

Abstract
To successfully colonize the intestine, bacteria must survive passage through the stomach. The permeability of the outer membrane renders the periplasm of Gram-negative bacteria vulnerable to stomach acid, which inactivates proteins. Here we report that the semipermeable nature of the outer membrane allows the development of a strong Donnan equilibrium across this barrier at low pH. As a result, when bacteria are exposed to conditions that mimic gastric juice, periplasmic chloride concentrations rise to levels that exceed 0.6 M. At these chloride concentrations proteins readily aggregate in vitro. The acid sensitivity of strains lacking acid-protective chaperones is enhanced by chloride, which suggests that these chaperones protect periplasmic proteins both from acidification and the accompanying accumulation of chloride. These results illustrate how organisms have evolved chaperones to respond to the substantial chemical threat imposed by otherwise innocuous chloride concentrations that are amplified to proteotoxic levels by low pH-induced Donnan equilibrium effects.
INTRODUCTION
Enteric bacteria, such as Escherichia coli, colonize the intestines of warm-blooded animals. To gain access to the intestinal tract, the bacteria must first transit the highly acidic environment of the stomach. Under fasting conditions, the pH of human gastric fluid is ~2.1,2 This low pH unfolds ingested proteins, making them susceptible to pepsin-mediated protein digestion; it also acts as an important barrier to infection by food-borne pathogens.1,3 The cytosol of E. coli, however, is protected from acidification by the relative impermeability of the inner membrane and through several amino acid-based decarboxylation systems that consume protons.4 In contrast, the outer membrane allows the free diffusion of molecules below ~600 Da via outer membrane porins,5 exposing the periplasm to stomach acid. Periplasmic proteins are thus subject to the powerful denaturant effects of acidification;6 acid-denatured proteins have been shown to form insoluble aggregates,7–12 rendering them non-functional.
The E. coli periplasmic proteome is protected from acid stress by HdeA and HdeB, two structurally related molecular chaperones that are important for the organism’s survival during acute acid stress. HdeA and HdeB bind to proteins that become unfolded under acid stress conditions, keeping them in a soluble form until the external pH approaches neutrality, as occurs when E. coli moves from the stomach to the intestines. Both chaperones exist as well-folded, chaperone-inactive dimers at neutral pH. Upon exposure to pH 2, the HdeA dimer rapidly converts into partially disordered, chaperone-active monomers.9 The HdeA monomers bind to proteins unfolded by the acid, effectively preventing them from aggregating.9 Upon return to neutral pH, HdeA slowly releases the bound proteins, allowing them to refold while minimizing the concentration of aggregation prone intermediates.13 HdeA then converts back into its chaperone-inactive dimer. HdeA and HdeB have non-overlapping pH optima for their chaperone activities, with HdeA operating during extreme acid stress (pH 1–3) and HdeB functioning under milder acid stress (pH 3–5).14 Unlike HdeA, HdeB remains dimeric even at the moderately acidic conditions where it is most active (pH 3–5). The HdeB dimer does, however, undergo a conformational rearrangement and its intrinsic dynamics change between neutral pH and pH 4; these changes might serve to activate HdeB’s chaperone function.14,15
Although it is has been assumed that periplasmic proteins aggregate under gastric fluid conditions,7–12 this supposition has not been directly verified. Surprisingly, we find that under in vitro conditions that mimic the pH and salt concentrations of gastric fluid, proteins remain highly soluble even though they are unfolded. However, we also find that the ionic conditions measured in the periplasm are radically different from those in gastric fluid. Chloride concentrations in the periplasm are at least 4-fold higher than those in the media, which we attribute to the development of a strong Donnan equilibrium at low pH. Under these high chloride concentrations, proteins rapidly aggregate in vitro. In vivo, however, they are protected from aggregation by HdeA and HdeB – apparently these chaperones are capable of protecting cells from the Donnan effect. Our findings show that enteric bacteria like E. coli have evolved mechanisms to protect proteins from the dual threat of acidification and the resulting massive accumulation of chloride in the periplasm that occurs when transiting the stomach.
RESULTS
Anions Cause Irreversible Protein Aggregation at Low pH
Biochemists frequently select buffer conditions that are optimal for the activity of their particular protein. The composition of these buffers varies widely and often contains ions that are not abundant within cells. In contrast, cells exert considerable effort to maintain pH and ion concentrations within very narrow ranges. As an example of the use of non-physiological buffers, prior in vitro studies on the chaperone activity of HdeA and HdeB, including our own studies, used buffers containing ammonium sulfate at concentrations ranging from 100–200 mM.6–11,14 However, both ammonium and sulfate ions are not present at anywhere near these concentrations in gastric fluid.16 Apparent disconnects between in vivo and in vitro buffer conditions are not unusual; scientists tend to use whatever buffer works for their particular system, often with little justification. The apparent disconnect was so large in our system that we decided to investigate the matter further. We thus tested in vitro protein aggregation and the action of these chaperones under buffer conditions that would seem to be more physiologically relevant. Human gastric fluid has a pH of ~2 (equivalent to 10 mM HCl) and contains, at most, 150 mM chloride with both sodium and potassium serving as counterions.16 We were surprised to find that in a buffer that simulates gastric fluid (10 mM HCl pH 2, 150 mM NaCl), the periplasmic protein maltose binding protein (MBP) fails to aggregate at all (Fig. 1a). MBP can even be concentrated to ~170 mg/mL without visible precipitation in this buffer. MBP at pH 2 is entirely monomeric but is still unfolded as judged by analytical ultracentrifugation and circular dichroism (Supplementary Fig. 1a, 1b). This complete lack of aggregation at pH 2 in the absence of ammonium sulfate is apparently a general phenomenon for proteins; exposing entire E. coli periplasmic extracts to pH 2 in buffers containing 150 mM ammonium sulfate induces widespread aggregation of most of the proteins in the extract, whereas if ammonium sulfate is omitted from the buffer, periplasmic proteins remain entirely soluble (Fig. 1b). Addition of HdeA suppresses the aggregation that occurs in ammonium sulfate at low pH. The aggregates of MBP that form at pH 2 in the presence of ammonium sulfate are irreversible; dialyzing the aggregates against neutral pH buffer without ammonium sulfate does not resolubilize the aggregates. Exchanging the ammonium cation for sodium had no impact on the aggregation of MBP at low pH, whereas exchanging the sulfate anion for chloride completely suppressed the aggregation (Fig. 1c). This indicates that the sulfate anion is largely responsible for inducing the aggregation of MBP at pH 2.
Figure 1. Sulfate Promotes the Acid-Induced Aggregation of Proteins.
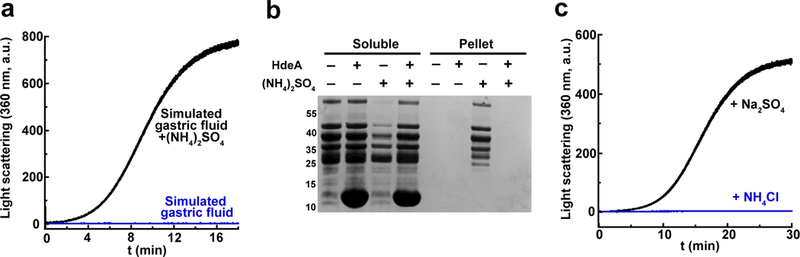
(a) 20 μM MBP was incubated in 10 mM HCl, pH 2, 150 mM NaCl (simulated gastric fluid) in the absence or presence of 150 mM ammonium sulfate. Aggregation, or lack thereof, was monitored over time by light scattering at 360 nm.
(b) SDS-PAGE analysis of the soluble and pellet fractions of 3 mg/mL periplasmic proteins from MG1655 E. coli that were incubated in simulated gastric fluid with or without ammonium sulfate, and in the absence or presence of 6 mg/mL purified HdeA at 37ºC for 1 hr. An uncropped gel can be seen in Supplementary Fig. 6.
(c) In vitro aggregation assays of 10 μM MBP in simulated gastric fluid supplemented with 150 mM sodium sulfate or ammonium chloride. These experiments were performed once.
Several reports have investigated the acid-induced folding of proteins.17–21 In these studies, addition of anions to proteins unfolded at low pH induced the formation of secondary structure in these proteins. This result was attributed to anions neutralizing the electrostatic repulsion that occurs between the positively charged basic amino acid side chains. Sulfate was found to be more potent than chloride in promoting the acid-induced folding. We wondered if the more biologically relevant chloride ion could also induce protein aggregation at low pH if it were present at concentrations higher than the 150 mM level found in gastric fluid. We therefore investigated the effect of sodium chloride on the in vitro aggregation at pH 2 of three periplasmic proteins from E. coli: MBP, OppA, and DppA (Fig. 2). All three proteins remained soluble below ~400 mM sodium chloride but aggregated extensively as the chloride concentration was raised above this threshold, indicating that chloride can indeed promote acid-induced protein aggregation if it is present at a high enough concentration. Using potassium chloride instead of sodium chloride produced similar results (Supplementary Fig. 1c). Periplasmic extracts showed extensive aggregation of most of the visible proteins when transferred into a pH 2 buffer that contains 500 mM sodium chloride, illustrating the generality of this chloride-dependent acid-induced aggregation (Supplementary Fig. 2a). Addition of HdeA completely suppressed the acid-induced aggregation of periplasmic proteins at high chloride concentrations. The same behavior occurs at higher periplasmic protein concentrations (30 mg/mL) and even occurs with cytosolic proteins (Supplementary Fig. 2b, 2c), indicating that HdeA is a general chaperone with far broader substrate specificity than previously reported.10,22–24
Figure 2. Chloride-Dependent Aggregation of Periplasmic Proteins.
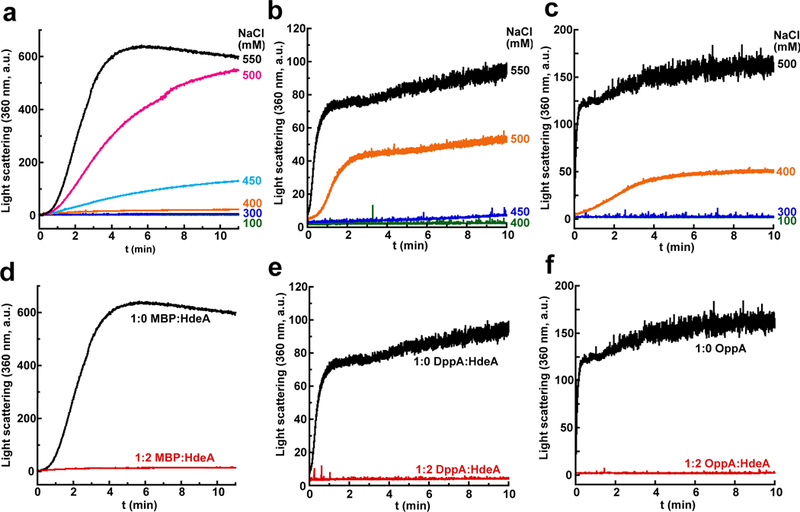
(a), (b), and (c) 20 μM MBP, 2 μM DppA, and 2 μM OppA, respectively, were incubated in 10 mM HCl, pH 2 containing various NaCl concentrations, and the amount of aggregation was monitored over time via light scattering at 360 nm.
(d)–(f) Addition of a two-fold molar excess of monomeric HdeA completely suppressed the aggregation of all three substrates at the highest chloride concentration tested. These experiments were performed once.
Chloride Accumulates Inside the Periplasm at pH 2
The chloride concentration in human gastric fluid ranges from 100–150 mM,25 yet perplexingly, our in vitro aggregation experiments indicate that proteins do not aggregate at these chloride concentrations at low pH. If this is the biologically relevant ion environment, then why are HdeA and HdeB even needed to prevent acid-mediated protein aggregation?
In an effort to resolve this apparent conundrum, we considered the fact that the periplasmic space is highly concentrated with various macromolecules (proteins, peptidoglycan, phospholipids) and that the presence of these macromolecules may substantially affect the solvent conditions present within the periplasm. Biological macromolecules contain numerous negatively charged carboxylic acid-based and positively charged amine-based functional groups at neutral pH. However, if the periplasm is plunged below pH ~4, the carboxylic acid groups will lose their negative charge due to protonation. Thus, macromolecules retained inside the periplasm will be positively charged. Given the prevalence of amine-based functional groups in biological macromolecules, we expect that these macromolecules will contain an abundance of positive charges at low pH. Since the outer membrane allows the free diffusion of ions smaller than ~600 Da, this expected abundance of trapped positively charged macromolecules inside the periplasm at low pH raises the possibility that anions from the gastric fluid could distribute unevenly across the membrane, accumulating within the periplasm to balance this positive charge – a phenomenon known as a Donnan equilibrium (Fig. 3a). Notably, a Donnan equilibrium across the outer membrane has already been observed at neutral pH, although at neutral pH there is an excess of negatively charged membrane-derived oligosaccharides inside the periplasm, which leads to accumulation of sodium ions within the periplasm.26,27 We were curious if a Donnan equilibrium could produce such an extensive accumulation of anions within the periplasm at low pH that it causes protein aggregation.
Figure 3. Acid Exposure Results in an Accumulation of Chloride Within the Periplasm Due to a Donnan Equilibrium.
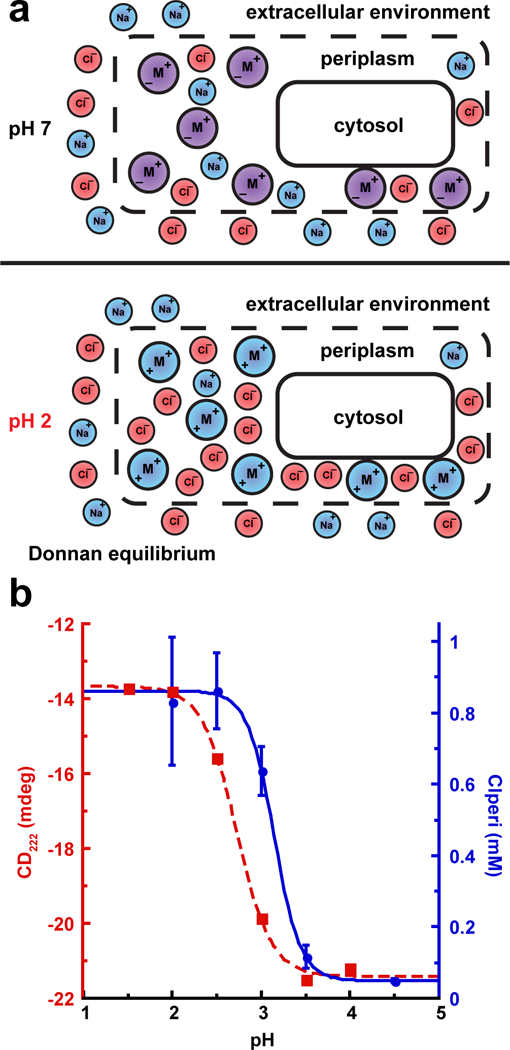
(a) Top: at neutral pH, the macromolecules (denoted as purple M circles) inside the periplasm contain a roughly even distribution of positively and negatively charged functional groups. At this pH, the concentration of sodium and chloride ions within the periplasm should be similar to that of the extracellular environment. Bottom: acid exposure protonates the acidic functional groups on the macromolecules. As a result, the macromolecules inside the periplasm only contain positive charges (denoted as blue M circles). The high density of positively charged macromolecules at low pH results in a Donnan equilibrium across the outer membrane, and chloride ions accumulate inside the periplasm above the extracellular concentration in order to maintain electroneutrality.
(b) The red squares show the secondary structure measured by circular dichroism of bulk proteins from periplasmic extracts as a function of pH. The blue circles show the measured Clperi values for the data in citrate buffer from Supplementary Table 1. The apparent pKa from fitting the pH dependent data was 2.7 ± 0.1 and 3.1 ± 0.1 for the CD and Clperi data, respectively. Values reported are the mean ± s.e.m. of the fit. n=3 independent samples and the error bars represent the mean ± s.d. for the Clperi measurements. Error bars for the CD data are too small to be visualized at this scale.
To measure the periplasmic chloride concentration at pH 2, we employed a solute partitioning method that uses several radiolabeled solutes that differ in their ability to penetrate into different compartments of E. coli.26–30 This technique allowed us to measure the absolute volume of the cytosol (Vcyto) and periplasm (Vperi) compartments of an E. coli sample, as well as the apparent volume of chloride within the periplasm (VClperi). The ratio of VClperi/Vperi then allows us to determine the fold change of the chloride concentration inside the periplasm relative to the chloride concentration in the external media (Clperi/Clo).26
We used this method to measure the volume of the different cellular compartments at pH 7 (as a reference) as well as at pH 2 under conditions that mimic those found in gastric fluid using potassium chloride (Table 1). We found that the periplasm shrinks at pH 2 relative to pH 7, constituting 28 ± 4% of the total cell volume at pH 2 versus 35 ± 5% at pH 7. Cryo-electron microscopy tilts demonstrated that the periplasm is visibly smaller at pH 2 than it is at pH 7 (Supplementary Video 1 and 2). Remarkably, however, the VClperi at pH 2 was 560% larger than VClperi at pH 7 (Table 1). The ratio of Clperi/Clo was determined to be 4.3 ± 0.3 when E. coli is exposed to conditions that mimic those present in gastric fluid, which corresponds to a ~0.69 M concentration of chloride inside the periplasm. This chloride concentration is well above the point at which proteins readily aggregate in vitro at pH 2 (Fig. 2). Periplasmic chloride accumulation was even more dramatic (Clperi/Clo of 8.3 ± 0.5) when cells were exposed to gastric fluid containing sodium chloride instead of potassium chloride (Supplementary Table 1). Bacteria use potassium, in part, to regulate osmotic pressure differences across the cellular envelope,31 possibly explaining the difference between measurements collected in a sodium versus potassium background at pH 2.
Table 1.
Compartment Volumes and Periplasmic Chloride for WT E. coli
| 20 mM KPO4, pH 7, 0.15 M KCl | 10 mM HCl, pH 2, 0.15 M KCl | |
|---|---|---|
| Vcell | 32.9 ± 1.4 | 38.5 ± 1.1 |
| Vcyto | 21.4 ± 1.3 | 27.7 ± 1.1 |
| Vperi | 11.5 ± 0.5 | 10.8 ± 0.4 |
| VClperi | 8.4 ± 0.6 | 47.0 ± 2.9 |
| [Clperi]/[Clo] | 0.7 ± 0.1 | 4.3 ± 0.3 |
| [Clperi] | 0.11 ± 0.02 M | 0.69 ± 0.05 M |
Values reported are the mean ± s.d. n = 3 independent samples
It is important to verify that the intracellular 36Cl in our pH 2 measurements is only in the periplasm. To test this, we performed rapid dilution-filtration experiments26 where E. coli pre-incubated with 36Cl was subject to a 100-fold dilution into unlabeled buffer followed by rapid filtration through a 0.45 um nitrocellulose filter. If the radiolabeled chloride was only present in the periplasm, it should rapidly diffuse out of the bacteria upon 100-fold dilution such that no radioactive chloride is retained in the bacteria collected by the filter. However, if the chloride entered the cytosol, it should not diffuse rapidly from the cells when diluted into the unlabeled buffer (since it would have to pass through inner membrane transporters) and should therefore be retained in the bacteria trapped on the filter. The amount of 36Cl retained in E. coli at low pH after rapid dilution-filtration was the same as the background, confirming that the chloride in our measurements was present in the periplasm – not in the cytosol.
Our prediction is that the accumulation of periplasmic chloride upon acid exposure is due to an abundance of positively charged proteins within the periplasm at low pH, which become positively charged due to protonation of their acidic side chains. If this is correct, the pH at which the transition occurs between when the periplasm repels chloride and when it begins accumulating chloride should occur at the average pKa of glutamate and aspartate carboxylate side chains, which for free amino acids is ~ 4. The local environment in folded proteins can alter this pKa; e.g., it will be lower than 4 when the carboxylate is involved in a salt bridge with a basic amino acid side chain. Protonation of glutamate and aspartate side chains also causes protein unfolding by disrupting salt bridges. If declining pH simultaneously causes both chloride accumulation and glutamate/aspartate protonation-mediated protein unfolding, both processes should parallel each other as the pH is lowered. As expected, we observed a correlation between the pH dependence of Clperi and in vitro unfolding of bulk periplasmic proteins (Fig. 3b). Thus, our results strongly suggest that chloride does indeed accumulate in the periplasm at low pH due to protonation of acidic protein side chains. Simple chemical considerations imply that this is likely due to the formation of a Donnan equilibrium.
Chloride Is Cytotoxic at Low pH to E. coli Lacking HdeA/B
Based on the observation that acid-induced protein aggregation depends on the chloride concentration, we predicted that the acid-induced lethality of HdeA/B null mutants should also be chloride dependent. We therefore exposed the WT E. coli strain MG1655 and an isogenic hdeAB mutant strain to a range of different chloride concentrations (10–500 mM) at pH 7 or pH 2 and measured their survival over time (Fig. 4a and Supplementary Fig. 3). As predicted, we found that the hdeAB null mutant strain is more chloride sensitive than the WT strain at pH 2, suggesting that HdeA/HdeB do indeed provide protection from chloride-induced protein aggregation at low pH. The known role of these acid-induced chaperones in protecting proteins from aggregation implies that they are acting by preventing cytotoxic protein aggregation that would otherwise occur inside the periplasm.
Figure 4. E. coli Lacking HdeA and HdeB Are More Sensitive to Acid Shock and Periplasmic Protein Aggregation in the Presence of Chloride.
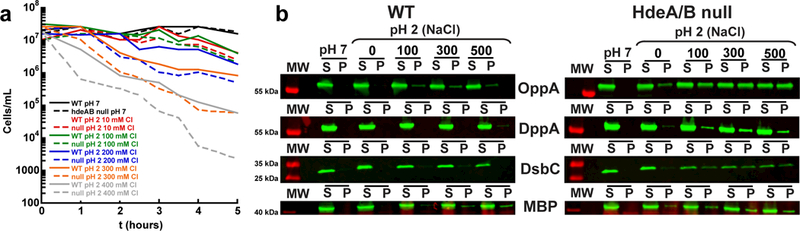
(a) Cell titers plotted against time for WT MG1655 E. coli or the hdeAB mutant MG1655 strain exposed to 20 mM NaPO4 pH 7, 150 mM NaCl, or 10 mM HCl, pH 2, 0.2 M sucrose with 0–400 mM NaCl in 100 mM increments at 37ºC. The 0.2 M sucrose was added to the pH 2 conditions to minimize the effect of osmotic stress on the cultures at low ionic strengths. The spotting assays used to determine the titer can be seen in Supplementary Fig. 3.
(b) Western blot analysis of the soluble (S) and pellet (P) fractions for four periplasmic proteins, OppA, DppA, DsbC and MBP, in WT or hdeAB null MG1655 E. coli that was exposed to 20 mM NaPO4 pH 7, 150 mM NaCl, or 10 mM HCl pH 2, 0.2 M sucrose with 0 M, 100 mM, 300 mM, or 500 mM NaCl. Uncropped blots can be seen in Supplementary Fig. 6. Each experiment was independently performed twice with similar results.
Chloride Causes Some Periplasmic Proteins to Aggregate at pH 2
We next investigated whether chloride induces the aggregation of proteins under acidic conditions in vivo. We exposed the WT and hdeAB mutant strains for one hour to conditions similar to those used in the survival assays and then transferred them to a neutral pH buffer. The neutral buffer halts the aggregation and prevents additional aggregation from occurring after lysis; aggregation is generally an irreversible process, particularly in the ATP-devoid environment of the periplasm. After lysing the cells through sonication, we used Western blotting to compare the amount of specific periplasmic proteins that remained soluble with the amount that aggregated in the pellet fraction. We chose this protocol over more standard periplasmic fractionation protocols because we reasoned that aggregates would fail to be released from the periplasm with the standard methods, which rely on selective permeabilization.32
The proteins we tested separated into three different categories. One class, which included OppA, DppA, DsbC and MBP, aggregated more extensively and in a chloride-dependent manner in the hdeAB mutant strain at low pH than in the WT strain (Fig. 4b). This result indicates that the aggregation of some proteins under acidic conditions in vivo is indeed dependent on the presence of chloride and that the acid-induced chaperones HdeA and HdeB likely play a role in protecting them. The second class, which included Skp, SurA, DegP and RBP, aggregated to a similar extent in both the mutant and the WT strain at pH 2, indicating that HdeA and HdeB did not protect them from chloride-induced aggregation (Supplementary Fig. 4a–4d). Finally, AppA did not exhibit any detectable chloride-dependent aggregation regardless of whether HdeA and HdeB were present, suggesting that this protein does not require these acid-responsive chaperones in order to stay soluble during acute acid stress (Supplementary Fig. 4e). Taken together, these results show that HdeA/HdeB can act to protect some proteins from chloride-mediated aggregation.
HdeA Fails to Refold Acid Denatured Client Proteins
As previously mentioned, most of the in vitro assays on HdeA’s chaperone activity were performed in the presence of sulfate. Experiments both from our lab and another lab showed that HdeA can improve the recovery of native substrate proteins from an acid-unfolded state during pH upshift from 2 to 7 (simulating passage from the stomach to the small intestine).10,13 These results were taken to indicate that HdeA plays an active role in restoring the native state of acid-unfolded substrates during pH neutralization. However, all of these experiments were performed with 0.1–0.2 M sulfate in the low pH buffer. Given our observation that sulfate induces the aggregation of proteins at low pH, another interpretation of these previously published results is that some of the substrate protein aggregated in the low pH buffer with sulfate prior to pH neutralization, eliminating the ability of the substrate to refold, and that HdeA prevented that aggregation at low pH. In other words, we wondered if the previously published experiments with sulfate might have been reporting on HdeA’s ability to prevent aggregation at low pH instead of an ability to actively refold proteins upon return to neutral pH. To test this model, we repeated the previously published malate dehydrogenase (MDH) refolding experiments13 in a condition in which MDH should not aggregate at all at low pH (8 mM phosphate buffer pH 2, 150 mM NaCl). If HdeA actually plays an active role in the refolding of MDH during pH neutralization as previously proposed, the yield of MDH activity should still increase over time in the presence of HdeA even under conditions where MDH does not aggregate at pH 2. However, if HdeA merely suppresses aggregation of MDH at low pH, HdeA should not affect the amount of active MDH recovered upon pH neutralization under these conditions. The results presented in Supplementary Fig. 5 point to the latter alternative: HdeA does not significantly enhance the yield of active MDH during refolding upon neutralization from a condition where MDH does not aggregate at low pH, indicating that HdeA is unable to assist the refolding of proteins during a pH upshift. HdeA only acts to prevent protein aggregation under acidic conditions.
DISCUSSION
Our experimental results indicate that chloride accumulates inside the periplasm of E. coli during acute acid stress to concentrations that exceed 0.6 M and that this build-up of chloride should, in chaperone-deficient cells, cause cytotoxic protein aggregation. Simple physical principles lead to this accumulation of chloride at low pH: the high concentration of positively charged macromolecules that are trapped inside the periplasmic compartment at low pH produces a Donnan equilibrium across the selectively permeable outer membrane. Anions accumulate in the periplasm in order to counterbalance the positively charged macromolecules trapped within this compartment at low pH, causing the proteins to aggregate. Enterobacteria have apparently responded to this threat by evolving the acid-activated chaperones HdeA and HdeB to protect their periplasmic proteins from the Donnan-mediated chloride accumulation that occurs at low pH.
Our chloride concentration calculations assume that an insignificant amount of radiolabeled chloride penetrates into the cytosol during the timeframe of our measurements. This assumption is supported by our rapid dilution-filtration experiments.26 However, it is now known that E. coli contain two ClC family H+/Cl– antiporters in the inner membrane, ClC-ec1 and ClCb. These antiporters play a role in E. coli’s ability to survive acid shock and the subsequent return to neutral pH.33–35 The presence of these H+/Cl– exchangers raises the possibility that the intracellular chloride measured in our solute partitioning experiments includes a cytosolic component. But because the cytoplasmic pH is maintained between 4.5 and 5.5 during acute acid stress (corresponding to a ~1000-fold inward-directed H+ gradient),35,36 the electrochemical H+ gradient thermodynamically favors extrusion of Cl– from the cytosol during acid shock. We and others postulate that Cl–/H+ antiporters instead contribute to acid survival and recovery by tuning the membrane potential.33,35 A direct test of Cl– accumulation in an E. coli strain with both H+/Cl– antiporters knocked out comes with an important caveat: the low (10%) survival rate of these bacteria may lead to breakdown of compartmentalization. In our hands, this strain also shows a mucoid phenotype when grown on LB agar, indicating that H+/Cl– antiporters may also have functions outside of acid survival. This mucoid phenotype may affect diffusion rates, possibly affecting quantification of compartment size. Even considering these important provisos, it is instructive that these bacteria still show a substantial accumulation of chloride within the periplasm when exposed to simulated gastric fluid (Supplementary Table 2).
Our results demonstrate that protein aggregation under acidic conditions is dependent on the anion concentration. As mentioned previously, it has been reported that anions can cause proteins unfolded by low pH to gain secondary structure by neutralizing the intramolecular repulsion of the positive charges present on an unfolded polypeptide.17–21 We propose that similar principles explain why anions induce the aggregation of proteins under acidic conditions (Fig. 5a). At relatively low anion concentrations, the positive charges on neighboring protein molecules repel each other through electrostatic repulsion. Thus, even though proteins unfolded by acid contain exposed hydrophobic patches, they cannot associate and aggregate due to the electrostatic repulsion from adjacent, positively charged portions of the unfolded polypeptide. However, at high anion concentrations, the intermolecular repulsion is minimized due to neutralization of the positive charges by solvent anions. This allows the hydrophobic effect to take over and for the proteins to associate into irreversible aggregates.
Figure 5. Mechanism of Chloride-Induced Protein Aggregation in Acid and its Effect on the E. coli Periplasm.
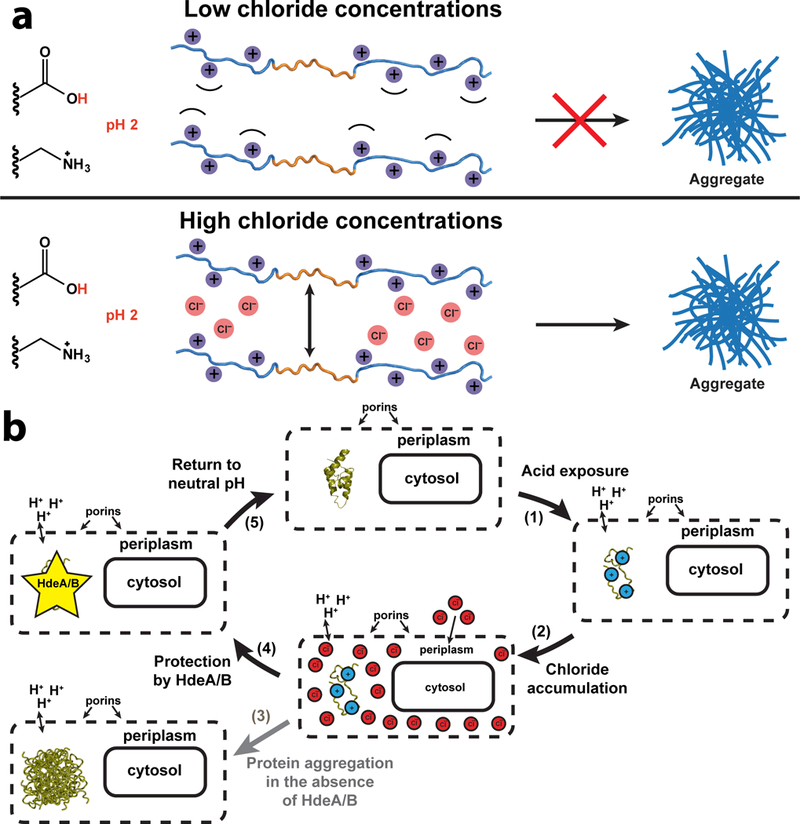
(a) Exposure of proteins to pH 2 protonates the side chains of acidic amino acids, causing the proteins to unfold. Top: at low chloride concentrations, the positive charges from the basic amino acids on neighboring protein molecules repel each other, preventing the intermolecular association of exposed hydrophobic stretches (depicted in orange) that would lead to aggregation. Bottom: at high chloride (or any anion) concentrations, the force of the electrostatic repulsion between positively charged protein molecules is minimized due to neutralization by the solvent anions. This allows the exposed hydrophobic regions to associate through the hydrophobic effect and for the acid-denatured proteins to aggregate.
(b) Model for the impact of acid stress on the E. coli periplasm. Clockwise from the top: (1) Because the outer membrane of E. coli contains porins, exposure to acute acid stress (e.g., in gastric fluid) acidifies the periplasmic space. Protonation of acidic side chains causes the periplasmic proteins to unfold and creates a high concentration of positively-charged macromolecules within the periplasm. (2) This generates a Donnan equilibrium across the outer membrane that causes the accumulation of chloride ions inside the periplasm. (3) In the absence of HdeA and HdeB, the high concentration of periplasmic chloride causes periplasmic proteins to associate into irreversible, toxic aggregates. (4) E. coli contains the acid stress chaperones, HdeA and HdeB, which bind to proteins unfolded by acid and prevent them from aggregating during acid stress. (5) Return to neutral pH (e.g., during transfer from the stomach to the small intestines) facilitates the release of unfolded proteins from HdeA and HdeB, allowing proteins to refold into their native structure and resume their normal functions.
In light of our findings, we present the following model to describe the biological consequences of acute acid stress on the periplasm of Gram-negative enteric microorganisms (Fig. 5b). Upon exposure to acute acid stress, the periplasmic space is directly exposed to the low pH environment of the extracellular media. The low pH protonates the aspartate and glutamate side chains on the proteins in the periplasm, disrupting salt bridges and leaving the protein entirely positively charged. This disruption of salt bridges and the repulsion of the charged groups cause the proteins to unfold. At the same time, a strong Donnan equilibrium develops across the outer membrane that causes the accumulation of large concentrations of chloride within the periplasm. This minimizes the electrostatic repulsion between positive charges on the proteins unfolded by the acid and normally would cause them to associate into irreversible cytotoxic aggregates. However, Gram-negative enteric bacteria have evolved the acid stress chaperones HdeA and HdeB, which are post-translationally activated by acid and bind to unfolded proteins in the periplasm, preventing them from aggregating. Once the bacteria are transferred to the more neutral pH environment of the intestines, HdeA and HdeB release their bound substrates so that they can refold back into their native conformations. We find that organisms have apparently evolved these chaperones, at least in part, to directly respond to the Donnan-mediated chloride accumulation that occurs at low pH.
Our findings also illustrate that in vivo ion conditions can deviate widely from those typically used in in vitro experiments. It is common practice to select buffer conditions that optimize the in vitro activity of proteins, often with little regard to its physiological relevance. Our work provides a caution that in vitro buffer selection should be carefully considered to make sure that it mimics the in vivo condition. Unexpectedly, the high ionic strength buffer previously optimized for the in vitro action of HdeA (8 mM phosphate buffer, pH 2, 150 mM KCl and 150 mM ammonium sulfate) 13 resembles the actual ionic strength that is present in vivo. Since sulfate is a mixture of monovalent and divalent ions at pH 2, it contributes substantially more to ionic strength than a purely monovalent ion like chloride. Thus, the buffer used in prior aggregation assays contains a total anionic charge similar to what we measured in the periplasm in vivo. This raises the provocative question whether other apparently non-physiological buffers optimized for in vitro activity may work well because they similarly mimic the cellular microscopic environment that the protein experiences in vivo.
The local conditions within the complex, subdivided and gel-like environment of the cell represent a much more heterogeneous environment than that which is found within the test tube. The ionic environment in vivo, for instance, is not as uniform as is widely assumed. It has long been known that counter ions in the vicinity of naked nucleic acids and other polyanions such as polyphosphate can reach high, even molar, concentrations.37 We recently reported that nucleic acids and polyphosphate are highly efficient in preventing the aggregation of proteins.38,39 Although we do not yet fully understand how these polyanions function as antiaggregation agents, it seems likely that their highly charged nature plays an important role. Simple biophysical principles dictate that high concentrations of counter ions exist in the vicinity of other highly charged surfaces. Our findings serve as a reminder that researchers investigating other fundamental processes, including protein folding, should consider and ideally measure these local concentration differences in vivo in order to better understand the process that they are studying.
ONLINE METHODS
Protein expression and purification
HdeA, OppA, and DppA were expressed and purified as described previously 9,40. The gene for E. coli MBP was cloned into a pET28a vector containing an N-terminal His-SUMO tag with a ULP1 cleavage site. The MBP expression plasmid was cloned into BL21 (DE3) and grown in 4 L TB media + 4% glucose at 37°C with shaking to an OD600 of 1.0. The temperature was then lowered to 20°C and induced with 100 μM IPTG. The culture was then grown overnight at 20°C. After harvesting, the cells were lysed at 4°C by sonication in 50 mM Tris-HCl, 400 mM NaCl, 15 mM imidazole, 10% glycerol, pH 8.0 (lysis buffer) with DNase I and cOmplete™ protease inhibitor cocktail. The lysate was cleared by centrifugation and the supernatant was loaded on two 5 mL HisTrap columns pre-equilibrated in lysis buffer. The columns were washed with 30 mL lysis buffer, and the MBP was then eluted in lysis buffer + 0.5 M imidazole. The MBP was then exchanged into 25 mM Tris-HCl, pH 8 + 0.2 M NaCl in the presence of ULP1 to cleave off the His-SUMO tag and was subsequently passed over the HisTrap column again in 25 mM Tris-HCl, pH 8 + 0.2 M NaCl to remove the His-SUMO tag. After exchanging into 25 mM Tris-HCl, pH 8 (Buffer A), the MBP was loaded onto two 5 mL HiTrap Q HP columns equilibrated in Buffer A at 4°C. MBP was eluted using an 18 column volume 0–0.3 M NaCl linear gradient in Buffer A. Fractions containing MBP were concentrated and then run over a HiLoad 16/600 Superdex 200 column in 40 mM HEPES-NaOH, pH 7.5, 100 mM NaCl at 4°C. Purified MBP was pooled, concentrated, and flash frozen in liquid nitrogen before storing at −80°C.
In vitro aggregation assays
Assay buffers generally contained 10 mM HCl, pH 2 supplemented with different salts. Gastric fluid was simulated using 10 mM HCl, pH 2, 150 mm NaCl. Assay buffer was equilibrated in a 1 mL fluorescence cuvette with stirring at 25°C for at least 15 min prior to adding MDH, MBP, OppA, or DppA. MDH, MBP, OppA, or DppA were then added from a 100x stock (in 10 mM HEPES, pH 7.5) to the cuvette, and aggregation was monitored by measuring light scattering at 360 nm over time using a Cary Eclipse fluorimeter. When testing the chaperone activity of HdeA, purified HdeA was included in the assay buffer prior to adding MDH, MBP, OppA or DppA.
Periplasmic extracts were generated from stationary phase MG1655 E. coli using polymyxin as described previously.32 Cytosolic protein extracts were generated by sonication of the spheroplasts remaining after the periplasmic extraction protocol. Periplasmic and cytosolic extracts were concentrated using 10 kDa cutoff centrifugal filters and dialyzed into 5 mM sodium phosphate buffer, pH 7 containing either 150 mM NaCl, 500 mM NaCl, or 150 mM NaCl and 150 mM ammonium sulfate. 3 or 30 mg/mL periplasmic or cytosolic proteins were then acidified to pH 2 by the addition of a small volume of 1–5 M phosphoric acid in the absence or presence of 6 mg/mL or 30 mg/mL HdeA, followed by incubation at 25°C for 1 hr. The samples were then separated into soluble and pellet fractions by centrifugation and visualized by SDS-PAGE followed by Coomassie staining.
Circular dichroism
Circular dichroism measurements were recorded on a Jasco J-1500 spectropolarimeter. 1 μM MBP in 10 mM sodium phosphate buffer at pH 2 or pH 7 was placed in a 1 cm quartz cuvette at 25°C, and the far-UV spectrum was recorded from 200–250 nm. For the pH titration of periplasmic proteins, 0.2 mg/mL periplasmic extract was placed in 10 mM phosphate buffer sodium phosphate buffer at pH 2, 2.5, 3, 3.5, or 4.5 and the far-UV CD spectrum was recorded from 200–250 nm. The pH dependent CD at 222 nm and Clperi/Clo data were fit to a modified Henderson-Hasselbalch equation
where Smin and Smax are the minimum and maximum signal values, respectively, n is the Hill coefficient and pKa is the apparent acid dissociation constant.
Analytical ultracentrifugation
Sedimentation velocity analysis of MBP was performed using a Beckman Proteome Lab XL-I analytical ultracentrifuge (Beckman Coulter, Indianapolis, IN) equipped with an AN50TI rotor. Samples containing 2 μM or 10 μM MBP in 50 mM potassium phosphate, pH 2, 100 mM NaCl were loaded into cells containing standard sector-shaped 2-channel Epon centerpieces with 1.2 cm path-length (Beckman Coulter, Indianapolis, IN) and allowed to equilibrate at 25°C for 1 hr in the non-spinning rotor prior to sedimentation. MBP was spun at 45,000 rpm and its sedimentation was monitored continuously by absorbance at 280 nm. Scans were collected every two minutes. The sedimentation velocity data was analyzed with the program SEDFIT (version 15.01b) 41. The sedimentation distribution plot was generated using the continuous c(s) distribution model, with a confidence level for the ME (Maximum Entropy) regularization of 0.95. The error of the molecular weights is given as standard deviation based on the individual molecular weights calculated by SEDFIT at 2 μM and 10 μM, respectively. Buffer density as well as viscosity was calculated using SEDNTERP (http://sednterp.unh.edu/).
Solute distribution measurements
Solute distributions were measured using the method of Stock et al.26 This method relies on the selective penetration of several radiolabeled chemicals into different cellular compartments of E. coli. 3H inulin is unable to cross the outer membrane and therefore is only present in the extracellular fluid trapped in an E. coli pellet. 14C sucrose can cross the outer membrane but not the inner membrane, so it is present in both the extracellular fluid and the periplasm. 3H water can cross all membranes and will be present in the cytosol, periplasm, and extracellular fluid. 36Cl, like sucrose, can penetrate the periplasm and extracellular fluid, but not the cytosol. However, since it is charged, it can accumulate within the periplasm if a Donnan equilibrium exists across the outer membrane.
In our hands, only the 14C sucrose obtained from PerkinElmer was free from contaminants and gave reliable compartment volumes. Commercially obtained 14C sucrose is usually contaminated with 14C glucose, which can enter the cytosol and introduce inaccuracies in the 14C sucrose measurements.26 Prior to performing any experiments, the 14C sucrose was purified by incubation with ~50 mg of E. coli in 20 mM sodium phosphate pH 7, 150 mM NaCl for 1 hr. The cells metabolize the 14C glucose, but leave the 14C sucrose in the supernatant. The supernatant was then used in all solute distribution measurements for 14C sucrose measurements.
MG1655 E. coli were grown overnight to stationary phase in 1 L LB supplemented with 20 mM glucose at 37°C. The glucose and stationary phase are necessary to upregulate the acid survival machinery of E. coli. Two aliquots equivalent to 2 mL OD600 = 10 were used for each measurement. The two aliquots were pelleted and washed twice in 1 mL of one of the buffers listed in Table 1, Supplementary Table 1 or Supplementary Table 2. The wash and incubation buffers at pH 4.5 or below were supplemented with 1 mM glutamic acid and 10 mM glucose, which are necessary for E. coli to survive acute acid stress.35 After washing, the aliquots were resuspended in 2 mL of the same wash buffer and were incubated for 30 min. Pairs of radiolabeled chemicals were then added to one of the aliquots (using pairs of radiolabels eliminates errors in compartment volume measurements introduced by residual supernatant in the pellet fractions). The radiolabeled pairs were 14C sucrose and 3H water (for measuring the cytosol), 14C sucrose and 3H inulin (for measuring the periplasm), or 36Cl and 3H inulin (for measuring the chloride within the periplasm). Within 5 min of adding the radiolabels, both aliquots were pelleted by centrifugation at 20,000 x g for 1 min. The supernatants were subsequently removed, and 100 μL of the radiolabeled supernatant was added to the unlabeled pellet. The pellets were then resuspended in 0.5 mL of 10 mM sodium phosphate, pH 7 and transferred to 10 mL scintillation fluid, and the radioactivity was assayed in a Hidex Triathler liquid scintillation counter. We then converted the radiolabel measurements to volumes (in μL) in the pellet for a given solute (Vs) using the formula:
where Cp is the counts per minute in the pellet sample and Cs is the counts per minute in 100 μL of the supernatant for a given solute. The volume of the cytosol (Vcyto), volume of the periplasm (Vperi), apparent volume of chloride within the periplasm (VClperi), and volume of the cell (Vcell) were calculated using the following equations:26
where Vwater is the volume of water, Vsucrose is the volume of sucrose, Vinulin is the volume of inulin, and VCl is the volume of chloride in the E. coli pellet. The chloride concentration within the periplasm can differ from the chloride concentration of the extracellular media if a Donnan equilibrium exists across the outer membrane. The fold change in periplasmic chloride concentration relative to the chloride concentration in the extracellular solution can be calculated using the ratio VClperi/Vperi.26 If, for example, a Donnan equilibrium does not exist across the outer membrane, VClperi should equal Vperi and VClperi/Vperi will be 1. If chloride accumulates inside the periplasm due to the presence of positively charged macromolecules, VClperi/Vperi > 1. Since we know the chloride concentration of the external solution (Clo), we can calculate the actual chloride concentration inside the periplasm by multiplying Clo by VClperi/Vperi.
Rapid dilution-filtration assays
Rapid dilution-filtration assays were performed as described previously.26 MG1655 E. coli were grown to stationary phase as described above for the solute distribution measurements. An aliquot equivalent to 2 mL of OD600 = 2.0 was thrice washed and resuspended in 2 mL 10 mM HCl, pH 2, 150 mM KCl, 10 mM glucose and 1 mM glutamic acid. 36Cl was added at a concentration where 100 uL of the suspension contained approximately 80,000 counts per minute. After incubating for 20 minutes, 100 uL of the suspension was diluted into 10 mL unlabeled 10 mM HCl, pH 2, 150 mM KCl, 10 mM glucose and 1 mM glutamic acid and the solution was immediately vacuum-filtered through a 2.5 cm 0.45 um Nitrocellulose membrane (Millipore cat. no. HAWP02500) using a Millipore XX2702550 vacuum manifold. The time between dilution and complete filtration was about 15 seconds. The filter was then resuspended in 10 mL scintillation fluid and the radioactivity was assayed in a Hidex Triathler liquid scintillation counter. Three replicates were performed. To determine the amount of 36Cl from residual liquid retained on the filter, the above dilution-filtration procedure was performed on a 2 mL sample of buffer containing 80,000 cpm 36Cl per 100 uL without E. coli.
Cryo-electron tomography
Samples were prepared as described above. After incubation at the respective pH for 30 minutes, the bacteria were applied to glow discharged C-Flat 2/2 holey carbon grids (EMS, Hatfield, PA) and flash frozen in liquid ethane using a Cryoplunge 3 (Gatan, Pleasanton, CA). Tilt series were collected on a Titan Krios (Thermo Fisher, Hillsboro, OR) equipped with an energy filter (Gatan, Pleasanton, CA) and a K2 direct electron camera (Gatan, Pleasanton, CA) using SerialEM.42 Tilts were collected from −60° to 60° in 2° steps with a total dose per tilt series below 120e/Å2, a pixel size of 5.09Å/px and a nominal defocus of −6μm. Tomograms were aligned in IMOD.43
Survival assays
WT MG1655 and ∆hdeAB MG1655 E. coli were grown overnight at 37°C to stationary phase in LB + 20 mM glucose. The cultures were then diluted to OD600 = 0.03 in 20 mM sodium phosphate, pH 7, 150 mM NaCl, or a variety of acid shock conditions (10 mM HCl, pH 2, 0.2 M sucrose, 10 mM glucose, 1 mM glutamic acid) containing 0, 100, 200, 300, or 400 mM NaCl. The sucrose was added to the acid shock conditions to minimize osmotic stress on the samples at low ionic strength. The shocked cultures were incubated at 37°C, and at the indicated time points, 5-fold serial dilutions were spotted on M9 minimal media agar plates to determine the titer for each condition and time point. Plates were incubated for 24 hr at 37°C.
Western blotting
Rabbit-derived antisera were used for blotting against OppA, DppA, Skp, SurA, DsbC, RBP, AppA, and DegP. Anti-mouse monoclonal antibody for MBP was obtained from New England Biolabs. 1:5000 dilutions of the primary antibody were used for RBP, Skp, and SurA; 1:10000 dilutions were used for DegP and MBP; 1:20000 dilutions were used for OppA, DppA, DsbC and AppA. All primary antibody incubations were done in TBS-T. Fluorescently-labeled anti-goat or anti-mouse IRDye 800 CW secondary antibodies from LI-COR Biosciences were used at 1:15000 dilutions. Imaging was performed using a LI-COR Odyssey Clx.
To prepare the samples, WT MG1655 and ∆ hdeAB MG1655 E. coli were grown overnight at 37°C to stationary phase in LB + 20 mM glucose. 1 mL aliquots were centrifuged for 1 min at 15,000 RCF and the spent media was removed. The pelleted cells were then resuspended in 1 mL 20 mM sodium phosphate, pH 7, 150 mM NaCl, or a variety of acid shock conditions (10 mM HCl, pH 2, 0.2 M sucrose, 10 mM glucose, 1 mM glutamic acid) containing 0, 100, 300, or 500 mM NaCl. The sucrose was added to the acid shock conditions to minimize osmotic stress on the samples at low ionic strength. The samples were incubated for 1 hr at room temperature, after which they were centrifuged again and resuspended in 200 μL 20 mM sodium phosphate, pH 7, 150 mM NaCl and placed on ice to halt the aggregation. After 10 min, the samples were lysed by sonication and then separated into soluble and pellet fractions by centrifugation.
MDH refolding assays
MDH refolding assays were performed as described previously,13 except in buffer lacking ammonium sulfate.
Statistics and reproducibility
None of the experiments required advanced statistical analysis. The number of times each experiment was repeated is given in the respective figure legend.
Supplementary Material
ACKNOWLEDGEMENTS
We thank P. Koldewey for providing technical expertise on the analytical ultracentrifugation experiments, R. Mitra for preliminary assistance with the CD experiments and C. Miller for the E. coli strain with both Cl–/H+ antiporters knocked out. The primary antibodies for detecting DsbC and AppA were a gift from M. Berkmen; the antibodies for detecting Skp, SurA and DegP were a gift from T. Silhavy; the antibody for detecting RBP was a gift from M. Ehrmann. We also thank the Purdue Cryo-EM facility, especially Seema Mattoo for arranging access and Thomas Klose for help with data collection. This work was funded by the National Institutes of Health (R01-GM102829 to J.C.A.B. and R00-GM111767 to R.B.S.) and an Alfred P. Sloan Research Fellowship (to R.B.S.). J.C.A.B is a Howard Hughes Medical Investigator.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
Data availability
All data used in this study are included in this published article (and its supplementary information files) and are available from the corresponding authors upon reasonable request
REFERENCES
- 1.Dressman JB et al. Upper gastrointestinal (GI) pH in young, healthy men and women. Pharmaceutical Research 7, 756–761 (1990). [DOI] [PubMed] [Google Scholar]
- 2.Russell TL et al. Upper gastrointestinal pH in seventy-nine healthy, elderly North American men and women. Pharmaceutical Research 10, 187–196 (1993). [DOI] [PubMed] [Google Scholar]
- 3.Smith J The role of gastric acid in preventing foodborne disease and how bacteria overcome acid conditions. J. Food Prot. 66, 1292–1303 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Kanjee U & Houry WA Mechanisms of acid resistance in Escherichia coli. Annu. Rev. Microbiol. 67, 65–81 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Nikaido H Molecular basis of bacterial outer membrane permeability. Microbiol Mol Biol Rev 67, 593–656 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malki A et al. Solubilization of protein aggregates by the acid stress chaperones HdeA and HdeB. J. Biol. Chem. 283, 13679–13687 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Hong W et al. Periplasmic protein HdeA exhibits chaperone-like activity exclusively within stomach pH range by transforming into disordered conformation. J. Biol. Chem. 280, 27029–27034 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Kern R, Malki A, Abdallah J, Tagourti J & Richarme G Escherichia coli HdeB is an acid stress chaperone. J. Bacteriol. 189, 603–610 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tapley TL et al. Structural plasticity of an acid-activated chaperone allows promiscuous substrate binding. Proc. Natl. Acad. Sci. 106, 5557–5562 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang M et al. A genetically incorporated crosslinker reveals chaperone cooperation in acid resistance. Nat. Chem. Biol. 7, 671–677 (2011). [DOI] [PubMed] [Google Scholar]
- 11.Zhang S et al. Comparative proteomics reveal distinct chaperone – client interactions in supporting bacterial acid resistance. Proc. Natl. Acad. Sci. 113, 10872–10877 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gajiwala KS & Burley SK HDEA, a periplasmic protein that supports acid resistance in pathogenic enteric bacteria. J. Mol. Biol. 295, 605–612 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Tapley TL, Franzmann TM, Chakraborty S, Jakob U & Bardwell JCA Protein refolding by pH-triggered chaperone binding and release. Proc. Natl. Acad. Sci. 107, 1071–1076 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dahl J-U et al. HdeB functions as an acid-protective chaperone in bacteria. J. Biol. Chem. 290, 65–75 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding J, Yang C, Niu X, Hu Y & Jin C HdeB chaperone activity is coupled to its intrinsic dynamic properties. Sci. Rep. 5, 16856 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meeroff JC, Rofrano JA & Meeroff M Electrolytes of the gastric juice in health and gastroduodenal diseases. Am. J. Dig. Dis. 18, 865–872 (1973). [DOI] [PubMed] [Google Scholar]
- 17.Goto Y & Fink AL Conformational states of beta-lactamase: molten-globule states at acidic and alkaline pH with high salt. Biochemistry 28, 945–952 (1989). [DOI] [PubMed] [Google Scholar]
- 18.Goto Y, Takahashi N & Fink AL Mechanism of acid-induced folding of proteins. Biochemistry 29, 3480–3488 (1990). [DOI] [PubMed] [Google Scholar]
- 19.Fink AL, Calciano LJ, Goto Y, Kurotsu T & Palleros DR Classification of acid denaturation of proteins: intermediates and unfolded states. Biochemistry 33, 12504–12511 (1994). [DOI] [PubMed] [Google Scholar]
- 20.Goto Y & Fink AL Phase diagram for acidic conformational states of apomyoglobin. J. Mol. Biol. 214, 803–805 (1990). [DOI] [PubMed] [Google Scholar]
- 21.Goto Y, Calciano LJ & Fink AL Acid-induced folding of proteins. Proc. Natl. Acad. Sci. 87, 573–577 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin S et al. Site-specific incorporation of photo-cross-linker and bioorthogonal amino acids into enteric bacterial pathogens. J. Am. Chem. Soc. 133, 20581–20587 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Yang Y et al. Genetically encoded protein photocrosslinker with a transferable mass spectrometry-identifiable label. Nat. Commun. 7, 12299 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang S et al. Comparative proteomics reveal distinct chaperone–client interactions in supporting bacterial acid resistance. Proc. Natl. Acad. Sci. 113, 10872–10877 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kararli TT Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm. Drug Dispos. 16, 351–380 (1995). [DOI] [PubMed] [Google Scholar]
- 26.Stock JB, Rauch B & Roseman S Periplasmic space in Salmonella typhimurium and Escherichia coli. J. Biol. Chem. 252, 7850–7861 (1977). [PubMed] [Google Scholar]
- 27.Sen K, Hellman J & Nikaido H Porin channels in intact cells of Escherichia coli are not affected by Donnan potentials across the outer membrane. J. Biol. Chem. 263, 1182–1187 (1988). [PubMed] [Google Scholar]
- 28.Cayley S, Lewis BA, Guttman HJ & Record MT Jr Characterization of the cytoplasm of Escherichia coli K-12 as a function of external osmolarity. Implications for protein-DNA interactions in vivo. J. Mol. Biol. 222, 281–300 (1991). [DOI] [PubMed] [Google Scholar]
- 29.Cayley S, Lewis BA & Record MT Jr Origins of the osmoprotective properties of betaine and proline in Escherichia coli K-12. J. Bacteriol. 174, 1586–1595 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cayley DS, Guttman HJ & Record MT Jr Biophysical characterization of changes in amounts and activity of Escherichia coli cell and compartment water and turgor pressure in response to osmotic stress. Biophys. J. 78, 1748–1764 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Epstein W The roles and regulation of potassium in bacteria. Prog. Nucleic Acid Res. Mol. Biol. 75, 293–320 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Quan S, Hiniker A, Collet J-F & Bardwell JCA Isolation of bacteria envelope proteins. Bact. Cell Surfaces Methods Protoc. 966, 53–75 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Iyer R, Iverson TM, Accardin A & Miller C A biological role for prokaryotic ClC chloride channels. Nature 419, 715–718 (2002). [DOI] [PubMed] [Google Scholar]
- 34.Accardi A & Miller C Secondary active transport mediated by a prokaryotic homologue of ClC Cl- channels. Nature 427, 803–807 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Richard H & Foster JW Escherichia coli glutamate- and arginine-dependent acid resistance systems increase internal pH and reverse transmembrane potential. J. Bacteriol. 186, 6032–6041 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang M et al. Biocompatible click chemistry enabled compartment-specific pH measurement inside E. coli. Nat. Commun. 5, 4981 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bloomfield VA, Crothers DM & Tinoco I Jr. Nucleic Acids: Structures, Properties and Functions. (University Science Books, 2000). [Google Scholar]
- 38.Gray MJ et al. Polyphosphate is a primordial chaperone. Mol. Cell 53, 689–699 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Docter BE, Horowitz S, Gray MJ, Jakob U & Bardwell JCA Do nucleic acids moonlight as molecular chaperones? Nucleic Acids Res. 44, 4835–4845 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lennon CW et al. Folding optimization in vivo uncovers new chaperones. J. Mol. Biol. 427, 2983–2994 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schuck P Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Kremer JR, Mastronarde DN & McIntosh JR Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71–76 (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.