High‐Grade Non‐Hodgkin's Lymphoma Study Group . Two‐weekly or 3 weekly CHOP chemotherapy with or without etoposide for the treatment of young patients with good‐prognosis (normal LDH) aggressive lymphomas: Results of the NHL‐B1 trial of the DSHNHL. Blood 2004;104:626–633. [DOI] [PubMed] [Google Scholar]
- 12. Gkotzamanidou M, Papadimitriou CA. Peripheral T‐cell lymphoma: The role of hematopoietic stem cell transplantation. Crit Rev Oncol Hematol 2014;89:248–261. [DOI] [PubMed] [Google Scholar]
- 13. Rodríguez J, Conde E, Gutiérrez A et al. The adjusted International Prognostic Index and beta‐2‐microglobulin predict the outcome after autologous stem cell transplantation in relapsing/refractory peripheral T‐cell lymphoma. Haematologica 2007;92:1067–1074. [DOI] [PubMed] [Google Scholar]
- 14.Reimer, Rüdiger T, Geissinger E et al. Autologous stem‐cell transplantation as first‐line therapy in peripheral T‐cell lymphomas: Results of a prospective multicenter study. J Clin Oncol 2009;27:106–113. [DOI] [PubMed] [Google Scholar]
- 15. Corradini P, Tarella C, Zallio F et al. Long‐term follow‐up of patients with peripheral T‐cell lymphomas treated up‐front with high dose chemotherapy followed by autologous stem cell transplantation. Leukemia 2006;20:1533–1538. [DOI] [PubMed] [Google Scholar]
- 16. Mercadal S, Briones J, Xicoy B et al. Intensive chemotherapy (high‐dose CHOP/ESHAP regimen) followed by autologous stem‐cell transplantation in previously untreated patients with peripheral T‐cell lymphoma. Ann Oncol 2008;19:958–963. [DOI] [PubMed] [Google Scholar]
- 17. Abramson JS, Feldman T, Kroll‐Desrosiers AR et al. Peripheral T‐cell lymphomas in a large US multicenter cohort: Prognostication in the modern era including impact of frontline therapy. Ann Oncol 2014;25:2211–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Corradini P, Dodero A, Zallio F et al. Graft‐versus‐lymphoma effect in relapsed peripheral T‐cell non‐Hodgkin's lymphomas after reduced‐intensity conditioning followed by allogeneic transplantation of hematopoietic cells. J Clin Oncol 2004;22:2172–2176. [DOI] [PubMed] [Google Scholar]
- 19. Kanakry JA, Kasamon YL, Gocke CD et al. Outcomes of related donor HLA‐identical or HLA‐haploidentical allogeneic blood or marrow transplantation for peripheral T cell lymphoma. Biol Blood Marrow Transplant 2013;19:602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kyriakou C, Canals C, Finke J et al. Allogeneic stem cell transplantation is able to induce long‐term remissions in angioimmunoblastic T‐cell lymphoma: A retrospective study from the lymphoma working party of the European Group for Blood and Marrow Transplantation. J Clin Oncol 2009;27:3951–3958. [DOI] [PubMed] [Google Scholar]
- 21. Ascani S, Zinzani PL, Gherlinzoni F et al. Peripheral T‐cell lymphomas. Clinico‐pathologic study of 168 cases diagnosed according to the R.E.A.L. Classification. Ann Oncol 1997;8:583–592. [DOI] [PubMed] [Google Scholar]
- 22. Arrowsmith ER, Macon WR, Kinney MC et al. Peripheral T‐cell lymphomas: Clinical features and prognostic factors of 92 cases defined by the revised European American lymphoma classification. Leuk Lymphoma 2003;44:241–249. [DOI] [PubMed] [Google Scholar]
- 23. Falini B, Pileri S, De Solas I et al. Peripheral T‐cell lymphoma associated with hemophagocytic syndrome. Blood 1990;75:434–444. [PubMed] [Google Scholar]
- 24. Barrington SF, Mikhaeel NG, Kostakoglu L et al. Role of imaging in the staging and response assessment of lymphoma: Consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J Clin Oncol 2014;32:3048–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. d'Amore F, Gaulard P, Trümper L et al.; ESMO Guidelines Committee. Peripheral T‐cell lymphomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol 2015;26(suppl 5):v108–v115. [DOI] [PubMed] [Google Scholar]
- 26. Ellin F, Landström J, Jerkeman M et al. Real‐world data on prognostic factors and treatment in peripheral T‐cell lymphomas: A study from the Swedish Lymphoma Registry. Blood 2014;124:1570–1577. [DOI] [PubMed] [Google Scholar]
- 27. Carson KR, Horwitz SM, Pinter‐Brown LC et al. A prospective cohort study of patients with peripheral T‐cell lymphoma in the United States. Cancer 2017;123:1174–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mahadevan D, Unger JM, Spier CM et al. Phase 2 trial of combined cisplatin, etoposide, gemcitabine, and methylprednisolone (PEGS) in peripheral T‐cell non‐Hodgkin lymphoma: Southwest Oncology Group Study S0350. Cancer 2013;119:371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mak V, Hamm J, Chhanabhai M et al. Survival of patients with peripheral T‐cell lymphoma after first relapse or progression: Spectrum of disease and rare long‐term survivors. J Clin Oncol 2013;31:1970–1976. [DOI] [PubMed] [Google Scholar]
- 30. Arkenau HT, Chong G, Cunningham D et al. Gemcitabine, cisplatin and methylprednisolone for the treatment of patients with peripheral T‐cell lymphome: The Royal Marsden Hospital experience. Hematologica 2007;92:271–272. [DOI] [PubMed] [Google Scholar]
- 31. Chen AI, McMillan A, Negrin RS et al. Long‐term results of autologous hematopoietic cell transplantation for peripheral T cell lymphoma: The Stanford experience. Biol Blood Marrow Transplant 2008;14:741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. d'Amore F, Relander T, Lauritzsen GF et al. Up‐front autologous stem‐cell transplantation in peripheral T‐cell lymphoma: NLG‐T‐01. J Clin Oncol 2012;30:3093–3099. [DOI] [PubMed] [Google Scholar]
- 33. Feyler S, Prince HM, Pearce R et al. The role of high‐dose therapy and stem cell rescue in the management of T‐cell malignant lymphomas: A BSBMT and ABMTRR study. Bone Marrow Transplant 2007;40:443–450. [DOI] [PubMed] [Google Scholar]
- 34. O'Connor OA, Pro B, Pinter‐Brown L et al. Pralatrexate in patients with relapsed or refractory peripheral T‐cell lymphoma: Results from the pivotal PROPEL study. J Clin Oncol 2011;29:1182–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shustov A, Pro B, Gisselbrecht C et al. Pralatrexate is effective as second‐line treatment following cyclophosphamide/doxorubicin/vincristine/prednisone (CHOP) failure in patients with relapsed or refractory peripheral T‐Cell lymphoma (PTCL). Blood 2010;116:4882A. [Google Scholar]
- 36. Couronné L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T‐cell lymphoma. N Engl J Med 2012;366:95–96. [DOI] [PubMed] [Google Scholar]
- 37. Palomero T, Couronné L, Khiabanian H et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet 2014;46:166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Quivoron C, Couronné L, Della Valle V et al. TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 2011;20:25–38. [DOI] [PubMed] [Google Scholar]
- 39. Odejide O, Weigert O, Lane AA et al. A targeted mutational landscape of angioimmunoblastic T‐cell lymphoma. Blood. 2014;123:1293–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cairns RA, Iqbal J, Lemonnier F et al. IDH2 mutations are frequent in angioimmunoblastic T‐cell lymphoma. Blood 2012;119:1901–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Coiffier B, Pro B, Prince HM et al. Results from a pivotal, open‐label, phase II study of romidepsin in relapsed or refractory peripheral T‐cell lymphoma after prior systemic therapy. J Clin Oncol 2012;30:631–636. [DOI] [PubMed] [Google Scholar]
- 42. Coiffier B, Pro B, Prince HM et al. Romidepsin for the treatment of relapsed/refractory peripheral T‐cell lymphoma: Pivotal study update demonstrates durable responses. J Hematol Oncol 2014;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.National Comprehensive Cancer Network . NCCN Clinical Practice Guidelines in Oncology: Non‐Hodgkin's Lymphomas Version 1.2017. Available at http://www.nccn.org/professionals/physician_gls/f_guidelines.asp. Accessed December 19, 2016.
- 44. O'Connor OA, Horwitz S, Masszi T et al. Belinostat in patients with relapsed or refractory peripheral T‐cell lymphoma: Results of the pivotal phase II BELIEF (CLN‐19) study. J Clin Oncol 2015;33:2492–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Francisco JA, Cerveny CG, Meyer DL et al. cAC10‐vcMMAE, an anti‐CD30‐monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003;102:1458–1465. [DOI] [PubMed] [Google Scholar]
- 46. Pro B, Advani R, Brice P et al. Brentuximab vedotin (SGN‐35) in patients with relapsed or refractory systemic anaplastic large‐cell lymphoma: Results of a phase II study. J Clin Oncol 2012;30:2190–2196. [DOI] [PubMed] [Google Scholar]
- 47. Pro B, Advani R, Brice P et al. Five‐year results of brentuximab vedotin in patients with relapsed or refractory systemic anaplastic large cell lymphoma. Blood 2017;130:2709–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Horwitz SM, Advani RH, Bartlett NL et al. Objective responses in relapsed T‐cell lymphomas with single‐agent brentuximab vedotin. Blood 2014;123:3095–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dupuis J, Morschhauser F, Ghesquières H et al. Combination of romidepsin with cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated patients with peripheral T‐cell lymphoma: A non‐randomised, phase 1b/2 study. Lancet Haematol 2015;2:e160–e165. [DOI] [PubMed] [Google Scholar]
- 50. Amengual JE, Lichtenstein R, Rojas C et al. The pralatrexate‐romidepsin doublet: A well tolerated and highly effective combination for patients with relapsed or refractory peripheral T‐cell lymphoma. Blood 2016;128:1824A. [Google Scholar]
- 51. Mehta‐Shah N, Lunning MA, Boruchov AM et al. A phase I/II trial of the combination of romidepsin and lenalidomide in patients with relapsed/refractory lymphoma and myeloma: Activity in T‐cell lymphoma. J Clin Oncol 2015;33(suppl 15):8521A. [Google Scholar]
- 52. Holkova B, Yazbeck V, Kmieciak M et al. A phase 1 study of bortezomib and romidepsin in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma, indolent B‐cell lymphoma, peripheral T‐cell lymphoma, or cutaneous T‐cell lymphoma. Leuk Lymphoma 2017;58:1349–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mehta‐Shah N, Moskowitz AJ, Lunning M et al. A phase Ib/IIa trial of the combination of romidepsin, lenalidomide and carfilzomib in patients with relapsed/refractory lymphoma shows complete responses in relapsed and refractory T‐cell lymphomas. Blood 2016;128:2991A. [Google Scholar]
- 54. Advani RH, Ansell SM, Lechowicz MJ et al. A phase II study of cyclophosphamide, etoposide, vincristine and prednisone (CEOP) alternating with pralatrexate (P) as front line therapy for patients with peripheral T‐cell lymphoma (PTCL): Final results from the T‐cell consortium trial. Br J Haematol 2016;172:535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shustov A, Johnston PB, Barta SK et al. Pralatrexate in combination with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) in previously untreated patients with peripheral T‐cell lymphoma (PTCL): A phase 1 dose‐escalation study. Blood 2017;130(suppl 1):818A. [Google Scholar]
- 56. Johnston PB, Cashen AF, Nikolinakos PG et al. Safe and effective treatment of patients with peripheral T‐cell lymphoma (PTCL) with the novel HDAC inhibitor, belinostat, in combination with CHOP: Results of the Bel‐CHOP phase 1 trial. Blood 2015;126:253A. [Google Scholar]
- 57. Fanale MA, Horwitz SM, Forero‐Torres A et al. Brentuximab vedotin in the front‐line treatment of patients with CD30+ peripheral T‐cell lymphomas: Results of a phase I study. J Clin Oncol 2014;32:3137–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fanale MA, Horwitz SM, Forero‐Torres A et al. Four‐year survival and durability results of brentuximab vedotin in combination with CHp in the frontline treatment of patients with CD30‐expressing peripheral t‐Cell Lymphomas. Blood 2016;128:2993A. [Google Scholar]
- 59. Damaj G, Gressin R, Bouabdallah K et al. Results from a prospective, open‐label, phase II trial of bendamustine in refractory or relapsed T‐cell lymphomas: The BENTLY trial. J Clin Oncol 2013;31:104–110. [DOI] [PubMed] [Google Scholar]
- 60. Toumishey E, Prasad A, Dueck G et al. Final report of a phase 2 clinical trial of lenalidomide monotherapy for patients with T‐cell lymphoma. Cancer 2015;121:716–723. [DOI] [PubMed] [Google Scholar]
- 61. Morschhauser F, Fitoussi O, Haioun C et al. A phase 2, multicentre, single‐arm, open‐label study to evaluate the safety and efficacy of single‐agent lenalidomide (Revlimid) in subjects with relapsed or refractory peripheral T‐cell non‐Hodgkin lymphoma: The EXPECT trial. Eur J Cancer 2013;49:2869–2876. [DOI] [PubMed] [Google Scholar]
- 62. Zinzani PL, Musuraca G, Tani M et al. Phase II trial of proteasome inhibitor bortezomib in patients with relapsed or refractory cutaneous T‐cell lymphoma. J Clin Oncol 2007;25:4293–4297. [DOI] [PubMed] [Google Scholar]
- 63. Kim SJ, Yoon DH, Kang HJ et al. Bortezomib in combination with CHOP as first‐line treatment for patients with stage III/IV peripheral T‐cell lymphomas: A multicentre, single‐arm, phase 2 trial. Eur J Cancer 2012;48:3223–3231. [DOI] [PubMed] [Google Scholar]
- 64. Enblad G, Hagberg H, Erlanson M et al. A pilot study of alemtuzumab (anti‐CD52 monoclonal antibody) therapy for patients with relapsed or chemotherapy‐refractory peripheral T‐cell lymphomas. Blood 2004;103:2920–2924. [DOI] [PubMed] [Google Scholar]
- 65. Gallamini A, Zaja F, Patti C et al. Alemtuzumab (Campath‐1H) and CHOP chemotherapy as first‐line treatment of peripheral T‐cell lymphoma: Results of a GITIL (Gruppo Italiano Terapie Innovative nei Linfomi) prospective multicenter trial. Blood 2007;110:2316–2323. [DOI] [PubMed] [Google Scholar]
- 66. Ogura M, Ishida T, Hatake K et al. Multicenter phase II study of mogamulizumab (KW‐0761), a defucosylated anti‐cc chemokine receptor 4 antibody, in patients with relapsed peripheral T‐cell lymphoma and cutaneous T‐cell lymphoma. J Clin Oncol 2014;32:1157–1163. [DOI] [PubMed] [Google Scholar]
- 67. Phillips AA, Fields P, Hermine O et al. A prospective, multicenter, randomized study of anti‐CCR4 monoclonal antibody mogamulizumab (moga) vs investigator's choice (IC) in the treatment of patients (pts) with relapsed/refractory (R/R) adult T‐cell leukemia‐lymphoma (ATL). J Clin Oncol 2016;34(suppl 15):7501A. [Google Scholar]
- 68. Kanagal‐Shamanna R, Lehman NL, O'Donnell JP et al. Differential expression of aurora‐A kinase in T‐cell lymphomas. Mod Pathol 2013;26:640–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Barr PM, Li H, Spier C et al. Phase II intergroup trial of alisertib in relapsed and refractory peripheral T‐cell lymphoma and transformed mycosis fungoides: SWOG 1108. J Clin Oncol 2015;33:2399–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zullo K, Guo Y, Cooke L et al. The investigational Aurora A kinase inhibitor alisertib exhibits broad activity in preclinical models of T‐cell lymphoma and is highly synergistic with romidepsin. Blood 2014;124:4493A. [Google Scholar]
- 71. Fanale MA, Hagemeister FB, Fayad L et al. A phase I trial of alisertib plus romidepsin for relapsed/refractory aggressive B‐ and T‐cell lymphomas. Blood 2014;124:1744A. [Google Scholar]
- 72. Mak V, Hamm J, Chhanabhai M et al. Survival of patients with peripheral T‐cell lymphoma after first relapse or progression: Spectrum of disease and rare long‐term survivors. J Clin Oncol 2013;31:1970–1976. [DOI] [PubMed] [Google Scholar]
- 73. Gambacorti Passerini C, Farina F, Stasia A et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase‐positive lymphoma patients. J Natl Cancer Inst 2014;106:djt378. [DOI] [PubMed] [Google Scholar]
- 74. Flinn IW, O'Brien S, Kahl B et al. Duvelisib, a novel oral dual inhibitor of PI3K‐δ,γ, is clinically active in advanced hematologic malignancies. Blood 2018;131:877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Moskowitz AJ, Koch R, Mehta‐Shah N et al. In vitro, in vivo, and parallel phase I evidence support the safety and activity of duvelisib, a PI3K‐δ,γ inhibitor, in combination with romidepsin or bortezomib in relapsed/refractory T‐cell lymphoma. Blood 2017;130(suppl 1):819A. [Google Scholar]
- 76. Lemonnier F, Couronné L, Parrens M et al. Recurrent TET2 mutations in peripheral T‐cell lymphomas correlate with TFH‐like features and adverse clinical parameters. Blood 2012;120:1466–1469. [DOI] [PubMed] [Google Scholar]
- 77. Sakata‐Yanagimoto M, Enami T, Yoshida K et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet. 2014;46:171–175. [DOI] [PubMed] [Google Scholar]
- 78. Delarue R, Dupuis J, Sujobert P et al. Treatment with hypomethylating agent 5‐azacytidine induces sustained response in angioimmunoblastic T cell lymphomas. Blood 2016;128:4164A. [DOI] [PubMed] [Google Scholar]
- 79. Carlo‐Stella C, Delarue R, Barde PJ et al. Clinical activity and safety of RP6530, a dual PI3Kδ/γ inhibitor, in patients with advanced hematologic malignancies: Final analysis of a phase 1 multicenter study. Blood 2016;128:3011A. 28007734 [Google Scholar]
- 80. Lesokhin AM, Ansell SM, Armand P et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: Preliminary results of a phase Ib study. J Clin Oncol 2016;34:2698–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological malignancies. Oncogene 2013;32:2601–2613. [DOI] [PubMed] [Google Scholar]
- 82. Küçük C, Jiang B, Hu X et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from γδ‐T or NK cells. Nat Commun 2015;6:6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Au WY, Weisenburger DD, Intragumtornchai T et al. Clinical differences between nasal and extranasal natural killer/T‐cell lymphoma: A study of 136 cases from the International Peripheral T‐Cell Lymphoma Project. Blood 2009;113:3931–3937. [DOI] [PubMed] [Google Scholar]
- 84. Li YX, Liu QF, Fang H et al. Variable clinical presentations of nasal and Waldeyer ring natural killer/T‐cell lymphoma. Clin Cancer Res 2009;15:2905–2912. [DOI] [PubMed] [Google Scholar]
- 85. Chan JK, Sin VC, Wong KF et al. Nonnasal lymphoma expressing the natural killer cell marker CD56: A clinicopathologic study of 49 cases of an uncommon aggressive neoplasm. Blood 1997;89:4501–4513. [PubMed] [Google Scholar]
- 86. Yamaguchi M, Suzuki R, Oguchi M et al. Treatments and outcomes of patients with extranodal natural killer/T‐cell lymphoma diagnosed between 2000 and 2013: A cooperative study in Japan. J Clin Oncol 2017;35:32–39. [DOI] [PubMed] [Google Scholar]
- 87. Zhang L, Jiang M, Xie L et al. Five‐year analysis from phase 2 trial of “sandwich” chemoradiotherapy in newly diagnosed, stage IE to IIE, nasal type, extranodal natural killer/T‐ cell lymphoma. Cancer Med 2016;5:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Suzuki R, Kwong Y, Maeda Y et al. 5‐year follow‐up of the SMILE phase II study for newly‐diagnosed stage IV, relapsed or refractory extra‐nodal NK/T‐cell lymphoma, nasal type. Hematol Oncol 2015;33(suppl 1):140A. [Google Scholar]
- 89. Li X, Cui Y, Sun Z et al. DDGP versus SMILE in newly diagnosed advanced natural killer/T‐cell lymphoma: A randomized controlled, multicenter, open‐label study in China. Clin Cancer Res 2016;22:5223–5228. [DOI] [PubMed] [Google Scholar]
- 90. Wang JH, Wang H, Wang YJ et al. Analysis of the efficacy and safety of a combined gemcitabine, oxaliplatin and pegaspargase regimen for NK/T‐cell lymphoma. Oncotarget 2016;7:35412–35422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yhim HY, Kim JS, Mun YC et al.; Consortium for Improving Survival of Lymphoma Study. Clinical outcomes and prognostic factors of up‐front autologous stem cell transplantation in patients with extranodal natural killer/T cell lymphoma. Biol Blood Marrow Transplant 2015;21:1597–1604. [DOI] [PubMed] [Google Scholar]
- 92. Kwong YL, Chan TSY, Tan D et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T‐cell lymphoma failing l‐asparaginase. Blood 2017;129:2437–2442. [DOI] [PubMed] [Google Scholar]
- 93. Wang H, Wang L, Liu WJ et al. High post‐treatment serum levels of soluble programmed cell death ligand 1 predict early relapse and poor prognosis in extranodal NK/T cell lymphoma patients. Oncotarget 2016;7:33035–33045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hari P, Raj RV, Olteanu H. Targeting CD38 in refractory extranodal natural killer cell‐T‐cell lymphoma. N Engl J Med 2016;375:1501–1502. [DOI] [PubMed] [Google Scholar]
- 95. Kim HK, Moon SM, Moon JH et al. Complete remission in CD30‐positive refractory extranodal NK/T‐cell lymphoma with brentuximab vedotin. Blood Res 2015;50:254–256. [DOI] [PMC free article] [PubMed] [Google Scholar]