

Abstract
While the application of early screening and HPV vaccines has reduced the incidence and mortality rates of cervical cancer, it remains the third most common carcinoma and fourth leading cause of cancer-associated death among women worldwide. The precise mechanisms underlying progression of cervical cancer are not fully understood at present. Here, we detected significant down-regulation of 15-hydroxyprostaglandin dehydrogenase (HPGD) in cervical cancer tissues. Overexpression of HPGD inhibited cervical cancer cell proliferation, migration and anchorage-independent growth to a significant extent. To clarify the mechanisms underlying HPGD down-regulation in cervical cancer, miRNA microarray, bioinformatics and luciferase reporter analyses were performed. HPGD was identified as a direct target of miR-146b-3p displaying up-regulation in cervical cancer tissues. Similar to the effects of HPGD overexpression, down-regulation of miR-146b-3p strongly suppressed proliferation, migration and anchorage-independent growth of cervical cancer cells. Furthermore, HPGD negatively regulated activities of STAT3 and AKT that promote cervical cancer cell proliferation. Notably, HPV oncogenes E6 and E7 were determined as potential contributory factors to these alterations. Our results collectively suggest that the HPGD/miR-146b-3p axis plays a significant role in cervical cancer and may serve as a potentially effective therapeutic target.
Bullet Points
HPGD inhibits cervical cancer cell proliferation, migration and anchorage-independent growth by negatively regulating STAT3 and AKT pathways.
miR-146b-3p directly targets HPGD.
Downregulation of HPGD and up-regulation of miR-146b-3p in cervical cancer are mediated by the high-risk HPVs E6 and E7.
Introduction
Cervical cancer, one of the most common gynecological tumors, is the fourth leading cause of cancer-associated deaths among women worldwide1. Cancer arising from the cervix is initially asymptomatic, later transforming into high-grade squamous intraepithelial lesions and invasive cancer2. A number of proteins reported to be abnormally expressed in cervical cancer may serve as potential prognostic markers and therapeutic targets for treatment, including Wnt family member 5A (Wnt5A)3, ATPase family AAA domain-containing protein 2 (ATAD2)4, sphingosine kinase 15 and erythropoietin-producing human hepatocellular carcinoma receptor B2 (EphB2)6. Transcription factor sex-determining region Y-box 9 protein (SOX9) is known to suppress cervical tumor growth through binding a specific promoter region of p21 (WAF1/CIP1) to block G1/S transition7. Neuroblastoma breakpoint family member 1 (NBPF1) inhibits cervical cancer invasion via regulating the PI3K/mTOR signaling pathway8.
Human papillomavirus (HPV) infection is predominantly responsible for the incidence of cervical cancer9. According to statistical analyses, among the 15 high-risk HPV types, HPV-16 and HPV-18 are responsible for 75% cervical cancer cases10,11. The HPV genome integrates into the host genome following which the viral oncogenes are constitutively expressed12. HPV oncoproteins E6 and E7 that are consistently overexpressed in cervical cancers activate the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) and Wnt/β-catenin/Notch pathways, inducing significant tumorigenic transformation13. Persistent infection of high-risk HPVs is necessary, but not sufficient to cause cervical cancer14–17. Additionally, long-term use of oral contraceptives18, certain sexually transmitted infections19, early sexual exposure, multiple partners, high parity, and smoking20 may contribute to the relative risk of developing cervical cancer.
15-Hydroxyprostaglandin dehydrogenase (HPGD) has been identified as a tumor suppressor in various malignancies, including gastrointestinal21, bladder22 and lung cancers23. Prostaglandins are involved in tumor progression through modulation of angiogenesis, migration, invasion, proliferation, and apoptosis24. A key enzyme participating in the metabolism of prostaglandins25, HPGD regulates their bioavailability through conversion into the biologically inactive forms, 13,14-dihydro-15-keto PGF2 and 13,14-dihydro-15-keto PGE226–28. HPGD additionally has the ability to inhibit prostaglandin-endoperoxide synthase 221,29–32, which catalyzes the metabolic conversion of arachidonic acid to prostaglandins33,34. While HPGD is clearly implicated in several malignancies, its potential association with cervical cancer development is unknown at present.
In this study, we analyzed cervical cancer and paracancerous tissues of patients using gene microarray. HPGD was significantly down-regulated in cervical cancer tissues, and conversely, overexpression of HPGD associated with dramatically reduced cancer cell proliferation, migration and anchorage-independent growth. miR-146b-3p, which directly targets HPGD, was up-regulated in cervical cancer tissues, contributing to enhanced cell proliferation, migration and anchorage-independent growth. Moreover, HPGD suppressed activation of STAT3 and AKT pathways and these alterations were potentially mediated by HPV oncoproteins E6 and E7.
Results
HPGD is down-regulated in cervical cancer tissues
To identify the critical genes correlated with cervical cancer development, three pairs of cervical cancer and paracancerous tissues were analyzed via gene microarray. In total, 990 genes were up-regulated and 1446 genes down-regulated by more than two fold in cervical cancer, compared with paracancerous tissues. Some of the altered genes are presented via hierarchical clustering (Fig. 1a). Among these, HPGD, known to be significantly associated with cancer development, drew our attention. Data from GO analyses indicated that HPGD is closely related to metabolic process, response to stimulus, protein binding, and catalytic activity (Fig. 1b, c). Validation was performed using 67 pairs of cervical cancer and adjacent non-cancerous tissues. As shown in Fig. 1d, in the majority of cervical cancer tissue specimens (60/67), HPGD expression was significantly lower relative to paracancerous tissues. Immunohistochemical staining consistently revealed lower numbers of HPGD-positive cells in cervical cancer than normal tissues (Fig. 1e).
Fig. 1. HPGD is down-regulated in cervical cancer tissues.

(a) A representative result of cervical cancer gene microarray analysis. (b, c) GO analysis of HPGD. (d) mRNA expression of HPGD in 67 pairs of cervical cancer and adjacent non-cancerous tissues determined via qPCR. (e) Immunohistochemical (IHC) staining of HPGD in cervical cancer tissues
HPGD overexpression inhibits cell proliferation, migration and anchorage-independent growth
In view of the decreased expression of HPGD in cervical cancer tissues, the cervical cancer cell lines, HeLa and SiHa, were transduced with lentivirus-HPGD (Fig. 2a, b), and the cell colony formation assay conducted. As shown in Fig. 2c, d, overexpression of HPGD induced a significant reduction in cell proliferation, compared with the control cell group. In the transwell migration assay, HPGD-infected HeLa and SiHa cells exhibited lower migration ability relative to control cells (Fig. 2e, f). The soft agar colony formation assay was adopted to determine the influence of HPGD on tumorigenicity in vitro. Notably, HPGD dramatically suppressed anchorage-independent growth of cervical cancer cells (Fig. 2g, h). Considering the association of HPGD with fatty acid metabolic processes in GO analyses, we further determined the expression patterns of four enzymes involved in de novo synthesis of fatty acids, specifically, ATP Citrate Lyase (ACLY), Fatty Acid Synthase (FAS), Fatty Acyl-CoA Elongase 6 (ELOVL6), and Stearoyl-CoA Desaturase-1 (SCD1), in HPGD-transduced HeLa and SiHa cells via RT-PCR. Transcription levels of all four fatty acid-related enzymes were down-regulated in the presence of HPGD, in strong agreement with GO data (Supplementary Figure 1).
Fig. 2. HPGD overexpression inhibits cell proliferation, migration and anchorage-independent growth.

(a) HeLa and SiHa cells were transduced with lentivirus empty vector (pCDH; left) and lentivirus-HPGD (HPGD; right), with representative images obtained under a light microscope (Phase; top) and fluorescent microscope (GFP; bottom) (Original magnification, × 100). Scale bar, 40 μm. (b) Western blot performed on cells treated as in (a) with the indicated antibodies. (c) Plate colony formation assay of cells treated as in (a). Scale bar, 1 cm. (d) Quantification of cell colony formation described in (c). (e) Transwell migration assay of cells treated as in (a). Scale bar, 40 μm. (f) Quantification of the results in (e). (g) Soft agar colony formation assay of cells treated as in (a). Scale bar, 40 μm. (h) Quantification of the results in (g). Quantified results represent means ± SD. Three independent experiments, each with quartic technical replicates, were performed. ** P < 0.01 and *** P < 0.001 for Student's t-test
HPGD is a direct target of miR-146b-3p
To identify the miRNAs directly targeting HPGD, we used microarray-based miRNA expression profiling for assessing differentially expressed miRNAs in cervical cancer and paracancerous tissues. Among the miRNAs up-regulated in cervical cancer tissues, seven were predicted to contain putative target sites in the 3' UTR or CDS of HPGD using bioinformatics analysis (Fig. 3a), which were selected for the luciferase reporter assay. As shown in Fig. 3b, miR-922, miR-146b-3p and miR-106b-3p significantly inhibited HPGD CDS reporter activity. To directly confirm the effects of these three miRNAs on HPGD protein, HEK 293T cells were co-transfected with miRNA mimics and the HPGD expression plasmid pCDH-HPGD CDS. Western blot experiments showed that only miR-146b-3p attenuated expression of HPGD in a dose-dependent manner (Fig. 3c–e). Additionally, miR-146b-3p decreased the CDS reporter activity of HPGD (Fig. 3f) and suppressed endogenous HPGD expression (Fig. 3g) in a dose-dependent manner. Mutagenesis of miR-146b-3p was subsequently conducted to further validate whether HPGD is a direct target (Fig. 3h). The mutant mimic did not affect CDS reporter activity (Fig. 3i) or protein levels of HPGD (Fig. 3j), confirming that miR-146b-3p directly targets HPGD.
Fig. 3. HPGD is directly targeted by miR-146b-3p.

(a) Representative results of cervical cancer miRNA microarray analysis. (b) Luciferase assay of HEK293T cells co-transfected with negative control miRNA (Neg. Ctrl.) or mimics of several miRNAs together with the pGL3-HPGD 3' UTR or pGL3-HPGD CDS luciferase reporter. (c) 293T cells were co-transfected with the 3 × Flag-HPGD construct and miR-922 mimic for 48 h. Cells were collected and immunoblotted with the indicated antibodies. (d) HEK293T cells were co-transfected with the 3 × Flag-HPGD construct and mimic of miR-146b-3p for 48 h. Cells were collected and immunoblotted with the indicated antibodies. (e) HEK293T cells were co-transfected with the 3 × Flag-HPGD construct along with pEGFP and a mimic of miR-106b-3p for 48 h. Cells were collected and immunoblotted with the indicated antibodies. (f) Luciferase assay of HEK293T cells co-transfected with pGL3-HPGD CDS reporter together with increasing amounts (5, 10, and 20 nM) of negative control miRNA (Neg. Ctrl.) or a mimic of miR-146b-3p (miR-146b-3p) for 24 h. (g) HeLa cells were transfected with increasing amounts (10, 20 and 50 nM) of miR-146b-3p mimic or negative control for 48 h, collected, and western blot performed with the indicated antibodies. (h) Putative binding site of miR-146b-3p in the CDS region of HPGD and mutagenesis of the target site in miR-146b-3p. (i) Western blot of HPGD expression in HeLa cells transfected with a negative control mimic (Neg. Ctrl; 20 nM), miR-146b-3p mimic (miR-146b-3p; 20 nM) or miR-146b-3p mutant mimic (miR-146b-3p mut; 20 nM) for 48 h. (j) Luciferase activity in HEK293T cells co-transfected with miR-146b-3p mimic (miR-146b-3p; 10 nM), miR-146b-3p mutant mimic (miR-146b-3p mut ; 10 nM) or a negative control (Neg. Ctrl; 10 nM) and the HPGD CDS reporter construct for 48 h. The quantified results are presented as means ± SD. Three independent experiments, each with four technical replicates, were performed. ** P < 0.01 for Student's t-test. n.s, not significant
miR-146b-3p inhibition attenuates cell proliferation, migration and anchorage-independent growth
miR-146b-3p expression was further examined in 23 paired clinical cervical cancer and normal tissues. miR-146b-3p levels were elevated in the majority of cervical cancer tissues (16/23) compared with paracancerous tissues (Fig. 4a). In situ hybridization confirmed the presence of higher amounts of miR-146b-3p in cervical cancer tissues (Fig. 4b). To further determine the potential contribution of miR-146b-3p to specific cell development processes, miR-146b-3p function was blocked with a specific inhibitor in SiHa cells and in cell colony formation, transwell migration and soft agar colony formation assays conducted. Our collective findings strongly suggest that miR-146b-3p inhibition leads to suppression of cell proliferation, migration and anchorage-independent growth (Fig. 4c–e).
Fig. 4. Inhibition of miR-146b-3p reduces cell proliferation, migration and anchorage-independent growth.

(a) Expression of miR-146b-3p in 23 pairs of cervical cancer and adjacent non-cancerous tissues determined via qPCR. (b) Fluorescence in situ hybridization for miR-146–3p in cervical cancer and adjacent non-cancerous tissues. (c) Plate colony formation assay of SiHa cells transfected with a miR-146b-3p inhibitor for 48 h. (d) Transwell migration assay for cells treated as in (c). (e) Soft agar colony formation assay of cells treated as in (c)
HPGD negatively regulates STAT3 and AKT signaling pathways
Several studies have demonstrated that abnormal activation of STAT3 and AKT signaling pathways promotes cervical cancer cell proliferation, migration and cell transformation35–37. Accordingly, we focused on whether STAT3 and AKT pathways are involved in HPGD-mediated inhibition of cell proliferation, migration and anchorage-independent growth. Overexpression of HPGD induced a decrease in phosphorylated STAT3 and AKT levels (Fig. 5a and 6). Consistently, miR-146b-3p inhibition suppressed STAT3 and AKT activation (Fig. 5b), indicating that HPGD negatively regulates STAT3 and AKT activities.
Fig. 5. HPGD negatively regulates STAT3 and AKT activities.
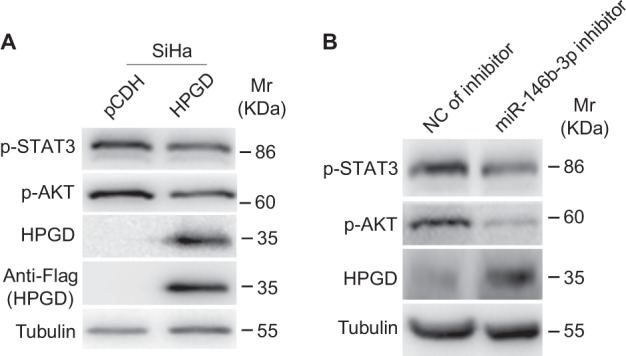
(a) Western blot of SiHa cells transfected with HPGD with the indicated antibodies. (b) Western blot of SiHa cells transduced with a miR-146b-3p inhibitor with the indicated antibodies
Fig. 6. HPV E6/E7 promotes miR-146b-3p-mediated inhibition of HPGD.

(a) Western blot analysis of HPGD protein in C33A, HeLa and SiHa cells. (b) qPCR showing miR-146b-3p expression in C33A, HeLa and SiHa cells. (c) qPCR showing E6/E7 expression in SiHa cells with E6/E7 knockdown. (d) qPCR showing miR-146b-3p expression in SiHa cells with E6/E7 knockdown. (e) Western blot analysis of SiHa cells with E6/E7 knockdown with the indicated antibodies
HPV E6/E7 promote miR-146b-3p-mediated inhibition of HPGD
To ascertain whether HPV infection plays a part in regulation of the miR-146b-3p/HPGD pathway, miR-146b-3p and HPGD expression patterns were examined in HPV-negative C33A, HPV18-positive HeLa and HPV16-positive SiHa cells. Our results showed a significant increase in miR-146b-3p and decrease in HPGD expression in HeLa and SiHa cells compared with C33A cells (Fig. 5a, b). The HPV oncoproteins E6 and E7 are the key factors that maintain the malignant phenotype of HPV-positive cancer cells38. To establish whether E6 and E7 exert regulatory effects on miR-146b-3p, these oncogenes were depleted with a mixture of siRNAs in SiHa cells (Fig. 5c), which induced a reduction in the miR-146b-3p level (Fig. 5d) and elevation of HPGD expression (Fig. 5e).
Discussion
The HPGD gene located on chromosome 4 encodes a protein that is widely distributed in various mammalian tissues39. HPGD was initially characterized as a key enzyme responsible for biological inactivation of prostaglandins25 and subsequently shown to be involved in a number of human malignancies22,40–42. miR-620 is reported to up-regulate PGE2 expression by directly targeting HPGD, contributing to tumor radiation resistance43. An earlier study by He et al. demonstrated that miR-21 participates in tumorigenesis via targeting the HPGD/PGE2 signaling pathway44. Moreover, omega-3 polyunsaturated fatty acids (ω-3 PUFA) induce HPGD expression through inhibition of miR-26a and miR-26b, leading to suppression of human cholangiocarcinoma cell growth45. HPGD has been shown to regulate the 15-keto-PGE2/peroxisome proliferator-activated receptor-γ/p21 (WAF1/Cip1) signaling pathway to suppress hepatocellular carcinoma growth46. However, the role of HPGD in cancer biology has not been adequately addressed to date. Data from the current study disclosed that HPGD expression is remarkably down-regulated in cervical cancer tissues and its overexpression suppresses cell proliferation, migration and anchorage-independent growth. Our findings support the potential value of HPGD as a prognostic and therapeutic target for cervical cancer management.
miRNAs (18–25 nucleotides in length) act as negative regulators of gene expression by binding mRNA sequences to inhibit translation or destabilization of transcripts47. Accumulating evidence has shown that aberrant expression of specific miRNAs contributes to progression of multiple tumors, including cervical cancer48. For example, miR-494 is up-regulated in cervical cancer cells and promotes proliferation by directly targeting phosphatase and tensin homolog (PTEN)49. Conversely, miR-342–3p acts as a tumor suppressor and inhibits proliferation, migration and invasion in cervical cell lines through effects on FOXM150. miR-491–5p is additionally down-regulated in cervical cancer tissues and suppresses cancer cell growth by targeting human telomerase reverse transcriptase (hTERT)51. On the other hand, miR-146b-3p is involved in progression and development of inflammation and arterial thrombosis52. Riesco-Eizaguirre et al. revealed abundant up-regulation of miR-146b-3p in papillary thyroid carcinoma, suggestive of its involvement in tumor progression53. In our investigation, frequent up-regulation of miR-146b-3p was evident in cervical cancer tissues and HPGD identified as its direct target. Moreover, suppression of miR-146b-3p inhibited cervical cancer cell proliferation, migration and anchorage-independent growth. The collective results indicate a critical role of miR-146b-3p in the pathogenesis of cervical cancer and support its application in tumor therapy.
Cervical cancer is responsible for 10–15% of cancer-related deaths in women worldwide, with HPV being the causative agent in over 99% cases. Although vaccines for HPVs and improvements in early screening have successfully reduced the mortality rate, cervical cancer is still considered a major public health problem in developing countries1,54. E6 and E7 encoded by high-risk HPVs are primary transforming viral proteins that regulate various cellular functions, such as proliferation, cell cycle, genomic instability and apoptosis55. Here, we further showed that knockdown of E6 and E7 trigger a decrease in miR-146b-3p and concomitant increase in HPGD. However, the precise underlying mechanisms remain to be established.
In conclusion, down-regulation of HPGD is associated with up-regulation of miR-146b-3p in cervical cancer. Moreover, overexpression of HPGD inhibits proliferation, migration and anchorage-independent growth in cervical cancer cells by inhibiting STAT3 and AKT activation. Our results clearly suggest that the miR-146b-3p/HPGD axis could serve as a promising prognostic and therapeutic target for cervical cancer management.
Materials and Methods
Cell culture transfection, reagents, and plasmids
HeLa, SiHa, C33A, and HEK293T cells were maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum at 37 °C with 5% CO2 in a humidified atmosphere. All cells were transfected using Lipofectamine 2000 Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. siRNAs, miRNA mimics and mutant miRNAs were synthesized by GenePharma (Suzhou, China) and miRNA inhibitors purchased from RiboBio (Guangzhou, China). The siRNA sequences are listed in Table 1. The respective HPGD 3’ UTR and HPGD CDS sequences were amplified via PCR and inserted into pGL3-Control plasmid (Promega, Madison, WI, USA). The 3 × Flag-HPGD construct was generated by inserting coding sequences into the lentiviral transferring plasmid pHAGE-CMV-MCSIzsGreen.
Table 1.
Sequences of siRNAs
| Gene | siRNA No. | Sequence of siRNA (5’ to 3’) |
|---|---|---|
| HPV16 E6/E7 | si1 | CCGGACAGAGCCCAUUACA |
| si2 | CACCUACAUUGCAUGAAUA | |
| si3 | CAACUGAUCUCUACUGUUA |
Antibodies and western blotting
Anti-Flag, anti-phospho-AKT and anti-phospho-STAT3 antibodies were obtained from Cell Signaling Technologies (Beijing, China). Anti-GAPDH, anti-HPGD, anti-HPV16 E7 and anti-α-tubulin antibodies were acquired from Santa Cruz Biotechnology (Dallas, TX, USA). Western blot analysis was conducted as described previously56,57.
Cell migration and plate colony formation assays
Cell migration and colony formation assays were conducted as described previously58–60.
Soft agar assay
Cells (1 × 104) suspended in 0.4% agarose (BD Biosciences) solution mixed with culture medium were seeded in six-well plates consisting of 0.8% agarose mixed with culture medium. Plates were incubated at 37 °C in the presence of 5% CO2 for 14 days. Five random fields were selected, photographed and cells were counted using NIH Image J software.
Luciferase reporter assay
The luciferase reporter assay was conducted using the Promega dual-luciferase reporter assay system as described previously61.
Reverse transcription and real-time quantitative PCR
RNA was extracted from cells using the RNA Isolator Total RNA Extraction Reagent (Vazyme Biotech Co., Ltd, Nanjing, China). Total RNA was reverse-transcribed with HiScript Q RT SuperMix (Vazyme Biotech Co., Ltd, Nanjing, China). Real-time quantitative PCR was performed using the AceQ qPCR SYBR Green Master Mix (Vazyme Biotech Co., Ltd, Nanjing, China). The primer sequences for PCR are presented in Table 2.
Table 2.
Sequences of specific primers for qPCR (F, Forward; R, Reverse)
| Target | Application | Primer |
|---|---|---|
| HPV16 E6/E7 | RT-qPCR | F: 5′- CAATGTTTCAGGACCCACAGG -3′ R: 5′- CTCACGTCGCAGTAACTGTTG -3′ |
| HPGD | RT-qPCR | F: 5′- CTCTGTTCATCCAGTGCGAT -3′ R: 5′- TCACTCCAGCATTATTGACCA -3′ |
| ACLY | RT-qPCR | F: 5′- CTCCTCTGCTCGATTATGCACT -3′ R: 5′- CTCCCGAGTAAAGGACCCACA -3′ |
| FAS | RT-qPCR | F: 5′- CGCCCACCTACGTACTGGCCTA -3′ R: 5′- GCTCCATGTCCGTGAACTGCT -3′ |
| ELOVL6 | RT-qPCR | F: 5′- TCTTCAGTATATTCGGTGCTC -3′ R: 5′- CTTAGCACAAATGCATAAGCC -3′ |
| SCD1 | RT-qPCR | F: 5′- CTACCTGCAAGTTCTACACC -3′ R: 5′- CAATGATCAGAAAGAGCCGTA -3′ |
| β-actin | RT-qPCR | F: 5′- TTGCCGACAGGATGCAGAAGGA -3′ R: 5′- AGGTGGACAGCGAGGCCAGGAT -3′ |
Immunohistochemistry (IHC)
Cervical cancer and normal tissue specimens provided by different hospitals were subjected to hematoxylin and eosin (H&E) and immunohistochemical staining. All human participants provided informed consent and all samples were anonymized. IHC was performed with specific antibodies as described previously60,62.
Fluorescence in situ hybridization
Fluorescence in situ hybridization was performed as described in a previous report63. The sequences of the miR-146b-3p and control probes were 5′- ACCAGAACTGAGTCCACAGGGC-3′ and 5′- ACCACA ACTGAGTCCAGAGGGC -3′, respectively. The 5′ and 3′ ends of the probes were modified with digoxigenin.
Statistical Analysis
All experiments were performed at least in triplicate, unless otherwise specified. Data are presented as means ± SD. Statistical analyses were conducted using the two-sided Student’s t-test. P values were calculated, and results considered statistically significant at P < 0.05.
Electronic supplementary material
Acknowledgements
We are grateful to members from Dr. Lu laboratory for helpful discussion. This work was supported by grants from National Natural Science Foundation of China (81730062 and 81761128003 to C. L.), the Start-up Fund Project of Nanjing Medical University (KY101RC1710 to C. L.), Application Research Project of Public Welfare Technology of Zhejiang Province (2016C33239 to S. Y.), Quzhou Science and Technology Bureau (2017G14 to S. Y.) and Science and Technology Development of Foundation of Nanjing Medical University (20182310 to W. L.).
Author Contributions
Conceived and designed the experiments: Chun Lu, Wan Li and Weifei Fan. Performed the experiments: Shuihong Yao, Jingyun Xu, Kaixuan Zhao and Pengxia Song. Provided the reagents: Qin Yan. Analyzed the data: Shuihong Yao and Wan Li. Wrote the paper: Wan Li and Chun Lu.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Shuihong Yao, Jingyun Xu and Kaixuan Zhao
Contributor Information
Weifei Fan, Phone: 86-25-83712838, Email: weifeifan1503@163.com.
Wan Li, Phone: 86-25-86869399, Email: wanli@njmu.edu.cn.
Chun Lu, Phone: 86-25-86869399, Email: clu@njmu.edu.cn.
Electronic supplementary material
Supplementary Information accompanies this paper at (10.1038/s41419-018-1059-y).
References
- 1.Small W, Jr., et al. Cervical cancer: A global health crisis. Cancer. 2017;123:2404–2412. doi: 10.1002/cncr.30667. [DOI] [PubMed] [Google Scholar]
- 2.Canavan TP, Doshi NR. Cervical cancer. Am. Fam. Physician. 2000;61:1369–1376. [PubMed] [Google Scholar]
- 3.Lin L, Liu Y, Zhao W, Sun B, Chen Q. Wnt5A expression is associated with the tumor metastasis and clinical survival in cervical cancer. Int. J. Clin. Exp. Pathol. 2014;7:6072–6078. [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng L, et al. Oncogene ATAD2 promotes cell proliferation, invasion and migration in cervical cancer. Oncol. Rep. 2015;33:2337–2344. doi: 10.3892/or.2015.3867. [DOI] [PubMed] [Google Scholar]
- 5.Kim HS, et al. Sphingosine kinase 1 is a reliable prognostic factor and a novel therapeutic target for uterine cervical cancer. Oncotarget. 2015;6:26746–26756. doi: 10.18632/oncotarget.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao Q, et al. EphB2 promotes cervical cancer progression by inducing epithelial-mesenchymal transition. Hum. Pathol. 2014;45:372–381. doi: 10.1016/j.humpath.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 7.Wang HY, Lian P, Zheng PSSOX9. a potential tumor suppressor in cervical cancer, transactivates p21WAF1/CIP1 and suppresses cervical tumor growth. Oncotarget. 2015;6:20711–20722. doi: 10.18632/oncotarget.4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qin Y, Tang X, Liu M. Tumor-Suppressor Gene NBPF1 Inhibits Invasion and PI3K/mTOR Signaling in Cervical Cancer Cells. Oncol. Res. 2016;23:13–20. doi: 10.3727/096504015X14410238486766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munoz N, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003;348:518–527. doi: 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- 10.Bosch FX, et al. Prevalence of human papillomavirus in cervical cancer: a worldwide perspective. International biological study on cervical cancer (IBSCC) Study Group. J. Natl. Cancer. Inst. 1995;87:796–802. doi: 10.1093/jnci/87.11.796. [DOI] [PubMed] [Google Scholar]
- 11.Ferlay J, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. 2015;136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 12.Woodman CB, Collins SI, Young LS. The natural history of cervical HPV infection: unresolved issues. Nat. Rev. Cancer. 2007;7:11–22. doi: 10.1038/nrc2050. [DOI] [PubMed] [Google Scholar]
- 13.Gupta S, Kumar P, Das BC. HPV: Molecular pathways and targets. Curr. Probl. Cancer. 2018;42:161–174. doi: 10.1016/j.currproblcancer.2018.03.003. [DOI] [PubMed] [Google Scholar]
- 14.zur Hausen H. Papillomaviruses and cancer: from basic studies to clinical application. Nat. Rev. Cancer. 2002;2:342–350. doi: 10.1038/nrc798. [DOI] [PubMed] [Google Scholar]
- 15.Brisson J, et al. Risk factors for cervical intraepithelial neoplasia: differences between low- and high-grade lesions. Am. J. Epidemiol. 1994;140:700–710. doi: 10.1093/oxfordjournals.aje.a117318. [DOI] [PubMed] [Google Scholar]
- 16.Millikan RC. Epidemiologic evidence showing that human papillomavirus infection causes most cervical intraepithelial neoplasia. J. Natl. Cancer. Inst. 1994;86:392–393. doi: 10.1093/jnci/86.5.392. [DOI] [PubMed] [Google Scholar]
- 17.Doorbar J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. 2006;110:525–541. doi: 10.1042/CS20050369. [DOI] [PubMed] [Google Scholar]
- 18.Moreno V, et al. Effect of oral contraceptives on risk of cervical cancer in women with human papillomavirus infection: the IARC multicentric case-control study. Lancet. 2002;359:1085–1092. doi: 10.1016/S0140-6736(02)08150-3. [DOI] [PubMed] [Google Scholar]
- 19.Smith JS, et al. Herpes simplex virus-2 as a human papillomavirus cofactor in the etiology of invasive cervical cancer. J. Natl. Cancer. Inst. 2002;94:1604–1613. doi: 10.1093/jnci/94.21.1604. [DOI] [PubMed] [Google Scholar]
- 20.Wyatt SW, Lancaster M, Bottorff D, Ross F. History of tobacco use among Kentucky women diagnosed with invasive cervical cancer: 1997-1998. J. Ky. Med. Assoc. 2001;99:537–539. [PubMed] [Google Scholar]
- 21.Yan M, et al. 15-Hydroxyprostaglandin dehydrogenase, a COX-2 oncogene antagonist, is a TGF-beta-induced suppressor of human gastrointestinal cancers. Proc..Natl. Acad. Sci. U.S.A. 2004;101:17468–17473. doi: 10.1073/pnas.0406142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gee JR, Montoya RG, Khaled HM, Sabichi AL, Grossman HB. Cytokeratin 20, AN43, PGDH, and COX-2 expression in transitional and squamous cell carcinoma of the bladder. Urol. Oncol. 2003;21:266–270. doi: 10.1016/S1078-1439(02)00271-5. [DOI] [PubMed] [Google Scholar]
- 23.Ding Y, Tong M, Liu S, Moscow JA, Tai HH. NAD + -linked 15-hydroxyprostaglandin dehydrogenase (15-PGDH) behaves as a tumor suppressor in lung cancer. Carcinogenesis. 2005;26:65–72. doi: 10.1093/carcin/bgh277. [DOI] [PubMed] [Google Scholar]
- 24.Kawahara K, Hohjoh H, Inazumi T, Tsuchiya S, Sugimoto Y. Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors. Biochim. Biophys. Acta. 2015;1851:414–421. doi: 10.1016/j.bbalip.2014.07.008. [DOI] [PubMed] [Google Scholar]
- 25.Tai HH, Cho H, Tong M, Ding Y. NAD + -linked 15-hydroxyprostaglandin dehydrogenase: structure and biological functions. Curr. Pharm. Des. 2006;12:955–962. doi: 10.2174/138161206776055958. [DOI] [PubMed] [Google Scholar]
- 26.Okita RT, Okita JR. Prostaglandin-metabolizing enzymes during pregnancy: characterization of NAD( + )-dependent prostaglandin dehydrogenase, carbonyl reductase, and cytochrome P450-dependent prostaglandin omega-hydroxylase. Crit. Rev. Biochem. Mol. Biol. 1996;31:101–126. doi: 10.3109/10409239609106581. [DOI] [PubMed] [Google Scholar]
- 27.Anggard E. The biological activities of three metabolites of prostaglandin E 1. Acta Physiol. Scand. 1966;66:509–510. doi: 10.1111/j.1748-1716.1966.tb03231.x. [DOI] [PubMed] [Google Scholar]
- 28.Kankofer M. The enzymes responsible for the metabolism of prostaglandins in bovine placenta. Prostaglandins Leukot. Essent. Fatty Acids. 1999;61:359–362. doi: 10.1054/plef.1999.0109. [DOI] [PubMed] [Google Scholar]
- 29.Backlund MG, et al. 15-Hydroxyprostaglandin dehydrogenase is down-regulated in colorectal cancer. J. Biol. Chem. 2005;280:3217–3223. doi: 10.1074/jbc.M411221200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Myung SJ, et al. 15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc..Natl. Acad. Sci. U.S.A. 2006;103:12098–12102. doi: 10.1073/pnas.0603235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan M, et al. 15-Hydroxyprostaglandin dehydrogenase inactivation as a mechanism of resistance to celecoxib chemoprevention of colon tumors. Proc..Natl. Acad. Sci. U.S.A. 2009;106:9409–9413. doi: 10.1073/pnas.0902367106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roberts HR, et al. Colon tumour cells increase PGE(2) by regulating COX-2 and 15-PGDH to promote survival during the microenvironmental stress of glucose deprivation. Carcinogenesis. 2011;32:1741–1747. doi: 10.1093/carcin/bgr210. [DOI] [PubMed] [Google Scholar]
- 33.Chan AT, Ogino S, Fuchs CS. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N. Engl. J. Med. 2007;356:2131–2142. doi: 10.1056/NEJMoa067208. [DOI] [PubMed] [Google Scholar]
- 34.Kraus S, Naumov I, Arber N. COX-2 active agents in the chemoprevention of colorectal cancer. Recent results in cancer research Fortschritte der Krebsforschung Progres dans les recherches sur le cancer. 2013;191:95–103. doi: 10.1007/978-3-642-30331-9_5. [DOI] [PubMed] [Google Scholar]
- 35.Zhang D, Sun G, Zhang H, Tian J, Li Y. Long non-coding RNA ANRIL indicates a poor prognosis of cervical cancer and promotes carcinogenesis via PI3K/Akt pathways. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2017;85:511–516. doi: 10.1016/j.biopha.2016.11.058. [DOI] [PubMed] [Google Scholar]
- 36.Zhao H, et al. BCL3 exerts an oncogenic function by regulating STAT3 in human cervical cancer. Onco. Targets Ther. 2016;9:6619–6629. doi: 10.2147/OTT.S118184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, et al. Down-regulation of microRNA-9 leads to activation of IL-6/Jak/STAT3 pathway through directly targeting IL-6 in HeLa cell. Mol. Carcinog. 2016;55:732–742. doi: 10.1002/mc.22317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoppe-Seyler K, Bossler F, Braun JA, Herrmann AL, Hoppe-Seyler F. The HPV E6/E7 Oncogenes: Key Factors for Viral Carcinogenesis and Therapeutic Targets. Trends Microbiol. 2018;26:158–168. doi: 10.1016/j.tim.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 39.Anggard E, Larsson C, Samuelsson B. The distribution of 15-hydroxy prostaglandin dehydrogenase and prostaglandin-delta 13-reductase in tissues of the swine. Acta Physiol. Scand. 1971;81:396–404. doi: 10.1111/j.1748-1716.1971.tb04914.x. [DOI] [PubMed] [Google Scholar]
- 40.Rao CV, et al. Enhancement of experimental colon cancer by genistein. Cancer Res. 1997;57:3717–3722. [PubMed] [Google Scholar]
- 41.Tong M, Tai HH. Induction of NAD( + )-linked 15-hydroxyprostaglandin dehydrogenase expression by androgens in human prostate cancer cells. Biochem. Biophys. Res. Commun. 2000;276:77–81. doi: 10.1006/bbrc.2000.3437. [DOI] [PubMed] [Google Scholar]
- 42.Quidville V, et al. Tumor growth inhibition by indomethacin in a mouse model of human medullary thyroid cancer: implication of cyclooxygenases and 15-hydroxyprostaglandin dehydrogenase. Endocrinology. 2004;145:2561–2571. doi: 10.1210/en.2003-0915. [DOI] [PubMed] [Google Scholar]
- 43.Huang X, et al. miR-620 promotes tumor radioresistance by targeting 15-hydroxyprostaglandin dehydrogenase (HPGD) Oncotarget. 2015;6:22439–22451. doi: 10.18632/oncotarget.4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He Q, et al. MicroRNA-21 regulates prostaglandin E2 signaling pathway by targeting 15-hydroxyprostaglandin dehydrogenase in tongue squamous cell carcinoma. BMC. Cancer. 2016;16:685. doi: 10.1186/s12885-016-2716-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yao L, et al. Omega-3 Polyunsaturated Fatty Acids Upregulate 15-PGDH Expression in Cholangiocarcinoma Cells by Inhibiting miR-26a/b Expression. Cancer Res. 2015;75:1388–1398. doi: 10.1158/0008-5472.CAN-14-2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu D, Han C, Wu T. 15-PGDH inhibits hepatocellular carcinoma growth through 15-keto-PGE2/PPARgamma-mediated activation of p21WAF1/Cip1. Oncogene. 2014;33:1101–1112. doi: 10.1038/onc.2013.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang YK, et al. MicroRNA-494 promotes cervical cancer proliferation through the regulation of PTEN. Oncol. Rep. 2015;33:2393–2401. doi: 10.3892/or.2015.3821. [DOI] [PubMed] [Google Scholar]
- 49.Ventura A, Jacks T. MicroRNAs and cancer: short RNAs go a long way. Cell. 2009;136:586–591. doi: 10.1016/j.cell.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li XR, et al. miR-342-3p suppresses proliferation, migration and invasion by targeting FOXM1 in human cervical cancer. FEBS Lett. 2014;588:3298–3307. doi: 10.1016/j.febslet.2014.07.020. [DOI] [PubMed] [Google Scholar]
- 51.Zhao Q, Zhai YX, Liu HQ, Shi YA, Li XB. MicroRNA-491-5p suppresses cervical cancer cell growth by targeting hTERT. Oncol. Rep. 2015;34:979–986. doi: 10.3892/or.2015.4013. [DOI] [PubMed] [Google Scholar]
- 52.Fulzele S, et al. MicroRNA-146b-3p regulates retinal inflammation by suppressing adenosine deaminase-2 in diabetes. Biomed. Res. Int. 2015;2015:846501. doi: 10.1155/2015/846501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riesco-Eizaguirre G, et al. The miR-146b-3p/PAX8/NIS Regulatory Circuit Modulates the Differentiation Phenotype and Function of Thyroid Cells during Carcinogenesis. Cancer Res. 2015;75:4119–4130. doi: 10.1158/0008-5472.CAN-14-3547. [DOI] [PubMed] [Google Scholar]
- 54.Casarin MR, Piccoli Jda C. [Education in health for prevention of uterine cervical cancer in women in Santo Angelo, state of Rio Grande do Sul, Brazil] Cien. Saude. Colet. 2011;16:3925–3932. doi: 10.1590/S1413-81232011001000029. [DOI] [PubMed] [Google Scholar]
- 55.Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat. Rev. Cancer. 2010;10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 56.Zeng Y, et al. Intracellular Tat of human immunodeficiency virus type 1 activates lytic cycle replication of Kaposi's sarcoma-associated herpesvirus: role of JAK/STAT signaling. J. Virol. 2007;81:2401–2417. doi: 10.1128/JVI.02024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qin D, et al. Induction of lytic cycle replication of Kaposi's sarcoma-associated herpesvirus by herpes simplex virus type 1: involvement of IL-10 and IL-4. Cell Microbiol. 2008;10:713–728. doi: 10.1111/j.1462-5822.2007.01079.x. [DOI] [PubMed] [Google Scholar]
- 58.Li W, et al. A viral microRNA downregulates metastasis suppressor CD82 and induces cell invasion and angiogenesis by activating the c-Met signaling. Oncogene. 2017;36:5407–5420. doi: 10.1038/onc.2017.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yao S, et al. MiRNA-891a-5p mediates HIV-1 Tat and KSHV Orf-K1 synergistic induction of angiogenesis by activating NF-kappaB signaling. Nucleic Acids Res. 2015;43:9362–9378. doi: 10.1093/nar/gkv988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xue M, et al. HIV-1 Nef and KSHV oncogene K1 synergistically promote angiogenesis by inducing cellular miR-718 to regulate the PTEN/AKT/mTOR signaling pathway. Nucleic Acids Res. 2014;42:9862–9879. doi: 10.1093/nar/gku583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li W, et al. The SH3BGR/STAT3 Pathway Regulates Cell Migration and Angiogenesis Induced by a Gammaherpesvirus MicroRNA. PLoS. Pathog. 2016;12:e1005605. doi: 10.1371/journal.ppat.1005605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hu M, et al. A KSHV microRNA Directly Targets G Protein-Coupled Receptor Kinase 2 to Promote the Migration and Invasion of Endothelial Cells by Inducing CXCR2 and Activating AKT Signaling. PLoS. Pathog. 2015;11:e1005171. doi: 10.1371/journal.ppat.1005171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li J, et al. Cell-specific detection of miR-375 downregulation for predicting the prognosis of esophageal squamous cell carcinoma by miRNA in situ hybridization. PLoS. ONE. 2013;8:e53582. doi: 10.1371/journal.pone.0053582. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.