

GPR40 modulates epileptic seizure and NMDA receptor function through the regulation of NR2A and NR2B surface expression.
Abstract
Epilepsy is a common neurological disease, and approximately 30% of patients do not respond adequately to antiepileptic drug treatment. Recent studies suggest that G protein–coupled receptor 40 (GPR40) is expressed in the central nervous system and is involved in the regulation of neurological function. However, the impact of GPR40 on epileptic seizures remains unclear. In this study, we first reported that GPR40 expression was increased in epileptic brains. In the kainic acid–induced epilepsy model, GPR40 activation after status epilepticus alleviated epileptic activity, whereas GPR40 inhibition showed the opposite effect. In the pentylenetetrazole-induced kindling model, susceptibility to epilepsy was reduced with GPR40 activation and increased with GPR40 inhibition. Whole-cell patch-clamp recordings demonstrated that GPR40 affected N-methyl-d-aspartate (NMDA) receptor–mediated synaptic transmission. Moreover, GPR40 regulated NR2A and NR2B expression on the surface of neurons. In addition, endocytosis of NMDA receptors and binding of GPR40 with NR2A and NR2B can be regulated by GPR40. Together, our findings indicate that GPR40 modulates epileptic seizures, providing a novel antiepileptic target.
INTRODUCTION
Epilepsy is a common, serious chronic neurological disease that affects >50 million people worldwide. Dozens of antiepileptic drugs (AEDs) have been developed and have good efficacy on epileptic seizures; nonetheless, approximately 30% of patients remain untreated and often experience side effects (1, 2). Most of these AEDs target neurotransmission, as the imbalance of excitatory and inhibitory neurotransmission is accepted as the common mechanism of various epileptic seizures. Neurotransmission is regulated by a variety of factors. Therefore, understanding the regulatory mechanisms of neurotransmission is necessary for exploring novel antiepileptic targets.
G protein–coupled receptor 40 (GPR40), which is also named free fatty acid receptor 1, is activated by medium- to long-chain saturated and polyunsaturated fatty acids (PUFAs) with carbon chain lengths greater than six, such as docosahexaenoic acid, elaidic acid, palmitic acid, arachidonic acid, and eicosapentaenoic acid (3–5). GPR40 is abundantly expressed in β cells in pancreatic islets, and many studies have confirmed its role in regulating insulin secretion (4). Accordingly, GPR40 agonists have been developed as potential antidiabetic drugs (6–8). Some evidence indicates a relationship between diabetes mellitus and epilepsy. Insulin deficiency–associated type 1 diabetes mellitus increases the risk of epilepsy (2) and shares some common genetic or autoimmune factors with epilepsy (9). Furthermore, some AEDs also exhibit antidiabetic effects; for example, topiramate can act both as an insulin secretagogue and as a sensitizer (10). Therefore, GPR40 may be involved in epilepsy. This hypothesis is also supported by recent evidence showing that GPR40 regulates neurological function. First, GPR40 was confirmed to be expressed in the central nervous system, including the cortex and hippocampus, which play key roles in epilepsy (11). Second, studies have shown that GPR40 regulates neuropathic pain in animal models (12, 13). Third, PUFAs, the natural ligands of GPR40, serve as crucial regulators of synaptic transmission via several mechanisms, including the regulation of membrane dynamics, the activation of synaptic receptors, and the modulation of the endocannabinoid system. Thus, PUFAs are essential for brain development and neurological function (14, 15). Moreover, studies have suggested a positive relationship between PUFAs and epileptic seizures (16). In light of this indirect evidence, we set out to study the role of GPR40 in epileptic seizures, which has not been studied to date.
In the present study, we first found that expression of GPR40 was up-regulated in the epileptic brains of patients and animal models. Animal behavior studies using two epilepsy models showed that GPR40 activation alleviated epileptic seizures and that GPR40 inhibition aggravated epileptic seizures. Patch-clamp recordings revealed that GPR40 mainly affected N-methyl-d-aspartate (NMDA) receptor (NMDAR)–mediated postsynaptic transmission. Moreover, we found that GPR40 regulated surface expression of NR2A and NR2B. The results of our study show that GPR40 can modulate seizure activity in epilepsy, providing a novel potential antiepileptic target.
RESULTS
GPR40 distribution in the epileptic brain
Previous studies have reported that GPR40 is expressed in the brains of healthy rodents and primates (3, 17). However, the distribution of GPR40 in the hippocampus, which is the primary zone closely associated with epilepsy, is poorly understood. First, we performed immunofluorescence to characterize the distribution of GPR40 in the hippocampus of healthy mice. We found GPR40 to be highly expressed in the lacunosum moleculare layer and in the pyramidal cell layer of the hippocampus, whereas it was not highly expressed in the dentate gyrus (fig. S1A).
We next evaluated the cellular localization of GPR40 in epileptic brain tissues compared with control brain tissues. Epileptic animal brain tissues were obtained from a model that was induced by a unilateral intrahippocampal injection of kainic acid (KA). The KA-induced temporal lobe epilepsy (TLE) model well recapitulated human TLE with typical hippocampal sclerosis (18, 19). First, GPR40 colocalized with microtubule-associated protein 2 (MAP2; a marker of dendrites) but not with glial fibrillary acidic protein (GFAP; a marker of astrocytes) in the hippocampus of both the KA-induced TLE mouse model and the healthy control, showing that GPR40 is expressed mainly in neurons but not in astrocytes (fig. S1). We also observed that GPR40 colocalized with postsynaptic density-95 (PSD95; a postsynaptic marker), indicating that the former is distributed in the postsynaptic area of excitatory neurons (fig. S1).
Furthermore, we confirmed the cellular localization characteristics of GPR40 in human tissues of temporal neocortices that were surgically obtained from patients with pharmacoresistant TLE and from patients suffering from head trauma without related neurological disease as a control group. GPR40 similarly colocalized with MAP2, but not with GFAP, and also colocalized with PSD95 (fig. S2).
Increased expression of GPR40 in the epileptic brain
We next compared the immunofluorescence intensities of GPR40 to determine expression levels in epileptic and nonepileptic tissues. Compared with nonepileptic tissues, GPR40 expression was significantly increased in epileptic tissues in the CA1 region of the mouse hippocampus and in the neocortices of TLE patients (figs. S1 to S3).
To further confirm changes in GPR40 expression in epileptic tissues, we next determined GPR40 protein levels by Western blotting in the epileptic brain tissues of a KA epilepsy model and human patients with TLE. Histologically, normal neocortical tissues obtained from patients who suffered from head trauma served as controls for the human epileptic brain tissues. In the mouse tissues, we first compared the expression levels of GPR40 in tissues from a traumatic brain injury (TBI) model with healthy controls and found no significant difference between these groups (Fig. 1, A and B). However, the TLE model showed significantly higher GPR40 expression in the cortex and hippocampus than in the two controls (Fig. 1, A and B). We also compared GPR40 protein levels in temporal neocortices surgically obtained from patients with pharmacoresistant TLE (n = 20) with control human tissues (n = 10). There were no significant differences in age or gender between the groups (P >0.05; table S1). GPR40 protein expression was significantly increased in the TLE patients compared with the controls (Fig. 1C). Together, GPR40 expression was increased in the epileptic brain, which suggests the possibility that GPR40 is involved in epilepsy.
Fig. 1. Western blot analysis of GPR40 expression in epilepsy.
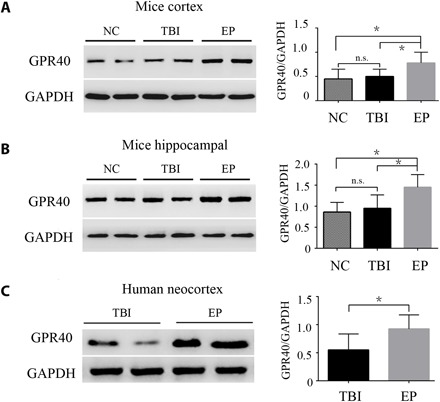
(A and B) Quantitative analysis of the GPR40/GAPDH ratio, showing that the expression of GPR40 did not change between TBI and normal control (NC) mice. Compared with controls for TBI and NC mice, GPR40 was increased in the cortex and hippocampus of the KA-induced epilepsy model (n = 6 in each group; *P < 0.05 versus controls, Student’s t test). (C) Quantitative analysis of the GPR40/GAPDH ratio, showing that GPR40 was up-regulated in human temporal neocortices from pharmacoresistant TLE patients (n = 20) compared with control individuals with head trauma (n = 10; Student’s t test, *P < 0.05). GAPDH, glyceraldehyde-3-phosphate dehydrogenase. n.s., not significant; EP, epilepsy.
GPR40 modulates epileptic seizure activity
Altered GPR40 expression may be an epiphenomenon or may play a causal role in epileptic seizures. Therefore, we investigated the effect of the GPR40 selective agonist GW9508 [1 μg in 0.5% dimethyl sulfoxide (DMSO) per mouse] and the selective antagonist GW1100 (5 μg in 0.5% DMSO per mouse) on regulating epileptic activity in the intrahippocampal KA-induced TLE model. Three days after status epilepticus (SE) induction, we administered an intracerebroventricular injection of 0.5% DMSO, GW9508, and GW1100 daily for seven consecutive days (Fig. 2A). One month after SE induction, we recorded seizure activity using local field potentials (LFPs). Consistent with other studies, frequent, repetitive seizure-like events (SLEs) were observed in all mice (Fig. 2, B and C). We analyzed the SLEs for a period of 30 min and found that the duration did not differ among the groups (Fig. 2E). However, compared with the DMSO control group, GW9508 decreased the number of SLEs and the total time spent in SLE during a 30-min period. GW1100 had the opposite effect (Fig. 2, D and F).
Fig. 2. GPR40 modulates epileptic seizure activity.
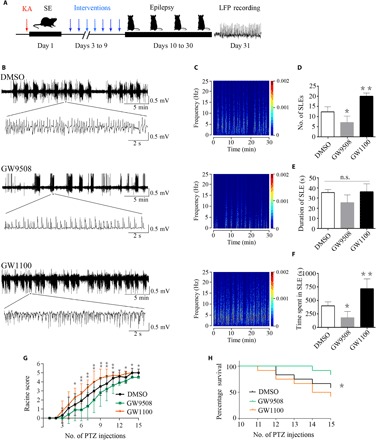
(A) Graphic representation of the experimental timeline in the KA experiment. Mice were treated daily with DMSO, GW9508, or GW1100 for seven consecutive days from the third to the ninth day after KA injection (n = 6 in each group). (B) Representative LFPs in the three groups. (C) Frequency spectrum corresponding to the LFPs in (B). (D) During 30 min, the number of SLEs was reduced in the GW9508 group and increased in the GW1100 group compared with the DMSO control group. (E) The duration of SLEs was not significantly changed between the groups. (F) The total time spent in SLEs during the 30 min was reduced in the GW9508 group and increased in the GW1100 group. For the analysis, n = 6 for each group. Error bars represent the means ± SEM; *P < 0.05, **P < 0.01 versus the DMSO group, one-way analysis of variance (ANOVA), followed by Tukey’s test. (G) In the PTZ kindling mouse model, seizure activity was suppressed in the GW9508 group and accelerated in the GW1100 group compared with the DMSO group after the fifth PTZ injection (n = 12 in each group). Error bars represent the means ± SEM; *P < 0.05, **P < 0.01, ANOVA. (H) Percentage survival over the number of PTZ injections (n = 12 in each group; *P < 0.05, log rank test).
The pentylenetetrazole (PTZ) kindling model is a classic model for studying antiepileptic effects. In our study, the kindling mice received an intracerebroventricular injection of 0.5% DMSO with GW9508 (1 μg in 0.5% DMSO per mouse each time) or GW1100 (5 μg in 0.5% DMSO per mouse each time) before each PTZ administration. The mice in all three groups showed progressively increased seizure scores. The GW9508-treated mice had lower seizure scores and the GW1100-treated mice had higher seizure scores compared with the DMSO group. After five PTZ injections, the differences among the groups were significant. During the kindling course, we observed that the susceptibility to epileptic seizure was reduced in the GW9508 group and increased in the GW1100 group compared with that in the DMSO group (Fig. 2G). After the establishment of generalized tonic-clonic seizures (GTCSs), mice are prone to GTCS-related death. After the 11th PTZ injection, the mice in the GW1100 group began to die of GTCSs. The survival rate over time was significantly different among the groups (Fig. 2H). Together, GW9508 has an antiepileptic effect, and GW1100 has a proepileptic effect.
GPR40 regulates NMDAR-mediated currents
The LFPs in the in vivo study reflect the synchronous activity of a population of neurons. To further understand the effect of GPR40 on neural activity, we performed whole-cell patch-clamp recordings of hippocampal CA1 pyramidal neurons. Neural activity at baseline and after treatment with the agonist GW9508 (20 μM) or the antagonist GW1100 (20 μM) was recorded in the Mg2+-free condition, which was used as an in vitro model of epileptiform activity (20).
To explore the impact of GPR40 on neuronal synaptic activity, we first monitored miniature excitatory postsynaptic currents (mEPSCs) and miniature inhibitory postsynaptic currents (mIPSCs), which reflect changes in excitatory and inhibitory synaptic transmission, respectively. Compared with the baseline condition, the frequency and amplitude of mEPSCs were reduced by GW9508 and increased by GW1100 (Fig. 3A). However, neither the frequency nor the amplitude of the mIPSCs was affected by either GW9508 or GW1100 (Fig. 3B). These results indicate that GPR40 mainly regulates excitatory synaptic transmission.
Fig. 3. GPR40 regulates NMDAR-mediated currents.
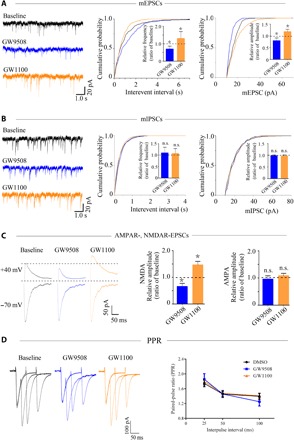
(A) Representative traces of mEPSCs from baseline and after interventions and summary of the frequency and amplitude of the mEPSCs. (B) Representative traces of mIPSCs from baseline and after interventions and summary of the frequency and amplitude of the mIPSCs. (C) Representative traces of NMDAR-mediated (+40 mV) and AMPAR-mediated (−70 mV) EPSCs from baseline and after interventions and summary of the EPSC amplitude. For the analysis, n = 6 in each group. The effect of each treatment was normalized to the baseline; error bars represent the means ± SEM; *P < 0.05 versus the baseline, paired t test. (D) Representative traces of the PPRs for AMPA-mediated EPSCs at three different interstimulus intervals and summary of the PPRs among the groups (n = 6 in each group). Error bars represent the means ± SEM; not significant, ANOVA analysis.
To determine whether the altered excitatory synaptic transmission was caused by AMPA or NMDA currents, we recorded the evoked EPSCs. Compared with the baseline condition, the amplitude of NMDAR-mediated EPSCs was reduced by GW9508 and increased by GW1100. In contrast, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPAR)–mediated EPSCs were unchanged by either treatment (Fig. 3C). To investigate whether the effect of GPR40 on NMDAR-mediated synaptic responses occurs presynaptically or postsynaptically, the paired pulse ratios (PPRs) for AMPA-mediated EPSCs at different intervals and NMDA-mediated EPSCs were recorded. No changes were found following either treatment, indicating that GPR40 may modulate glutamatergic transmission through a postsynaptic rather than a presynaptic mechanism (Fig. 3D and fig. S4).
Considering the difference in mEPSC frequency without changes in the PPR between the groups, we further examined whether GPR40 affects spine density on primary apical dendrites in cultured hippocampal neurons. Representative images of dendritic spines at DIV16 (16 days in vitro) neurons are shown in fig. S5. Compared with the DMSO group, the dendritic spine density was significantly increased by GW1100 and reduced by GW9508. This result suggests that the change in the number of synapses caused by GPR40 modulation may be responsible for the corresponding mEPSCs frequency change.
GPR40 regulates the function of NR2A and NR2B
We found that NMDAR-mediated synaptic responses were regulated by GPR40. NMDARs are heterotetrameric assemblies of NR1 and NR2/3 subunits, among which the NR2 subunit plays an essential role in the biophysical and pharmacological properties of NMDARs (21). In addition, studies have suggested that NR2 may play a more important role in epilepsy (22, 23). Accordingly, we assessed the effect of GPR40 on NR2A and NR2B regulation.
Our behavioral experiments revealed that GPR40 modulated epileptic seizures in both the KA and PTZ kindling models. Therefore, using hippocampal tissues from these models, we determined the cell surface and total expression of NR2A and NR2B. Compared with the DMSO groups, no changes in NR2A or NR2B total expression were observed in either the GW9508 or the GW1100 group for both models (Fig. 4, A to D). However, compared with the DMSO group, the ratio of surface to total expression of both NR2A and NR2B was reduced in the GW9508 group, and an opposite effect was observed in the GW1100 group (Fig. 4, A to D). These results indicate that GPR40 affected surface expression of NR2A and NR2B in the hippocampus.
Fig. 4. Cell surface and total expression of NR2A and NR2B in hippocampal tissues from GPR40-treated mice during epilepsy.
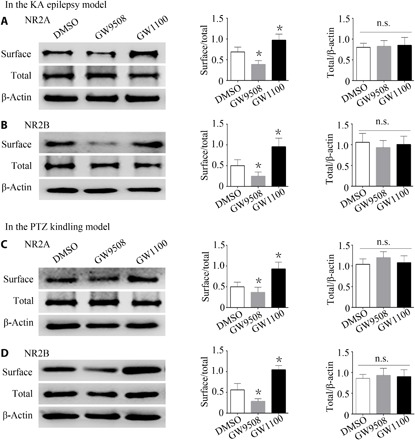
(A and B) Representative images of surface and total NR2A and NR2B expression in the KA model, summary of the normalized total NR2A values (NR2A/β-actin) and NR2B values (NR2B/β-actin), and the ratio of surface to total NR2A and NR2B expression. (C and D) Representative images of surface and total NR2A and NR2B expression in the PTZ kindling model, summary of the normalized total NR2A values (NR2A/β-actin) and NR2B values (NR2B/β-actin), and the ratio of surface to total NR2A and NR2B expression. For the analysis, n = 5 in each group. Error bars represent the means ± SEM; *P < 0.05 versus the DMSO group, one-way ANOVA, followed by Tukey’s test.
To further determine the mechanism by which GPR40 regulates the level of surface NMDARs, we examined the NMDAR-mediated excitatory postsynaptic currents (NMDA-EPSCs) in CA1 hippocampal neurons from acute brain slices by blocking exocytosis or endocytosis. As shown in Fig. 3C, GW9508 decreased NMDA-EPSC amplitudes, whereas GW1100 increased these amplitudes. This effect was not blocked by application of the exocytosis blocker tetanus toxin (TeTx; 0.1 μM; Sigma-Aldrich) but was blocked by the endocytosis blocker dynasore (80 mM; Abcam; Fig. 5, A to D). The results suggest that NMDAR endocytosis is involved in the effect of GPR40 on neural activity.
Fig. 5. GPR40 regulates NR2A/NR2B endocytosis.
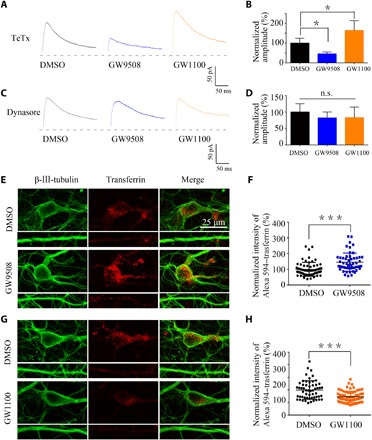
(A to D) Representative traces of evoked NMDAR-EPSCs after treatment with dynasore (80 mM) or TeTx (0.1 μM; Sigma-Aldrich) and the summary of the amplitude. For the analysis, n = 5 in each group. Error bars represent the means ± SEM; *P < 0.05 versus the DMSO group, one-way ANOVA followed by Tukey’s test. (E and F) Representative confocal images showing uptake of Alexa 594–transferrin (40 mg/ml for 30 min at 37°C) by neurons after treatment with GW9508 (20 μM) compared with 0.1% DMSO and summary of the normalized intensity of Alexa 594–transferrin (DMSO, n = 63; GW9508, n = 54; ***P < 0.001, Student’s t test). (G and H) Representative confocal images of the GW1100 (20 μM) group and the 0.1% DMSO group and summary of the results (DMSO, n = 56; GW9508, n = 69; ***P < 0.001, Student’s t test).
To further confirm the effect of GPR40 on endocytosis, we conducted a transferrin uptake assay using cultured neurons after application of GW9508 or GW1100 for 20 min. We found that GW9508 increased uptake of fluorescently conjugated transferrin compared with the DMSO group and that GW1100 had the opposite effect (Fig. 5, E to H). Together, GPR40 regulates NMDAR endocytosis.
GPR40 regulates its interaction with NR2
Receptor endocytosis is a complicated and well-regulated process that may involve interactions with other proteins (24, 25). Both GPR40 and NMDARs are membrane proteins. An immunofluorescence experiment showed that GPR40 colocalized with PSD95, which is a scaffolding protein that can anchor NMDARs. Thus, we sought to determine whether GPR40 interacts with NMDARs. We tested binding between GPR40 and NR2A or NR2B in vivo using coimmunoprecipitation experiments. The results showed that GPR40 coimmunoprecipitated with NR2A and NR2B. We also validated this result by performing a reciprocal experiment using anti-NR2A and anti-NR2B antibodies, and the results indicate that GPR40 and NR2 subunits physically interact (Fig. 6, A and B).
Fig. 6. GPR40 interacts with NR2A and NR2B.
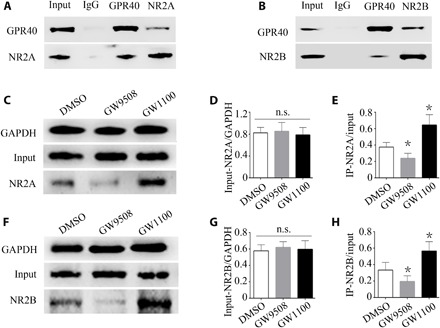
(A) Reciprocal coimmunoprecipitation between GPR40 and NR2A using anti-NR2A or anti-GPR40 antibodies. IgG, immunoglobulin G. (B) Reciprocal coimmunoprecipitation between GPR40 and NR2B using anti-NR2B or anti-GPR40 antibodies demonstrating the binding of GPR40 and NR2B. (C and F) Quantitative coimmunoprecipitation for detecting binding between GPR40 and NR2A/NR2B. (D) Quantitative analysis showing that the input of NR2A/GAPDH was not changed by GPR40 intervention. (E) Quantitative analysis of NR2A coimmunoprecipitation using the anti-NR2A antibody, showing that the level of coimmunoprecipitated NR2A was increased by GPR40 inhibition and decreased by GPR40 activation. (G) Quantitative analysis showing that the input of NR2B/GAPDH was not changed by GPR40 intervention. (H) Quantitative analysis of NR2B coimmunoprecipitation using the anti-NR2B antibody, showing that the level of coimmunoprecipitated NR2B was increased by GPR40 inhibition and decreased by GPR40 activation. For the analysis, n = 5 in each group. Error bars represent the means ± SEM; *P < 0.05 versus the DMSO group, one-way ANOVA, followed by Tukey’s test.
We next investigated whether GPR40 activation or inhibition can affect this interaction of GPR40 with NR2A or NR2B. In cultured primary mouse hippocampal neurons, GPR40 activation or inhibition did not alter the initial protein levels of solubilized NR2A and NR2B (Fig. 6, D and G). However, GPR40 activation decreased the level of coimmunoprecipitated NR2A, whereas GPR40 inhibition increased the level (Fig. 6E). Similarly, activation of GPR40 decreased the level of coimmunoprecipitated NR2B, whereas GPR40 inhibition increased this level (Fig. 6H). This result indicates that GPR40 activation or inhibition regulates interactions between GPR40 and NR2A/NR2B.
DISCUSSION
In the present study, we first found GPR40 to be up-regulated in epileptic brain tissue, and we report a novel finding that GPR40 modulates epileptic seizure activity in two mouse models of epilepsy. Furthermore, we found that GPR40 can regulate NMDAR-mediated neurotransmission. These effects may be due to GPR40-mediated regulation of NR2A/NR2B receptor endocytosis.
In the mouse hippocampus, GPR40 was highly expressed in the pyramidal cell layer and lacunosum moleculare layer of the CA1 and CA3 regions, which are the primary regions of epileptic pathological alterations (26). Immunofluorescent labeling of the human epileptic temporal lobe tissue adjacent to the sclerous hippocampus also showed GPR40 to be expressed in neurons but not in astrocytes. The protein level of GPR40 was increased in both human epileptic tissue and in the KA-injected hippocampus. These characteristics of GPR40 expression indicate that GPR40 may be involved in epilepsy.
The pathophysiological alterations that occur in epilepsy include pathogenic injuries that aggravate epileptic seizure activity and protective factors that prevent seizure activity (27, 28). Therefore, enhancing protective factors may suppress seizure activity. In our study, using the post-SE epilepsy model induced by KA and the PTZ kindling model, we demonstrated that GPR40 plays an important role in modulating epileptic activity.
Altered expression of a protein in a disease state may be a detrimental pathogenic factor, a compensatory protective factor, or an unrelated simultaneous epiphenomenon (29). Although alleviating pathogenic processes is a good target for preventing and treating epilepsy, enhancing protective factors is a less frequently studied option but may also be a good strategy for disease modification. The antiepileptic effect of GPR40 activation in combination with the increased expression of GPR40 indicates a compensatory and protective effect of GPR40 in epilepsy. This is consistent with the results from a previous study on pain reporting that GPR40 expression is increased in pain models and that a GPR40 agonist has antiallodynic and antihyperalgesic effects (13). Several other pieces of evidence also indicate that GPR40 may be a protective factor in brain function or neurological disease. The natural ligand of GPR40, PUFAs, is essential for brain development and nervous system function (15). Furthermore, PUFAs have been shown to exhibit antiseizure properties in animal studies, although this effect has not been consistently confirmed in human trials (30).
The effect of GPR40 in neuropathological conditions is only beginning to be elucidated. Regarding the mechanism of epilepsy, both transcriptomics and specific functional gene group studies have provided insight into the genes involved in synaptic transmission or the regulation of synaptic transmission (27). Therefore, we focused on the effects of GPR40 on synaptic transmission. We found that GPR40 activation and inhibition led to a decrease and increase in mEPSC frequency, respectively. These observations are consistent with findings showing that GPR40 agonists decrease spontaneous EPSC frequency in mechanical and thermal nociceptive models (13). Epileptic activity reflects a hyperexcitable state, which is caused by the imbalance of excitatory versus inhibitory cells or effects (31, 32). Our finding that GPR40 intervention did not affect mIPSCs suggests that GPR40 activation leads to a hypoexcitable state by decreasing excitatory effects. Furthermore, regarding excitatory synaptic transmission, we found that GPR40 mainly affected NMDAR-mediated but not AMPAR-mediated postsynaptic currents, which is consistent with human hippocampal studies suggesting that the primary pathological alteration of epilepsy is associated with physiologically active NMDARs (33).
In the present study, modulation of GPR40 caused changes in mEPSC frequency and amplitude without alteration of the PPR of the AMPA-EPSCs at different intervals. The PPR of AMPA-EPSCs is a sensitive index of presynaptic release probability (34, 35). In general, the amplitude of mEPSC discharge reflects the postsynaptic receptor effect, and frequency changes are associated with a presynaptic effect (36). Dendritic spines comprise the majority of excitatory synapses on hippocampal pyramidal neurons, and changes in spine density can lead to alterations in mEPSC frequency (37). Similar findings have also been reported by Gomez et al. (38). We further verified that GPR40 can alter the number of dendritic spines, which may be responsible for the changes in mEPSC frequency observed in the present study.
The molecular mechanism by which GPR40 regulates NMDAR-mediated excitatory synaptic transmission is unknown. Fundamentally, surface expression of NMDARs is essential for NMDAR-mediated postsynaptic responses, and NMDAR mislocalization contributes to the pathology of epilepsy (39). Hence, we mainly investigated the distribution of NR2 and found that cell surface expression of NR2A and NR2B was reduced in mice treated with the GPR40 agonist. Total NR2A and NR2B expression was unchanged, hinting at the possibility that GPR40-mediated regulation of NMDAR-mediated excitatory synaptic transmission involves regulation of NMDAR surface expression. Overall, the mechanism underlying NMDAR surface expression is not fully understood. Nonetheless, we demonstrated that GPR40 regulates NMDAR endocytosis. In addition, protein-protein interactions contribute to NMDAR surface expression (24). In our study, reciprocal coimmunoprecipitation revealed that GPR40 interacts with NR2. Furthermore, GPR40 activation decreased the binding of GPR40 with NR2A and NR2B, whereas GPR40 inhibition increased binding, suggesting that regulation of GPR40 binding to NR2 is a possible mechanism by which GPR40 regulates epileptic activity and neural excitability.
In conclusion, our findings demonstrate that GPR40 activation can alleviate epileptic seizures in animal models and NMDAR-mediated postsynaptic transmission, providing a novel antiepileptic target. The molecular mechanism of GPR40 action was preliminarily revealed to be the regulation of NR2A and NR2B surface expression, although further mechanisms remain to be explored.
MATERIALS AND METHODS
Animals
Adult male C57BL/6 mice weighing 26 to 30 g from the Laboratory Animal Center of Chongqing Medical University were housed in a standardized environment with a 12-hour light/dark cycle and controlled temperature at 22°C with food and water available ad libitum. All procedures used on mice were approved by the Commission of Chongqing Medical University for Ethics of Experiments on Animals and were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
KA model, drug administration, and LFP recordings
The chronic mouse model of spontaneous seizures induced by intrahippocampal administration of KA has been extensively described (18, 40). Mice were anesthetized and mounted on a stereotaxic apparatus (RWD Life Science Co. Ltd., Shenzhen, China). Using a 0.5-μl syringe (Hamilton, Reno, NV), 1.0 nmol of KA (Sigma-Aldrich Co., St. Louis, USA) in 50 nl of saline was injected into the right hippocampus [anteroposterior (AP), −1.6 mm; mediolateral (ML), −1.5 mm; dorsoventral (DV), −1.5 mm] over 3 min (40). The syringe was maintained in situ for an additional 5 min to minimize reflux along the injection trace. For intracerebroventricular injection of reagents, a guide cannula (0.64 mm outer diameter and 0.35 mm inner diameter; RWD Life Science Co. Ltd., Shenzhen, China) was implanted into the lateral ventricle (AP, 0.5 mm; ML, 1.0 mm; DV, 2.3 mm). For LFP recording, two stainless steel screws were implanted in the anterior cranium as ground screws, and a platinum-iridium alloy microwire (25 μm in diameter; Plexon, Hong Kong) was implanted into the right dorsal hippocampus (AP, 1.6 mm; ML, 1.6 mm; DV, 1.5 mm). The guide cannula, the microwire, and a U-shaped frame for holding the head were cemented to the skull.
Two hours after KA injection, nonconvulsive SE was terminated using diazepam. Three days after SE induction, the mice began receiving daily intracerebroventricular injections of GW9508 (1 μg per mouse; Selleck Chemicals), GW1100 (5 μg per mouse; Millipore), or vehicle (0.5% DMSO) for seven consecutive days through the implanted guide cannula. The doses were based on previous studies (12, 41).
One month after SE induction, the LFP recording was performed for 2 hours as previously described (40). Briefly, the head of the awake mouse was fixed to minimize behavioral state–induced LFPs changes. LFPs were recorded using an MAP data acquisition system (Plexon, Dallas, TX). The signals were filtered (0.1 to 500 Hz), preamplified (1000×), and digitized at 4 kHz.
The LFPs data were inspected using NeuroExplorer (Nex Technologies, Littleton, MA). A cluster of spontaneous paroxysmal discharges with a high-amplitude spike activity above 2 SDs from the baseline and a frequency above 1 Hz was defined as an SLE if it lasted for 5 s or more. SLEs were also confirmed by spectrogram analysis.
PTZ kindling model
Mice received intraperitoneal injections of PTZ (35 mg/kg; Sigma-Aldrich Co., St. Louis, USA) every other day for 30 days. GW9508 (1 μg per mouse), GW1100 (5 μg per mouse), or vehicle (0.5% DMSO) was intracerebroventricularly administered before PTZ injection. Behavioral seizures were scored according to the Racine scale: grade 0, no response; grade 1, staring and reduced locomotion; grade 2, activation of extensors and rigidity; grade 3, repetitive head and limb movements; grade 4, sustained rearing with clonus; and grade 5, GTCS with loss of posture and death (42).
TBI model
TBI was induced in mice using a controlled cortical impact (CCI) device TBI-0310 (Precision Systems and Instrumentation, Fairfax, VA, USA), as described previously (43). Mice were anesthetized with 3% isoflurane in 67% N2O/30% O2 until they were nonresponsive to the tail-pinch test. Then, 1.5% isoflurane was used to maintain anesthesia. The diameter of the impactor was 3 mm. The machine was set at a velocity of 5.0 m/s, a depth of 2.0 mm, and a dwell time of 100 ms, which produced a moderate contusion in the right cortex and underlying hippocampus, with pronounced behavioral deficits but virtually no mortality, thereby mimicking moderate TBI in humans. Mice were maintained at 36° to 37°C throughout the procedure. At 24 hours after CCI, mice were sacrificed under deep anesthesia, and the brain tissues around the injury site were subsequently harvested for Western blotting.
Whole-cell patch-clamp recordings
Whole-cell patch-clamp recordings were conducted as previously described in our laboratory (44). Male C57BL/6 mice (7 to 8 weeks old) were anesthetized and sacrificed for brain slice preparation. Transverse hippocampal slices (400 μm) were prepared (Leica VT1200S; Nussloch, Germany) in a cold sterile slice solution [2 mM CaCl2, 2 mM MgCl2, 2.5 mM KCl, 26 mM NaHCO3, 1.25 mM KH2PO4, 10 mM glucose, and 220 mM sucrose (pH 7.4) bubbled with 95% O2⁄5% CO2] and transferred to a storage chamber containing Mg2+-free artificial cerebrospinal fluid [ACSF; 125 mM NaCl, 2.5 mM KCl, 2 mM CaCl2, 26 mM NaHCO3, 1.25 mM KH2PO4, and 25 mM glucose (pH 7.4) bubbled with 95% O2/5% CO2] at 34°C for a recovery period of 1 hour before recording. For recordings, the slices were fully submerged in the same flowing Mg2+-free ACSF (4 ml/min) at room temperature (RT) (20 to 25°C).
For mIPSC recordings, glass pipette electrodes with a resistance of 3 to 5 megohms were filled with the following internal solution: 100 mM CsCl, 10 mM Hepes, 1 mM MgCl2, 1 mM EGTA, 5 mM MgATP, 0.5 mM Na3GTP, 12 mM phosphocreatine, and 30 mM N-methyl-d-glucamine (NMG) (pH 7.4), 280 to 290 mOsm. Voltage-clamp recordings were performed at a holding potential of −70 mV, and mIPSCs were recorded in the presence of 20 μM 6,7-dinitroquinoxaline-2,3(1H,4H)-dione (DNQX), 50 μM dl-2-amino-5-phosphonovaleric acid (D-APV), and 1 μM tetrodotoxin (TTX).
For mEPSC recordings, glass pipette electrodes with a resistance of 3 to 6 megohms were filled with the internal solution containing 130 mM CsMeSO4, 10 mM CsCl2, 10 mM Hepes, 4 mM NaCl, 1 mM MgCl2, 1 mM EGTA, 5 mM MgATP, 0.5 mM Na3GTP, 12 mM phosphocreatine, and 5 mM NMG (pH 7.4), 280 to 290 mOsm. Membrane potential was held at −70 mV in voltage-clamp mode, and mEPSCs were recorded in the presence of 1 μM TTX and 100 μM picrotoxin (PTX) to block GABAA (γ-aminobutyric acid type A).
To evaluate NMDAR- and AMPAR-mediated EPSCs, evoked currents were generated using a 400-μs pulse at a rate of 0.1 Hz (intensity, 50 to 200 μA) delivered by a stimulation isolation unit using an S48 pulse generator (AstroMed). A bipolar stimulating electrode was positioned in the Schaffer collaterals. The evoked currents were measured in the presence of 100 μM PTX and collected at two holding potentials. At −70 mV, with application of the NMDAR-selective antagonist D-APV (50 μM), the peak amplitude of the evoked EPSCs was identified as the AMPAR-mediated current. At +40 mV and in the presence of the AMPAR-selective antagonist DNQX (20 μM), the amplitude of the evoked EPSC at 50 ms poststimulus was identified as the NMDAR-mediated current.
To record the PPRs of the NMDA-EPSCs, EPSCs were evoked by stimulating the Schaffer collaterals–CA1 pathway at a holding potential of +40 mV in the presence of 100 μM PTX and 20 μM DNQX. Each stimulation was 0.1 Hz for 40 μs, and paired pulses were delivered at a 50-ms interpulse interval. To record the PPRs of the AMPA-EPSCs, EPSCs were evoked at a holding potential of −70 mV in the presence of 100 μM PTX and 50 μM D-APV. The intervals of paired stimulations were set at 25, 50, and 100 ms. The values of the ratios were defined as [p2/p1], where p1 and p2 are the amplitudes of the EPSCs evoked by the first and second pulses, respectively (34, 35). To clarify the mechanism by which GPR40 affects NMDAR surface expression levels, the exocytosis blocker TeTx (0.1 μM; Sigma-Aldrich) and the endocytosis blocker dynasore (80 mM; Abcam) were added to the internal solution for NMDA-EPSC recordings (45, 46).
All recordings were initially conducted in Mg2+-free ACSF with 0.1% DMSO. After the baseline data were recorded, GW9508 (20 μM) or GW1100 (20 μM) in 0.1% DMSO was added to the perfusate. Signals were acquired using a MultiClamp 700B amplifier (Axon, USA), filtered at 2 kHz and digitized at 10 kHz, followed by recording using pClamp 9.2 software (Molecular Devices, Sunnyvale, CA, USA). The Mini Analysis Program (Synaptosoft, Leonia, NJ) was used to analyze synaptic activity. In whole-cell recording experiments, the series resistance was kept below 20 megohms and was not compensated. Cells were rejected if series resistance fluctuated by more than 25% of the initial value. Data were collected after the current was stable for 5 to 15 min.
Human brain tissue collection
The collection of human epileptic brain tissue was previously described (47). Briefly, 20 neocortical tissues from pharmacoresistant TLE patients and 10 histological normal neocortical tissues from head trauma patients were surgically obtained from The First Affiliated Hospital of Chongqing Medical University. This process was in accordance with the Declaration of Helsinki and approved by the Ethics Committee of The First Affiliated Hospital of Chongqing Medical University. Before surgery, a written informed consent form was signed by the patients or their lineal relatives. All TLE patients received optimal medical treatment with three or more first-line AEDs without good seizure control. Before surgery, all patients underwent a comprehensive presurgical assessment, including an elaborate history and neurological examination, electroencephalogram studies, neuropsychological testing, and neuroimaging studies. Table S1 summarizes the clinical features of the TLE patients (7 males and 13 females; mean age, 22.5 ± 1.7 years; range, 9 to 39 years; epilepsy course, 9.5 ± 0.9 years). For controls, 10 histologically normal neocortical tissues were obtained from patients who were treated for increased intracranial pressure due to head trauma. No patients exhibited signs of central nervous system disease, and no patients had received AEDs before head trauma. The control patients did not undergo early seizure after head trauma. Before surgery, all patients underwent a comprehensive presurgical assessment, including an elaborate history, a neurological examination, and neuroimaging studies. Temporal neocortex tissues were taken from these patients only for decompression treatment purposes. Brain tissues that were relatively normal upon pathological examination were used as control human tissues. Table S2 summarizes the clinical features of the TBI patients (six males and four females; mean age, 23.4 ± 3.7 years; range, 8 to 40 years; Glasgow Coma Scale, 6.7 ± 0.40; time to surgical intervention, 16.4 ± 1.10 hours). No significant differences in age and sex were detected between the TLE patients and the control group (P >0.05).
Hippocampal neuronal culture
Hippocampal neurons were prepared from C57BL/6 mouse embryos at gestational day 18. First, brains were removed from mice and placed in 60-mm dishes with dissection medium. After dissection of the hippocampus from the brain, the hippocampi were collected and digested in a 60-mm dish containing 3 ml of 0.25% trypsin solution (Invitrogen) for 15 min at 37°C with gentle shaking every 5 min. The dissociated cells were plated at a density of 65 cells/mm2 on 18-mm coverslips in a six-well dish that contained Dulbecco’s modified Eagle’s medium basic [20% fetal bovine serum, 2 mM glutamine, and penicillin-streptomycin (100 units/ml); all from Invitrogen] and maintained in a 37°C/5% CO2 cell culture incubator. After 4 hours, the medium was replaced with Neurobasal medium containing 2% B27 supplement, 1% antibiotic, and 0.25% glutamine (Invitrogen). Thereafter, half of the Neurobasal medium was gently aspirated from each well and replaced every 3 days with an equal volume of fresh Neurobasal medium containing 2% B27 supplement, 1% antibiotic, and 0.25% glutamine. To determine the effect of GPR40 on the binding of GPR40 and NR2A/NR2B, the cultured neurons were treated with GW9508 (20 μM), GW1100 (20 μM), or 0.1% DMSO for 60 min before the neurons were lysed.
Biochemical measurement of surface-expressed receptors
Biochemical measurement of surface-expressed receptors was performed as previously described (44). Briefly, brain tissues were washed in wash solution and homogenized in permeabilization buffer. According to the instructions for the Mem-PER Plus Membrane Protein Extraction Kit for Soft Tissue (Thermo Fisher Scientific, Waltham, MA), the homogenate was incubated at 4°C for 10 min and centrifuged at 16,000g at 4°C for 15 min, and supernatant fractions were collected. A sample of 15 μg of protein from the supernatant was used to determine total protein expression. To determine surface expression, 150 μg of protein from the supernatant was incubated with 100 μl of 50% NeutrAvidin agarose (Pierce Chemical Co.) at 4°C overnight, and the bound proteins were resuspended in SDS sample buffer and boiled for 10 min. Western blotting was performed to detect both total and surface protein expression.
Western blotting and coimmunoprecipitation
Hippocampal neurons and brain tissue samples were collected for Western blot analysis. Protein samples were separated by 10% SDS–polyacrylamide gel electrophoresis gels and electrotransferred onto 0.45-μm polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). The membranes were blocked with 5% nonfat dry milk in tris-buffered saline with Tween (TBST) at RT for 1 hour and then incubated with rabbit anti-GPR40 (1:500; Santa Cruz, CA), anti-NR2B (1:1000; Abcam, Cambridge, UK), anti-NR2A (1:1000; Abcam), mouse anti-GAPDH (Proteintech), or mouse anti–β-actin (Proteintech) antibodies overnight at 4°C. The blots were washed three times and incubated for 1 hour with horseradish peroxidase (HRP)–conjugated anti-mouse or anti-rabbit secondary antibodies (1:5000; Proteintech) in TBST with 5% milk. The blots were then washed in TBST, and bands were visualized using an enhanced chemiluminescence reagent (Thermo, Marina, CA, USA) and the Fusion FX5 image analysis system (Vilber Lourmat, Marne-la-Vallée, France). Relative protein levels were determined by normalization to the GAPDH or β-actin signal using Quantity One software (Bio-Rad, CA, USA).
For coimmunoprecipitation, protein extracts from mouse hippocampal tissues or cultured hippocampal neurons were mixed with immunoprecipitation lysis buffer and incubated with 2 μl of rabbit IgG (Abcam), 4 μl of GPR40, 4 μl of NR2A, or 4 μl of NR2B antibodies overnight at 4°C followed by incubation with 20 μl of protein A/G agarose beads (Santa Cruz) for 2 h at 4°C. The protein-bead complex was then washed five times and collected by centrifugation. The protein samples were mixed with 2× loading buffer and subjected to Western blot analysis with GPR40, NR2A, or NR2B primary antibodies and HRP conjugate mouse anti-rabbit IgG (Conformation Specific, L27A9, Cell Signaling).
Immunofluorescence staining
Immunofluorescence was performed as previously described by our laboratory (48). Briefly, tissue sections were fixed in 4% polyformaldehyde for 1 min, washed three times with phosphate-buffered saline (PBS), permeabilized with 0.4% Triton X-100 for 30 min, and blocked with goat serum working liquid (Wuhan Boster Biological Technology, Wuhan, China) for 1 hour. The sections were then incubated overnight with mixed primary antibodies at 4°C, washed in PBS to remove unbound primary antibodies, and incubated with secondary antibodies in the dark at RT for 1 hour. The sections were counterstained with 4′,6 diamidino-2-phenylindole (Sigma-Aldrich) for 5 min and washed with PBS. The primary antibodies included rabbit anti-GPR40 (1:50; Santa Cruz, CA), guinea pig anti-MAP2 antibody (1:200; Synaptic Systems, Goettingen, Germany), mouse anti-GFAP antibody (1:100; Proteintech, China), and mouse anti-PSD95 antibody (1:400; Merck Millipore, USA). The fluorophore-conjugated secondary antibodies used were goat anti-guinea pig Alexa Fluor 650 (1:100; Abcam, USA), goat anti-rabbit Alexa Fluor 488 (1:100; Wuhan Boster Biological Technology, China), and goat anti-mouse Alexa Fluor 549 (1:100; Wuhan Boster Biological Technology, China). Images were captured by confocal laser scanning microscopy (Leica, Wetzlar, Germany). The fluorescence intensity was analyzed using Image-Pro Plus 6.0, and colocalization analyses were performed using ZEISS.
Spine density analysis
Neurons at DIV10 were transfected with PCAGGS-IRES-eGFP. At DIV16, neurons were first treated with 0.1% DMSO, GW9508 (20 μM), or GW1100 (20 μM) for 30 min and then washed, fixed, stained, and mounted. To label the dendrites and spines, fixed neurons were incubated with rabbit anti–green fluorescent protein (GFP) primary antibody (Invitrogen) and subsequently with fluorophore-conjugated goat anti-rabbit Alexa Fluor 488 secondary antibody. Alexa Fluor 488 images were captured on a confocal laser scanning microscope (Leica, Wetzlar, Germany). The number of spines was counted in a blinded manner, and dendrite length was measured in Image-Pro Plus 6.0.
Transferrin uptake assay
This experiment was carried out as described previously (49). Briefly, neurons were incubated at 37°C under neurobasal conditions with B27, l-Glu, and penicillin-streptomycin. At DIV14, the neurons were first treated with 0.1% DMSO, GW9508 (20 μM), or GW1100 (20 μM) for 20 min and then incubated with Alexa Fluor 594–transferrin conjugate (40 μg/ml; Invitrogen) for 30 min at 37°C. The neurons were then washed, fixed, stained, and mounted. To mark the neurons, fixed neurons were incubated with the primary mouse anti–β-III-tubulin antibody (Sigma-Aldrich) and, subsequently, with a fluorophore-conjugated secondary goat anti-mouse Alexa Fluor 488 antibody. Alexa Fluor 488 and Alexa Fluor 594 images were captured by confocal laser scanning microscopy (Leica, Wetzlar, Germany).
Image analysis was performed using Image-Pro Plus 6.0. The region of interest (ROI) of the cell was manually drawn on the Alexa Fluor 488 channel of the image and then transferred to the Alexa Fluor 594 channel. Finally, the mean pixel density of the ROI was measured.
Statistical analyses
To analyze measurement data (means ± SEM), Student’s t test was used for comparisons between two groups, and one-way ANOVA, followed by Tukey’s test, was used for comparisons between three groups. As the patch-clamp recordings were performed before and after treatment, the effect of each treatment was normalized to the baseline and analyzed using the paired t test. The χ2 test was used to compare sex differences between the epileptic patients and controls. The Mantel-Cox log rank test was used to compare percent survival over time. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank all the patients and their families for their participation in this study. Funding: This work was funded by the National Natural Science Foundation of China (grant number 81671301 to X.W. and grant number 31500821 to B.Z.). Author contributions: Y.Y. and X.T. designed and performed all experiments, analyzed data, and wrote the manuscript. F.Z. and Y.Z. participated in the behavioral experiments and analyzed data. D.X. and X.L. carried out the whole-cell patch-clamp recordings and analyzed data. B.Z., Y.M., Y.L., and X.X. participated in the molecular biology experiments and neuron cultures. B.Z. and X.W. designed all experiments, reviewed all data, and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/10/eaau2357/DC1
Fig. S1. Immunofluorescent labeling of GPR40 in nonepileptic brain tissues.
Fig. S2. Immunofluorescent labeling of GPR40 in epileptic tissues.
Fig. S3. Immunofluorescence intensity of GPR40 in epileptic and nonepileptic brain tissues.
Fig. S4. GPR40 has no effect on PPR for NMDA-mediated EPSCs.
Fig. S5. GPR40 regulates spine density.
Table S1. Clinical characteristics of epileptic patients in this study.
Table S2. Clinical characteristics of control individuals with brain trauma.
REFERENCES AND NOTES
- 1.Centers for Disease Control and Prevention , Epilepsy in adults and access to care—United States, 2010. MMWR Morb. Mortal. Wkly. Rep. 61, 909–913 (2012). [PubMed] [Google Scholar]
- 2.Fiest K. M., Sauro K. M., Wiebe S., Patten S. B., Kwon C.-S., Dykeman J., Pringsheim T., Lorenzetti D. L., Jetté N., Prevalence and incidence of epilepsy: A systematic review and meta-analysis of international studies. Neurology 88, 296–303 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Briscoe C. P., Tadayyon M., Andrews J. L., Benson W. G., Chambers J. K., Eilert M. M., Ellis C., Elshourbagy N. A., Goetz A. S., Minnick D. T., Murdock P. R., Sauls H. R. Jr, Shabon U., Spinage L. D., Strum J. C., Szekeres P. G., Tan K. B., Way J. M., Ignar D. M., Wilson S., Muir A. I., The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 278, 11303–11311 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Itoh Y., Kawamata Y., Harada M., Kobayashi M., Fujii R., Fukusumi S., Ogi K., Hosoya M., Tanaka Y., Uejima H., Tanaka H., Maruyama M., Satoh R., Okubo S., Kizawa H., Komatsu H., Matsumura F., Noguchi Y., Shinohara T., Hinuma S., Fujisawa Y., Fujino M., Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature 422, 173–176 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Kotarsky K., Nilsson N. E., Flodgren E., Owman C., Olde B., A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem. Biophys. Res. Commun. 301, 406–410 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Yazaki R., Kumagai N., Shibasaki M., Enantioselective synthesis of a GPR40 agonist AMG 837 via catalytic asymmetric conjugate addition of terminal alkyne to α,β-unsaturated thioamide. Org. Lett. 13, 952–955 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Yazaki R., Kumagai N., Shibasaki M., Cooperative activation of alkyne and thioamide functionalities; direct catalytic asymmetric conjugate addition of terminal alkynes to α,β-unsaturated thioamides. Chem. Asian J. 6, 1778–1790 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Walsh S. P., Severino A., Zhou C., He J., Liang G.-B., Tan C. P., Cao J., Eiermann G. J., Xu L., Salituro G., Howard A. D., Mills S. G., Yang L., 3-Substituted 3-(4-aryloxyaryl)-propanoic acids as GPR40 agonists. Bioorg. Med. Chem. Lett. 21, 3390–3394 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Tricò D., Herzog R. I., Metabolic brain adaptations to recurrent hypoglycaemia may explain the link between type 1 diabetes mellitus and epilepsy and point towards future study and treatment options. Diabetologia 60, 938–939 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khanna V., Arumugam S., Roy S., Mittra S., Bansal V. S., Topiramate and type 2 diabetes: An old wine in a new bottle. Expert Opin. Ther. Targets 12, 81–90 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Zamarbide M., Etayo-Labiano I., Ricobaraza A., Martínez-Pinilla E., Aymerich M. S., Luis Lanciego J., Pérez-Mediavilla A., Franco R., GPR40 activation leads to CREB and ERK phosphorylation in primary cultures of neurons from the mouse CNS and in human neuroblastoma cells. Hippocampus 24, 733–739 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Nakamoto K., Nishinaka T., Sato N., Aizawa F., Yamashita T., Mankura M., Koyama Y., Kasuya F., Tokuyama S., The activation of supraspinal GPR40/FFA1 receptor signalling regulates the descending pain control system. Br. J. Pharmacol. 172, 1250–1262 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karki P., Kurihara T., Nakamachi T., Watanabe J., Asada T., Oyoshi T., Shioda S., Yoshimura M., Arita K., Miyata A., Attenuation of inflammatory and neuropathic pain behaviors in mice through activation of free fatty acid receptor GPR40. Mol. Pain 11, 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan M. Z., He L., The role of polyunsaturated fatty acids and GPR40 receptor in brain. Neuropharmacology 113, 639–651 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Bazinet R. P., Layé S., Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 15, 771–785 (2014). [DOI] [PubMed] [Google Scholar]
- 16.DeGiorgio C. M., Miller P. R., Harper R., Gornbein J., Schrader L., Soss J., Meymandi S., Fish oil (n-3 fatty acids) in drug resistant epilepsy: A randomised placebo-controlled crossover study. J. Neurol. Neurosurg. Psychiatry 86, 65–70 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Ma D., Lu L., Boneva N. B., Warashina S., Kaplamadzhiev D. B., Mori Y., Nakaya M.-A., Kikuchi M., Tonchev A. B., Okano H., Yamashima T., Expression of free fatty acid receptor GPR40 in the neurogenic niche of adult monkey hippocampus. Hippocampus 18, 326–333 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Riban V., Bouilleret V., Pham-Lê B. T., Fritschy J.-M., Marescaux C., Depaulis A., Evolution of hippocampal epileptic activity during the development of hippocampal sclerosis in a mouse model of temporal lobe epilepsy. Neuroscience 112, 101–111 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Bouilleret V., Ridoux V., Depaulis A., Marescaux C., Nehlig A., Le Gal La Salle G., Recurrent seizures and hippocampal sclerosis following intrahippocampal kainate injection in adult mice: Electroencephalography, histopathology and synaptic reorganization similar to mesial temporal lobe epilepsy. Neuroscience 89, 717–729 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Mody I., Lambert J. D., Heinemann U., Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J. Neurophysiol. 57, 869–888 (1987). [DOI] [PubMed] [Google Scholar]
- 21.Lau C. G., Zukin R. S., NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 8, 413–426 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Yourick D. L., Repasi R. T., Rittase W. B., Staten L. D., Meyerhoff J. L., Ifenprodil and arcaine alter amygdala-kindling development. Eur. J. Pharmacol. 371, 147–152 (1999). [DOI] [PubMed] [Google Scholar]
- 23.Sprengel R., Suchanek B., Amico C., Brusa R., Burnashev N., Rozov A., Hvalby O., Jensen V., Paulsen O., Andersen P., Kim J. J., Thompson R. F., Sun W., Webster L. C., Grant S. G., Eilers J., Konnerth A., Li J., McNamara J. O., Seeburg P. H., Importance of the intracellular domain of NR2 subunits for NMDA receptor function in vivo. Cell 92, 279–289 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Lee F. J., Xue S., Pei L., Vukusic B., Chéry N., Wang Y., Wang Y. T., Niznik H. B., Yu X. M., Liu F., Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell 111, 219–230 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Yang K., Jackson M. F., MacDonald J. F., Recent progress in understanding subtype specific regulation of NMDA receptors by G Protein Coupled Receptors (GPCRs). Int. J. Mol. Sci. 15, 3003–3024 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bronen R. A., Cheung G., Charles J. T., Kim J. H., Spencer D. D., Spencer S. S., Sze G., McCarthy G., Imaging findings in hippocampal sclerosis: Correlation with pathology. AJNR Am. J. Neuroradiol. 12, 933–940 (1991). [PMC free article] [PubMed] [Google Scholar]
- 27.Pitkänen A., Lukasiuk K., Mechanisms of epileptogenesis and potential treatment targets. Lancet Neurol. 10, 173–186 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Pitkänen A., Lukasiuk K., Molecular and cellular basis of epileptogenesis in symptomatic epilepsy. Epilepsy Behav. 14 (suppl. 1), 16–25 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Dalby N. O., Mody I., The process of epileptogenesis: A pathophysiological approach. Curr. Opin. Neurol. 14, 187–192 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Taha A. Y., Burnham W. M., Auvin S., Polyunsaturated fatty acids and epilepsy. Epilepsia 51, 1348–1358 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Rubenstein J. L. R., Merzenich M. M., Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2, 255–267 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDonald J. W., Garofalo E. A., Hood T., Sackellares J. C., Gilman S., McKeever P. E., Troncoso J. C., Johnston M. V., Altered excitatory and inhibitory amino acid receptor binding in hippocampus of patients with temporal lobe epilepsy. Ann. Neurol. 29, 529–541 (1991). [DOI] [PubMed] [Google Scholar]
- 33.Ghasemi M., Schachter S. C., The NMDA receptor complex as a therapeutic target in epilepsy: A review. Epilepsy Behav. 22, 617–640 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Clark K. A., Randall A. D., Collingridge G. L., A comparison of paired-pulsed facilitation of AMPA and NMDA receptor-mediated excitatory postsynaptic currents in the hippocampus. Exp. Brain Res. 101, 272–278 (1994). [DOI] [PubMed] [Google Scholar]
- 35.Zinebi F., Russell R. T., McKernan M., Shinnick-Gallagher P., Comparison of paired-pulse facilitation of AMPA and NMDA synaptic currents in the lateral amygdala. Synapse 42, 115–127 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Oberlander J. G., Woolley C. S., 17β-Estradiol acutely potentiates glutamatergic synaptic transmission in the hippocampus through distinct mechanisms in males and females. J. Neurosci. 36, 2677–2690 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sala C., Piëch V., Wilson N. R., Passafaro M., Liu G., Sheng M., Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31, 115–130 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Gomez A. M., Froemke R. C., Burden S. J., Synaptic plasticity and cognitive function are disrupted in the absence of Lrp4. eLife 3, e04287 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frasca A., Aalbers M., Frigerio F., Fiordaliso F., Salio M., Gobbi M., Cagnotto A., Gardoni F., Battaglia G. S., Hoogland G., Di Luca M., Vezzani A., Misplaced NMDA receptors in epileptogenesis contribute to excitotoxicity. Neurobiol. Dis. 43, 507–515 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Sada N., Lee S., Katsu T., Otsuki T., Inoue T., Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 347, 1362–1367 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Nakamoto K., Aizawa F., Nishinaka T., Tokuyama S., Regulation of prohormone convertase 2 protein expression via GPR40/FFA1 in the hypothalamus. Eur. J. Pharmacol. 762, 459–463 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Racine R. J., Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 32, 281–294 (1972). [DOI] [PubMed] [Google Scholar]
- 43.Jiang Y., Brody D. L., Administration of COG1410 reduces axonal amyloid precursor protein immunoreactivity and microglial activation after controlled cortical impact in mice. J. Neurotrauma 29, 2332–2341 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu X., Hu Y., Xiong Y., Li Z., Wang W., Du C., Yang Y., Zhang Y., Xiao F., Wang X., Association of microtubule dynamics with chronic epilepsy. Mol. Neurobiol. 53, 5013–5024 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Bu Y., Wang N., Wang S., Sheng T., Tian T., Chen L., Pan W., Zhu M., Luo J., Lu W., Myosin IIb-dependent regulation of actin dynamics is required for N-methyl-D-aspartate receptor trafficking during synaptic plasticity. J. Biol. Chem. 290, 25395–25410 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang W.-T., Pan G.-Q., Zhang Z.-Y., Suo Z.-W., Yang X., Hu X.-D., Ht31 peptide inhibited inflammatory pain by blocking NMDA receptor-mediated nociceptive transmission in spinal dorsal horn of mice. Neuropharmacology 89, 290–297 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y., Chen G., Gao B., Li Y., Liang S., Wang X., Wang X., Zhu B., NR4A1 knockdown suppresses seizure activity by regulating surface expression of NR2B. Sci. Rep. 6, 37713 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiong Y., Zhang Y., Zheng F., Yang Y., Xu X., Wang W., Zhu B., Wang X., Expression of Glypican-4 in the brains of epileptic patients and epileptic animals and its effects on epileptic seizures. Biochem. Biophys. Res. Commun. 478, 241–246 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Royle S. J., Bright N. A., Lagnado L., Clathrin is required for the function of the mitotic spindle. Nature 434, 1152–1157 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/10/eaau2357/DC1
Fig. S1. Immunofluorescent labeling of GPR40 in nonepileptic brain tissues.
Fig. S2. Immunofluorescent labeling of GPR40 in epileptic tissues.
Fig. S3. Immunofluorescence intensity of GPR40 in epileptic and nonepileptic brain tissues.
Fig. S4. GPR40 has no effect on PPR for NMDA-mediated EPSCs.
Fig. S5. GPR40 regulates spine density.
Table S1. Clinical characteristics of epileptic patients in this study.
Table S2. Clinical characteristics of control individuals with brain trauma.