

Abstract
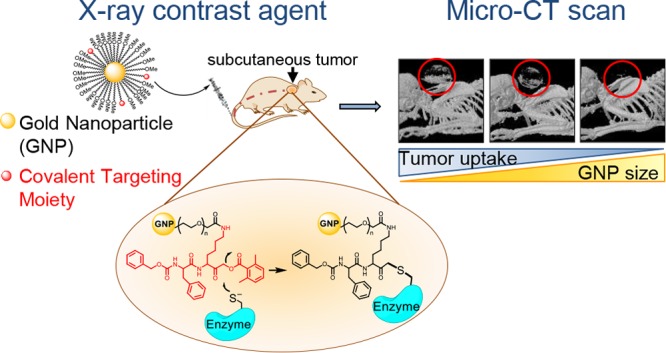
X-ray CT instruments are among the most available, efficient, and cost-effective imaging modalities in hospitals. The field of CT molecular imaging is emerging which relies mainly on the detection of gold nanoparticles and iodine-containing compounds directed to tagging a variety of abundant biomolecules. Here for the first time we attempted to detect enzymatic activity, while the low sensitivity of CT scanners to contrast reagents made this a challenging task. Therefore, we developed a new class of nanosized cathepsin-targeted activity-based probes (ABPs) for functional CT imaging of cancer. ABPs are small molecules designed to covalently modify enzyme targets in an activity-dependent manner. Using a CT instrument, these novel probes enable detection of the elevated cathepsin activity within cancerous tissue, thus creating a direct link between biological processes and imaging signals. We present the generation and biochemical evaluation of a library of ABPs tagged with different sized gold nanoparticles (GNPs), with various ratios of cathepsin-targeting moiety and a combination of different polyethylene glycol (PEG) protective layers. The most potent and stable GNP-ABPs were applied for noninvasive cancer imaging in mice. Surprisingly, detection of CT contrast from the tumor had reverse correlation to GNP size and the amount of targeting moiety. Interestingly, TEM images of tumor sections show intercellular lysosomal subcellular localization of the GNP-ABPs. In conclusion, we demonstrate that the covalent linkage is key for detection using low sensitive imaging modalities and the utility of GNP-ABPs as a promising tool for enzymatic-based CT imaging.
Introduction
One of the leading causes of death in the Western world is still cancer. Regardless of tremendous progress in the pharmaceutical and nanomaterial chemistry, early and accurate detection of the disease remains a major challenge.1 The ability to visualize, characterize, and quantify specific proteins and genes and evaluate molecular pathways within the living organism will certainly improve the diagnosis and prognosis of various conditions. Traditional pathological evaluation is often done by microscopy, in which excised tissues are typically examined to characterize histological changes within the disease process. Molecular imaging, in contrast, targets distinct molecular pathways in vivo, providing noninvasive visual and quantitative information for diverse applications.2
Computed tomography (CT) is an imaging method that uses X-rays to create cross-sectional images of the body. CT visualizes the internal structures of the body providing three-dimensional (3D) anatomic details and is routinely used for diagnosis of diseases, prediction of therapy, and treatment assessment. Currently, it is one of the most commonly used noninvasive clinical imaging modalities owing to its wide availability, high efficiency, and cost effectiveness.3,4 Although it has high spatial resolution, the main drawback of this modality for molecular imaging remains the low sensitivity and difficulty to distinguish subtle changes in soft tissue, particularly tumors which have similar X-ray absorption as their surroundings. We strongly believe that exogenous CT detectable agents that enhance tumor contrast will lead to better characterization and detection of cancer.5
A wide variety of CT contrast agents have been developed in the past decade which focus mainly on iodine-containing probes and gold nanoparticles (GNPs).6 Although iodine-based molecules are clinically used, they have some limitations, such as fast renal clearance and renal toxicity, making them incompatible for image-guided surgery (in humans) or preclinical studies by micro-CT. Additionally, blood pool agents, such as iodine-based probes, exhibit fast body dissipation, leading to uniform enhancement of the entire body. In the case of CT imaging contrast enhancement, GNPs attenuate X-rays more efficiently than iodine by many orders of magnitude. This is due not only to the much stronger attenuation per atom (gold vs iodine) but also to the large number of atoms per gold nanoparticle. In addition, a previous study showed a very high LD50 for gold nanoparticles of 3.2 g Au/kg,7 which is a great advantage for CT imaging where high dosages of a contrast agent are required. Most of the nanoparticle-based contrast agents accumulate in the tumor via passive targeting due to enhanced permeability and retention (EPR) effect over the course of hours to days.8 Nevertheless, no optimal particle size or surface charge was identified for maximizing this effect due to the significant variability between tumors. Nanoparticles in the range of 10 to 100 nm showed the highest passive tumor penetration in animal models with minimal sublethal toxicity effect.9,10 However, to achieve functional imaging using CT instruments, active targeting of nanoparticles is obligatory.11 Targeting probes to specific pathologies can significantly reduce the injection dose and thus minimize side effects and toxicity. As recently revealed, many techniques are used for GNP surface modification for multiple tumor markers such as antibodies, peptides, or small molecules.12 In general, the targeting efficacy of functionalized nanoparticles depends on the nature of the ligand, the selected coupling reaction, and the ligand surface density.
In this study, we evaluate three different sizes of GNPs stabilized by polyethylene glycol (PEG) coating conjugated via an Au–S bond, providing a stable CT tag. The tag was easily and reproducibly conjugated to a targeting moiety, an active site inhibitor specifically targeted to cathepsin proteases, thus generating an activity-based probe (ABP). ABPs are tagged molecules that are designed to covalently modify enzyme targets in an activity-dependent manner thus reporting their activity. To this end, ABPs have been developed for a number of enzyme families, including proteases, kinases, phosphatases, glycosidases ect.13 Proteases recognize their substrate target by the amino acid sequence around their cleavage site; in most cases the recognition is up to four amino acids on each side of the scissile bond (the target site for cleavage). The covalent nature of ABPs is generated by an electrophile termed “warhead” which is placed close to the recognition element. Several cysteine protease ABPs include an acyloxymethyl ketone (AOMK) chemical group as their warhead, because it is highly selective for cysteine proteases.14
Cysteine cathepsins are highly upregulated in a wide range of cancer processes by mechanisms ranging from gene amplification to post transcriptional modification. Within the tumor microenvironment, a large variety of cell types highly express cathepsins including fibroblasts, T cells, endothelial cells, osteoclasts, neutrophils, and predominantly macrophages.15 We have previously reported on cathepsin fluorescent ABPs that allow for visualization of active cathepsins B, L, and S in tumor-bearing mice.16,17 These ABPs were based on the covalent and specific cathepsin inhibitor GB111-NH2 as their targeting moiety.18 Cathepsins B, L, and S are highly abundant within intracellular endolysosomal vesicles and are constitutively expressed, while cathepsin B can reach a concentration as high as 1 mM.19 Cathepsin activity is highly elevated in metastatic tumors, where they increase motility and invasion of cancer cells and mediate degradation of the extracellular matrix as well as angiogenesis.20 Furthermore, many studies link the high cathepsin activity with prognostic and predictive factors in human cancers, prominently breast, colorectal, pancreatic, ovarian, and lung cancers. Cathepsin expression is commonly elevated in tumors compared to normal tissue; thus, recently cysteine cathepsins were targeted for anticancer strategies within the tumor microenvironment.15 On the basis of this evidence, we hypothesized that cathepsin activity will be sufficiently elevated in tumors, allowing for its detection by GNP-labeled ABPs, overcoming the low sensitivity of CT scanners.
Here we explore the newly generated cathepsin-targeted GNP-ABPs for their potency, overall permeability, and imaging capabilities. We based the GNP probes on our published potent and selective GB111-NH2 cathepsin inhibitor. Our novel probes are beneficial because they retain the contrast element at the desired site, increasing the imaging sensitivity. Moreover, reporting on the levels of activated cathepsins may reveal critical information regarding disease progression, enabling more accurate diagnoses and better targeted treatments using highly abundant CT scanners.
Experimental Section
Synthesis of GB111-NH2
The synthesis was performed as previously reported16 and is described in detail in Figure S1.
Synthesis of Ac-DWK-amide
Ac-DWK-amide was prepared on Rink resin (Advanced Chemtech) using the standard solid-phase peptide synthesis method (detailed description in Figure S2).
GNPs stabilized in citrate buffer were purchased from Cytodiagnostic Inc., at three different sizes: 10, 30, 100 nm. A protective layer of PEG was absorbed on the surface of the GNPs at different molar ratios and different lengths consisting of a mixture of thiol-polyethylene-glycol (mPEG-SH) and a heterofunctional thiol- polyethylene-acid (SH-PEG-COOH) (Creative PEGWorks, Winston Salem, NC). In a typical process, 11 μL of PEG mixture solution was added to 1.0 mL of GNP solution (OD 1) at different concentrations to obtain the following final molar concentrations of PEG: 0.01, 0.1, 1, 10 mM. The reaction mixture was vortexed immediately and then incubated at room temperature for 2 h. After PEGylation, each solution was centrifuged at 14000, 9400, 8600 rpm (10, 30, 100 nm, respectively) for 20 min and redispersed with Milli-Q water by gentle shaking. This process was repeated three times. From the resulting solution, a small part was analyzed by measuring absorption spectra, hydrodynamic size, and zeta potential. The mixture was stored at 4 °C. PEGylation at 0.01 mM final concentration was found to be most efficient and stable.
Conjugation of GB111-NH2 to PEGylated Gold Nanoparticles
PEG-GNPs were washed in 1 mL of DMF:DDW (1:10) several times. EDC (1.3 μmol, 0.21 mg) and NHS (1.3 μmol, 0.13 mg, 10 equiv relative to the PEG-COOH moiety) were dissolved together and added to the PEG-GNP solution for 30 min at room temperature, providing active sites on GNPs that underwent an amidation reaction with GB111-NH2. The amount of peptide added was 0.38 mg (75 nmol) per 2.5 mg of molecular gold (1:10 ratio to activated moiety). The mixture was incubated overnight at room temperature followed by centrifugation and DMF washes to remove unbound peptides in the solution.
Conjugation of Ac-DWK-amide to PEGylated Gold Nanoparticles
The conjugation was performed in a similar fashion to that of GB111-NH2 described above.
Chemical Evaluation
Dynamic light scattering (DLS) and ζ potential measurements were conducted in DDW using a Zetasizer nano-ZSP (Malvern). Transmission electron microscopy (TEM) analysis was performed using JEOL JEM-1400Plus by applying ∼10 μL of samples resuspended in DDW to a 200- or 400-mesh copper grid covered by carbon-stabilized Formvar film (SPI, West Chester, PA). The samples were dried overnight before scans were performed at different kV levels.
Thermogravimetric analysis (TGA), to establish the mass ratio between organic moiety (PEG) and gold atoms, was performed using a Mettler Toledo instrument. Temperature program: heat from 30 °C to 450 °C at a rate of 10 °C/min in nitrogen atmosphere with a purge rate of 20 mL/min.
Recombinant Cathepsin Labeling
Recombinant human cathepsin B, 0.7 μg in reaction buffer (50 mM acetate, 2 mM DTT, and 5 mM MgCl2 at pH 5.5), was treated at room temperature (RT) with GNP-PEG-GB111 baring 0.1–20 μM inhibitor moiety (GB111), or vehicle for 1 h. GB123 (1 μM),17 a fluorescent labeled cathepsin ABP, was added to samples for 30 min at RT. The reaction was stopped by addition of sample buffer 4× (40% glycerol, 0.2 M Tris/HCl 6.8, 20% β-mercaptoethanol, 12% SDS, and 0.4 mg/mL bromophenol blue), samples were then boiled, separated on a 12.5% SDS gel, and scanned for fluorescence with a Typhoon scanner FLA 9500, excitation/emission 635/670 nm.
Evaluation of Probes Permeability to Intact Cells. Competition Assay
NIH-3T3 cells (1 × 105 cells/well) were seeded in a 12-well plate 1 day before treatment. Cells were treated with vehicle or probe containing indicated concentrations of GB111 moiety that were predissolved in 0.1% DMF, 0.9% DDW. After 4 or 24 h of probe incubation, residual cathepsin activity was labeled with GB123 (1 μM). Cells were washed with PBS and lysed by addition of sample buffer. Lysates were boiled for 10 min and centrifuged. Protein content was determined and equal amounts were separated by 12.5% SDS gel. Residual labeled proteases in cells were visualized by scanning the gel with a Typhoon scanner FLA 9500, excitation/emission wavelengths of 635/670 nm.
Cell Viability Assay
NIH-3T3 mouse fibroblast cells (5 × 103, 24 h; 3 × 103, 48 h) and 4T1 murine mammary carcinoma cells (6 × 103, 24 h; 4 × 103, 48 h) were seeded in a 96-well plate and incubated for 24 h for cell attachment. GNP-ABPs or control GNPs, dispersed in fresh culture media, were added at equivalent concentrations to each well (200 μL) and incubated for 24 or 48 h at 37 °C. The medium was discarded to remove free particles in the solution, and cell survival was determined by standard methylene blue assay.
Animal Care
All animals were maintained on a 12:12 h light/dark cycle under fixed conditions of temperature (23 °C) and humidity (50%), with free access to food and water. All experimental procedures were approved by the Animal Care Committee of Hebrew University of Jerusalem and in accordance with the NIH Guide for the Care and Use of Laboratory Animals.
In Vivo Imaging
4T1 cells were grown to subconfluency, followed by detachment with trypsin, spin down, and resuspension in 0.5% BSA in sterile PBS and 25% matrigel. Cells (1 × 106 per spot in a total volume of 20 μL) were injected subcutaneously into the back of 3–4 week-old male BALB/c mice under isoflurane anesthesia. Tumors were typically established 9–11 days after cells injections, and compounds were injected intravenously via the tail vein as follows: in a total volume of 200 μL from stock solution of 25 mg/mL (5 mg of GNP per mouse). Mice anesthetized with isoflurane were imaged prior to injection and at 24, 48, and 72 h postinjection. The number of mice used is indicated in the figures. A micro-CT scanner (Skyscan High Resolution Model 1176) equipped with 64 detectors with a nominal resolution of 35 μm was used (0.2 mm aluminum filter, a tube voltage of 40 kV and 500 mA). Reconstruction was performed using SkyScanNRecon software. Ring artifact reduction, Gaussian smoothing (3%), and beam hardening correction (25%) were applied. Volume-rendered three-dimensional (3D) images were generated using SkyScan CT-Volume (“CTVol”) software. To quantify the exact amount of GNPs that reached the tumor tissue at each time point, we analyzed the tissue absorbance and GNP absorbance for the same ROI apart. The number of voxels for soft tissue absorbance was characterized by low energies and hence was defined by us as 25–255 in gray scale, while gold (and bone) was 53–255 for higher energies. The minimum threshold for gold detection was derived from a prescan of the mouse, showing no contrast in voxels in the tumor at that grayscale index. After the total voxel number was derived from the image for each range, the labeling percentage of voxels per tumor was calculated (% labeling = num. of vox (53–255)/num. of vox (25–255)).
In vivo imaging with inhibitor pretreatment: tumor-bearing mice were injected intraperitoneally (IP) with 40 mg/kg cathepsin inhibitor GB111-NH2 (10% DMSO, 15% Tween 80 and 75% PBS) 5 h before T-GNPs injection and an additional dose of 20 mg/kg 48 h postinjection of T-GNP. Mice in all other GNP treatment groups (Targeted (T), Non-Targeted (NT) and Non-Specific (NS)) received saline solution injections (IP) at the same time points (5 and 48 h).
Flame Atomic Absorption Spectroscopy
Flame atomic absorption spectroscopy (FAAS, SpectrAA 140, Agilent Technologies) was used to determine amounts of gold in the investigated organs. The tissues obtained from the experimental animals were melted with aqua regia (a mixture of nitric acid and hydrochloric acid in a volume ratio of 1:3). The acidic liquids were then evaporated, DDW was added to a final volume of 7 mL and then solution was filtered. An Au wavelength lamp was used to determine the gold concentration in the samples in comparison to a calibration curve with known gold concentrations (commonly: 1, 2, 5, and 10 mg/L). Each sample was analyzed in triplicate, averages and standard deviations were calculated. The measured amount of gold was divided by the organs’ weight.
Preparation of Tissues for Transmission Electron Microscopy
Tissue was dissected from animals into PBS (phosphate-buffered saline, pH 7.4) and fixed in 2% paraformaldehyde, 2.5% glutaraldeyde in 0.1 M cacodylate buffer (pH 7.4) for several hours at RT. The tissues were then rinsed four times, 10 min each, in cacodylate buffer, postfixed and stained with 1% osmium tetroxide, 1.5% potassium ferricyanide in 0.1 M cacodylate buffer for 1 h. Tissues were then washed four times in cacodylate buffer followed by dehydration in increasing concentrations of ethanol consisting of 30%, 50%, 70%, 80%, 90%, 95%, for 10 min each step followed by 100% anhydrous ethanol three times, 20 min each, and propylene oxide twice, for 10 min each. Following dehydration, the tissues were infiltrated with increasing concentrations of agar 100 resin in propylene oxide, consisting of 25, 50, 75, and 100% resin for 16 h each step. The tissues were then embedded in fresh resin and allowed to polymerize in an oven at 60 °C for 48 h. Embedded tissues in blocks were sectioned with a diamond knife on an LKB 3 microtome, and ultrathin sections (80 nm) were collected onto 200 mesh, thin-bar copper grids. The sections on grids were sequentially stained with uranyl acetate for 5 min and lead citrate for 2 min and viewed with a Tecnai 12 TEM at 100 kV (Phillips, Eindhoven, The Netherlands) equipped with a MegaView II CCD camera and Analysis version 3.0 software (SoftImaging System GmbH, Münstar, Germany).
Preparation of Tumor Tissue for Pathological Evaluation
To evaluate the tumor model, 65% of the resected tumors obtained from an in vivo experiment were arbitrarily collected and fixed with 4% paraformaldehyde at 4 °C, transferred to 10% formalin neutral buffer solution, and embedded in paraffin. Sections (10 μM thick) were obtained, stained by using hematoxylin and eosin, and examined under a light microscope.
Statistics
Statistical analyses for in vitro experiments were performed using Excel Software, and error bars represent standard deviation. A two-tailed t test was performed to determine significance when comparing the means of two groups. Alternatively, one-way ANOVA with Dunett’s correction for multiple comparisons was performed using GraphPad Prism software to compare the means of gold accumulation measured by FAAS as indicated in the figure legend. Independent experiments were conducted with a minimum of two biological replicates per condition to allow for statistical comparison. Repeated measures analysis of variance (ANOVA) was performed in SPSS and applied to examine effects of T-GNP-ABP uptake in vivo and tumor volume containing gold due to time (in four levels), size (three levels), and targeting (targeted vs nontargeted). In addition, in the cases of significant differences between more than two categories, posthoc pairwise comparisons (with Bonferroni correction) were conducted. The alpha level was set at p < 0.0083.
Results
Micro-CT Sensitivity Evaluation
To choose the optimal contrasting agent as an ABP tag, we first evaluated the detection level of the various elements using dual energy micro-CT. Scanning each contrast agent separately demonstrated linearity of the contrast with concentrations, thus enabling us to determine both the lowest concentrations detected in DDW as well as the attenuation efficiency (derived from the attenuation slope). While the minimal concentration of iodine and gadolinium detected was lower, the GNPs showed X-ray attenuation three times more efficiently, making gold an excellent tag for our probes. This is due not only to the much stronger attenuation per atom but also to the large number of atoms per GNP, Figure S3.
GNP-ABP Synthesis and Characterization
In our attempt to generate efficient GNP-ABPs we first designed a library of cathepsin-targeted nanoparticles with different particle sizes and coatings that were evaluated for various characteristics such as potency, cell permeability, and in vivo properties. We selected three different sizes of GNPs to be linked to the targeting moiety: 10, 30, and 100 nm which eventually enabled us to validate the influence of particle size on the uptake profile. As the targeting moiety of the ABP, we selected GB111-NH2, a potent and selective cathepsin B, L, and S inhibitor successfully used in our previous reports.18 GB111-NH2 was synthesized using a combination of solution and solid-phase chemistry as we previously published,16Figure 1A. To generate the GNP-ABP first, a protective layer of PEG with different lengths and different molar ratios of capped mPEG-SH and a heterofunctional COOH-PEG-SH were absorbed on the surface of the GNPs, Figure 1B. The monofunctional PEG (mPEG-), capped with a methoxy group, was used for stability and biocompatibility of the particles21 while the COOH-PEG moiety provided a chemical handle for further binding of GB111-NH2. The most stable coating was achieved with 0.01 mM final molar concentration of PEG mixture. Overall, we generated a library of GNP-ABPs containing combinations of different percentages of targeting moiety: 10%, 50%, and 100% on the particle surface with different PEG lengths (Figure 1C, i–vi). A control probe was synthesized for each size of GNP, lacking the targeting moiety (Figure 1C, vii). After synthesis, spectroscopy measurements were taken to characterize the GNP-ABPs, including transmission electron microscopy (TEM), light absorption, size, and zeta potential, with the TEM images revealing that the gold core remained intact. The absorbance peak changed within 2–4 nm from the parent GNP core indicating aggregation did not occur. The hydrodynamic diameter (HD) measured by dynamic light scattering (DLS) revealed 10–30 nm increase in the HD after PEGylation and targeting moiety coupling. In addition, each step of the chemical modification and stability was confirmed by zeta potential measurements showing a decrease in the negative charge. Correlation between all methods provided reliable analysis of our probes, confirming GNP-ABP formation and gold core stability, Figure 1D–F. We measured the stability of a selected GNP-ABP, DTS-iv, and found it exhibited good stability in 10% DMF:DDW over 10 weeks, Figure S4. Furthermore, particles could be easily resuspended without notable aggregation, with no change in surface potential and size detected. To proceed with biochemical evaluation of the GNP-ABPs, a quantitative analysis to determine the amount of targeting moiety on each particle was performed. Thermogravimetric analysis (TGA) was used to establish the mass ratio between organic moiety (PEG) and gold atoms. Because PEG has a simple linear-chain-bond structure, thermal dissociation occurs in a narrow temperature range, providing a reliable feature to measure its quantity. We thus measured the weight differences between GNP-PEGs before and after thermal dissociation. Calculations enabled assessment of the average PEG density being 0.21 PEG/nm2 on the GNP which corresponds to the low PEG density particle group (density lower than 1 PEG/nm2) having a “mushroom” configuration. This result is consistent with the low concentration of PEG mixture taken for the process.22,23
Figure 1.

Chemical synthesis and evaluation of gold nanoparticle activity-based probes. (A) Synthesis of GB111-NH2 as published in ref (16). Lysine bromomethyl ketone was reacted with dimethylbenzoic acid (DMBA) with KF, the Boc protecting group was removed, and the compound was loaded on resin to give 2. The Fmoc protecting group was removed with DEA and phenylalanine capped with Cbz was coupled, generating GB111-NH2 (3). (B) Three different sizes of GNPs were coupled to combinations of different lengths of PEG protective layers, by PEG absorption on the surface of the GNPs. Different percentages of PEG-COOH moiety were used, providing reactive sites on GNPs that were bound to GB111-NH2 (3), to generate T-GNP-ABP. Nonfunctional PEG capped with a methoxy group (OMe) was used for stability. (C) Schematic diagram representing various derivatives of GNP-ABPs that were generated. Characterization of selected GNP-ABPs: (D) TEM images of 10 nm, 30 nm, 100 nm GNP-ABPs. (E) Visible spectra absorbance of GNP-ABPs exhibit a unique wavelength: before (bare GNP), red; after chemical modifications, blue. (F) Size (DLS) and zeta-potential measurements of T-GNP-ABPs represent the hydrodynamic diameter and surface charge. Correlation was found between all methods.
Biochemical Evaluation of GNP-ABPs
To determine the potency of various derivatives, we performed competitive inhibition of recombinant human cathepsins B. Indicated concentrations of the various 30 nm GNP-ABPs were added to cathepsin B followed by labeling of the residual activity with GB123, a Cy5 fluorescent cathepsin ABP.17 The detection of the residual cathepsin B activity was visualized by fluorescent scanning of the samples run on SDS PAGE, demonstrating reactivity of the 10% and 100% targeting probes (DTS-i, -iii, -iv) each to different extents, Figure 2A. Surprisingly, derivatives containing 50% of targeting moiety on the surface (DTS-ii, -v) showed no potency at all. We then focused on derivative DTS-iv and investigated the inhibition of recombinant cathepsin B using different sized GNPs; we found an interesting correlation of potency with size, the potency being reduced with an increase in GNP particle size, Figure 2B. Evaluation of the probes’ cell permeability and capability of labeling cellular cathepsin enzymes were performed using competitive inhibition assay by incubating selected compounds with NIH-3T3 or 4T1 cells for 4 and 24 h. Next, residual cathepsin activity was labeled by GB123. To ensure that the binding is dependent on protease activity, a nontargeted control (NT-GNP) was applied as well (DTS-vii). Cell lysates were separated by SDS PAGE and scanned for fluorescence, Figure 2C. Results for NIH-3T3 cells after 24 h incubation and 4T1 cells after 4 and 24 h incubation are presented in Figure S5. Encouragingly, all 10 nm T-GNP-ABPs derivatives (carrying targeting moiety) tested showed high cell permeability and inhibition of cathepsin activity; DTS-iii however was less potent than DTS-iv. Within the 30 nm probes, DTS-i and DTS-iv (consisting of 10% GB111) showed a clear preference for shorter PEG (3 kDa) as a targeting moiety linker. Both 10 and 30 nm T-GNP-ABP were more potent than the 100 nm probes. Similar data were observed for 24 h probe incubation, Figure S5. Dose–response inhibition of cathepsin activity was detected with targeted GNPs, revealing a size-dependent pattern. Again, derivative DTS-iv exhibited the highest potency and therefore was selected to continue to in vivo evaluation. Importantly, because no inhibition was detected by the control DTS-vii, we concluded that the binding of the probes occurred in an activity-dependent manner. Prior to examining the GNP-ABPs in non-invasive imaging experiments, their cytotoxicity was evaluated on NIH-3T3 and 4T1 cells, selected as models for normal and cancer cells, respectively. We defined the targeted derivative DTS-iv, T-GNP-ABP, and the non-targeted control DTS-vii, NT-GNP. Viability was determined by methylene blue assay after treating the cells with increasing concentrations of T-GNP-ABP or NT-GNP for 24 and 48 h. Only minor cytotoxicity was detected with 10 nm T-GNP-ABP at the highest concentration tested for 48 h while, surprisingly, notable cytotoxicity was detected with 10 nm NT-GNP at 0.3 mg/mL at both 24 and 48 h, Figure 2D. We thus concluded that working concentrations of ∼0.15 mg/mL Au should be safe for animal use.
Figure 2.
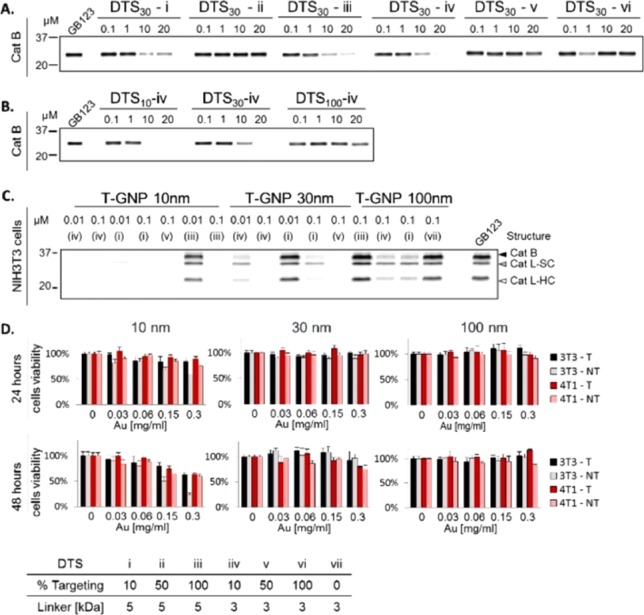
Biochemical evaluations of GNP-ABPs. (A) Competitive inhibition of recombinant cathepsin B by the 30 nm GNP-ABP derivatives (Figure 1). Indicated probe concentrations were incubated with recombinant cathepsin B for 1 h followed by 30 min labeling of the residual activity with GB123, a fluorescent ABP.17 Samples were separated on SDS gel and scanned for fluorescence at ex/em 635/670 nm. (B) Competitive inhibition of recombinant cathepsin B by DTS-iv derivatives of different particle sizes, size dependent inhibition was observed. (C) Inhibition of endogenous cathepsin activity in intact NIH-3T3 cells treated with probes in growth media for 4 h followed by labeling of residual enzyme activity by GB123. Control DTS-vii (NT-GNP) was applied. Whole cells lysates were separated on SDS gel and scanned for fluorescence. Highly potent inhibition of cathepsin activity was detected with DTS-i and DTS-iv probes. A size-dependent pattern was observed: the smallest compounds showed the highest potency. (D) NIH-3T3 and 4T1 cell viability was determined by methylene blue assay after incubation with various concentrations of DTS-iv (T) or DTS-vii (NT) for 24 or 48 h. Only minor cytotoxicity was observed with 10 nm T-GNP at the highest concentration tested. T, targeted; NT, non-targeted. Data are presented as mean ± SD (n = 6).
Non-invasive Imaging of Cancer in a Tumor Mouse Module
We continue with in vivo experiments to evaluate the efficacy of the ABP targeting methodology compared to passive accumulation of GNP alone. For this reason, we took a syngeneic mouse model in which 4T1 cells were injected subcutaneously to the back of male BALB/c mice. After tumors were established, each mouse received an intravenous (iv) injection (to the tail) of 5 mg of T-GNP-ABP (DTS-iv) or NT-GNP (DTS-vii) compounds of various sizes. Mice were scanned at 24, 48, and 72 h post-injection by a micro-CT scanner.
To overcome the diversity in tumor size over the experiment duration, we first measured the voxels specific to gold attenuation within the tumor (out of the full attenuation range) and then measured the total range of attenuations of the tissue (including gold) for the same ROI. We divided the gold voxels by the total voxels giving the percentage of gold-containing voxels within each tumor; see Experimental Section for details. Similar to the in vitro data, a marked size-dependent accumulation of both targeted and nontargeted GNPs was observed at 72 h post-injection as presented in volume-rendered 3D images. In general, accumulation of NT-GNPs in tumor tissues is thought to occur because of the EPR effect, slightly enhancing the CT signal.24 Nevertheless, a pronounced enhancement of the CT signal was observed in tumors from T-GNP-ABP injected mice (10 and 30 nm) as compared to NT-GNP controls, Figure 3A. The 2D axial cross-section images revealed a similar pattern with a uniform accumulation of T-GNP-ABPs in the peripheral region of the tumor, presumably due to high interstitial pressure of the solid tumors,25 or higher cathepsin activity in that tumor region,20Figure 3B. Furthermore, a distinct signal was already detected 24 h post-injection and became highly elevated at 72 h post-probe injection, excluding all 100 nm GNPs. The 100 nm GNPs exhibited poor active and passive accumulation in the tumor through all time points, Figure 3C. While pronounced signals were detected in the tumors of mice injected with 10 and 30 nm GNP-ABPs, we also observed signals from the digestive system at all time points. This signal most likely results from the high mineral content of the chow as it was detected in the prescan images, Figure 3D. Overall, a clear size-dependent accumulation was seen with the highest signal enhancement detected with 10 nm T-GNP-ABP and lowest tumor uptake with 100 nm.
Figure 3.

Non-invasive micro-CT imaging of tumor bearing mice. (A) CT scans of mice 72 h postinjection of 10, 30, and 100 nm T or NT GNPs (5 mg of gold per mouse). Representative CT images of volume-rendered 3D images represent X-ray absorption of bones and gold, circles indicate tumor location. (B) 2D axial cross-section images of the same mice as in A. Accumulation of T-GNP-ABPs at the tumor led to significant signal enhancement (shown in red circles). The highest enhancement was obtained with 10 nm T-GNP-ABPs. (C) Time course of all T-GNP-ABP and NT-GNP compounds. Images were acquired prior to probe injection (0 h) and 24, 48, and 72 h postprobe injection. (D) Prescan image indicates that contrast from the digestive system is attributed to the high mineral content of the chow.
Biodistribution of GNP-ABPs
GNP biodistribution was monitored to follow pharmacokinetics throughout the body and its accumulation in the tumors. After the last time point, the mice were sacrificed and then tumors and other selected organs were analyzed by flame atomic absorption spectroscopy (FAAS), determining the gold content in each tissue, Figure 4A. The T-GNP-ABPs and the control NT-GNPs showed uptake in the spleen, kidney, and liver, indicating both passive and active uptake. As previously published, contrast agents larger than 6 nm avoid renal clearance and hence are excreted from the blood pool by phagocytic cells in the reticuloendothelial system, occurring primarily in the liver and spleen and thus leading to accumulation of contrast agents in those organs.11 Based on FAAS analysis, GNP-ABP excretion was detected over a 96 h period. The GNP-ABPs all showed fecal excretion, with the 10 nm showing the highest clearance with a total 3.3% injected dose (ID), 30 nm 2.3% ID, and 100 nm only 1.5% ID, Figure S6. Despite the targeting of our GNPs, the maximal tumor uptake was lower than in the liver or the spleen. Tumor uptake was prevalently stronger for T-GNP-ABPs than the NT-GNPs; this finding was consistent both in the FAAS analysis and average tumor uptake extracted from CT images, Figure 4A. A strong size-dependent uptake in favor of small GNPs was found in tumor tissue both in the T-GNP-ABPs and NT-GNPs. In repeated measures ANOVA, comparing the changes in CT attenuation derived by time after administration, particle size and targeting, a clear significant statistical difference between the targeted group and the non-targeted group in all sizes together was found (Wilk’s lambda P value = 0.034). However, when analyzing the 100 nm particles alone, there was no statistical difference; therefore, we conclude that this effect is driven by the 10 and 30 nm size particles and not from the 100 nm particles. In a repeated measures ANOVA, a statistical significant difference was also found between the following parameters: time and size, Wilk’s lambda P value = 0.001; time and targeting, Wilk’s lambda P value = 0.034, and a trend when comparing time, size, and targeting, Wilk’s lambda P value = 0.08. Furthermore, over 6% of the total tumor volume contained detectable gold content both at 48 and 72 h postinjection of either 10 or 30 nm T-GNP-ABP while the maximal detectable gold content impressively reached over 10% at 72 h post 10 nm T-GNP-ABPs injection, Figure 4B.
Figure 4.

T-GNP-ABPs uptake in vivo is size dependent. Quantification of gold attenuation in CT scans vs gold concentration in main organs. (A) Top: Percentage of tumor voxels containing gold out of the total voxel in the tumors extracted from CT images of indicated times after iv injection of T-GNP-ABP or NT-GNP. Repeated measures ANOVA showed a significant (Wilk’s lambda P value = 0.034) difference between targeted and non-targeted groups across all measured sizes. Bottom: Average distribution of gold in the main organs 72 h post-injection of T-GNP-ABP or NT-GNP detected by FAAS. T-Test analysis revealed that both 10 and 30 nm T-GNP show higher accumulation in tumors compared to NT-GNP; P values are indicated. (B) Top: Comparison of different sized T-GNP-ABP. The percentage of tumor volume containing gold was analyzed from CT scans post-injection as in A; the highest signal was detected with the 10 nm T-GNP-ABP. The gold signal in the tumor increased over time and became highly elevated at 72 h post-injection. Repeated measures ANOVA showed a significant difference between time and size (Wilk’s lambda P value = 0.001). Bottom: Gold accumulation in major organs detected by FAAS as in A. Similar size-dependent accumulation was observed both by CT and FAAS. One-way ANOVA showed a statistical significant difference as indicated. The number in brackets represent the number of mice used. Bars represent standard error.
Since we detected high gold content in the tumors we attempted to visualize the cellular localization of GNP within the cells. Tumors were resected 72 h post iv injection of each gold-containing compound and then subjected to TEM analysis. GNPs were clearly visualized in the lysosomes of tumor cells from 10 and 30 nm T-GNP-ABP injected mice, which is supported by the known cathepsin lysosomal cellular location.26,27 Small amounts of 10 and 30 nm NT-GNPs were also observed in the cells, most likely due to passive uptake. The 100 nm compounds, however, showed low tumor cell presence with no difference observed between passive and active uptake, possibly due to low permeability and extensive spleen accumulation leading to short blood circulation, hence a low availability of GNPs, Figure 5. The TEM results further support the CT attenuation increase derived from T-GNP-ABPs that were detected by the CT scans localized within the tumor cells.
Figure 5.
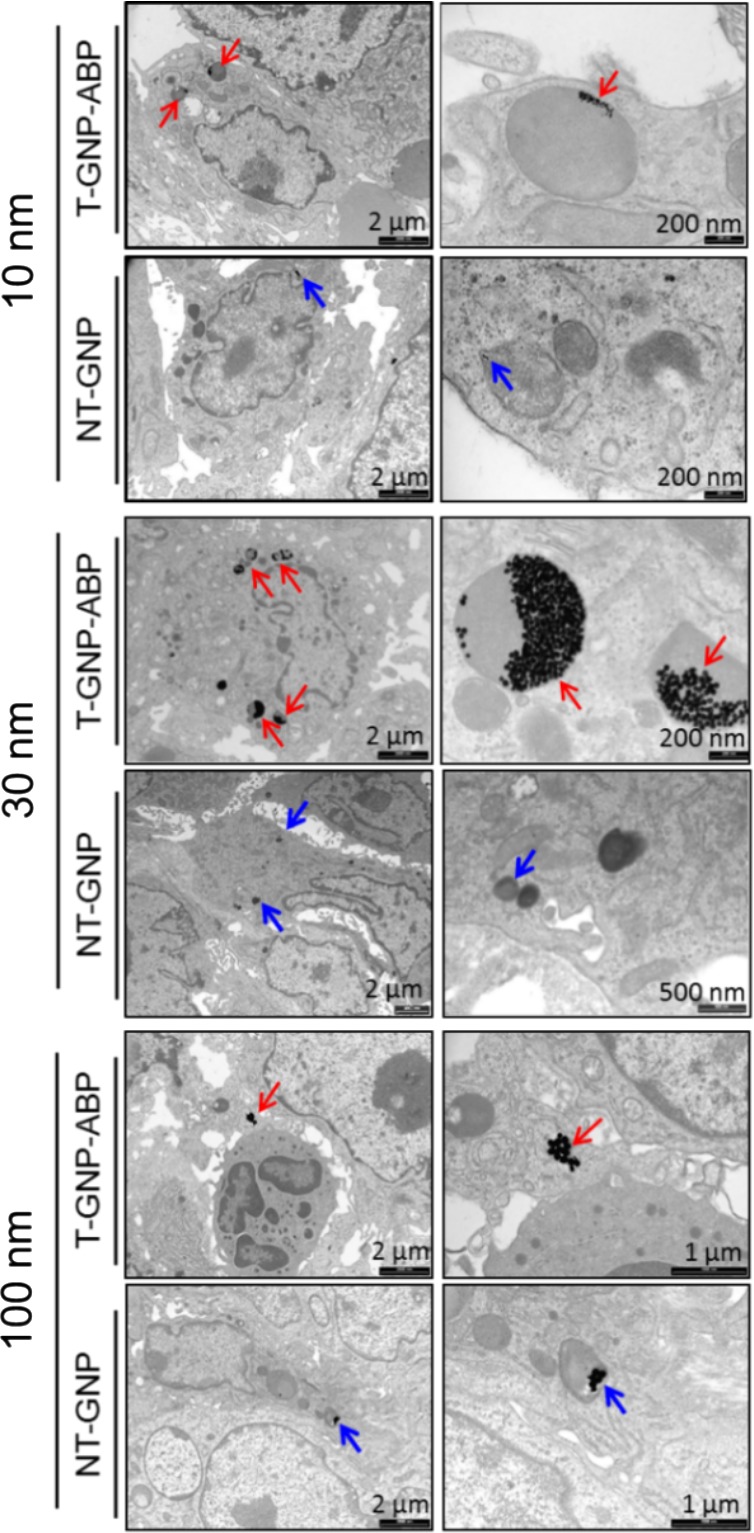
TEM imaging of tumor tissue. TEM images of a section of tumor tissue taken from a tumor-bearing mouse 72 h post iv injection of 10 or 30 or 100 nm T-GNP-ABP or NT controls. Gold particles are marked with red arrows (T) and blue arrows (NT). Tumor uptake of T-GNP-ABP was clearly visualized for 10, 30, and 100 nm. Lower amounts of NT-GNPs were observed in 10 and 30 nm; for the 100 nm, very few GNPs were detected with minor differences between T and NT compounds. Images depict localization of 10 and 30 nm T-GNP-ABP within the endosome/lysosome of tumor cells. Scale bars are marked.
Evaluation of T-GNP-ABP Uptake Efficiency
To further confirm that the signal detected and measured in the tumors was due to targeting moiety contribution and its covalent ability to target protease activity, we designed an additional control probe (in addition to the NT-GNP; DTS-vii). A non-specific GNP (NS-GNP; DTS-viii) bearing a short peptide, Ac-DWK-amide (that lacks the ability to target cathepsin B, L, or S), was conjugated to 3 kDa PEG of a 30 nm GNP via an amide bond. This control was generated to mimic the surface potential of our leading compound T-GNP (DTS-iv); see schematic presentation in Figure 6A.
Figure 6.
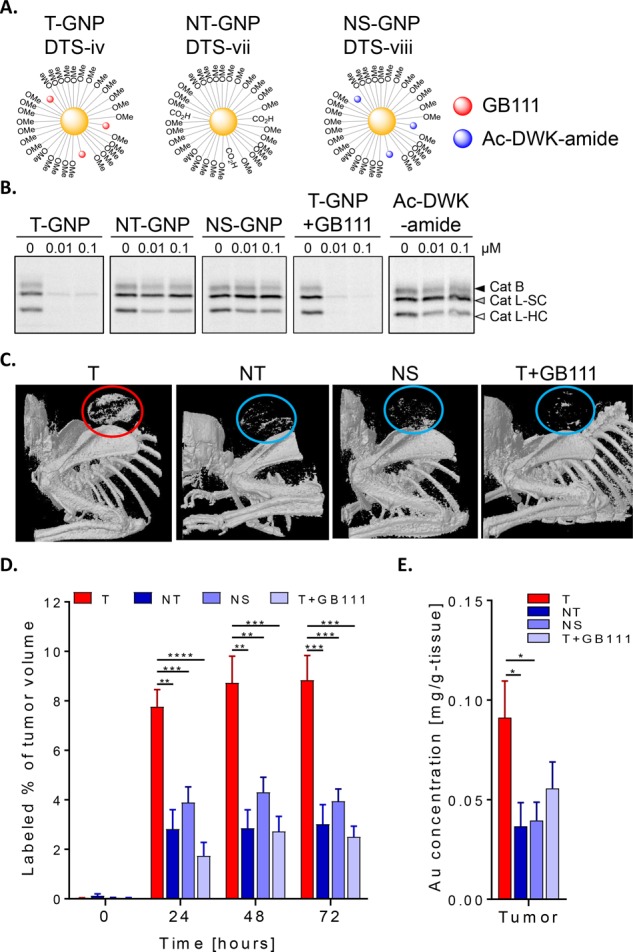
Covalent targeting increases tumor uptake. (A) Schematic diagram representing GNP-ABPs DTS-iv (T-GNP-ABP) and control NT-GNP DTS-vii as in Figure 1C along with DTS-viii, a non-specific (NS-GNP) control bearing off-target short peptide Ac-DWK-amide. (B) Cathepsin activity within intact 4T1 cells was examined after GNP treatment in growth media for 24 h, followed by labeling of residual enzyme activity by GB123. In addition to T-GNP, NT-GNP, and NS-GNP, cells treated with the cathepsin inhibitor GB111-NH2 (1 μM) 2 h prior to T-GNP-ABP addition (T+GB111) were examined. Whole cells lysates were separated on SDS gel and scanned for fluorescence as in Figure 2. Highly potent inhibition of cathepsin activity was detected after treatment with T-GNP-ABP and T+GB111 whereas NT and NS showed no cathepsin B or L inhibition. (C) Representative micro-CT scans (volume-rendered 3D images) of tumor-bearing mice 72 h postinjection of indicated GNPs (30 nm) is shown, circles indicate tumor location). Accumulation of T-GNP-ABPs at the tumor led to significant signal enhancement (shown in red circles) in all time points, indicating the efficacy of covalent targeting of our probe. (D) Percentage of tumor voxels containing gold out of the total voxel in the tumors extracted from CT images of indicated times after iv injections of indicated GNPs. Statistical analyses were performed by repeated measures ANOVA of the targeting group relative to all negative controls combined (NS, NT, and T + inhibitor), followed by Bonferroni posthoc tests. A significant difference was found with a Wilk’s lambda P value <0.001. A graph depicting the statistical differences between the marginal means relative to the time is shown in Figure S8. (E) Average accumulation of gold in tumors 72 h postinjection of indicated GNPs detected by FAAS, one-way ANOVA P* < 0.05. The number in brackets represent the number of mice used. Data are represented as mean ± SE.
Moreover, we applied an additional control group that included pretreating mice with the inhibitor, GB111-NH2, to block the cathepsin activity prior to T-GNP-ABP (30 nm) administration (T-GNP+GB111). To confirm our approach, we first evaluated endogenous cathepsin inhibition by the T-GNP compared to the new controls in 4T1 cells by competitive inhibition with fluorescent GB123, Figure 6B. Encouragingly, 30 nm NT-GNPs and NS-GNP derivatives showed no inhibition of cathepsin B as well as the Ac-DWK-amide peptide alone. The T-GNPs and the pretreated (T+GB111) group showed similar inhibition efficiency as expected. Importantly, non-invasive CT imaging was studied in 4T1 tumor-bearing mice to evaluate the efficacy of the ABPs’ covalent targeting methodology compared to all three control groups (NT, NS, and T+GB111) in a fashion similar to that in Figure 3. The inhibitor pretreated group received the GB111-NH2 inhibitor by intraperitoneal injections 5 h before T-GNPs and an additional dose 48 h post-injection of T-GNP. This inhibitor treatment regimen was chosen because of the expected difference in pharmacokinetics, between the small molecule inhibitor (GB111-NH2) and the nanosize probe. Mice were scanned at 0, 24, 48, and 72 h post T-GNP injection by a Micro-CT scanner. In correlation with our in vitro experiment, a significant CT signal was detected in the tumor of mice injected with T-GNP-ABPs while all three control groups showed lower X-ray absorption, Figure 6C. We excluded that the differences in gold uptake was a result of tumor heterogeneity by pathological evaluation of the extracted tumors. All tumors demonstrated similar histological features without necrotic tissue except for one tumor (from the non-targeted group), Figure S7. Quantification of the gold attenuation in the CT scans as well as the FAAS analysis confirmed significant reduction in signal in the mice treated with the control systems, Figure 6D,E. These results ensure that the signal in T-GNP-injected mice was a result of the covalent modification of cathepsin activity. Overall, our data supports our hypothesis that cathepsin activity sufficiently elevated in tumors allows for its detection by T-GNP-ABPs.
Discussion
The cysteine cathepsins are a family of proteases that, besides their main function of degrading proteases, play a critical role in several types of cancer such as melanoma, colorectal, glioma, breast, and lung.28 Some members of the cathepsin family have redundant functions and some members, such as cathepsin B and L, have unique functions that play different roles in tumorigenic processes such as angiogenesis, metastasis, and invasion. The involvement of active proteases in various diseases led to the development of several protease-targeting clinical drugs, laying the basis for proteases as valid targets in pathological conditions. Here we exploited the highly overexpressed cysteine cathepsins to develop activity-dependent probes for molecular imaging of cancer. We described the development of a series of cysteine cathepsin-targeted ABPs linked to different sized GNPs coated with various combinations of PEG protecting layers. We showed the covalent binding of the T-GNP-ABPs to recombinant cathepsin and their ability to label and inhibit endogenous cathepsins within intact cells. After validating that T-GNP-ABPs have minimal cytotoxicity, we showed a size-dependent uptake in subcutaneous tumors where the smallest particles showed significantly high accumulation, up to 12%, of the tumor volume (Figure 4), thus enabling micro-CT detection and overcoming its low sensitivity. The pronounced tumor accumulation was corroborated by FAAS studies and TEM images that clearly showed lysosomal localization within the cells. The significantly high amounts of T-GNP-ABP found in cancerous tissue are a combination of both the EPR effect and the covalent nature of the probes bound to the locally, highly expressed cathepsins. Covalent suicide inhibitors, however, often raise a potential problem, lacking signal amplification because the target protease is inhibited upon binding. Nevertheless, the T-GNP-ABPs elegantly showed the potential use of the ABP methodology for functional CT imaging of cancer. Furthermore, by remaining bound to the target enzyme, a direct link between imaging signals and the biological process is made. One might suggest that smaller particles cannot produce efficient contrast enhancement due to lower amounts of gold atoms per voxel; however, our data suggest a higher accumulation of the smaller particles thus generating significant CT signals. This might be explained by the fact that smaller particles are engulfed more intensively to the lysosome compartment, leading to higher local concentrations of our probe and hence higher contrast.29,30 Active targeting of GNPs has grown extensively in the past few years, due to the ease of surface activation, with many pathological conditions being examined and evaluated by non-invasive CT imaging including multiple tumor receptors, lymph nodes, inflammation, and atherosclerosis.12 Furthermore, inherent targeting of high-density lipoprotein particles31 and cell labeling by GNPs were also successfully accomplished.32 However, to the best of our knowledge there have been no reports on enzyme activity monitoring by the CT technique, most likely due to low sensitivity concerns. The goals of this work were to evaluate whether enzymatic activity can be detected by micro-CT, to assess the additional effect of protease targeting strategy compared to passive accumulation, and to select the optimal GNP size for visualization in vivo. We showed that cathepsin activity is sufficiently elevated in cancer to allow for its detection by T-GNP-ABPs, generating additional contrast above the signal generated by passive uptake of similar sized GNPs. To generate functional compounds, various protective layers were used in our chemical scaffolds designed from two lengths of PEG, 3 and 5 kDa, in various ratios of reactive vs capped PEG. The derivatives were studied in terms of their potency toward recombinant and endogenous cathepsins, and our results show that probes with 10% targeting moiety (GB111) linked to the shorter PEG (DTS-iv) possess high potency in all GNP sizes. The suggestive “mushroom” configuration may explain this enhanced potency of DTS-iv when GB111 is linked to the shorter PEG, enabling its exposure, rather than to the longer PEG that may be folded. As suspected, the similarity in size between GB111 and its PEG linker is advantageous.33 The biodistribution studies of targeted GNP-ABPs reveal a size-dependent tumor uptake. Apart from the tumor, the spleen, liver, and kidneys exhibited considerable amounts of GNP-ABPs yet with different biodistributions for each GNP size. Additionally, to take into account tumor variability and potential clinical applications, evaluation of the 10 and 30 nm T-GNP-ABPs should be considered in other tumor models. As previously suggested by S. Hirn et al., negatively charged monosulfonated triphenylphosphine GNPs (TPPMS-GNPs) exhibited a hepatobiliary fecal clearance pathway from rat liver into the small intestine, showing an inverse linear relationship to their diameter.9 The excretion kinetics assessment of our probes follows a similar tendency. Furthermore, the excretion of GNP-ABPs was slow but steady, thus partially reducing the concern of long-term toxicity in the liver. Nevertheless, the 10 nm GNP-ABPs showed the highest liver uptake, as well as some toxicity in intact cells (Figure 3D), raising the necessity to better understand long-term effects.
Conclusion
We found our covalent GNP-ABPs as promising new tools for functional imaging of specific protease activity in vivo using the CT instrument. The covalent linkage is key for detection using the low sensitive imaging modalities. CT provides 3D anatomic details of the body; thus, it is widely used in clinical and preclinical practice for diagnosis of disease, prediction of therapy, and treatment assessment. We screened and evaluated several types of GNP tags for most efficient and selective targeting of cysteine cathepsins. Tumor accumulation of the targeted GNP-ABPs is determined by the delicate balance between the targeting moieties’ features and particle size. We show that the chemical structure of the particle strongly influences their biological behavior. We found our lead compounds DTS-iv 10 and 30 nm to be highly effective for tumor imaging, its small size enabling excretion mainly through the hepatobiliary system, which appears to be a sufficient way to remove metal nanoparticles from the body. Still, the main limitation of our technology is the enhanced absorption in organs such as the liver, kidney, and spleen where there is high cathepsin activity, making imaging of tumors in these organs challenging at this point. However, further developments in CT technology could improve the sensitivity and specificity limitations, expanding the potential utility of our reagents. Additionally, Bindin et al. analyzed different stages of breast, lung, and cervical tumors using multiplex zymography of elevated activities of cathepsins K, L, and S.34 Thus, real-time measurements of tumor growth, metastasis formation, and cancer staging could potentially be performed in large living organisms using our method of cathepsin activity measurements.
Moreover, after calibration of our system, we suggest our platform could be adopted to target additional types of pathologies with high cathepsin activity such as atherosclerosis35 and arthritis.36 The strategy described here overcomes a major limitation of signal detection in deep tissue that still exists with fluorescent protease substrates and fluorescent activity-based probes that have been extensively reported during the past decade. Thus, the ABPs reported here have the potential to be developed for clinical use.
Acknowledgments
This work was supported by the European Research Council ERC #337238 (GB), Grass Center for Drug Design and Synthesis of Novel Therapeutics (GB) Rothschild Caesarea Foundation (DT). We thank Prof. Boris Turk from J. Stefan Institute, Ljubljana, Slovenia, for providing recombinant cathepsins. We thank Dr. Menachem Motiei and Oshra Betzer from Bar Ilan University for guidance on CT and FAAS, Dr. Yossi Tam from the Institute for Drug Research, the Hebrew University of Jerusalem, for the use of metabolic cages, Dr. Yael Friedmann from the Bio-Imaging Unit at The Institute of Life Sciences, The Hebrew University of Jerusalem for electron microscopy work, and Eduard Berenshtein from the Electron Microscopy Lab of the Hebrew University Core Research Facility for assistance in TEM scans. We thank Ami Altman from Phillips healthcare for crucial advice on CT methodologies.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b05817.
Figure S1. Chemical synthesis of GB111-NH2. Figure S2. Chemical synthesis of acetylated aspartic acid-tryptophan-lysine-amide. Figure S3. Detection level of various contrast agents by micro-CT. Figure S4. Characterization of GNP-ABPs. Figure S5. Biochemical evaluation of GNP-ABPs. Figure S6. Targeted GNP excretion is size dependent in vivo. Figure S7. Pathological sections of tumor tissue. Figure S8. Estimated marginal means of measures (PDF)
Author Contributions
The manuscript was written through contributions of all authors, and all authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Siegel R. L.; Miller K. D.; Jemal A. Ca-Cancer J. Clin. 2016, 66, 7. 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- van Duijnhoven S. M.; Robillard M. S.; Langereis S.; Grull H. Contrast Media Mol. Imaging 2015, 10, 282. 10.1002/cmmi.1636. [DOI] [PubMed] [Google Scholar]
- Lee N.; Choi S. H.; Hyeon T. Adv. Mater. 2013, 25, 2641. 10.1002/adma.201300081. [DOI] [PubMed] [Google Scholar]
- Goodman L. R. Thorac. Surg. Clin. 2010, 20, 1. 10.1016/j.thorsurg.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Lusic H.; Grinstaff M. W. Chem. Rev. 2013, 113, 1641. 10.1021/cr200358s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan P.; Kumar A.; Kumar S.; Chaudhary R. K.; Gulyas B. Acta Biomater. 2016, 41, 1. 10.1016/j.actbio.2016.06.003. [DOI] [PubMed] [Google Scholar]
- Hainfeld J. F.; Slatkin D. N.; Focella T. M.; Smilowitz H. M. Br. J. Radiol. 2006, 79, 248. 10.1259/bjr/13169882. [DOI] [PubMed] [Google Scholar]
- Maeda H.; Wu J.; Sawa T.; Matsumura Y.; Hori K. J. Controlled Release 2000, 65, 271. 10.1016/S0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- Hirn S.; Semmler-Behnke M.; Schleh C.; Wenk A.; Lipka J.; Schaffler M.; Takenaka S.; Möller W.; Schmid G.; Simon U.; Kreyling W. G. Eur. J. Pharm. Biopharm. 2011, 77, 407. 10.1016/j.ejpb.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z.; Al Zaki A.; Hui J. Z.; Muzykantov V. R.; Tsourkas A. Science 2012, 338, 903. 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton J. R.; West J. L.; Badea C. T. Front. Pharmacol. 2015, 6, 256. 10.3389/fphar.2015.00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Anton N.; Zuber G.; Vandamme T. Adv. Drug Delivery Rev. 2014, 76, 116. 10.1016/j.addr.2014.07.013. [DOI] [PubMed] [Google Scholar]
- Fonović M.; Bogyo M. Expert Rev. Proteomics 2008, 5, 721. 10.1586/14789450.5.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewings D. S.; Flygare J. A.; Wertz I. E.; Bogyo M. FEBS J. 2017, 284, 1540. 10.1111/febs.14016. [DOI] [PubMed] [Google Scholar]
- Olson O. C.; Joyce J. A. Nat. Rev. Cancer 2015, 15, 712. 10.1038/nrc4027. [DOI] [PubMed] [Google Scholar]
- Ben-Nun Y.; Merquiol E.; Brandis A.; Turk B.; Scherz A.; Blum G. Theranostics 2015, 5, 847. 10.7150/thno.10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum G.; von Degenfeld G.; Merchant M. J.; Blau H. M.; Bogyo M. Nat. Chem. Biol. 2007, 3, 668. 10.1038/nchembio.2007.26. [DOI] [PubMed] [Google Scholar]
- Blum G.; Mullins S. R.; Keren K.; Fonovic M.; Jedeszko C.; Rice M. J.; Sloane B. F.; Bogyo M. Nat. Chem. Biol. 2005, 1, 203. 10.1038/nchembio728. [DOI] [PubMed] [Google Scholar]
- Turk B.; Turk D.; Turk V. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 2000, 1477, 98. 10.1016/S0167-4838(99)00263-0. [DOI] [PubMed] [Google Scholar]
- Tan G. J.; Peng Z. K.; Lu J. P.; Tang F. Q. World J. Biol. Chem. 2013, 4, 91. 10.4331/wjbc.v4.i4.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreaden E. C.; Austin L. A.; Mackey M. A.; El-Sayed M. A. Ther. Delivery 2012, 3, 457. 10.4155/tde.12.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson T. A.; Joshi P. P.; Sokolov K. ACS Nano 2012, 6, 9182. 10.1021/nn3035155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Paholak H.; Ito M.; Sansanaphongpricha K.; Qian W.; Che Y.; Sun D. Nanotechnology 2013, 24, 355101. 10.1088/0957-4484/24/35/355101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreaden E. C.; Alkilany A. M.; Huang X.; Murphy C. J.; El-Sayed M. A. Chem. Soc. Rev. 2012, 41, 2740. 10.1039/C1CS15237H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trédan O.; Galmarini C. M.; Patel K.; Tannock I. F. J. Natl. Cancer. Inst. 2007, 99, 1441. 10.1093/jnci/djm135. [DOI] [PubMed] [Google Scholar]
- Sloane B.; Dunn J.; Honn K. Science 1981, 212, 1151. 10.1126/science.7233209. [DOI] [PubMed] [Google Scholar]
- Brix K.; Dunkhorst A.; Mayer K.; Jordans S. Biochimie 2008, 90, 194. 10.1016/j.biochi.2007.07.024. [DOI] [PubMed] [Google Scholar]
- Mohamed M. M.; Sloane B. F. Nat. Rev. Cancer 2006, 6, 764. 10.1038/nrc1949. [DOI] [PubMed] [Google Scholar]
- Oh E.; Delehanty J. B.; Sapsford K. E.; Susumu K.; Goswami R.; Blanco-Canosa J. B.; Dawson P. E.; Granek J.; Shoff M.; Zhang Q.; Goering P. L.; Huston A.; Medintz I. L. ACS Nano 2011, 5, 6434. 10.1021/nn201624c. [DOI] [PubMed] [Google Scholar]
- Agudo-Canalejo J.; Lipowsky R. ACS Nano 2015, 9, 3704. 10.1021/acsnano.5b01285. [DOI] [PubMed] [Google Scholar]
- Cormode D. P.; Roessl E.; Thran A.; Skajaa T.; Gordon R. E.; Schlomka J. P.; Fuster V.; Fisher E. A.; Mulder W. J.; Proksa R.; Fayad Z. A. Radiology 2010, 256, 774. 10.1148/radiol.10092473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Chhour P.; Hsu J.; Litt H. I.; Ferrari V. A.; Popovtzer R.; Cormode D. P. Bioconjugate Chem. 2017, 28, 1581. 10.1021/acs.bioconjchem.7b00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S. S.; Aher N.; Patil R.; Khandare J. J. Drug Delivery 2012, 2012, 103973. 10.1155/2012/103973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Platt M. O. J. Transl. Med. 2011, 9, 109. 10.1186/1479-5876-9-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss-Sadan T.; Gotsman I.; Blum G. FEBS J. 2017, 284, 1455. 10.1111/febs.14043. [DOI] [PubMed] [Google Scholar]
- Fonovic M.; Turk B. Proteomics: Clin. Appl. 2014, 8, 416. 10.1002/prca.201300085. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.