

Abstract
Polycystic ovary syndrome (PCOS) is a heterogeneous endocrine disorder affecting women of reproductive age. The origin of PCOS is still not clear and appears to be a function of gene x environment interactions. This review addresses the current knowledge of the genetic and developmental contributions to the etiology of PCOS, the ovarian and extra-ovarian mediators of PCOS and the gaps and key challenges that need to be addressed in the diagnosis, treatment and prevention of PCOS.
Keywords: Polycystic ovary syndrome, Ovary, Follicle, Granulosa cells, Developmental programming, Androgen, Insulin, Sheep
INTRODUCTION
Polycystic ovarian syndrome (PCOS) is the most common infertility disorder affecting 5–20% of women in their reproductive age (Azziz, et al. 2016). Stein and Leventhal described this condition for the first time in 1935 when they identified in 7 patients enlarged ovaries in association with menstrual disturbances (most notably amenorrhea), sterility, pain, or hyperandrogenism (Stein and Leventhal 1935). Since then PCOS has been recognized as a heterogeneous condition with various consensus meetings establishing diagnostic criteria for the identification and treatment of this condition. The 1990 United States National Institutes of Health (NIH) diagnostic criteria includes clinical and/or biochemical signs of hyperandrogenism and menstrual dysfunctions (Zawadzki and Dunaif 1992), while the 2003 Rotterdam consensus requires meeting two of the following features: Clinical and/or biochemical hyperandrogenism, oligo-ovulation or polycystic ovaries (The Rotterdam ESHRE/ASRM‐sponsored PCOS consensus workshop group 2004). The androgen excess society in 2006 also proposed that all features such as clinical and/or biochemical hyperandrogenism and ovarian dysfunction including oligo/anovulation and/or polycystic ovaries should be considered as diagnostic features of PCOS (Azziz, et al. 2009). All these criteria, regardless of which source, required exclusion of disorders that mimic similar symptoms such as thyroid dysfunction, hyperprolactinaemia, adrenal hyperplasia, androgen-secreting tumors, among others. In spite of these, confusion regarding the use of different criteria in diagnosis and research continued, which prompted NIH through its Consensus Development Programs to organize an Evidence-Based Methodology PCOS Workshop. This workshop panel recommended adoption of the broader 2003 Rotterdam criteria with sub-classification of the PCOS sub-phenotypes as depicted in Table 1 (National Institues of Health 2012). All in all, PCOS is a complex condition manifesting a wide variety of dysfunctions in addition to those considered as diagnostic criteria. These include i) cardiometabolic dysfunctions such as basal and glucose-stimulated hyperinsulinemia independent of their body mass index (Dunaif, et al. 1989), insulin resistance (DeUgarte, et al. 2005; Dunaif, et al. 2001), and increased risk for cardiovascular diseases (Cobin 2013; Legro, et al. 2001), ii) depression (Cooney and Dokras 2017), iii) gestational diabetes and pre-eclampsia, and iv) poor birth outcomes such as small or large for gestational age newborns, congenital abnormalities and perinatal mortality (Boomsma, et al. 2006; Doherty, et al. 2015; Qin, et al. 2013). This review aims to summarize the potential causes with focus on the ovarian and extra-ovarian factors implicated in the pathogenesis of PCOS.
Table 1:
Different PCOS sub-phenotypes for diagnostic criteria recommended by the expert panel at the NIH’s Evidence-Based Methodology PCOS Workshop
| Symptoms | Sub-phenotype A | Sub-phenotype B | Sub-phenotype C | Sub-phenotype D |
|---|---|---|---|---|
| Clinical & Biochemical signs of hyperandrogenism | + | + | + | - |
| Ovulatory dysfunctions | + | + | - | + |
| Polycystic ovary morphology (PCOM) | + | - | + | + |
ORIGIN OF PCOS
Because of the heterogeneous nature of this condition, the etiopathology of PCOS is still not clearly identified. Factors ranging from genetic to environmental and/ or the interaction between them have been proposed to have a role in the origin of PCOS.
Genetic basis in the origin of PCOS
The observations that there is a high amount of familial aggregation among first degree female relatives of women with PCOS (Legro, et al. 1998a) coupled with heritability score of 0.79 for PCOS phenotype in the Dutch twin study (Vink, et al. 2006) provide credence for the notion that PCOS is heritable (Franks, et al. 1997). Among the diagnostic characteristics of PCOS, evidence for heritability is strongest for hyperandrogenism (Legro et al. 1998a; Legro, et al. 1998b). Nonetheless, considering the heterogeneity in PCOS phenotypes and the varying attributes it is now not believed to be a monogenic disease (Fenichel, et al. 2017). In support of this, mutations or polymorphism in several genes have been identified (Dunaif 2016). Both individual gene analysis and genome-wide association studies (GWAS) have identified mutations or polymorphisms in follicle stimulating hormone receptor (FSHR), luteinizing hormone receptor (LHCGR), DENN/MADD domain containing 1A (DENND1A), RAB5B, member RAS oncogene family (RAB5B), and thyroid adenoma associated (THADA) gene loci in Han Chinese and American or European Caucasian individuals with PCOS (Eriksen, et al. 2013; Goodarzi, et al. 2012; Ha, et al. 2015; Hayes, et al. 2015; Louwers, et al. 2013). While mutations, polymorphisms and splice variants in genes such as follistatin, fibrilin 3, cytochrome p450 side-chain cleavage (CYP11A), insulin receptor (INSR), hydroxysteroid dehydrogenase (HSD) 17B5, HSD17B6 and androgen receptor have also been linked to PCOS, such observations have not been confirmed in large populations or in multiple ethnicities (Azziz 2016; Dunaif 2016; Puttabyatappa, et al. 2017a). The degree to which each of these genes contribute to the final reproductive and metabolic phenotype of PCOS women remains unclear. For instance, while overexpression and silencing of the polymorphic variant of the DENND1A in the human theca cells, the main source of testosterone for the hyperandrogenic phenotype, has been shown to increase and decrease testosterone synthesis, respectively (McAllister, et al. 2014), to what extent alterations in this is linked to other PCOS symptoms is unclear.
Genetic mouse models involving many of the gene variants linked to PCOS are limited. A transgenic mouse overexpressing the DENND1A variant identified in PCOS patients resulted in a hyperandrogenic state with no impact on fertility (Modi 2016); detailed phenotypic assessment of PCOS characteristics is not available for this model. Transgenic mouse models overexpressing LH beta subunit while manifesting chronically elevated LH / testosterone levels and infrequent ovulation produced a cystic, tumorogenic ovarian phenotype (Risma, et al. 1995). While FSH deficiency and polymorphism in FSHR has been linked to PCOS (Dolfin, et al. 2011), mouse models of FSH deficiency although infertile fail to manifest a hyperandrogenic or multifollicular ovarian phenotype (Barnett, et al. 2006; Burns and Matzuk 2002). Prevention of the development of PCOS phenotype that normally follow prenatal androgen treatment in global, brain or theca cell-specific AR knock mice emphasizes a role for AR (Walters and Handelsman 2017). This observation is consistent with polymorphisms in AR gene being linked to PCOS (Wang, et al. 2015). These findings from transgenic models combined with the <10% heritability estimate of PCOS-linked loci identified through GWAS (Azziz 2016; Moore and Campbell 2017) suggest involvement of additional loci and factors. Several transgenic models are available that link other loci (nerve growth factor, plasminogen activator inhibitor 1, estrogen receptor alpha) to PCOS characteristics (Barnett et al. 2006; Walters, et al. 2012). However these loci have not been substantiated in large PCOS cohort studies.
Developmental basis in the origin of PCOS
The observation that individuals born with low birth weight are at high risk for manifestation of cardiometabolic disorders during adulthood led to the developmental origin of health and disease (DOHaD) hypothesis by Barker (Gluckman and Hanson 2004). According to this hypothesis, early fetal exposure to stressors can induce physiological adaptations that fail and manifest as disease during adulthood. The findings that girls born either small or large for their gestational age are at increased risk for developing PCOS during reproductive life (Melo, et al. 2010; Mumm, et al. 2013), suggests PCOS could also have developmental basis. Additional support for this premise comes from reports of 1) PCOS phenotype in offspring exposed to excess androgen in utero, which occur in conditions such as congenital adrenal hyperplasia (Hague, et al. 1990), congenital virilizing tumors (Barnes, et al. 1994), and loss of function mutations in aromatase (Morishima, et al. 1995) or sex hormone-binding globulin gene (SHBG) (Hogeveen, et al. 2002), 2) elevated second trimester amniotic fluid testosterone levels in PCOS women (Palombo, et al. 2012) and 3) increased anogenital distance (Barret, et al. 2018; Wu, et al. 2017) and 2nd to 4th finger (2D:4D) ratio (Palombo, et al. 2012), biomarkers of prenatal androgen exposure in offspring of PCOS women. The developmental origins of PCOS theory is also supported by studies in murine, rodent, sheep and monkey models, which show that administration of steroids such as testosterone, dihydrotestosterone or estradiol valerate or steroid synthesis inhibitors during the perinatal period induce the development of PCOS like phenotype (Abbott, et al. 2005; Maliqueo, et al. 2014; Padmanabhan and Veiga-Lopez 2013). For instance, the prenatal testosterone-treated female sheep, the animal model our group works with, manifests fetal growth restriction and is born with low birth weight (Manikkam, et al. 2004; Steckler, et al. 2005). As these animals age they manifest disruptions in neuroendocrine steroid feedback, increased pituitary sensitivity to gonadotropin-releasing hormone, LH hyper-secretion, functional hyperandrogenism, multifollicular ovarian morphology, oligo- / an-ovulation and insulin resistance. In addition to meeting the diagnostic criteria proposed for PCOS by all agencies, these animals manifest cardiometabolic disruptions such as that seen in women with PCOS (Cardoso, et al. 2015; Padmanabhan and Veiga-Lopez 2013, 2014). Similarly, developmental exposure of sheep to bisphenol A (BPA, an environmental endocrine disrupting compound (EDC) results in low birth weight (Savabieasfahani, et al. 2006) offspring, which during adulthood manifest hypothalamic, pituitary and ovarian changes that mimic the PCOS phenotype (Veiga-Lopez, et al. 2014a). Human studies also point to an association between BPA and hyperandrogenism (Rutkowska and Rachon 2014).
While the findings from animal and human disease models suggest that inappropriate developmental exposure to native and environmental steroidal mimics during critical windows of differentiation can result in a PCOS phenotype, this does not necessarily imply that this is the basis for the etiology of human PCOS. For instance, although cordocentesis studies have found 40% of human female fetuses during the second trimester have male circulating levels of testosterone (Beck-Peccoz, et al. 1991), the prevalence rate for PCOS is far less, only 5–20%. Similarly, the prevalence of PCOS in females who are co-twin with male is not different compared to females born as part of same-sex twin pairs or singletons (Kuijper, et al. 2009). These observations are at odds with the premise that developmental exposure to excess steroids, by itself, can explain the etiology of PCOS.
Gene-environment interaction
The evidence accumulated so far suggests that the pathogenesis of PCOS is likely complex and quite possibly involve gene x environment interaction. Such an interaction would not only explain the prevalence estimate of PCOS (5–20%) relative to number of fetuses getting exposed to excess testosterone (40%), but also the differing phenotypic manifestation of PCOS phenotypes. Phenotypic differences in neuroendocrine, ovarian, and metabolic defects in animal models following prenatal androgen excess can originate from differences in the timing, duration, and degree of exposure. However, the fact that phenotypic differences are also evident amongst animals subjected to identical exposure paradigms highlights contribution from individual genetic susceptibility to such developmental insults. Such gene x environment interactions are likely mediated by epigenetic mechanisms (Barros and Offenbacher 2009), involving changes in DNA methylation, histone acetylation, and non-coding RNA expression (Amaral and Mattick 2008; Kim, et al. 2009). As such the epigenome provides a means to translate the information captured from the environment to heritable changes by turning on or off gene expression patterns.
From a PCOS perspective, epigenetic changes in the androgen receptor gene (Hickey, et al. 2006) and increased presence of markers of epigenomic alterations in the whole blood, ovarian and adipose tissues from PCOS women have been reported (Jones, et al. 2015; Li, et al. 2016). Epigenetic modifications have also been observed in prenatal androgenized animal models of PCOS. These include changes in 163 and 325 methylated loci in infant and adult visceral adipose tissues of prenatally androgenized rhesus macaques (Xu, et al. 2011), changes in expression of miR-497 and miR-15b in fetal ovaries from prenatal testosterone-treated sheep (Luense, et al. 2011) and hypomethylation of five CpG sites of AR and one single CpG site in Cyp11a1 (CpG +953) in ovaries of prenatal testosterone-treated rats (Xia, et al. 2015). In this context it is important to recognize that sex hormones (estrogens and androgens), which are perturbed in PCOS women, are known activators of epigenetic mechanisms (Crews, et al. 2014; Hunter, et al. 2015).
Severity of PCOS phenotype
While genetic and developmental insults induce disruptions early in life, various postnatal events such as diet, lifestyle, disease states, stress and environmental exposures can have continuing influence on the disease state. Recently the idea that these postnatal factors may act as ‘second hit’ to either maintain, unmask or amplify the severity of disease phenotype programmed by genetic or developmental insults is gaining prominence. Such a phenomenon has been described in the pathogenesis of schizophrenia and cancer (Bayer, et al. 1999; Tang and Ho 2007). Because PCOS symptoms do not become apparent till puberty, postnatal factors can serve as a second hit in terms of the manifestation or amplification of the severity of the PCOS phenotype. For instance, LH excess and metabolic disruptions that originate from the disruption of the fetal hypothalamic-pituitary-ovarian axis by the maternal hyperandrogenic condition (first hit) themselves can serve as a second hit contributing to the severity of offspring’s reproductive and metabolic phenotype (Bremer 2010). Additionally, as 38 to 88% of PCOS patients have obesity with associated metabolic disorders such as insulin resistance (Diamanti-Kandarakis and Dunaif 2012), hyperinsulinemia due to insulin resistance can act as a second hit. Hyperinsulinemia increases theca cell androgen production (Nahum, et al. 1995), which when associated with reduced levels of SHBG - a buffer to sequester free testosterone, can induce hyperandrogenemia (Nestler, et al. 1991). Studies in animal models provide support for the two hit hypothesis in PCOS pathogenesis. In the prenatal testosterone-treated sheep model of PCOS phenotype, postnatal obesity has been found to act as a second hit to increase the severity of PCOS-like symptoms (Steckler, et al. 2009). Although not tested, PCOS phenotype has also been postulated to involve both the prenatal testosterone exposure and postnatal adiposity in rhesus macaques (Abbott, et al. 1998).
MEDIATORS OF PCOS
PCOS patients manifest disruptions at both ovarian and extra-ovarian levels. Ovarian changes that contribute to the diagnostic criteria of PCOS include multifollicular appearance, hyperandrogenism, oligo/an-ovulation and luteal defects. The extra-ovarian changes, although not part of diagnostic criteria, include LH hypersecretion with increased LH/FSH ratio at the neuroendocrine level and hyperinsulinemia, hyperglycemia, dyslipidemia and altered adipokine secretion at the metabolic level. The ovarian and extra-ovarian factors that contribute towards the phenotypic manifestation of PCOS are discussed in detail below.
Ovarian mediators of PCOS
Women with PCOS are characterized by multifollicular ovarian morphology and ovarian enlargement (an increase in ovarian area and volume). Polycystic (multifollicular) ovarian morphology (PCOM) is a diagnostic criterion for the diagnosis of PCOS as defined by the 2003 Rotterdam consensus and the 2006 Androgen Excess & PCOS Society criteria and recent NIH consensus meeting (Azziz et al. 2016; Dewailly, et al. 2014). Earlier ovarian studies in humans were confined to wedge resection or postmortem tissues and these studies have shown that the polycystic ovary (PCO, a misnomer terminology) appearance results from presence of 10–12 growing follicles that measure < 10mm in size along with increase in stromal hypertrophy (Hughesdon 1982). With advance in non-invasive imaging tools such as transvaginal ultrasound, follicle count thresholds for distinguishing PCOM ovaries from normal ovaries have changed over time. The most recent recommendation by the task force of the Androgen Excess and the Polycystic Ovary Syndrome Society (AE-PCOS) is a threshold setting of >25 follicles, when scanned with transducer frequency ≥8 MHz (Dewailly et al. 2014).
Most studies addressing follicular number and distribution have involved single time point scanning or histological observations with postmortem ovaries. These studies have concluded ovaries of PCOS women are characterized not only by increased number of antral follicles (Dewailly et al. 2014), but also a reduction in number of primordial follicles (Webber, et al. 2003) and follicular arrest (Franks, et al. 2008). The PCOM phenotype in PCOS women can therefore arise from 1) enhanced recruitment and 2) follicular persistence due to arrest in follicular development, premature luteinization, and reduced state of atresia (Franks et al. 2008). Additionally, women with PCOS undergoing in vitro fertilization have been found to produce more number of oocytes, which are often of poor quality thus contributing to the reduced fertilization, cleavage and implantation rates (Qiao and Feng 2011). The potential ovarian mediators of each of these aspects of ovarian disruptions are discussed below and summarized in Table 2 and Figure 1.
Table 2:
Ovarian factors that contribute to PCOS phenotype
| Factor | Changes observed in PCOS patients (References) | Ovary-specific mechanisms | Role |
|---|---|---|---|
| Follicular activation/recruitment | |||
| Anti-Mullerian Hormone (AMH) | Low AMH expression in primordial and transitional follicles (Stubbs et al 2005) | Inhibits follicular activation | Inhibits activation |
| Fibrillin 3 (FBN3) | Polymorphisms in D19S884 allele 8 that maps to fibrillin 3 (FBN3) gene (Urbanek et al 1999) | Regulates bioavailability of TGF family members | May contribute to activation of follicles |
| Androgens | General and ovarian hyperandrogeneism (Reviewed in Franks et al 2008 & Dewailly et al. 2016) | Stimulates AKT pathway that leads to inactivation of FOXO3 | Increases activation |
| Follicular persistence/arrest | |||
| Androgens | General and ovarian hyperandrogeneism (Reviewed in Dewailly et al. 2016) | Contributes to increase FSHR and LHR expression that leads to induction of premature luteinization | Causes arrest |
| Estrogens | Increased or low follicular fluid levels (Reviewed in Franks et al. 2008) | Increased levels reduce pituitary FSH secretion & low levels can inhibit follicular growth. | Contributes to arrest |
| Activin | Decreased follicular fluid and serum levels (Norman et al. 2001) | Promotes pituitary FSH secretion and ovarian FSH action. | Prevents arrest |
| Follistatin | Increased follicular fluid and serum levels (Erickson et al. 1995) | Inhibits activin | Contributes to arrest |
| Inhibin | Decreased inhibin A and B forms in follicular fluid levels; Inhibits activins (Magoffin and Jakimiuk 1998; Welt et al. 2005) | Decreases pituitary FSH secretion | Causes arrest |
| AMH | Increased antral follicular fluid levels (Fallat et al. 1997; Desforges-Bullet, et al. 2010) | Reduces sensitivity to FSH | Causes arrest |
| Insulin-like growth factor (IGF) | Increased follicular fluid levels (Eden et al. 1990) | In excess levels may inhibit follicular growth | Contributes to arrest |
| IGF binding proteins (IGFBP) | Decreased IGFBP1 and 4 follicular fluid levels (Holly et al 1990; Cataldo & Giudice 1992) | Regulates IGF bioavailability | Contributes to arrest |
| Epidermal growth factor (EGF) | Increased follicular fluid levels (Volpe et al. 1991) | Induces premature luteinization | Causes arrest |
| Nerve growth factor (NGF) | High levels in follicular fluid (Dissen et al. 2009) | Unknown | Causes arrest |
| Vascular endothelial growth factor (VEGF) | High levels in follicular fluid Increased granulosa cell and reduced theca cell protein content (Savchev et al. 2010 (20697141); Ferrara et al. 2003) | Induces premature luteinization | Causes arrest |
| Endocrine gland-derived VEGF (EB-VEGF) | Increased expression in stromal theca-interna cells(Ferrara et al. 2003) | Promotes VEGF expression | Contributes to arrest |
| Fibroblast growth factor (FGF) | Low levels in follicular fluid levels (Hammadeh et al. 2003 (12846675) Elevated levels in follicular fluid (Artini et al. 2006) |
Induces premature luteinization | Causes arrest |
| Adiponectin | Low levels of high molecular weight form in follicular fluid; decreased adiponectin and its receptor expression in granulosa cells. (Artimani et al. 2016) | Promotes theca cell androgen synthesis | Causes arrest |
| Tumor necrosis factor alpha (TNF) | High in follicular fluid (Amato et al. 2003) | Participates in granulosa cell differentiation, proliferation and apoptosis | Contributes to arrest |
| Interleukins (IL) | High follicular fluid IL6 and 13 and low follicular fluid IL12 (Amato et al. 2003) | Reduces estradiol synthesis | Contributes to arrest |
| Matrix metalloproteinase (MMP) | No change in MMP2 and 9 activities in follicular fluid (Lahav-Baratz et al. 2003); Higher follicular fluid content of MMP2 and 9 and increased expression of MMP9 in granulosa cells (Shalev et al. 2001) | Induces premature luteinization | Contributes to arrest |
| Tissue inhibitors of MMP (TIMP) | Low TIMP1 levels in follicular fluid (Lahav-Baratz et al. 2003); No change in expression in granulosa cells (Shalev et al. 2001). | Regulates MMP function | Contributes to arrest |
| Fas | Decreased serum and follicular fluid levels of soluble Fas Promotes apoptosis (Onalan et al. 2005) |
Reduced levels decrease atresia | Contributes to arrest |
| MicroRNAs | miR-320a down-regulated in cumulus cells – targets RUNX2 which regulates steroidogenesis and granulosa cell differentiation (Zhang et al 2017) miR-483 down-regulated in ovarian cortex – targets IGF1 (Xiang et al. 2016) miR-145 down-regulated in granulosa cells – targets insulin receptor substrate 1 (IRS1) that regulates granulosa cell proliferation (Cai et al. 2017) Decreased follicular fluid levels of miR-93 and miR-21 – targets TGF beta family members (Naji et al. 2017) miRNAs 200a-3p, 10b-3p, 200b-3p, 29c-3p, 99a-3p, and 125a-5p are elevated & miR-105–3p is decreased in follicular fluid – Many of these are associated with androgen synthesis (Xue et al., 2017) Decreased miR-145 and miR-182 in granulosa cells & Increased miR-182 in follicular fluid (Naji et al 2018) |
Variable mechanism depending on their gene targets | Variable outcomes depending on their gene targets |
| WNT/frizzled/β-catenin | Frizzled 3 expression increased in cumulus cells (Qaio et al. 2017) | Decreases estradiol synthesis | Causes arrest |
| Forkhead box O3 transcription factor (FOXO3) | Increased FOXO3 mRNA and protein and decreased phospho FOXO3 in granulosa cells from antral follicles (Mikaeili et al. 2016) |
Promotes apoptosis of granulosa cells, participates in follicular atresia | Causes follicular persistence |
| Oocyte quality | |||
| Growth differentiation factor 9 (GDF9) | Reduced cumulus cells and normal oocyte expression of mRNA (Zhao et al. 2010) | Participates in oocyte maturation | Promotes oocyte quality |
| Bone morphogenetic protein 15 (BMP15) | No change in oocyte or cumulus expression of mRNA (Zhao et al. 2010) | Participates in oocyte maturation | Promotes oocyte quality |
| Brain-derived neurotropic factor (BDNF) | Increased follicular fluid levels (Johnstone et al. 2008) | Unknown | Promotes oocyte quality |
| FGF | Low levels in follicular fluid levels (Hammadeh et al. 2003 (12846675)) Elevated levels in follicular fluid (Artini et al. 2006) |
Unknown | Reduces oocyte quality |
| EGF | Increased follicular fluid levels (Volpe et al. 1991) | Involved in oocyte maturation | May promote oocyte quality |
| IGF | Increased follicular fluid levels (Eden et al. 1990) | May be involved in oocyte maturation | May promote oocyte quality |
| VEGF | High levels in follicular fluid (Savchev et al. 2010) |
Stimulates maturation of oocytes | Promotes oocyte quality |
| AMH | Increased antral follicular fluid levels (Fallat et al. 1997; Desforges-Bullet, et al. 2010) | May participate in oocyte maturation | Reduces oocyte quality |
| IL | High follicular fluid IL6 and 13 and low follicular fluid IL12 (Amato et al. 2003) | May participate in oocyte maturation | Reduces oocyte quality |
| Leukemia inhibiting factor (LIF) | Low follicular fluid levels (Lédée-Bataille et al. 2001) | Promotes oocyte maturation | Promotes oocyte quality |
| TNF | High follicular fluid (Amato et al. 2003) | Decreases oocyte maturation and induces abnormal chromosomal alignment and cytoskeleton structure in oocyte | Reduces oocyte quality |
| Corticotrophin-releasing hormones (CRF) | Low follicular fluid levels (Mastorakos et al. 1994) | Unknown | Promotes oocyte quality |
Figure 1:

Schematic showing the stages of follicular development in normal ovary.
Follicular activation/recruitment
Ovaries have finite number of primordial follicles at birth that form the ovarian follicular pool or reserve where they remain in a quiescent state. The transition of quiescent primordial follicles in the ovary to primary follicles that initiates the growing phase is referred to as follicular activation or recruitment (Figure 1). During each reproductive cycle, a few follicles from the primordial pool undergo either activation / recruitment, of which only a few undergo further follicular development into preantral follicles with others undergoing atresia through programmed cell death (Pru and Tilly 2001). The follicular activation process is tightly controlled by paracrine and autocrine factors such as transforming growth factor (TGF) family members TGFα, TGFβ, bone morphogenetic protein 4 (BMP4), and anti-Mullerian hormone (AMH), growth factors or cytokines such as kit ligand (KITL), fibroblast growth factor (FGF), and leukemia inhibitory factor (LIF) and steroid hormones (Skinner 2005). These growth factors either activate (KITL, FGF, TGFα, LIF, and BMP4) or inhibit (AMH and TGFβ) the activation process so the balance between them likely determine the direction of the activation process (Gougeon 2010). Additionally, presence of factors that regulate the bioavailability of these growth factors, for example fibrillins that sequester TGF family members (Bastian, et al. 2016), can also govern the follicular activation process. Genetic studies showing increased follicular activation in protein kinase B (AKT) inhibitor, phosphatase and tensin homolog (PTEN) (Reddy, et al. 2008), and AKT-dependent transcription factor forkhead box O3a (FOXO3A) (Castrillon, et al. 2003) knockout mouse have shed light on these cell signaling pathways. Androgens can also induce follicular activation via stimulation of AKT signaling and inhibition of FOXO3A protein (Lebbe and Woodruff 2013). In addition, anti-apoptotic proteins such as B-cell lymphoma-2 (BCL2) promote follicular survival and increase the rate of activation, while pro-apoptotic proteins such as BCL2-associated X protein (BAX) decrease the rate activation by increasing follicular atresia (Hussein 2005).
The suggestion of increased follicular recruitment in PCOS came from Initial histological studies by Hughesdon (Hughesdon 1982) and later confirmed by others (Maciel, et al. 2004; Webber et al. 2003), which indicated that ovaries from PCOS women have increased number of primary follicles. Although, the reason for such increase is still not clear, the identification that one of the PCOS susceptibility locus D19S884 allele 8 that maps to fibrillin 3 (FBN3) gene (Urbanek, et al. 1999) sheds some light on ovarian factors involved during early follicular development. Considering that: 1) the expression of FBN3 is restricted to perifollicular stromal area of the follicles transitioning from primordial to primary stage (Jordan, et al. 2010), 2) its expression is highest in the first trimester fetal ovaries (Hatzirodos, et al. 2011), and 3) fibrillins can sequester TGFs (Bastian et al. 2016), FBN3 has the potential to alter the follicular developmental trajectory by altering bioavailability of TGF members. In addition, lower expression of AMH in primordial and primordial to primary transitional follicles in ovaries from PCOS women compared with normal women (Stubbs, et al. 2005) coupled with findings of increased follicular activation evidenced in AMH null mouse (Durlinger, et al. 1999), and the ability of AMH to inhibit the number of early growing follicles from mouse ovaries cultured in vitro (Durlinger et al. 1999) indicate lower AMH expression to be conducive to follicular activation in the PCOS ovary. As such, the lower number of atretic early growing follicles in PCOS ovarian tissue (Webber, et al. 2007) in the face of low AMH levels could promote follicular activation / recruitment and survival thus increasing the cohort of early growing follicles available for further differentiation (Figure 2) (Franks et al. 2008).
Figure 2:

Schematic showing directionality of changes in factors contributing to disruptions in the follicular activation/recruitment in PCOS women and animal models of PCOS.
Support for enhanced recruitment also comes from prenatal testosterone-treated Suffolk (Smith, et al. 2009; Steckler et al. 2005) and Poll Dorset sheep models of PCOS, which manifest reduced primordial follicles with corresponding increase in growing follicles. In addition to this morphological evidence,1) reduced number of early growing follicles staining positively for the follicular activation inhibitor, AMH, in ovaries from both of these prenatal testosterone-treated sheep breeds (Bull, et al. 2004; Veiga-Lopez, et al. 2012), 2) increased presence of AR protein in the fetal granulosa and stromal cells of the prenatal testosterone-treated Suffolk sheep (Ortega, et al. 2009) indicative of increased androgen signaling, and 3) reduced expression of pro-apoptotic protein BAX in granulosa cells of fetal primordial and primary follicles (Salvetti, et al. 2012) are all supportive of increased follicular activation (Figure 2). All in all, studies with sheep models of PCOS phenotype, as was the case with human PCOS provide support for involvement of paracrine factors and apoptotic machinery in increasing follicular recruitment.
Follicular persistence
The activated primary follicles differentiate and grow to become pre-antral follicles. In vitro studies in non-human primates have found that AMH supports preantral follicular growth (Xu, et al. 2018). As such AMH appears to have opposing effects on primordial and preantral follicles, namely one of inhibition on primordial to primary transition but stimulation on survival and growth of preantral follicles. Pre-antral follicles continue to develop under the stimulation of FSH into antral follicles that transition through small to large antral follicle stages. The fate of antral follicles is to either undergo atresia or mature to become preovulatory follicles that ovulate in response to LH surge and luteinize (Figure 1). As such, the persistence of small antral follicles leading to the PCOM can arise from arrest in antral follicular growth, premature luteinization and reduced rate of atresia.
i. Arrest in antral follicular development:
Because FSH is a major regulator of antral follicular development, reduction in factors that promote sensitivity of antral follicles to FSH such as activin and insulin-like growth factor (IGF) or increase in factors that inhibits sensitivity to FSH such as inhibins, follistatin and AMH (Knight, et al. 2012; Mazerbourg, et al. 2003; Visser and Themmen 2014) or IGF binding proteins (IGFBP) that regulate bioavailability of IGFs (Kwintkiewicz and Giudice 2009) can arrest follicular growth. Other factors that can contribute to follicular growth arrest include epidermal growth factor (EGF), nerve growth factor (NGF) and tumor necrosis factor alpha (TNF) (Jonard and Dewailly 2004).
Lower levels of activin (Norman, et al. 2001) coupled with higher follistatin (Erickson, et al. 1995) and AMH levels in follicular fluid of women with PCOS (Desforges-Bullet, et al. 2010; Fallat, et al. 1997) are consistent with reduced FSH sensitivity and growth arrest (Figure 3). In addition, higher antral follicular fluid levels of EGFs (Volpe, et al. 1991), NGF (Dissen, et al. 2009) and TNF (Amato, et al. 2003) also contribute to antral follicular growth arrest / follicular persistence. While the high levels of IGF1 found in PCOS follicles (Eden, et al. 1990) are inconsistent with growth arrest, their action may have been offset by parallel increases in IGF binding proteins (IGFBP) (Cataldo and Giudice 1992; Holly, et al. 1990).
Figure 3:
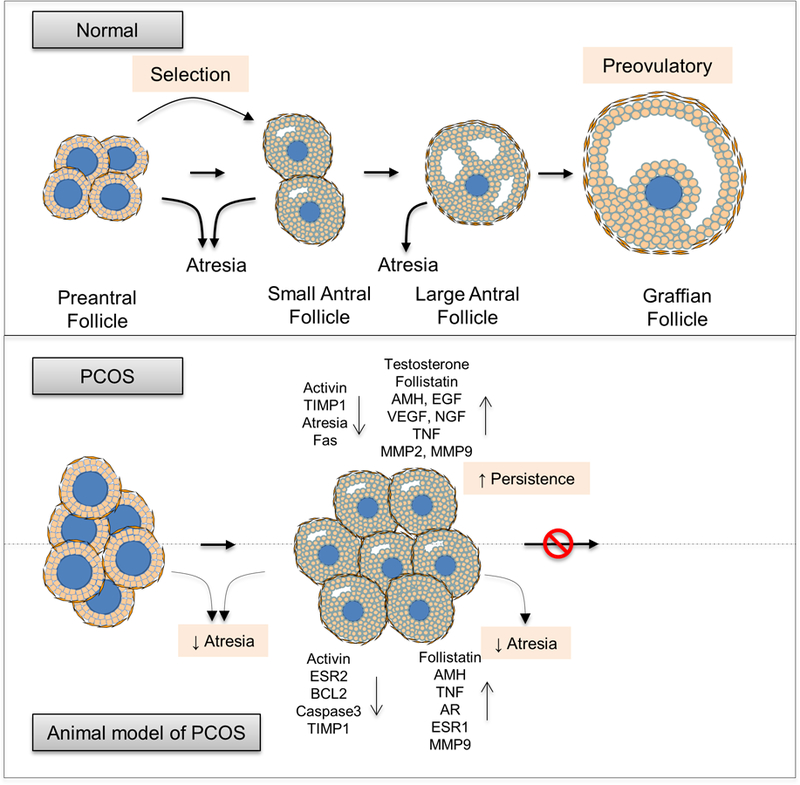
Schematic showing directionality of changes in factors contributing to follicular persistence in PCOS women and animal models of PCOS.
The prenatal T-treated sheep model of PCOS, only model where serial ultrasonographic scans have been performed for over 25 days, has provided evidence in support of antral follicles surviving for longer periods (Manikkam, et al. 2006; Veiga-Lopez, et al. 2014b) without further growth, supportive of follicular arrest and persistence. Intra-follicular changes observed in prenatal testosterone-treated sheep namely increased follistatin and reduced activin βB mRNA levels (West, et al. 2001) and increased AMH protein content in granulosa cells (Veiga-Lopez et al. 2012) and TNF in the theca cells (Puttabyatappa, et al. 2017b) of antral follicles, parallel what has been observed in PCOS women. Altered steroid receptor balance with an increase in estrogen receptor alpha (ESR1) and androgen receptor (AR) and decrease in estrogen receptor beta (ESR2) protein content in granulosa cells of antral follicles (Ortega et al. 2009) leading to reduced estrogen action are consistent with impaired antral follicle growth (Volpe et al. 1991). These findings are consistent with increase in negative mediators of follicular FSH sensitivity and reduced antral follicle growth contributing to the follicular arrest in prenatal testosterone-treated sheep (Figure 3).
ii. Premature luteinization:
The growth of the antral follicles that develop from small to large antral and preovulatory stage terminates with the follicle differentiating under the influence of ovulatory gonadotropin surge into corpus luteum through a process known as luteinization. The intra-ovarian factors that promote luteinization are androgens (Nielsen, et al. 2011), adiponectin (Palin, et al. 2012), proteases such as matrix metalloproteases (MMP), growth factors such as epidermal growth factors (EGF), vascular endothelial growth factors (VEGF), FGFs and cytokines such as interleukins and LIF (Giovanni Artini, et al. 2007). The increases in these factors during the small antral stage can induce premature luteinization (Dewailly, et al. 2016; Franks et al. 2008) thus contributing to the follicular persistence.
Thecal androgen excess (Gilling-Smith, et al. 1994), premature expression of LH receptors possibly the consequence of the hyperandrogenic status (Dewailly et al. 2016; Willis, et al. 1998), increased follicular fluid EGF (Volpe et al. 1991) and VEGF (Ferrara, et al. 2003) and increased granulosa cell expression of VEGF and endocrine gland-derived VEGF (EB-VEGF) (Ferrara et al. 2003), increased MMP2 and 9 and reduced tissue inhibitor of MMPs (TIMP1) content in the follicular fluid and increased MMP9 but not TIMP1 expression in granulosa cells (Lahav-Baratz, et al. 2003; Shalev, et al. 2001) and increase in cytokines interleukin 6 and TNF (Amato et al. 2003) evidenced in women with PCOS are all consistent with premature luteinization and follicular persistence.
Similarly, the increases in androgen receptor (Ortega et al. 2009), VEGF and its receptor VEGFR3 (Ortega, et al. 2015), and increase in MMP9 with reduction in TIMP1 and matrix proteins laminin B and collagen protein observed in antral follicles of prenatal testosterone-treated sheep (Puttabyatappa et al. 2017b) are consistent with premature luteinization in this animal model of PCOS.
iii. Follicular atresia:
Follicular development involves cyclical recruitment of a cohort of small antral follicles out of which one of them (in monoovulatory and several in litter-bearing species) emerge to be dominant that goes on to ovulate while the rest of them undergo atresia through apoptosis (Figure 1) (Pru and Tilly 2001). Increased expression of growth factors such as EGF and IGF that promote cell survival (Homburg and Amsterdam 1998) and loss of balance between pro-apoptotic proteins such as Fas and BCL2 associated X, apoptosis regulator (BAX) and anti-apoptotic proteins such as BCL2 (Escobar, et al. 2011) can disrupt the atretic process contributing to follicular persistence and accumulation leading to the PCOM phenotype.
Consistent with the decreased atresia providing a basis for accumulation of follicles leading to PCOM phenotype DNA fragmentation, soluble Fas (sFas) and soluble Fas ligand (sFasL), markers of apoptosis were lower in granulosa cells from PCOS patients (Onalan, et al. 2005). As such, low follicular fluid levels of soluble Fas (Onalan et al. 2005) coupled with high levels of growth factors EGF (Volpe et al. 1991) and IGF1 (Eden et al. 1990) in PCOS patients are consistent with reduced atresia and increased follicular survival leading to persistence.
Similarly, imbalance in opposing proliferative and apoptotic signals represented as an increase in expression of the proliferative marker PCNA and decrease in BCL2 and activated caspase-3 in granulosa cells (Salvetti et al. 2012) coupled with increased TNF in theca cells of antral follicles (Puttabyatappa et al. 2017b) in the sheep model of PCOS phenotype are also consistent with reduced atresia contributing to accumulation of antral follicles.
Oocyte quality
Ovarian folliculogenesis and oogenesis occurs in parallel with follicular somatic cells and oocytes influencing each other during the developmental process. Although oocyte meiotic arrest occurs during early stages of follicle formation, oocyte secreted factors influence follicle growth (Eppig 2001). While, the granulosa cell secretions provide nutrient support and maintain meiotic arrest till induction of ovulation (Albertini 2015), factors produced by follicular somatic cells such as EGFs (Hsieh, et al. 2009), IGFs (Wang and Sun 2007), FGFs (Artini, et al. 2006), brain-derived neurotropic factor (BDNF) (Kawamura, et al. 2005), VEGF (Luo, et al. 2002) and LIF (Ledee-Bataille, et al. 2001) and oocyte derived factors GDF9 and BMP15 (Hussein, et al. 2006) are associated with good oocyte quality. Factors such as androgens (Teissier, et al. 2000), AMH (Fallat et al. 1997), TNF (Ma, et al. 2010) and cytokine IL6, IL12 and IL13 (Amato et al. 2003; Gazvani, et al. 2000) and corticotrophin-releasing hormones (CRF) (Mastorakos, et al. 1994) are associated with poor oocyte quality.
Several studies have elaborated on the poor oocyte quality of women with PCOS (Franks, et al. 2003; Qiao and Feng 2011). For instance, ovulation induction through stimulation cycles in anovulatory PCOS have been shown to produce increased number of oocytes (Qiao and Feng 2011) but of smaller oocyte size (Marquard, et al. 2011). Excess ovarian androgen (Gilling-Smith et al. 1994), and follicular fluid FGF (Artini et al. 2006), TNF, IL6 and IL13 (Amato et al. 2003) and low follicular fluid levels of LIF (Ledee-Bataille et al. 2001) and CRF (Mastorakos et al. 1994) and reduced expression of GDF9 in cumulus cells (Zhao, et al. 2010) of PCOS women (Table 2) are consistent with poor oocyte quality (Qiao and Feng 2011). On the contrary, increased follicular fluid content of EGF (Volpe et al. 1991), VEGF (Ferrara et al. 2003), IGF1 (Eden et al. 1990) and BDNF (Johnstone, et al. 2008) are inconsistent with poor oocyte quality in PCOS patients. These data indicate that the balance between factors that promote oocyte quality and those that reduce quality may be shifted to the negative side in women with PCOS thus contributing to reduced oocyte competence and fertility outcomes in PCOS patients (Franks, et al. 1998; Franks et al. 2003). Differential expression of genes in oocytes from PCOS women indicative of defects in meiosis or early embryonic development (Wood, et al. 2007) provide further support for this premise. Additionally, fertilization rates in PCOS women in IVF settings have yielded variable results ranging from no effect to higher rates of fertilization (Sermondade, et al. 2013). While the pregnancy rates among PCOS women undergoing IVF have been found to be similar to the non-PCOS women undergoing IVF (Heijnen, et al. 2006), various meta-analysis have shown that infants born to women with PCOS are at higher risk for preterm birth, low birth weight, perinatal mortality, congenital abnormalities, and likelihood of birth by Caesarean section, (Boomsma et al. 2006; Qin et al. 2013). These findings raise concerns regarding the quality of oocytes used in IVF settings.
While the factors that regulate oocyte maturation and competency have not been well characterized in most animal models of PCOS, reduced oocyte competency have been reported in prenatally androgenized rhesus macaques (Dumesic, et al. 2002). In the sheep model of PCOS, oocyte competence has not been directly examined but mating studies showing reduced pregnancy rate (Steckler, et al. 2007) suggest poor oocyte quality may also be a contributing factor in this model. An increase in VEGF (Ortega et al. 2015), which parallel what is seen in women with PCOS (Ferrara et al. 2003), is not consistent with poor oocyte quality, however, it’s beneficial effect appears to be offset by the increase in granulosa cell AMH (Veiga-Lopez et al. 2012) and thecal cell TNF (Puttabyatappa et al. 2017b).
Extra-ovarian mediators of PCOS
In addition to the ovarian mediators discussed above, disruptions in extra-ovarian mediators (Figure 4) such as neuroendocrine secretions from the hypothalamo-pituitary axis that act on the ovary to regulate ovarian steroidogenesis, follicular development, ovulation and corpus luteum can play an important role in the pathogenesis of PCOS. The main neuroendocrine changes observed in PCOS women are high LH and subnormal FSH levels leading to higher LH/FSH ratio, the consequence of disruptions in steroid negative feedback mechanisms and reduced pituitary sensitivity to GnRH (Balen, et al. 1993; Gill and Hall 2014; McCartney, et al. 2002; Taylor, et al. 1997). In addition, metabolic factors such as insulin and adiponectin can also influence ovarian function with the former serving as a co-gonadotropin and the latter influencing sensitivity to insulin. Consistent with this, hyperinsulinemia is the main metabolic abnormality observed in majority of the patients with PCOS (Diamanti-Kandarakis and Dunaif 2012) along with hyperglycemia, dyslipidemia and hypoadiponectinemia (Churchill, et al. 2015; Palin et al. 2012). While ovary is the major contributor for the hyperandrogenic state, increased circulating adrenal androgen dehydroepiandrosterone sulfate (DHEAS) levels present in 15 to 45% in women with PCOS (Luque-Ramírez and Escobar-Morreale 2016) indicates adrenal factors also play an important role in the development of PCOS. Further, growing evidence also point to role of other factors, such as environmental chemicals and lifestyle, in the manifestation of PCOS.
Figure 4:

Schematic showing extra-ovarian factors contributing to ovarian disruptions in PCOS women and animal models of PCOS. Images used in this figure were sourced from www.pixabay.com, www.publicdomainvectors.org, commons.wikimedia.org and www.openclipart.org.
Neuroendocrine mediators
The ovarian function is tightly regulated by the integration of the HPO axis through feed-forward and negative and positive feedback loops. The hypothalamic secretion of GnRH on the pituitary and anterior pituitary secretion of FSH and LH on the ovary form the feed forward response. The ovarian steroids androgens, estradiol and progesterone, and peptide hormones such as inhibin and follistatin produced by the ovary and the pituitary provide the negative feedback loop to the hypothalamo-pituitary axis. The mid-cycle rise in estradiol drives the positive feedback at the level of the hypothalamus and pituitary to cause surge release of LH that triggers ovulation. Disruptions in the feed-forward and feedback mechanism will influence LH and FSH, the main hypothalamo-pituitary regulators of ovary, which in turn will have a negative impact on ovarian function.
Neuroendocrine studies carried out in PCOS women provide evidence in support of abnormalities in the hypothalamic-pitutary axis contributing to the pathogenesis of PCOS. The increase in frequency of GnRH release assessed using LH as a bioassay (Pagan, et al. 2006) and exaggerated LH response to exogenous GnRH (Gill and Hall 2014) together appear to underlie the LH hypersecretion in women with PCOS. The increases in GnRH / LH in turn appear to be a function of reduced sensitivity to steroid negative feedback (Burt Solorzano, et al. 2012; Marshall and Eagleson 1999). Findings that high concentrations of estradiol and progesterone are required to reduce pulsatile LH release (Pastor, et al. 1998) and steroid sensitivity can be restored with androgen antagonist flutamide treatment (Eagleson, et al. 2000) do provide evidence in support of compromised steroid negative feedback sensitivity in PCOS patients. While pituitary levels of activin and follistatin are not known, the observations that PCOS patients have low circulating levels of activin (Norman et al. 2001) and increased follistatin (Erickson et al. 1995) are consistent with the low FSH levels.
Analogous scenario also exists in prenatal testosterone-treated animal models (Abbott, et al. 2016; Cardoso et al. 2015). For instance, consistent with disruptions in the feedback mechanisms at pituitary and hypothalamic levels, prenatal testosterone treated sheep manifest increased pituitary sensitivity to GnRH (Manikkam, et al. 2008), reduced sensitivity to estradiol negative (Sarma, et al. 2005; Wood and Foster 1998), estradiol positive (Sharma, et al. 2002; Unsworth, et al. 2005; Wood and Foster 1998), and progesterone negative feedback (Robinson, et al. 1999; Veiga-Lopez, et al. 2009). Neuroanatomical studies support neuropeptide imbalance in the KNDy (kisspeptin, neurokinin B and dynorphin) neurons reflected as reduced inhibitory (dynorphin) and no change in stimulatory (kisspeptin) (Cheng, et al. 2010) neuropeptides as a potential mediator of the decreased ability of progesterone to exert negative feedback effect on GnRH / LH secretion. Similar disruptions in estradiol and progesterone feedbacks have also been observed in prenatal testosterone-treated macaque (Abbott et al. 2016) and rodent (Moore, et al. 2015) models. Furthermore, the protection from development of PCOS like phenotype induced by prenatal dihydrotestosterone (DHT) treatment in brain-specific AR knockout mouse (Caldwell, et al. 2017) indicates a neuroendocrine role for androgens in disrupting the HPO axis.
Metabolic mediators
Insulin is the major metabolic hormone that regulates glucose homeostasis and lipid metabolism in the body. When cell or tissue requires excess insulin to respond normally, it develops insulin resistance and as pancreatic beta cells respond with production of more insulin compensatory hyperinsulinemia develops. Hyperinsulinemia can induce hyperandrogenism by either directly stimulating ovarian androgen production (Hernandez, et al. 1988), or indirectly through 1) enhancement of gonadotropin secretion from pituitary (Adashi, et al. 1981), 2) intensifying gonadotropin action at the ovary (Cara and Rosenfield 1988) or 3) increasing bioavailability of androgens through inhibition of liver sex hormone binding globulin (SHBG) production (Nestler et al. 1991). In addition, hyperinsulinemic state together with hyperandrogenimia can increase FSH induction of LH receptor in granulosa cells of antral follicles and also LH action to induce premature luteinization (Willis, et al. 1996). Hyperinsulinemia and insulin resistance evidenced in metabolic diseases are commonly associated with inflammatory state, hypoadiponectinemia and dyslipidemia, which can indirectly influence ovarian function by modulating insulin and gonadotropin action (De Leo, et al. 2016; Macut, et al. 2013; Moran, et al. 2015; Palin et al. 2012; Vassilatou 2014).
Although 60–70% of women with PCOS are obese or overweight (Moran et al. 2015; Naderpoor, et al. 2015) and obesity is associated with insulin resistance (Esser, et al. 2014) the observation that majority of lean women with PCOS also manifest insulin resistance (Yildizhan, et al. 2016) support a role for hyperinsulinemia in the manifestation of PCOS. The increased prevalence of chronic low-grade inflammation (Boots and Jungheim 2015), dyslipidemia (Couto Alves, et al. 2017), hypoadiponectemia (Manneras-Holm, et al. 2011) and NAFLD (Makri and Tziomalos 2017) in PCOS patients, features that can negatively affect HPO axis, are also consistent with the role of metabolic factors contributing to the development of PCOS.
Similar to women with PCOS, prenatal testosterone treated sheep also manifest reduced peripheral insulin sensitivity and hyperinsulinemia (DeHaan, et al. 1990; Hansen, et al. 1995; Padmanabhan, et al. 2010; Recabarren, et al. 2005), dyslipidemia (Puttabyatappa et al. 2017a; Veiga-Lopez, et al. 2013) and hepatic lipid accumulation (Puttabyatappa et al. 2017a). Hyperinsulinemia with increased adiposity is also a feature of prenatal testosterone treated macaque (Abbott et al. 1998) and rodent models (Roland, et al. 2010). Hypoadiponectemia, a feature seen in women with PCOS, was observed in DHT-treated mouse model (Benrick, et al. 2017) but not in prenatal testosterone treated sheep (Puttabyatappa et al. 2017a).
Adrenal mediators
Although ovarian theca cell androgen production is a major source of hyperandrogenemia, adrenal production of DHEAS accounts for hyperandrogenism in about 15–45% of the women with PCOS (Luque-Ramírez and Escobar-Morreale 2016). The presence of high DHEAS among sisters and daughters of patients with PCOS suggests that adrenal hyperandrogenism (AH) may be an inherited trait (Yildiz et al. 2016; Maliqueo et al. 2009). The cause for AH among PCOS patients is either adrenal hyperresponsiveness to adrenocorticotropic hormone (ACTH) (Moran et al. 2005) or increased ACTH drive to adrenal due to reduced negative feedback stemming from decreased hyperinsulinemia-induced hepatic cortisol regeneration (Rodin et al. 1994).
Most of the data on the developmental programming of AH has come from prenatal androgen treated monkeys. Prenatally androgenized female rhesus macaques show enhanced basal and adrenocorticotropic hormone (ACTH)-stimulated adrenal DHEA production suggesting adrenal defect in this model (Zhou et al. 2005). Hyperinsulinemia and increased adiposity observed in this model has been proposed as the potential mediator of AH (Abbott et al. 2009) and the observation that insulin sensitizer pioglitzone normalizes DHEAS response to ACTH stimulation confirms such assertions (Zhou et al. 2007).
Other mediators
In addition to ovarian and extra-ovarian mediators, other factors that can influence the ovarian function include environmental chemicals and lifestyle. Environmental cues play an important role in the regulation of ovarian function and its influence is integrated through the HPO axis (Vermeulen 1993). Environmental chemicals especially endocrine disruptors with steroid potential such as phthalates and bisphenol A (BPA) can alter the HPO functions by disrupting the steroidal feedbacks at the hypothalamus and pituitary level and steroid action at the level of the ovary (Gore, et al. 2015; Peretz, et al. 2014). Both under- and over-nutrition, through production of stress steroids (Whirledge and Cidlowski 2010), sex steroids (Mossa, et al. 2013; Whyte, et al. 2007) or metabolic factors (Duque-Guimaraes and Ozanne 2013), can have a bearing on ovarian functions (Evans and Anderson 2017). Lifestyle factors such as a socioeconomic status, neighborhood one lives in, stress and sedentariness can also impact the ovarian function through adverse health behavior that can lead to excessive weight gain, activation of the stress hormone axis or increased exposure to environmental chemicals (Beydoun and Wang 2010; Nelson, et al. 2012; Rosmond 2005).
The identification of environmental chemicals with steroidogenic potential such as perfluorooctanoate, polychlorinated biphenyls (PCBs), pesticides, polycyclic aromatic hydrocarbons and BPA (Kandaraki, et al. 2011; Vagi, et al. 2014) in PCOS women raise concerns regarding risks posed by these chemicals in development of PCOS. In support of this increased level of BPA has been associated with hyperandrogenic status (Rutkowska and Rachon 2014). Likewise, overnutrition and sedentary lifestyle likely contribute to the observation that 60–70% of women with PCOS are obese. A contributory role for nutrition is emphasized by the fact that dietary changes and weight loss ameliorates PCOS symptoms (Huber-Buchholz, et al. 1999; Merkin, et al. 2016).
Although these observations from prospective and retrospective studies in humans suggest a role for environmental and nutritional factors in the development of PCOS, causative role for these come from studies in animal models. For example, prenatal BPA treatment in sheep induced neuroendocrine and ovarian changes that mimic PCOS patients (Savabieasfahani et al. 2006; Veiga-Lopez et al. 2014a). Similarly, postnatal overfeeding was found to amplify the reproductive phenotype of the sheep model of PCOS, leading to anovulation (Steckler et al. 2009).
Ovulatory and luteal defects
An estimated 40% of PCOS patients manifest infertility that arise due to infrequent or absent ovulation or luteal phase deficiency (Boutzios, et al. 2013; Teede, et al. 2010). Majority of the anovulatory PCOS phenotype is associated with accumulation of 2–8 mm antral follicles that fail to undergo follicle selection and dominance but persist due to excess production of AMH or premature acquisition of LH receptors in the granulosa cells. Normalization of HPO axis or metabolic functions has been shown to be effective in achieving ovulation (Legro 2016). Because PCOS patients have generally high LH and low FSH, utilization of estrogen receptor antagonists or aromatase inhibitors that reduce negative feedback of the estrogens on pituitary FSH secretion or exogenous FSH supplementation aid in ovulation induction (Jayasena and Franks 2014; Legro 2016). Improving metabolic functions through administration of metformin, thiazolidinediones or lifestyle improvements such a dietary change, exercise and bariatric surgery have also been shown to improve ovulation induction (Balen, et al. 2016). Infertility due to luteal phase deficiency is also evident in ovulatory PCOS patients who have low levels of progesterone during early luteal phase (Joseph-Horne, et al. 2002). This low progesterone levels may arise due to abnormal synthesis of progesterone by granulosa cells (Doldi, et al. 1998). Because high concentrations of progesterone are required to reduce LH release in PCOS patients (Pastor et al. 1998) supplementation of progesterone is a therapeutic approach to help normalize LH secretion and promote implantation (Unfer, et al. 2005).
In the sheep model of PCOS, similar rescue of ovarian function by exogenous gonadotropin administration (Steckler, et al. 2008), cyclic progesterone supplementation (Manikkam et al. 2006) or gestational rosiglitazone (an insulin sensitizing thiazolidinedione) treatment (Veiga-Lopez, et al. 2010) or normalization of LH hypersecretion by postnatal rosiglitazone intervention (Cardoso, et al. 2016) emphasize the role played by steroidal and metabolic factors in the development and maintenance of the pathology. The specific changes in intra- and extra-ovarian factors that contribute to the success of these interventions on follicular selection and dominance needs further investigation.
CONCLUSIONS
As discussed above, multiple factors that impact neuroendocrine, ovarian and metabolic functions are involved in the development of PCOS phenotype. The ovarian defects associated with PCOS are impacted by various intra- and extra-ovarian factors that influence follicular developmental process at multiple steps leading to increased recruitment, failure to achieve dominance or undergo atresia resulting in antral follicular developmental arrest. Differences in these multitudes of factors among the subtypes of PCOS (Table 1) may shed light on the mediators of phenotypic differences in these sub classes. However, several challenges still exist in 1) understanding the etiology of various subtypes of PCOS and lifelong consequences of PCOS, 2) developing interventions to prevent transmission of PCOS traits, 3) identifying optimal treatment strategies, and 4) complete phenotyping of the male counterpart of PCOS.
Development of preventive and intervention strategies require complete understanding of the underlying ovarian and extra-ovarian mechanisms contributing to the development of the PCOS phenotype. While genetic studies explain only a small percentage of PCOS prevalence, disease gene mapping in a larger sample size that includes women from multiple ethnic groups to identify other loci involved in the development of PCOS are required. In terms of using animal models to probe underlying mechanisms and identifying additional ovarian and extra-ovarian factors, there is a great need to expand studies to precocial species that have similar developmental trajectory of organ systems as in humans.
In view of the potential for PCOS traits to be passed on to subsequent generations and the findings of microRNA expression (Table 2) and epigenetic changes in PCOS patients (Jones et al. 2015; Li et al. 2016) and animal models of PCOS (Luense et al. 2011; Xu et al. 2011), it is important to determine if transmission across generations involve transgenerational transmission of PCOS traits or they merely reflect repetitive multigenerational transfer of traits due to programmed manifestation of hyperandrogenism and hyperinsulinemia that serve as repetitive programmers from generation to generation. For instance, PCOS women during pregnancy manifest hyperandrogenemia and hyperinsulinemia (Sir-Petermann, et al. 2009) that could program PCOS phenotype in the genetically-susceptible offspring. Studies in animal models also support that multi- and trans-generational transfer is possible. Studies in rodents have provided evidence of transgenerational transfer of reproductive and metabolic traits following exposure to experimental chemicals (Guerrero-Bosagna 2016). To what extent insulin sensitivity changes of prenatal testosterone-treated F1 female sheep that are evident also in the F2 female sheep offspring (Burns, et al. 2016) reflect repetitive hyperandrogenic status remains to be determined. Therefore, in addition to long term follow-up of offspring of PCOS women to determine which of the PCOS traits get passed on, studies across several generations that include careful phenotyping of each generation are required in animal models to determine the mechanisms by which traits are passed on from generation to generation.
Important are also studies targeted towards development of optimal interventions for not just therapeutic but also preventative strategies. Continued efforts to identify factors and mechanisms involved in the origin of PCOS would be of immense benefit in this regard. In humans, safe intervention strategies that improve inflammatory, oxidative stress, and dyslipidemic state and/or reduce exposure to environmental endocrine disrupting chemicals are needed to prevent the ovarian and extra-ovarian pathologies that contribute to the development of PCOS phenotype and improve women’s reproductive health. Animal models are a great resource in this regard for assessing the effectiveness of such interventions before their implementation in patients.
An emerging area of concern is also the long term health of the PCOS women. As they age, PCOS women have shown improvements in menstrual cyclicity (Elting, et al. 2000), hyperandrogenemia and insulin resistance (Brown, et al. 2011). Although perimenopausal women are found to have increased prevalence of hypertension and diabetes mellitus (Dahlgren, et al. 1992), a study with small sample size found the prevalence of cardiovascular diseases did not differ between general population and women previously diagnosed with PCOS during postmenopausal period (Merz, et al. 2016). Studies with larger sample size are needed to determine if women with PCOS manifest higher prevalence of chronic cardiometabolic complications such as obesity, diabetes, and cardiovascular diseases during later life and the contributing factors.
Although, PCOS is generally known as a reproductive disorder in women, the metabolic complications characteristic of PCOS women are also found in male relatives and offspring of PCOS women. The observations that male relatives of PCOS women have premature male baldness (Ferriman and Purdie 1979) and endocrine changes such as increased dehydroepiandrosterone sulfate (DHEAS), increased AMH, low SHBG, insulin resistance and abnormal gonadotropin secretion (Cannarella, et al. 2018) support a male PCOS-like phenotype. Studies in prenatal T-treated male sheep also found reduced sperm cell count (Recabarren, et al. 2008) and motility as well as testicular defects (Rojas-Garcia, et al. 2010). Therefore, detailed characterization of the male complement of PCOS phenotype are required.
In conclusion, the different phenotypes of PCOS that contributes to the heterogeneous nature of this syndrome presents challenges in understanding the disease development and treatment. Meeting these challenges through research in humans and animal models will help in developing successful strategies to not only treat but also to prevent the development of PCOS in subsequent generations.
Acknowledgments
Funding support: NIH: P01 HD044232
Footnotes
Disclosure statement: The authors have nothing to disclose
REFERENCES
- Abbott DH, Barnett DK, Bruns CM & Dumesic DA 2005. Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome? Hum Reprod Update 11 357–374. [DOI] [PubMed] [Google Scholar]
- Abbott DH, Dumesic DA, Eisner JR, Colman RJ & Kemnitz JW 1998. Insights into the development of polycystic ovary syndrome (PCOS) from studies of prenatally androgenized female rhesus monkeys. Trends Endocrinol Metab 9 62–67. [DOI] [PubMed] [Google Scholar]
- Abbott DH, Levine JE & Dumesic DA 2016. Translational Insight Into Polycystic Ovary Syndrome (PCOS) From Female Monkeys with PCOS-like Traits. Curr Pharm Des 22 5625–5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adashi EY, Hsueh AJ & Yen SS 1981. Insulin enhancement of luteinizing hormone and follicle-stimulating hormone release by cultured pituitary cells. Endocrinology 108 1441–1449. [DOI] [PubMed] [Google Scholar]
- Albertini DF 2015. Chapter 2 - The Mammalian Oocyte. In Knobil and Neill’s Physiology of Reproduction (Fourth Edition), pp 59–97. San Diego: Academic Press. [Google Scholar]
- Amaral PP & Mattick JS 2008. Noncoding RNA in development. Mamm Genome 19 454–492. [DOI] [PubMed] [Google Scholar]
- Amato G, Conte M, Mazziotti G, Lalli E, Vitolo G, Tucker AT, Bellastella A, Carella C & Izzo A 2003. Serum and follicular fluid cytokines in polycystic ovary syndrome during stimulated cycles. Obstet Gynecol 101 1177–1182. [DOI] [PubMed] [Google Scholar]
- Artimani T, Saidijam M, Aflatoonian R, Ashrafi M, Amiri I, Yavangi M, SoleimaniAsl S, Shabab N, Karimi J, Mehdizadeh M 2015. Downregulation of adiponectin system in granulosa cells and low levels of HMW adiponectin in PCOS. J Assist Reprod Genet 33 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artini PG, Monti M, Matteucci C, Valentino V, Cristello F & Genazzani AR 2006. Vascular endothelial growth factor and basic fibroblast growth factor in polycystic ovary syndrome during controlled ovarian hyperstimulation. Gynecol Endocrinol 22 465–470. [DOI] [PubMed] [Google Scholar]
- Azziz R 2016. PCOS in 2015: New insights into the genetics of polycystic ovary syndrome. Nat Rev Endocrinol 12 74–75. [DOI] [PubMed] [Google Scholar]
- Azziz R, Carmina E, Chen Z, Dunaif A, Laven JS, Legro RS, Lizneva D, Natterson-Horowtiz B, Teede HJ & Yildiz BO 2016. Polycystic ovary syndrome. Nat Rev Dis Primers 2 16057. [DOI] [PubMed] [Google Scholar]
- Azziz R, Carmina E, Dewailly D, Diamanti-Kandarakis E, Escobar-Morreale HF, Futterweit W, Janssen OE, Legro RS, Norman RJ, Taylor AE, et al. 2009. The Androgen Excess and PCOS Society criteria for the polycystic ovary syndrome: the complete task force report. Fertil Steril 91 456–488. [DOI] [PubMed] [Google Scholar]
- Balen AH, Morley LC, Misso M, Franks S, Legro RS, Wijeyaratne CN, Stener-Victorin E, Fauser BC, Norman RJ & Teede H 2016. The management of anovulatory infertility in women with polycystic ovary syndrome: an analysis of the evidence to support the development of global WHO guidance. Hum Reprod Update 22 687–708. [DOI] [PubMed] [Google Scholar]
- Balen AH, Tan SL & Jacobs HS 1993. Hypersecretion of luteinising hormone: a significant cause of infertility and miscarriage. Br J Obstet Gynaecol 100 1082–1089. [DOI] [PubMed] [Google Scholar]
- Barnes RB, Rosenfield RL, Ehrmann DA, Cara JF, Cuttler L, Levitsky LL & Rosenthal IM 1994. Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. J Clin Endocrinol Metab 79 1328–1333. [DOI] [PubMed] [Google Scholar]
- Barnett KR, Schilling C, Greenfeld CR, Tomic D & Flaws JA 2006. Ovarian follicle development and transgenic mouse models. Hum Reprod Update 12 537–555. [DOI] [PubMed] [Google Scholar]
- Barrett ES, Hoeger KM, Sathyanarayana S, Abbott DH, Redmon JB, Nguyen RHN, Swan SH 2018. Anogenital distance in newborn daughters of women with polycystic ovary syndrome indicates fetal testosterone exposure. J Dev Orig Health Dis 10.1017/S2040174417001118. [DOI] [PMC free article] [PubMed]
- Barros SP & Offenbacher S 2009. Epigenetics: connecting environment and genotype to phenotype and disease. J Dent Res 88 400–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastian NA, Bayne RA, Hummitzsch K, Hatzirodos N, Bonner WM, Hartanti MD, Irving-Rodgers HF, Anderson RA & Rodgers RJ 2016. Regulation of fibrillins and modulators of TGFbeta in fetal bovine and human ovaries. Reproduction 152 127–137. [DOI] [PubMed] [Google Scholar]
- Bayer TA, Falkai P & Maier W 1999. Genetic and non-genetic vulnerability factors in schizophrenia: the basis of the “two hit hypothesis”. J Psychiatr Res 33 543–548. [DOI] [PubMed] [Google Scholar]
- Beck-Peccoz P, Padmanabhan V, Baggiani AM, Cortelazzi D, Buscaglia M, Medri G, Marconi AM, Pardi G & Beitins IZ 1991. Maturation of hypothalamic-pituitary-gonadal function in normal human fetuses: circulating levels of gonadotropins, their common alpha-subunit and free testosterone, and discrepancy between immunological and biological activities of circulating follicle-stimulating hormone. J Clin Endocrinol Metab 73 525–532. [DOI] [PubMed] [Google Scholar]
- Benrick A, Chanclon B, Micallef P, Wu Y, Hadi L, Shelton JM, Stener-Victorin E & Wernstedt Asterholm I 2017. Adiponectin protects against development of metabolic disturbances in a PCOS mouse model. Proc Natl Acad Sci U S A 114 E7187–E7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beydoun MA & Wang Y 2010. Pathways linking socioeconomic status to obesity through depression and lifestyle factors among young US adults. J Affect Disord 123 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boomsma CM, Eijkemans MJ, Hughes EG, Visser GH, Fauser BC & Macklon NS 2006. A meta-analysis of pregnancy outcomes in women with polycystic ovary syndrome. Hum Reprod Update 12 673–683. [DOI] [PubMed] [Google Scholar]
- Boots CE & Jungheim ES 2015. Inflammation and Human Ovarian Follicular Dynamics. Semin Reprod Med 33 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutzios G, Karalaki M & Zapanti E 2013. Common pathophysiological mechanisms involved in luteal phase deficiency and polycystic ovary syndrome. Impact on fertility. Endocrine 43 314–317. [DOI] [PubMed] [Google Scholar]
- Bremer AA 2010. Polycystic ovary syndrome in the pediatric population. Metab Syndr Relat Disord 8 375–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown ZA, Louwers YV, Fong SL, Valkenburg O, Birnie E, de Jong FH, Fauser BC & Laven JS 2011. The phenotype of polycystic ovary syndrome ameliorates with aging. Fertil Steril 96 1259–1265. [DOI] [PubMed] [Google Scholar]
- Bull L, Stubbs S, Birch R, Robinson J, Themmen A, Visser J, Groome N, Hardy K & Franks S 2004. Reduced expression of anti-Mullerian hormone (AMH) protein in the androgenised sheep ovary
- Burns A, Cardoso RC, Puttabyatappa M, Herkimer C, Padmanabhan V 2016. Developmental programming: multigenerational effects of gestational exposure to excess testosterone on insulin sensitivity in female sheep Abstract Endocrine Society’s 98th Annual Meeting and Expo, April 1–4, 2016 Boston, MA 10.1210/endo-meetings.2016.RE.5.SUN-160. [DOI] [Google Scholar]
- Burns KH & Matzuk MM 2002. Minireview: genetic models for the study of gonadotropin actions. Endocrinology 143 2823–2835. [DOI] [PubMed] [Google Scholar]
- Burt Solorzano CM, Beller JP, Abshire MY, Collins JS, McCartney CR & Marshall JC 2012. Neuroendocrine dysfunction in polycystic ovary syndrome. Steroids 77 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai G, Ma X, Chen B, Huang Y, Liu S, Yang H, Zou W 2017. MicroRNA-145 Negatively regulates cell proliferation through targeting IRS1 in isolated ovarian granulosa cells from patients with polycystic ovary syndrome. Reprod Sci 24 902–910. [DOI] [PubMed] [Google Scholar]
- Caldwell ASL, Edwards MC, Desai R, Jimenez M, Gilchrist RB, Handelsman DJ & Walters KA 2017. Neuroendocrine androgen action is a key extraovarian mediator in the development of polycystic ovary syndrome. Proc Natl Acad Sci U S A 114 E3334–E3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannarella R, Condorelli RA, Mongioi LM, La Vignera S & Calogero AE 2018. Does a male polycystic ovarian syndrome equivalent exist? J Endocrinol Invest 41 49–57. [DOI] [PubMed] [Google Scholar]
- Cara JF & Rosenfield RL 1988. Insulin-like growth factor I and insulin potentiate luteinizing hormone-induced androgen synthesis by rat ovarian thecal-interstitial cells. Endocrinology 123 733–739. [DOI] [PubMed] [Google Scholar]
- Cardoso RC, Burns A, Moeller J, Skinner DC & Padmanabhan V 2016. Developmental Programming: Insulin Sensitizer Prevents the GnRH-Stimulated LH Hypersecretion in a Sheep Model of PCOS. Endocrinology 157 4641–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso RC, Puttabyatappa M & Padmanabhan V 2015. Steroidogenic versus Metabolic Programming of Reproductive Neuroendocrine, Ovarian and Metabolic Dysfunctions. Neuroendocrinology 102 226–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castrillon DH, Miao L, Kollipara R, Horner JW & DePinho RA 2003. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science 301 215–218. [DOI] [PubMed] [Google Scholar]
- Cataldo NA & Giudice LC 1992. Follicular fluid insulin-like growth factor binding protein profiles in polycystic ovary syndrome. J Clin Endocrinol Metab 74 695–697. [DOI] [PubMed] [Google Scholar]
- Cheng G, Coolen LM, Padmanabhan V, Goodman RL & Lehman MN 2010. The kisspeptin/neurokinin B/dynorphin (KNDy) cell population of the arcuate nucleus: sex differences and effects of prenatal testosterone in sheep. Endocrinology 151 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill SJ, Wang ET & Pisarska MD 2015. Metabolic consequences of polycystic ovary syndrome. Minerva Ginecol 67 545–555. [PubMed] [Google Scholar]
- Cobin RH 2013. Cardiovascular and metabolic risks associated with PCOS. Intern Emerg Med 8 Suppl 1 S61–64. [DOI] [PubMed] [Google Scholar]
- Cooney LG & Dokras A 2017. Depression and Anxiety in Polycystic Ovary Syndrome: Etiology and Treatment. Curr Psychiatry Rep 19 83. [DOI] [PubMed] [Google Scholar]
- Couto Alves A, Valcarcel B, Makinen VP, Morin-Papunen L, Sebert S, Kangas AJ, Soininen P, Das S, De Iorio M, Coin L, et al. 2017. Metabolic profiling of polycystic ovary syndrome reveals interactions with abdominal obesity. Int J Obes (Lond) 41 1331–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews D, Gillette R, Miller-Crews I, Gore AC & Skinner MK 2014. Nature, nurture and epigenetics. Mol Cell Endocrinol 398 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren E, Johansson S, Lindstedt G, Knutsson F, Oden A, Janson PO, Mattson LA, Crona N & Lundberg PA 1992. Women with polycystic ovary syndrome wedge resected in 1956 to 1965: a long-term follow-up focusing on natural history and circulating hormones. Fertil Steril 57 505–513. [DOI] [PubMed] [Google Scholar]
- De Leo V, Musacchio MC, Cappelli V, Massaro MG, Morgante G & Petraglia F 2016. Genetic, hormonal and metabolic aspects of PCOS: an update. Reprod Biol Endocrinol 14 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaan K, Berger L, Bechtel P, Kesler D, McKeith F & Thomas D 1990. Effect of prenatal testosterone treatment on nitrogen utilization and endocrine status of ewe lambs. Journal of animal science 68 4100–4108. [DOI] [PubMed] [Google Scholar]
- Desforges-Bullet V, Gallo C, Lefebvre C, Pigny P, Dewailly D & Catteau-Jonard S 2010. Increased anti-Mullerian hormone and decreased FSH levels in follicular fluid obtained in women with polycystic ovaries at the time of follicle puncture for in vitro fertilization. Fertil Steril 94 198–204. [DOI] [PubMed] [Google Scholar]
- DeUgarte CM, Bartolucci AA & Azziz R 2005. Prevalence of insulin resistance in the polycystic ovary syndrome using the homeostasis model assessment. Fertil Steril 83 1454–1460. [DOI] [PubMed] [Google Scholar]
- Dewailly D, Lujan ME, Carmina E, Cedars MI, Laven J, Norman RJ & Escobar-Morreale HF 2014. Definition and significance of polycystic ovarian morphology: a task force report from the Androgen Excess and Polycystic Ovary Syndrome Society. Hum Reprod Update 20 334–352. [DOI] [PubMed] [Google Scholar]
- Dewailly D, Robin G, Peigne M, Decanter C, Pigny P & Catteau-Jonard S 2016. Interactions between androgens, FSH, anti-Mullerian hormone and estradiol during folliculogenesis in the human normal and polycystic ovary. Hum Reprod Update 22 709–724. [DOI] [PubMed] [Google Scholar]
- Diamanti-Kandarakis E & Dunaif A 2012. Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocr Rev 33 981–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dissen GA, Garcia-Rudaz C, Paredes A, Mayer C, Mayerhofer A & Ojeda SR 2009. Excessive ovarian production of nerve growth factor facilitates development of cystic ovarian morphology in mice and is a feature of polycystic ovarian syndrome in humans. Endocrinology 150 2906–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty DA, Newnham JP, Bower C & Hart R 2015. Implications of polycystic ovary syndrome for pregnancy and for the health of offspring. Obstet Gynecol 125 1397–1406. [DOI] [PubMed] [Google Scholar]
- Doldi N, Gessi A, Destefani A, Calzi F & Ferrari A 1998. Polycystic ovary syndrome: anomalies in progesterone production. Hum Reprod 13 290–293. [DOI] [PubMed] [Google Scholar]
- Dolfin E, Guani B, Lussiana C, Mari C, Restagno G & Revelli A 2011. FSH-receptor Ala307Thr polymorphism is associated to polycystic ovary syndrome and to a higher responsiveness to exogenous FSH in Italian women. J Assist Reprod Genet 28 925–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumesic DA, Schramm RD, Peterson E, Paprocki AM, Zhou R & Abbott DH 2002. Impaired developmental competence of oocytes in adult prenatally androgenized female rhesus monkeys undergoing gonadotropin stimulation for in vitro fertilization. The Journal of Clinical Endocrinology & Metabolism 87 1111–1119. [DOI] [PubMed] [Google Scholar]
- Dunaif A 2016. Perspectives in Polycystic Ovary Syndrome: From Hair to Eternity. J Clin Endocrinol Metab 101 759–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunaif A, Segal KR, Futterweit W & Dobrjansky A 1989. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes 38 1165–1174. [DOI] [PubMed] [Google Scholar]
- Dunaif A, Wu X, Lee A & Diamanti-Kandarakis E 2001. Defects in insulin receptor signaling in vivo in the polycystic ovary syndrome (PCOS). American Journal of Physiology-Endocrinology and Metabolism 281 E392–E399. [DOI] [PubMed] [Google Scholar]
- Duque-Guimaraes DE & Ozanne SE 2013. Nutritional programming of insulin resistance: causes and consequences. Trends Endocrinol Metab 24 525–535. [DOI] [PubMed] [Google Scholar]
- Durlinger AL, Kramer P, Karels B, de Jong FH, Uilenbroek JT, Grootegoed JA & Themmen AP 1999. Control of primordial follicle recruitment by anti-Mullerian hormone in the mouse ovary. Endocrinology 140 5789–5796. [DOI] [PubMed] [Google Scholar]
- Eagleson CA, Gingrich MB, Pastor CL, Arora TK, Burt CM, Evans WS & Marshall JC 2000. Polycystic ovarian syndrome: evidence that flutamide restores sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab 85 4047–4052. [DOI] [PubMed] [Google Scholar]
- Eden JA, Jones J, Carter GD & Alaghband-Zadeh J 1990. Follicular fluid concentrations of insulin-like growth factor 1, epidermal growth factor, transforming growth factor-alpha and sex-steroids in volume matched normal and polycystic human follicles. Clin Endocrinol (Oxf) 32 395–405. [DOI] [PubMed] [Google Scholar]
- Elting MW, Korsen TJ, Rekers-Mombarg LT & Schoemaker J 2000. Women with polycystic ovary syndrome gain regular menstrual cycles when ageing. Hum Reprod 15 24–28. [DOI] [PubMed] [Google Scholar]
- Eppig JJ 2001. Oocyte control of ovarian follicular development and function in mammals. Reproduction 122 829–838. [DOI] [PubMed] [Google Scholar]
- Erickson GF, Chung DG, Sit A, DePaolo LV, Shimasaki S & Ling N 1995. Follistatin concentrations in follicular fluid of normal and polycystic ovaries. Hum Reprod 10 2120–2124. [DOI] [PubMed] [Google Scholar]
- Eriksen MB, Nielsen MF, Brusgaard K, Tan Q, Andersen MS, Glintborg D & Gaster M 2013. Genetic alterations within the DENND1A gene in patients with polycystic ovary syndrome (PCOS). PLoS One 8 e77186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar M, Vázquez-Nin G & Echeverría O 2011. Follicular Cells. In Cell Death in Mammalian Ovary, pp 185–200: Springer; Netherlands. [Google Scholar]
- Esser N, Legrand-Poels S, Piette J, Scheen AJ & Paquot N 2014. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract 105 141–150. [DOI] [PubMed] [Google Scholar]
- Evans MC & Anderson GM 2017. Neuroendocrine integration of nutritional signals on reproduction. J Mol Endocrinol 58 R107–R128. [DOI] [PubMed] [Google Scholar]
- Fallat ME, Siow Y, Marra M, Cook C & Carrillo A 1997. Mullerian-inhibiting substance in follicular fluid and serum: a comparison of patients with tubal factor infertility, polycystic ovary syndrome, and endometriosis. Fertil Steril 67 962–965. [DOI] [PubMed] [Google Scholar]
- Fenichel P, Rougier C, Hieronimus S & Chevalier N 2017. Which origin for polycystic ovaries syndrome: Genetic, environmental or both? Ann Endocrinol (Paris) 78 176–185. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Frantz G, LeCouter J, Dillard-Telm L, Pham T, Draksharapu A, Giordano T & Peale F 2003. Differential expression of the angiogenic factor genes vascular endothelial growth factor (VEGF) and endocrine gland-derived VEGF in normal and polycystic human ovaries. Am J Pathol 162 1881–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferriman D & Purdie AW 1979. The inheritance of polycystic ovarian disease and a possible relationship to premature balding. Clin Endocrinol (Oxf) 11 291–300. [DOI] [PubMed] [Google Scholar]
- Franks S, Gharani N, Waterworth D, Batty S, White D, Williamson R & McCarthy M 1997. The genetic basis of polycystic ovary syndrome. Hum Reprod 12 2641–2648. [DOI] [PubMed] [Google Scholar]
- Franks S, Mason H, White D & Willis D 1998. Etiology of anovulation in polycystic ovary syndrome. Steroids 63 306–307. [DOI] [PubMed] [Google Scholar]
- Franks S, Roberts R & Hardy K 2003. Gonadotrophin regimens and oocyte quality in women with polycystic ovaries. Reprod Biomed Online 6 181–184. [DOI] [PubMed] [Google Scholar]
- Franks S, Stark J & Hardy K 2008. Follicle dynamics and anovulation in polycystic ovary syndrome. Hum Reprod Update 14 367–378. [DOI] [PubMed] [Google Scholar]
- Gazvani MR, Bates M, Vince G, Christmas S, Lewis-Jones DI & Kingsland C 2000. Follicular fluid concentrations of interleukin-12 and interleukin-8 in IVF cycles. Fertil Steril 74 953–958. [DOI] [PubMed] [Google Scholar]
- Gill S & Hall JE 2014. The Hypothalamic–Pituitary Axis in PCOS. In Polycystic Ovary Syndrome, pp 81–93: Springer. [Google Scholar]
- Gilling-Smith C, Willis DS, Beard RW & Franks S 1994. Hypersecretion of androstenedione by isolated thecal cells from polycystic ovaries. J Clin Endocrinol Metab 79 1158–1165. [DOI] [PubMed] [Google Scholar]
- Giovanni Artini P, Monteleone P, Parisen Toldin MR, Matteucci C, Ruggiero M, Cela V & Genazzani AR 2007. Growth factors and folliculogenesis in polycystic ovary patients. Expert Review of Endocrinology & Metabolism 2 215–223. [DOI] [PubMed] [Google Scholar]
- Gluckman PD & Hanson MA 2004. The developmental origins of the metabolic syndrome. Trends Endocrinol Metab 15 183–187. [DOI] [PubMed] [Google Scholar]
- Goodarzi MO, Jones MR, Li X, Chua AK, Garcia OA, Chen YD, Krauss RM, Rotter JI, Ankener W, Legro RS, et al. 2012. Replication of association of DENND1A and THADA variants with polycystic ovary syndrome in European cohorts. J Med Genet 49 90–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore AC, Chappell VA, Fenton SE, Flaws JA, Nadal A, Prins GS, Toppari J & Zoeller RT 2015. EDC-2: The Endocrine Society’s Second Scientific Statement on Endocrine-Disrupting Chemicals. Endocr Rev er20151010. [DOI] [PMC free article] [PubMed]
- Gougeon A 2010. Human ovarian follicular development: from activation of resting follicles to preovulatory maturation. Ann Endocrinol (Paris) 71 132–143. [DOI] [PubMed] [Google Scholar]
- Guerrero-Bosagna C 2016. Chapter 21 - Transgenerational Epigenetic Inheritance: Past Exposures, Future Diseases A2 - Rosenfeld, Cheryl S. In The Epigenome and Developmental Origins of Health and Disease, pp 425–437. Boston: Academic Press. [Google Scholar]
- Ha L, Shi Y, Zhao J, Li T & Chen ZJ 2015. Association Study between Polycystic Ovarian Syndrome and the Susceptibility Genes Polymorphisms in Hui Chinese Women. PLoS One 10 e0126505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hague WM, Adams J, Rodda C, Brook CG, de Bruyn R, Grant DB & Jacobs HS 1990. The prevalence of polycystic ovaries in patients with congenital adrenal hyperplasia and their close relatives. Clin Endocrinol (Oxf) 33 501–510. [DOI] [PubMed] [Google Scholar]
- Hansen L, Drackley J, Berger L & Grum D 1995. Prenatal androgenization of lambs: I. Alterations of growth, carcass characteristics, and metabolites in blood. Journal of animal science 73 1694–1700. [DOI] [PubMed] [Google Scholar]
- Hatzirodos N, Bayne RA, Irving-Rodgers HF, Hummitzsch K, Sabatier L, Lee S, Bonner W, Gibson MA, Rainey WE, Carr BR, et al. 2011. Linkage of regulators of TGF-beta activity in the fetal ovary to polycystic ovary syndrome. FASEB J 25 2256–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MG, Urbanek M, Ehrmann DA, Armstrong LL, Lee JY, Sisk R, Karaderi T, Barber TM, McCarthy MI, Franks S, et al. 2015. Genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat Commun 6 7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijnen EM, Eijkemans MJ, Hughes EG, Laven JS, Macklon NS & Fauser BC 2006. A meta-analysis of outcomes of conventional IVF in women with polycystic ovary syndrome. Hum Reprod Update 12 13–21. [DOI] [PubMed] [Google Scholar]
- Hernandez ER, Resnick CE, Holtzclaw WD, Payne DW & Adashi EY 1988. Insulin as a regulator of androgen biosynthesis by cultured rat ovarian cells: cellular mechanism(s) underlying physiological and pharmacological hormonal actions. Endocrinology 122 2034–2043. [DOI] [PubMed] [Google Scholar]
- Hickey TE, Legro RS & Norman RJ 2006. Epigenetic modification of the X chromosome influences susceptibility to polycystic ovary syndrome. J Clin Endocrinol Metab 91 2789–2791. [DOI] [PubMed] [Google Scholar]
- Hogeveen KN, Cousin P, Pugeat M, Dewailly D, Soudan B & Hammond GL 2002. Human sex hormone-binding globulin variants associated with hyperandrogenism and ovarian dysfunction. J Clin Invest 109 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holly JM, Eden JA, Alaghband-Zadeh J, Carter GD, Jemmott RC, Cianfarani S, Chard T & Wass JA 1990. Insulin-like growth factor binding proteins in follicular fluid from normal dominant and cohort follicles, polycystic and multicystic ovaries. Clin Endocrinol (Oxf) 33 53–64. [DOI] [PubMed] [Google Scholar]
- Homburg R & Amsterdam A 1998. Polysystic ovary syndrome--loss of the apoptotic mechanism in the ovarian follicles? J Endocrinol Invest 21 552–557. [DOI] [PubMed] [Google Scholar]
- Hsieh M, Zamah AM & Conti M 2009. Epidermal growth factor-like growth factors in the follicular fluid: role in oocyte development and maturation. Semin Reprod Med 27 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber-Buchholz MM, Carey DG & Norman RJ 1999. Restoration of reproductive potential by lifestyle modification in obese polycystic ovary syndrome: role of insulin sensitivity and luteinizing hormone. J Clin Endocrinol Metab 84 1470–1474. [DOI] [PubMed] [Google Scholar]
- Hughesdon PE 1982. Morphology and morphogenesis of the Stein-Leventhal ovary and of so-called “hyperthecosis”. Obstet Gynecol Surv 37 59–77. [DOI] [PubMed] [Google Scholar]
- Hunter RG, Gagnidze K, McEwen BS & Pfaff DW 2015. Stress and the dynamic genome: Steroids, epigenetics, and the transposome. Proc Natl Acad Sci U S A 112 6828–6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein MR 2005. Apoptosis in the ovary: molecular mechanisms. Hum Reprod Update 11 162–177. [DOI] [PubMed] [Google Scholar]
- Hussein TS, Thompson JG & Gilchrist RB 2006. Oocyte-secreted factors enhance oocyte developmental competence. Dev Biol 296 514–521. [DOI] [PubMed] [Google Scholar]
- Jayasena CN & Franks S 2014. The management of patients with polycystic ovary syndrome. Nat Rev Endocrinol 10 624–636. [DOI] [PubMed] [Google Scholar]
- Johnstone E, Shelly W, Mellon S & Cedars M 2008. Brain-derived neurotrophic factor is elevated in follicular fluid of women with PCOS. Fertility and sterility 90 S256. [Google Scholar]
- Jonard S & Dewailly D 2004. The follicular excess in polycystic ovaries, due to intra-ovarian hyperandrogenism, may be the main culprit for the follicular arrest. Hum Reprod Update 10 107–117. [DOI] [PubMed] [Google Scholar]
- Jones MR, Brower MA, Xu N, Cui J, Mengesha E, Chen YD, Taylor KD, Azziz R & Goodarzi MO 2015. Systems Genetics Reveals the Functional Context of PCOS Loci and Identifies Genetic and Molecular Mechanisms of Disease Heterogeneity. PLoS Genet 11 e1005455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan CD, Bohling SD, Charbonneau NL & Sakai LY 2010. Fibrillins in adult human ovary and polycystic ovary syndrome: is fibrillin-3 affected in PCOS? Journal of Histochemistry & Cytochemistry 58 903–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph-Horne R, Mason H, Batty S, White D, Hillier S, Urquhart M & Franks S 2002. Luteal phase progesterone excretion in ovulatory women with polycystic ovaries. Hum Reprod 17 1459–1463. [DOI] [PubMed] [Google Scholar]
- Kandaraki E, Chatzigeorgiou A, Livadas S, Palioura E, Economou F, Koutsilieris M, Palimeri S, Panidis D & Diamanti-Kandarakis E 2011. Endocrine disruptors and polycystic ovary syndrome (PCOS): elevated serum levels of bisphenol A in women with PCOS. J Clin Endocrinol Metab 96 E480–484. [DOI] [PubMed] [Google Scholar]
- Kawamura K, Kawamura N, Mulders SM, Sollewijn Gelpke MD & Hsueh AJ 2005. Ovarian brain-derived neurotrophic factor (BDNF) promotes the development of oocytes into preimplantation embryos. Proc Natl Acad Sci U S A 102 9206–9211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JK, Samaranayake M & Pradhan S 2009. Epigenetic mechanisms in mammals. Cell Mol Life Sci 66 596–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight PG, Satchell L & Glister C 2012. Intra-ovarian roles of activins and inhibins. Mol Cell Endocrinol 359 53–65. [DOI] [PubMed] [Google Scholar]
- Kuijper EA, Vink JM, Lambalk CB & Boomsma DI 2009. Prevalence of polycystic ovary syndrome in women from opposite-sex twin pairs. J Clin Endocrinol Metab 94 1987–1990. [DOI] [PubMed] [Google Scholar]
- Kwintkiewicz J & Giudice LC 2009. The interplay of insulin-like growth factors, gonadotropins, and endocrine disruptors in ovarian follicular development and function. Semin Reprod Med 27 43–51. [DOI] [PubMed] [Google Scholar]
- Lahav-Baratz S, Kraiem Z, Shiloh H, Koifman M, Ishai D & Dirnfeld M 2003. Decreased expression of tissue inhibitor of matrix metalloproteinases in follicular fluid from women with polycystic ovaries compared with normally ovulating patients undergoing in vitro fertilization. Fertil Steril 79 567–571. [DOI] [PubMed] [Google Scholar]
- Lebbe M & Woodruff TK 2013. Involvement of androgens in ovarian health and disease. Mol Hum Reprod 19 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledee-Bataille N, Lapree-Delage G, Taupin JL, Dubanchet S, Taieb J, Moreau JF & Chaouat G 2001. Follicular fluid concentration of leukaemia inhibitory factor is decreased among women with polycystic ovarian syndrome during assisted reproduction cycles. Hum Reprod 16 2073–2078. [DOI] [PubMed] [Google Scholar]
- Legro RS 2016. Ovulation induction in polycystic ovary syndrome: Current options. Best Pract Res Clin Obstet Gynaecol 37 152–159. [DOI] [PubMed] [Google Scholar]
- Legro RS, Driscoll D, Strauss JF 3rd, Fox J & Dunaif A 1998a. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc Natl Acad Sci U S A 95 14956–14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legro RS, Kunselman AR & Dunaif A 2001. Prevalence and predictors of dyslipidemia in women with polycystic ovary syndrome. Am J Med 111 607–613. [DOI] [PubMed] [Google Scholar]
- Legro RS, Spielman R, Urbanek M, Driscoll D, Strauss JF3rd & Dunaif A 1998b. Phenotype and genotype in polycystic ovary syndrome. Recent Prog Horm Res 53 217–256. [PubMed] [Google Scholar]
- Li S, Zhu D, Duan H & Tan Q 2016. The epigenomics of polycystic ovarian syndrome: from pathogenesis to clinical manifestations. Gynecol Endocrinol 32 942–946. [DOI] [PubMed] [Google Scholar]
- Louwers YV, Stolk L, Uitterlinden AG & Laven JS 2013. Cross-ethnic meta-analysis of genetic variants for polycystic ovary syndrome. J Clin Endocrinol Metab 98 E2006–2012. [DOI] [PubMed] [Google Scholar]
- Luense LJ, Veiga-Lopez A, Padmanabhan V & Christenson LK 2011. Developmental programming: gestational testosterone treatment alters fetal ovarian gene expression. Endocrinology 152 4974–4983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo H, Kimura K, Aoki M & Hirako M 2002. Effect of vascular endothelial growth factor on maturation, fertilization and developmental competence of bovine oocytes. J Vet Med Sci 64 803–806. [DOI] [PubMed] [Google Scholar]
- Luque-Ramírez M, Escobar-Morreale HF 2016. Adrenal Hyperandrogenism and Polycystic Ovary Syndrome. Curr Pharm Des 22 5588–5602. [DOI] [PubMed] [Google Scholar]
- Ma CH, Yan LY, Qiao J, Sha W, Li L, Chen Y & Sun QY 2010. Effects of tumor necrosis factor-alpha on porcine oocyte meiosis progression, spindle organization, and chromosome alignment. Fertil Steril 93 920–926. [DOI] [PubMed] [Google Scholar]
- Maciel GA, Baracat EC, Benda JA, Markham SM, Hensinger K, Chang RJ & Erickson GF 2004. Stockpiling of transitional and classic primary follicles in ovaries of women with polycystic ovary syndrome. J Clin Endocrinol Metab 89 5321–5327. [DOI] [PubMed] [Google Scholar]
- Macut D, Bjekic-Macut J & Savic-Radojevic A 2013. Dyslipidemia and oxidative stress in PCOS. Front Horm Res 40 51–63. [DOI] [PubMed] [Google Scholar]
- Makri E & Tziomalos K 2017. Prevalence, etiology and management of non-alcoholic fatty liver disease in patients with polycystic ovary syndrome. Minerva Endocrinol 42 122–131. [DOI] [PubMed] [Google Scholar]
- Maliqueo M, Benrick A & Stener-Victorin E 2014. Rodent models of polycystic ovary syndrome: phenotypic presentation, pathophysiology, and the effects of different interventions. Semin Reprod Med 32 183–193. [DOI] [PubMed] [Google Scholar]
- Maliqueo M, Sir-Petermann T, Pérez V, Echiburú B, de Guevara AL, Gálvez C, Crisosto N, Azziz R 2009. Adrenal function during childhood and puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab 94 3282–3288. [DOI] [PubMed] [Google Scholar]
- Magoffin DA & Jakimiuk AJ 1998Inhibin A, inhibin B and activin A concentration in follicular fluid from women with polycystic ovary syndrome. Hum Reprod 13 2693–2698. [DOI] [PubMed] [Google Scholar]
- Manikkam M, Crespi EJ, Doop DD, Herkimer C, Lee JS, Yu S, Brown MB, Foster DL & Padmanabhan V 2004. Fetal programming: prenatal testosterone excess leads to fetal growth retardation and postnatal catch-up growth in sheep. Endocrinology 145 790–798. [DOI] [PubMed] [Google Scholar]
- Manikkam M, Steckler TL, Welch KB, Inskeep EK & Padmanabhan V 2006. Fetal programming: prenatal testosterone treatment leads to follicular persistence/luteal defects; partial restoration of ovarian function by cyclic progesterone treatment. Endocrinology 147 1997–2007. [DOI] [PubMed] [Google Scholar]
- Manikkam M, Thompson RC, Herkimer C, Welch KB, Flak J, Karsch FJ & Padmanabhan V 2008. Developmental programming: impact of prenatal testosterone excess on pre- and postnatal gonadotropin regulation in sheep. Biol Reprod 78 648–660. [DOI] [PubMed] [Google Scholar]
- Manneras-Holm L, Leonhardt H, Kullberg J, Jennische E, Oden A, Holm G, Hellstrom M, Lonn L, Olivecrona G, Stener-Victorin E, et al. 2011. Adipose tissue has aberrant morphology and function in PCOS: enlarged adipocytes and low serum adiponectin, but not circulating sex steroids, are strongly associated with insulin resistance. J Clin Endocrinol Metab 96 E304–311. [DOI] [PubMed] [Google Scholar]
- Marquard KL, Stephens SM, Jungheim ES, Ratts VS, Odem RR, Lanzendorf S & Moley KH 2011. Polycystic ovary syndrome and maternal obesity affect oocyte size in in vitro fertilization/intracytoplasmic sperm injection cycles. Fertil Steril 95 2146–2149, 2149 e2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JC & Eagleson CA 1999. Neuroendocrine aspects of polycystic ovary syndrome. Endocrinol Metab Clin North Am 28 295–324. [DOI] [PubMed] [Google Scholar]
- Mastorakos G, Scopa CD, Vryonidou A, Friedman TC, Kattis D, Phenekos C, Merino MJ & Chrousos GP 1994. Presence of immunoreactive corticotropin-releasing hormone in normal and polycystic human ovaries. J Clin Endocrinol Metab 79 1191–1197. [DOI] [PubMed] [Google Scholar]
- Mazerbourg S, Bondy CA, Zhou J & Monget P 2003. The insulin-like growth factor system: a key determinant role in the growth and selection of ovarian follicles? a comparative species study. Reprod Domest Anim 38 247–258. [DOI] [PubMed] [Google Scholar]
- McAllister JM, Modi B, Miller BA, Biegler J, Bruggeman R, Legro RS & Strauss JF, 3rd 2014. Overexpression of a DENND1A isoform produces a polycystic ovary syndrome theca phenotype. Proc Natl Acad Sci U S A 111 E1519–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney CR, Eagleson CA & Marshall JC 2002. Regulation of gonadotropin secretion: implications for polycystic ovary syndrome. Semin Reprod Med 20 317–326. [DOI] [PubMed] [Google Scholar]
- Melo AS, Vieira CS, Barbieri MA, Rosa ESAC, Silva AA, Cardoso VC, Reis RM, Ferriani RA, Silva-de-Sa MF & Bettiol H 2010. High prevalence of polycystic ovary syndrome in women born small for gestational age. Hum Reprod 25 2124–2131. [DOI] [PubMed] [Google Scholar]
- Merkin SS, Phy JL, Sites CK & Yang D 2016. Environmental determinants of polycystic ovary syndrome. Fertil Steril 106 16–24. [DOI] [PubMed] [Google Scholar]
- Merz CN, Shaw LJ, Azziz R, Stanczyk FZ, Sopko G, Braunstein GD, Kelsey SF, Kip KE, Cooper-DeHoff RM, Johnson BD, et al. 2016. Cardiovascular Disease and 10-Year Mortality in Postmenopausal Women with Clinical Features of Polycystic Ovary Syndrome. J Womens Health (Larchmt) 25 875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikaeili S, Rashidi BH, Safa M, Najafi A, Sobhani A, Asadi E, Abbasi M 2016. Altered FoxO3 expression and apoptosis in granulosa cells of women with polycystic ovary syndrome. Arch Gynecol Obstet 294 185–192. [DOI] [PubMed] [Google Scholar]
- Modi BP 2016. Genetic and epigenetic mechanisms of complex reproductive disorders Richmond, VA: Virginia Commonwealth University. [Google Scholar]
- Moore AM & Campbell RE 2017. Polycystic ovary syndrome: Understanding the role of the brain. Front Neuroendocrinol 46 1–14. [DOI] [PubMed] [Google Scholar]
- Moore AM, Prescott M, Marshall CJ, Yip SH & Campbell RE 2015. Enhancement of a robust arcuate GABAergic input to gonadotropin-releasing hormone neurons in a model of polycystic ovarian syndrome. Proc Natl Acad Sci U S A 112 596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran LJ, Norman RJ & Teede HJ 2015. Metabolic risk in PCOS: phenotype and adiposity impact. Trends Endocrinol Metab 26 136–143. [DOI] [PubMed] [Google Scholar]
- Moran C, Reyna R, Boots LS, Azziz R 2005. Adrenocortical hyperresponsiveness to corticotropin in polycystic ovary syndrome patients with adrenal androgen excess. Fertil Steril 81 126–131. [DOI] [PubMed] [Google Scholar]
- Morishima A, Grumbach MM, Simpson ER, Fisher C & Qin K 1995. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab 80 3689–3698. [DOI] [PubMed] [Google Scholar]
- Mossa F, Carter F, Walsh SW, Kenny DA, Smith GW, Ireland JL, Hildebrandt TB, Lonergan P, Ireland JJ & Evans AC 2013. Maternal undernutrition in cows impairs ovarian and cardiovascular systems in their offspring. Biol Reprod 88 92. [DOI] [PubMed] [Google Scholar]
- Mumm H, Kamper-Jorgensen M, Nybo Andersen AM, Glintborg D & Andersen M 2013. Birth weight and polycystic ovary syndrome in adult life: a register-based study on 523,757 Danish women born 1973–1991. Fertil Steril 99 777–782. [DOI] [PubMed] [Google Scholar]
- Naderpoor N, Shorakae S, Joham A, Boyle J, De Courten B & Teede HJ 2015. Obesity and polycystic ovary syndrome. Minerva Endocrinol 40 37–51. [PubMed] [Google Scholar]
- Nahum R, Thong KJ & Hillier SG 1995. Metabolic regulation of androgen production by human thecal cells in vitro. Hum Reprod 10 75–81. [DOI] [PubMed] [Google Scholar]
- Naji M, Nekoonam S, Aleyasin A, Arefian E, Mahdian R, Azizi E, Shabani Nashtaei M, Amidi F 2018. Expression of miR-15a, miR-145, and miR-182 in granulosa-lutein cells, follicular fluid, and serum of women with polycystic ovary syndrome (PCOS). Arch Gynecol Obstet 297 221–231. [DOI] [PubMed] [Google Scholar]
- Naji M, Aleyasin A, Nekoonam S, Arefian E, Mahdian R, Amidi F 2017. Differential Expression of miR-93 and miR-21 in granulosa cells and follicular fluid of polycystic ovary syndrome associating with different phenotypes. Sci Rep 7 14671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Institues of Health. Evidence-based Methodology Workshop on Polycystic Ovary Syndrome - Final Panel Report. 2012.
- Nelson JW, Scammell MK, Hatch EE & Webster TF 2012. Social disparities in exposures to bisphenol A and polyfluoroalkyl chemicals: a cross-sectional study within NHANES 2003–2006. Environ Health 11 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler JE, Powers LP, Matt DW, Steingold KA, Plymate SR, Rittmaster RS, Clore JN & Blackard WG 1991. A direct effect of hyperinsulinemia on serum sex hormone-binding globulin levels in obese women with the polycystic ovary syndrome. J Clin Endocrinol Metab 72 83–89. [DOI] [PubMed] [Google Scholar]
- Nielsen ME, Rasmussen IA, Kristensen SG, Christensen ST, Mollgard K, Wreford Andersen E, Byskov AG & Yding Andersen C 2011. In human granulosa cells from small antral follicles, androgen receptor mRNA and androgen levels in follicular fluid correlate with FSH receptor mRNA. Mol Hum Reprod 17 63–70. [DOI] [PubMed] [Google Scholar]
- Norman RJ, Milner CR, Groome NP & Robertson DM 2001. Circulating follistatin concentrations are higher and activin concentrations are lower in polycystic ovarian syndrome. Hum Reprod 16 668–672. [DOI] [PubMed] [Google Scholar]
- Onalan G, Selam B, Baran Y, Cincik M, Onalan R, Gunduz U, Ural AU & Pabuccu R 2005. Serum and follicular fluid levels of soluble Fas, soluble Fas ligand and apoptosis of luteinized granulosa cells in PCOS patients undergoing IVF. Hum Reprod 20 2391–2395. [DOI] [PubMed] [Google Scholar]
- Ortega HH, Salvetti NR & Padmanabhan V 2009. Developmental programming: prenatal androgen excess disrupts ovarian steroid receptor balance. Reproduction 137 865–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega HH, Veiga-Lopez A, Sreedharan S, del Lujan Velazquez MM, Salvetti NR & Padmanabhan V 2015. Developmental Programming: Does Prenatal Steroid Excess Disrupt the Ovarian VEGF System in Sheep? Biol Reprod 93 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan V & Veiga-Lopez A 2013. Sheep models of polycystic ovary syndrome phenotype. Mol Cell Endocrinol 373 8–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan V & Veiga-Lopez A 2014. Reproduction Symposium: developmental programming of reproductive and metabolic health. J Anim Sci 92 3199–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan V, Veiga-Lopez A, Abbott D, Recabarren S & Herkimer C 2010. Developmental programming: impact of prenatal testosterone excess and postnatal weight gain on insulin sensitivity index and transfer of traits to offspring of overweight females. Endocrinology 151 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagan YL, Srouji SS, Jimenez Y, Emerson A, Gill S & Hall JE 2006. Inverse relationship between luteinizing hormone and body mass index in polycystic ovarian syndrome: investigation of hypothalamic and pituitary contributions. J Clin Endocrinol Metab 91 1309–1316. [DOI] [PubMed] [Google Scholar]
- Palin MF, Bordignon VV & Murphy BD 2012. Adiponectin and the control of female reproductive functions. Vitam Horm 90 239–287. [DOI] [PubMed] [Google Scholar]
- Palomba S, Marotta R, Di Cello A, Russo T, Falbo A, Orio F, Tolino A, Zullo F, Esposito R, La Sala GB 2012. Pervasive developmental disorders in children of hyperandrogenic women with polycystic ovary syndrome: a longitudinal case-control study. Clin Endocrinol (Oxf) 77 898–904. [DOI] [PubMed] [Google Scholar]
- Pastor CL, Griffin-Korf ML, Aloi JA, Evans WS & Marshall JC 1998. Polycystic ovary syndrome: evidence for reduced sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab 83 582–590. [DOI] [PubMed] [Google Scholar]
- Peretz J, Vrooman L, Ricke WA, Hunt PA, Ehrlich S, Hauser R, Padmanabhan V, Taylor HS, Swan SH, VandeVoort CA, et al. 2014. Bisphenol a and reproductive health: update of experimental and human evidence, 2007–2013. Environ Health Perspect 122 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pru JK & Tilly JL 2001. Programmed cell death in the ovary: insights and future prospects using genetic technologies. Mol Endocrinol 15 845–853. [DOI] [PubMed] [Google Scholar]
- Puttabyatappa M, Andriessen V, Mesquitta M, Zeng L, Pennathur S & Padmanabhan V 2017a. Developmental Programming: Impact of Gestational Steroid and Metabolic Milieus on Mediators of Insulin Sensitivity in Prenatal Testosterone-Treated Female Sheep. Endocrinology. [DOI] [PMC free article] [PubMed]
- Puttabyatappa M, Burns A, Martin JD, Mesquita M, Veiga-Lopez A & Padmanabhan V 2017b. Developmental Programming: Gestational Exposure to Excess Testosterone Alters Expression of Ovarian Matrix Metalloproteases and Their Target Proteins. Reprod Sci 10.1177/1933719117697127. [DOI] [PMC free article] [PubMed]
- Qiao GY, Dong BW, Zhu CJ, Yan CY, Chen BL 2017. Deregulation of WNT2/FZD3/β-catenin pathway compromises the estrogen synthesis in cumulus cells from patients with polycystic ovary syndrome. Biochem Biophys Res Commun 493 847–854. [DOI] [PubMed] [Google Scholar]
- Qiao J & Feng HL 2011. Extra- and intra-ovarian factors in polycystic ovary syndrome: impact on oocyte maturation and embryo developmental competence. Hum Reprod Update 17 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin JZ, Pang LH, Li MJ, Fan XJ, Huang RD & Chen HY 2013. Obstetric complications in women with polycystic ovary syndrome: a systematic review and meta-analysis. Reprod Biol Endocrinol 11 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recabarren SE, Padmanabhan V, Codner E, Lobos A, Durán C, Vidal M, Foster DL & Sir-Petermann T 2005. Postnatal developmental consequences of altered insulin sensitivity in female sheep treated prenatally with testosterone. American Journal of Physiology-Endocrinology and Metabolism 289 E801–E806. [DOI] [PubMed] [Google Scholar]
- Recabarren SE, Rojas-Garcia PP, Recabarren MP, Alfaro VH, Smith R, Padmanabhan V & Sir-Petermann T 2008. Prenatal testosterone excess reduces sperm count and motility. Endocrinology 149 6444–6448. [DOI] [PubMed] [Google Scholar]
- Reddy P, Liu L, Adhikari D, Jagarlamudi K, Rajareddy S, Shen Y, Du C, Tang W, Hamalainen T, Peng SL, et al. 2008. Oocyte-specific deletion of Pten causes premature activation of the primordial follicle pool. Science 319 611–613. [DOI] [PubMed] [Google Scholar]
- Risma KA, Clay CM, Nett TM, Wagner T, Yun J & Nilson JH 1995. Targeted overexpression of luteinizing hormone in transgenic mice leads to infertility, polycystic ovaries, and ovarian tumors. Proc Natl Acad Sci U S A 92 1322–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JE, Forsdike RA & Taylor JA 1999. In utero exposure of female lambs to testosterone reduces the sensitivity of the gonadotropin-releasing hormone neuronal network to inhibition by progesterone. Endocrinology 140 5797–5805. [DOI] [PubMed] [Google Scholar]
- Rodin A, Thakkar H, Taylor N, Clayton R 1994. Hyperandrogenism in polycystic ovary syndrome. Evidence of dysregulation of 11 beta-hydroxysteroid dehydrogenase. N Engl J Med 330 460–465. [DOI] [PubMed] [Google Scholar]
- Rojas-Garcia PP, Recabarren MP, Sarabia L, Schon J, Gabler C, Einspanier R, Maliqueo M, Sir-Petermann T, Rey R & Recabarren SE 2010. Prenatal testosterone excess alters Sertoli and germ cell number and testicular FSH receptor expression in rams. Am J Physiol Endocrinol Metab 299 E998–E1005. [DOI] [PubMed] [Google Scholar]
- Roland AV, Nunemaker CS, Keller SR & Moenter SM 2010. Prenatal androgen exposure programs metabolic dysfunction in female mice. J Endocrinol 207 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosmond R 2005. Role of stress in the pathogenesis of the metabolic syndrome. Psychoneuroendocrinology 30 1–10. [DOI] [PubMed] [Google Scholar]
- Rutkowska A & Rachon D 2014. Bisphenol A (BPA) and its potential role in the pathogenesis of the polycystic ovary syndrome (PCOS). Gynecol Endocrinol 30 260–265. [DOI] [PubMed] [Google Scholar]
- Salvetti NR, Ortega HH, Veiga-Lopez A & Padmanabhan V 2012. Developmental programming: impact of prenatal testosterone excess on ovarian cell proliferation and apoptotic factors in sheep. Biol Reprod 87 22, 21–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarma HN, Manikkam M, Herkimer C, Dell’Orco J, Welch KB, Foster DL & Padmanabhan V 2005. Fetal programming: excess prenatal testosterone reduces postnatal luteinizing hormone, but not follicle-stimulating hormone responsiveness, to estradiol negative feedback in the female. Endocrinology 146 4281–4291. [DOI] [PubMed] [Google Scholar]
- Savabieasfahani M, Kannan K, Astapova O, Evans NP & Padmanabhan V 2006. Developmental programming: differential effects of prenatal exposure to bisphenol-A or methoxychlor on reproductive function. Endocrinology 147 5956–5966. [DOI] [PubMed] [Google Scholar]
- Savchev SI, Moragianni VA, Senger D, Penzias AS, Thornton K, Usheva A 2010. Follicular fluid-specific distribution of vascular endothelial growth factor isoforms and sFlt-1 in patients undergoing IVF and their correlation with treatment outcomes. Reprod Sci 17 1036–1042. [DOI] [PubMed] [Google Scholar]
- Sermondade N, Dupont C, Massart P, Cedrin-Durnerin I, Levy R & Sifer C 2013. [Impact of polycystic ovary syndrome on oocyte and embryo quality]. Gynecol Obstet Fertil 41 27–30. [DOI] [PubMed] [Google Scholar]
- Shalev E, Goldman S & Ben-Shlomo I 2001. The balance between MMP-9 and MMP-2 and their tissue inhibitor (TIMP)-1 in luteinized granulosa cells: comparison between women with PCOS and normal ovulatory women. Mol Hum Reprod 7 325–331. [DOI] [PubMed] [Google Scholar]
- Sharma TP, Herkimer C, West C, Ye W, Birch R, Robinson JE, Foster DL & Padmanabhan V 2002 Fetal programming: prenatal androgen disrupts positive feedback actions of estradiol but does not affect timing of puberty in female sheep. Biol Reprod 66 924–933. [DOI] [PubMed] [Google Scholar]
- Sir-Petermann T, Codner E, Perez V, Echiburu B, Maliqueo M, Ladron de Guevara A, Preisler J, Crisosto N, Sanchez F, Cassorla F, et al. 2009. Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab 94 1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner MK 2005. Regulation of primordial follicle assembly and development. Human Reproduction Update 11 461–471. [DOI] [PubMed] [Google Scholar]
- Smith P, Steckler TL, Veiga-Lopez A & Padmanabhan V 2009. Developmental programming: differential effects of prenatal testosterone and dihydrotestosterone on follicular recruitment, depletion of follicular reserve, and ovarian morphology in sheep. Biol Reprod 80 726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steckler T, Roberts E, Doop D, Lee T & Padmanabhan V 2007. Developmental programming in sheep: administration of testosterone during 60–90 days of pregnancy reduces breeding success and pregnancy outcome. Theriogenology 67 459–467. [DOI] [PubMed] [Google Scholar]
- Steckler T, Wang J, Bartol FF, Roy SK & Padmanabhan V 2005. Fetal programming: prenatal testosterone treatment causes intrauterine growth retardation, reduces ovarian reserve and increases ovarian follicular recruitment. Endocrinology 146 3185–3193. [DOI] [PubMed] [Google Scholar]
- Steckler TL, Herkimer C, Dumesic DA & Padmanabhan V 2009. Developmental programming: excess weight gain amplifies the effects of prenatal testosterone excess on reproductive cyclicity—implication for polycystic ovary syndrome. Endocrinology 150 1456–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steckler TL, Lee JS, Ye W, Inskeep EK & Padmanabhan V 2008. Developmental programming: exogenous gonadotropin treatment rescues ovulatory function but does not completely normalize ovarian function in sheep treated prenatally with testosterone. Biol Reprod 79 686–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein IF & Leventhal ML 1935. Amenorrhea associated with bilateral polycystic ovaries. American Journal of Obstetrics and Gynecology 29 181–191. [Google Scholar]
- Stubbs SA, Hardy K, Da Silva-Buttkus P, Stark J, Webber LJ, Flanagan AM, Themmen AP, Visser JA, Groome NP & Franks S 2005. Anti-mullerian hormone protein expression is reduced during the initial stages of follicle development in human polycystic ovaries. J Clin Endocrinol Metab 90 5536–5543. [DOI] [PubMed] [Google Scholar]
- Tang WY & Ho SM 2007. Epigenetic reprogramming and imprinting in origins of disease. Rev Endocr Metab Disord 8 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AE, McCourt B, Martin KA, Anderson EJ, Adams JM, Schoenfeld D & Hall JE 1997. Determinants of abnormal gonadotropin secretion in clinically defined women with polycystic ovary syndrome. J Clin Endocrinol Metab 82 2248–2256. [DOI] [PubMed] [Google Scholar]
- Teede H, Deeks A & Moran L 2010. Polycystic ovary syndrome: a complex condition with psychological, reproductive and metabolic manifestations that impacts on health across the lifespan. BMC Med 8 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teissier MP, Chable H, Paulhac S & Aubard Y 2000. Comparison of follicle steroidogenesis from normal and polycystic ovaries in women undergoing IVF: relationship between steroid concentrations, follicle size, oocyte quality and fecundability. Hum Reprod 15 2471–2477. [DOI] [PubMed] [Google Scholar]
- The Rotterdam ESHRE/ASRM‐sponsored PCOS consensus workshop group 2004. Revised 2003 consensus on diagnostic criteria and long‐term health risks related to polycystic ovary syndrome (PCOS). Hum Reprod 19 41–47. [DOI] [PubMed] [Google Scholar]
- Unfer V, Casini ML, Marelli G, Costabile L, Gerli S & Di Renzo GC 2005. Different routes of progesterone administration and polycystic ovary syndrome: a review of the literature. Gynecol Endocrinol 21 119–127. [DOI] [PubMed] [Google Scholar]
- Unsworth WP, Taylor JA & Robinson JE 2005. Prenatal programming of reproductive neuroendocrine function: the effect of prenatal androgens on the development of estrogen positive feedback and ovarian cycles in the ewe. Biol Reprod 72 619–627. [DOI] [PubMed] [Google Scholar]
- Urbanek M, Legro RS, Driscoll DA, Azziz R, Ehrmann DA, Norman RJ, Strauss JF3rd, Spielman RS & Dunaif A 1999. Thirty-seven candidate genes for polycystic ovary syndrome: strongest evidence for linkage is with follistatin. Proc Natl Acad Sci U S A 96 8573–8578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagi SJ, Azziz-Baumgartner E, Sjodin A, Calafat AM, Dumesic D, Gonzalez L, Kato K, Silva MJ, Ye X & Azziz R 2014. Exploring the potential association between brominated diphenyl ethers, polychlorinated biphenyls, organochlorine pesticides, perfluorinated compounds, phthalates, and bisphenol A in polycystic ovary syndrome: a case-control study. BMC Endocr Disord 14 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilatou E 2014. Nonalcoholic fatty liver disease and polycystic ovary syndrome. World J Gastroenterol 20 8351–8363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Lopez A, Astapova OI, Aizenberg EF, Lee JS & Padmanabhan V 2009. Developmental programming: contribution of prenatal androgen and estrogen to estradiol feedback systems and periovulatory hormonal dynamics in sheep. Biol Reprod 80 718–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Lopez A, Beckett EM, Abi Salloum B, Ye W & Padmanabhan V 2014a. Developmental programming: prenatal BPA treatment disrupts timing of LH surge and ovarian follicular wave dynamics in adult sheep. Toxicol Appl Pharmacol 279 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Lopez A, Lee JS & Padmanabhan V 2010. Developmental programming: insulin sensitizer treatment improves reproductive function in prenatal testosterone-treated female sheep. Endocrinology 151 4007–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Lopez A, Moeller J, Patel D, Ye W, Pease A, Kinns J & Padmanabhan V 2013. Developmental programming: impact of prenatal testosterone excess on insulin sensitivity, adiposity, and free fatty acid profile in postpubertal female sheep. Endocrinology 154 1731–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Lopez A, Wurst AK, Steckler TL, Ye W & Padmanabhan V 2014b. Developmental programming: postnatal estradiol amplifies ovarian follicular defects induced by fetal exposure to excess testosterone and dihydrotestosterone in sheep. Reprod Sci 21 444–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Lopez A, Ye W & Padmanabhan V 2012. Developmental programming: prenatal testosterone excess disrupts anti-Müllerian hormone expression in preantral and antral follicles. Fertility and sterility 97 748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen A 1993. Environment, human reproduction, menopause, and andropause. Environ Health Perspect 101 Suppl 2 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink JM, Sadrzadeh S, Lambalk CB & Boomsma DI 2006. Heritability of polycystic ovary syndrome in a Dutch twin-family study. J Clin Endocrinol Metab 91 2100–2104. [DOI] [PubMed] [Google Scholar]
- Visser JA & Themmen AP 2014. Role of anti-Mullerian hormone and bone morphogenetic proteins in the regulation of FSH sensitivity. Mol Cell Endocrinol 382 460–465. [DOI] [PubMed] [Google Scholar]
- Volpe A, Coukos G, D’Ambrogio G, Artini PG & Genazzani AR 1991. Follicular fluid steroid and epidermal growth factor content, and in vitro estrogen release by granulosa-luteal cells from patients with polycystic ovaries in an IVF/ET program. Eur J Obstet Gynecol Reprod Biol 42 195–199. [DOI] [PubMed] [Google Scholar]
- Walters KA, Allan CM & Handelsman DJ 2012. Rodent models for human polycystic ovary syndrome. Biol Reprod 86 149, 141–112. [DOI] [PubMed] [Google Scholar]
- Walters KA & Handelsman DJ 2017. Role of androgens in the ovary. Mol Cell Endocrinol. [DOI] [PubMed]
- Wang F, Pan J, Liu Y, Meng Q, Lv P, Qu F, Ding GL, Klausen C, Leung PC, Chan HC, et al. 2015. Alternative splicing of the androgen receptor in polycystic ovary syndrome. Proc Natl Acad Sci U S A 112 4743–4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q & Sun QY 2007. Evaluation of oocyte quality: morphological, cellular and molecular predictors. Reprod Fertil Dev 19 1–12. [DOI] [PubMed] [Google Scholar]
- Webber LJ, Stubbs S, Stark J, Trew GH, Margara R, Hardy K & Franks S 2003. Formation and early development of follicles in the polycystic ovary. Lancet 362 1017–1021. [DOI] [PubMed] [Google Scholar]
- Webber LJ, Stubbs SA, Stark J, Margara RA, Trew GH, Lavery SA, Hardy K & Franks S 2007. Prolonged survival in culture of preantral follicles from polycystic ovaries. J Clin Endocrinol Metab 92 1975–1978. [DOI] [PubMed] [Google Scholar]
- Welt CK, Taylor AE, Fox J, Messerlian GM, Adams JM, Schneyer AL 2005. Follicular arrest in polycystic ovary syndrome is associated with deficient inhibin A and B biosynthesis. J Clin Endocrinol Metab 90 5582–5587. [DOI] [PubMed] [Google Scholar]
- West C, Foster DL, Evans NP, Robinson J & Padmanabhan V 2001. Intra-follicular activin availability is altered in prenatally-androgenized lambs. Mol Cell Endocrinol 185 51–59. [DOI] [PubMed] [Google Scholar]
- Whirledge S & Cidlowski JA 2010. Glucocorticoids, stress, and fertility. Minerva Endocrinol 35 109–125. [PMC free article] [PubMed] [Google Scholar]
- Whyte J, Alexenko A, Davis A, Ellersieck M, Fountain E & Rosenfeld C 2007. Maternal diet composition alters serum steroid and free fatty acid concentrations and vaginal pH in mice. Journal of Endocrinology 192 75–81. [DOI] [PubMed] [Google Scholar]
- Willis D, Mason H, Gilling-Smith C & Franks S 1996. Modulation by insulin of follicle-stimulating hormone and luteinizing hormone actions in human granulosa cells of normal and polycystic ovaries. J Clin Endocrinol Metab 81 302–309. [DOI] [PubMed] [Google Scholar]
- Willis DS, Watson H, Mason HD, Galea R, Brincat M & Franks S 1998. Premature response to luteinizing hormone of granulosa cells from anovulatory women with polycystic ovary syndrome: relevance to mechanism of anovulation. J Clin Endocrinol Metab 83 3984–3991. [DOI] [PubMed] [Google Scholar]
- Wood JR, Dumesic DA, Abbott DH, Strauss JF 3rd 2007. Molecular abnormalities in oocytes from women with polycystic ovary syndrome revealed by microarray analysis. J Clin Endocrinol Metab 92 705–713. [DOI] [PubMed] [Google Scholar]
- Wood RI & Foster DL 1998. Sexual differentiation of reproductive neuroendocrine function in sheep. Rev Reprod 3 130–140. [DOI] [PubMed] [Google Scholar]
- Wu Y, Zhong G, Chen S, Zheng C, Liao D, Xie M 2017. Polycystic ovary syndrome is associated with anogenital distance, a marker of prenatal androgen exposure. Hum Reprod 32 937–943. [DOI] [PubMed] [Google Scholar]
- Xia Y, Shen S, Zhang X, Deng Z, Xiang Z, Wang H, Yi L, Gao Q, Wang Y 2015. Epigenetic pattern changes in prenatal female Sprague-Dawley rats following exposure to androgen. Reprod Fertil Dev 28 1414–1423. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Song Y, Li Y, Zhao D, Ma L, Tan L 2016. miR-483 is down-regulated in polycystic ovarian syndrome and inhibits KGN cell proliferation via targeting Insulin-Like Growth Factor 1 (IGF1). Med Sci Monit 22 3383–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Xu F, Lawson MS, Tkachenko OY, Ting AY, Kahl CA, Park BS, Stouffer RR, Bishop CV 2018. Anti-Müllerian hormone is a survival factor and promotes the growth of rhesus macaque preantral follicles during matrix-free culture. Biol Reprod 98 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, Kwon S, Abbott DH, Geller DH, Dumesic DA, Azziz R, Guo X & Goodarzi MO 2011. Epigenetic mechanism underlying the development of polycystic ovary syndrome (PCOS)-like phenotypes in prenatally androgenized rhesus monkeys. PLoS One 6 e27286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Lv J, Xu P, Gu L, Cao J, Xu L, Xue K, Li Q 2017. Identification of microRNAs and genes associated with hyperandrogenism in the follicular fluid of women with polycystic ovary syndrome. J Cell Biochem 10.1002/jcb.26531. [DOI] [PubMed]
- Yildizhan B, Anik Ilhan G & Pekin T 2016. The impact of insulin resistance on clinical, hormonal and metabolic parameters in lean women with polycystic ovary syndrome. J Obstet Gynaecol 36 893–896. [DOI] [PubMed] [Google Scholar]
- Yildiz BO, Goodarzi MO, Guo X, Rotter JI, Azziz R 2006. Heritability of dehydroepiandrosterone sulfate in women with polycystic ovary syndrome and their sisters. Fertil Steril 86 1688–1693. [DOI] [PubMed] [Google Scholar]
- Zawadzki J & Dunaif A 1992. Current Issues in Endocrinology and Metabolism: Polycystic Ovary Syndrome Blackwell Scientific Publications; Cambridge, MA [Google Scholar]
- Zhao SY, Qiao J, Chen YJ, Liu P, Li J & Yan J 2010. Expression of growth differentiation factor-9 and bone morphogenetic protein-15 in oocytes and cumulus granulosa cells of patients with polycystic ovary syndrome. Fertil Steril 94 261–267. [DOI] [PubMed] [Google Scholar]
- Zhang CL, Wang H, Yan CY, Gao XF, Ling XJ 2017. Deregulation of RUNX2 by miR-320a deficiency impairs steroidogenesis in cumulus granulosa cells from polycystic ovary syndrome (PCOS) patients. Biochem Biophys Res Commun 482 1469–1476. [DOI] [PubMed] [Google Scholar]
- Zhou R, Bird IM, Dumesic DA, Abbott DH 2005. Adrenal hyperandrogenism is induced by fetal androgen excess in a rhesus monkey model of polycystic ovary syndrome. J Clin Endocrinol Metab 90 6630–6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Bruns CM, Bird IM, Kemnitz JW, Goodfriend TL, Dumesic DA, Abbott DH 2007. Pioglitazone improves insulin action and normalizes menstrual cycles in a majority of prenatally androgenized female rhesus monkeys. Reprod Toxicol 23 438–448. [DOI] [PMC free article] [PubMed] [Google Scholar]