

Summary
Although essential for epigenetic inheritance, the transfer of parental histone (H3-H4)2 tetramers that contain epigenetic modifications to replicating DNA strands is poorly understood. Here, we show that the Mcm2-Ctf4-Polα axis facilitates the transfer of parental (H3-H4)2 tetramers to lagging-strand DNA at replication forks. Mutating the conserved histone-binding domain of the Mcm2 subunit of the CMG (Cdc45-MCM-GINS) DNA helicase, which translocates along the leading-strand template, results in a marked enrichment of parental (H3-H4)2 on leading-strand, due to the impairment of the transfer of parental (H3-H4)2 to lagging strands. Similar effects are observed in Ctf4 and Polα primase mutants that disrupt the connection of the CMG helicase to Polα that resides on lagging strand template. Our results support a model whereby parental (H3-H4)2 complexes displaced from nucleosomes by DNA unwinding at replication forks are transferred by the CMG-Ctf4-Polα complex to lagging-strand DNA for nucleosome assembly at the original location.
Graphical Abstract
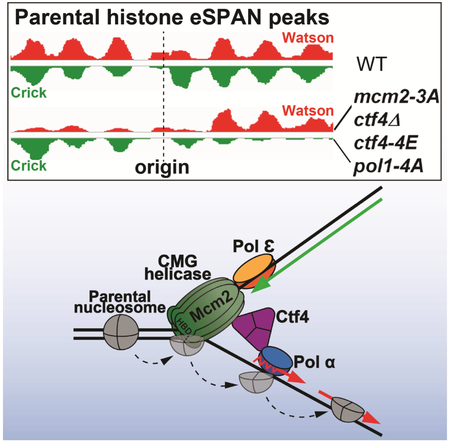
eTOC blurb
How parental histone H3-H4 tetramers are transferred to replicating DNA strands for epigenetic inheritance remains largely unknown. Gan and al. show that parental H3-H4 tetramers bind to Mcm2, which travel along the leading-strand template, and are then transferred to the lagging strand by the Mcm2-Ctf4-Polα complex for nucleosome assembly.
Introduction
Chromatin, an organized complex of DNA, RNA and proteins, encodes epigenetic information and maintains genome integrity. The basic repeating unit of chromatin is the nucleosome, consisting of 147 base-pairs of DNA wrapped around a histone octamer, comprised of one histone (H3-H4)2 tetramer and two histone H2A-H2B dimers (Luger et al., 1997). Histone posttranslational modifications have a profound impact on gene expression during the response to environmental stimuli and development (Campos et al., 2014; Martin and Zhang, 2007). Recently, it has been shown that some of these modifications are inherited during mitotic cell division and meiosis (Coleman and Struhl, 2017; Laprell et al., 2017; Ragunathan et al., 2015; Wang and Moazed, 2017). DNA as well as DNA methylation is inherited through a “semi-conservative mechanism,” and many proteins participate in this process (Bell and Labib, 2016; Burgers and Kunkel, 2017). However, how parental histones that carry epigenetic modifications are propagated to daughter cells remains largely unknown (Groth et al., 2007; Ransom et al., 2010; Serra-Cardona and Zhang, 2018). Moreover, it has become increasingly clear that alterations in chromatin states drive tumorigenesis in many organs (Chan et al., 2013; Lewis et al., 2013). Therefore, it is important to determine how chromatin states are inherited during mitotic cell divisions.
The “first step” in the transmission of chromatin states to daughter cells is the formation of nucleosomes on replicating DNA. To allow DNA replication machinery access to nucleosomal DNA, nucleosomes ahead of DNA replication forks are first disassembled likely by the action of the replicative helicase in association with chromatin remodeling complexes and histone chaperones (Devbhandari et al., 2017; Kurat et al., 2017). Immediately following DNA replication, nascent DNA is assembled into nucleosomes via two distinct pathways, the transfer of parental histones disassembled through the passage of replication forks and de novo deposition of newly synthesized histones onto replicating DNA strands. It is known that nucleosomal H2A-H2B dimers can rapidly exchange with cytoplasmic H2A-H2B during the cell cycle (Xu et al., 2010). Therefore, it is thought that the transfer of parental (H3-H4)2 tetramers and the deposition of new (H3-H4)2 tetramers are the rate-limiting steps in nucleosome formation during DNA replication. Over the years, various studies have shown that histone chaperones and histone modifications regulate the de novo deposition of new H3-H4. However, the molecular mechanisms governing the transfer of parental histone (H3-H4)2 tetramers to replicating DNA strands remain enigmatic (Groth et al., 2007; Ransom et al., 2010; Serra-Cardona and Zhang, 2018).
Eukaryotic DNA replication initiates from multiple replication origins during the S phase of the cell cycle (Bell and Dutta, 2002; Bell and Labib, 2016; Burgers and Kunkel, 2017). During G1-phase, the origin recognition complex (ORC) loads an inactive double-hexamer of the Mcm2-7 proteins around double-stranded DNA (dsDNA) at origins. Subsequently, the activation of CDK (cyclin-dependent kinases) and DDK (Dbf4-dependent kinase) in S-phase leads to splitting of the MCM2-7 double hexamer and recruitment of the Cdc45 protein and the GINS complex, thereby producing two active CMG helicases at a pair of bi-directional replication forks. Each CMG helicase travels along the template of the leading strand (Fu et al., 2011; Yu et al., 2014), unwinding the parental dsDNA template and producing single-strand DNA (ssDNA) that is stabilized by the heterotrimeric complex RPA. Subsequently, DNA polymerase α (Pol α) synthesizes primers that are used by Pol ε for synthesis of the leading strand and by Pol δ for lagging strand synthesis.
Many factors assemble around the CMG helicase to form the replisome. Whereas Pol ε binds directly to the CMG helicase and couples the rate of fork progression to leading strand synthesis (Yeeles, et al., 2017;(Langston et al., 2014), Pol α is linked to the helicase by the Ctf4 adaptor protein (Gambus et al., 2009; Simon et al., 2014; Villa et al., 2016; Zhu et al., 2007) and is enriched on lagging-strand template (Yu et al., 2014). Although Ctf4 was originally thought to facilitate primer synthesis by Pol α at replication forks, Ctf4 is dispensable for efficient lagging strand DNA synthesis in a reconstituted in vitro DNA replication system based on purified budding yeast proteins (Yeeles et al., 2017), suggesting that tethering Polα by Ctf4 to the CMG helicase may have other functions in chromatin replication.
Genetic evidence accumulated over the years indicates a tight connection between the DNA replication machinery and the inheritance of chromatin states. For instance, mutations in many DNA replication machinery components, including Pol α and Pol ε (Ehrenhofer-Murray et al., 1999; Nakayama et al., 2001; Zhang et al., 2000), affect transcriptional silencing at heterochromatin loci, suggesting that the replisome has a role in the inheritance of chromatin states. Consistent with this idea, nucleosome assembly is tightly coupled to lagging strand synthesis (Smith and Whitehouse, 2012). Moreover, the Mcm2 subunit of the CMG helicase, contains a conserved histone-binding domain (Foltman et al, 2013), which in human cells was shown to bind specifically to H3-H4. Mutations in the histone-binding domain of budding yeast Mcm2 (e.g. mcm2-3A that replaces conserved tyrosines with alanines) result in a loss of heterochromatin silencing at sub-telomeres and mating type loci (Foltman et al., 2013). Therefore, it was proposed that the histone-binding activity of Mcm2 functions in the transfer of parental histones, consistent with the association of Mcm2 with histone proteins released from chromatin by nuclease digestion (Foltman et al., 2013). In addition, human Mcm2 was proposed to function as a chaperone for both parental and newly synthesized H3-H4 (Huang et al., 2015; Richet et al., 2015). However, the mechanism by which Mcm2 functions in nucleosome assembly is not well understood.
We have shown recently that Dpb3 and Dpb4, two auxiliary and non-essential subunits of Pol ε, facilitate the transfer of parental histones (H3-H4)2 to leading strands (Yu et al., 2018), implying that other proteins likely help the transfer of parental (H3-H4)2 tetramers to lagging strands to maintain chromatin states during DNA replication. Here, we report the finding that, surprisingly, Mcm2 facilitates the transfer of parental H3-H4 to lagging strands, in association with the Ctf4 adaptor protein and Polα axis, which are linked together within the replisome to form the Mcm2-Ctf4-Polα axis.
Results
Mutating the H3-H4 binding domain of Mcm2 impairs the transfer of H3K4me3-H4 to lagging strands
To understand how Mcm2 is involved in nucleosome assembly, we analyzed the impact of the histone-binding defective mcm2-3A allele on nucleosome assembly (Figure S1A and Figure 1A), using surrogate marks of newly synthesized histones (H3 lysine 56 acetylation: H3K56ac) and of parental histones (tri-methylation of H3 lysine 4: H3K4me3) as reported previously (Yu et al., 2018). Briefly, G1 arrested yeast cells were released into medium containing BrdU (to label newly synthesized DNA) and hydroxyurea (HU) for 45 minutes. HU has no apparent effect on the initiation of DNA replication from early replication origins, but slows DNA synthesis and thereby enables monitoring of histone segregation during early S phase (Liu et al., 2017; Yu et al., 2018). To map nucleosomes at single nucleosome resolution, chromatin from early S phase cells was digested with micrococcal nuclease (MNase), which cleaves DNA between nucleosomes, and analyzed by deep sequencing (MNase-ssSeq). The digested chromatin was also used for chromatin immunoprecipitation (ChIP) using antibodies against H3K56ac and H3K4me3 and subsequent strand-specific sequencing (ChIP-ssSeq) and eSPAN (enrichment and Sequencing of Protein Associated Nascent DNA; Figure S1A), which detects a protein including histone enrichment at leading or lagging strands of DNA replication forks (Yu et al., 2014). The mcm2-3A mutant had no apparent effect on overall nucleosome occupancy and positioning surrounding early DNA replication origins based on MNase-ssSeq, H3K56ac and H3K4me3 ChIP-ssSeq datasets (Figure S1B-E), suggesting that Mcm2 is unlikely to be involved in nucleosome assembly of new H3-H4 in budding yeast.
Figure 1. Mutation of the histone-binding domain of Mcm2 impairs the transfer of parental histone H3-H4 to lagging strands.

(A) Graphic explanation of the strand bias calculation of the eSPAN procedure based on a hypothetical enrichment of new and parental (H3-H4)2 tetramers at the lagging and leading strands, respectively (B) Snapshot of BrdU IP-ssSeq, H3K56ac, and H3K4me3 eSPAN peaks at individual nucleosomes surrounding the ARS1309 origin in mcm2-3A mutant cells. (C) Heatmap representing the bias ratio and pattern of H3K56ac eSPAN peaks in mcm2-3A mutant cells at each of the 20 individual nucleosomes surrounding each of the 134 early DNA replication origins ranked from top to bottom based on replication efficiency. Individual nucleosomes are represented by a circle and their positions are indicated (−10 to +10). Each row represents the average log2 Watson/Crick ratio of H3K56ac eSPAN sequence reads at one origin. (D) The average bias ratio of H3K56ac eSPAN peaks in wild type and mcm2-3A mutant cells at each of the 20 individual nucleosomes of the 134 early replication origins. Data are represented as mean ± SEM from three independent experiments. (E-F) H3K4me3 eSPAN peaks at newly replicated chromatin show a marked leading strand bias in mcm2-3A mutant cells compared to wild type cells. (E) Data are represented as mean ± SEM from two independent experiments. (G) Top: heatmap representing the relative levels of H3K4me3 at leading and lagging strands, calculated using the formula shown in Figure S2A, in mcm2-3A mutant cells compared to wild type cells at each of the 134 individual origins coded in with color. Bottom: the average of relative amount of H3K4me3 at lagging and leading strands of 134 replication origins in mcm2-3A cells compared to wild type cells. See also Figure S1.
We recently showed that parental histones are assembled onto nascent DNA with a slight preference for the lagging strand, whereas assembly of new histones shows a slight preference for the leading strand (Yu et al, 2018). Inspection of H3K56ac and H3K4me3 eSPAN in mcm2-3A cells at individual origins such as ARS1309 revealed, unexpectedly, that H3K56ac eSPAN peaks showed a strongly enhanced lagging strand bias, whereas H3K4me3 eSPAN peaks showed a strong bias towards leading strands (Figure 1B). These mutant bias patterns of H3K56ac and H3K4me3 eSPAN peaks were opposite to those observed in wild type cells (Figure 1 C-F) as well as opposite to those observed in dpb3Δ cells defective in the transfer of parental (H3-H4)2 to leading strands (Yu et al., 2018). To analyze the bias ratio and pattern of H3K56ac and H3K4me3 eSPAN peaks quantitatively, we calculated the log2 ratio of sequence reads of the Watson over Crick strand at 20 individual nucleosomes surrounding each of the 134 early replication origins, which spans the replicated region (about 2 kb in each direction) under the experimental conditions. We observed a strong lagging- and leading-strand bias for the H3K56ac and H3K4me3 eSPAN peaks, respectively, in mcm2-3A cells at individual nucleosomes surrounding each of the 134 early replication origins (Figure 1C-F), except at the +1 and −1 nucleosomes of the H3K4me3 eSPAN peaks (Figure 1E-F). More remarkably, the bias ratio of both the H3K56ac and the H3K4me3 eSPAN peaks in mcm2-3A cells was increased dramatically compared to the mild bias observed in wild type cells (Figure 1D and 1F). These results demonstrate that the mcm2-3A mutation radically impacts the distribution of parental and new H3-H4 at replicating DNA strands, while having little impact on the overall nucleosome occupancy on replicated chromatin.
The average bias ratio of the histone eSPAN peaks at 134 early replication origins, defined by log2 ratio of sequence reads of the Watson over the Crick strand, reflects the relative amount of histones on the leading strand vs the corresponding lagging strand (Yu et al., 2014; Yu et al., 2018). Therefore, two models could explain the changes in H3K4me3 eSPAN peak bias in mcm2-3A mutant cells compared to wild type cells. First, more H3K4me3-H4 (parental) tetramers might be deposited onto leading strands in mcm2-3A cells. Second, less H3K4me3-H4 (parental) tetramers might be transferred to lagging strands compared to wild type cells. To differentiate between these two possibilities, we calculated the relative amounts of H3K4me3 in mcm2-3A cells on leading and lagging strand compared to wild type cells, using the formula in Figure S2A (bottom panel). We observed that H3K4me3 on lagging strands was dramatically reduced, whereas H3K4me3 on leading strands was increased in mcm2-3A cells compared to wild type, with the reduction of H3K4me3 on lagging strands being more pronounced than the increase of H3K4me3 on leading strands (Figure 1G). A similar analysis of H3K56ac eSPAN indicated that the mcm2-3A mutation had a minor impact on the relative amount of H3K56ac compared to wild type (Figure S2B). These results indicate that the primary defect observed in mcm2-3A cells is the impairment of the parental (H3-H4)2 transfer onto lagging strands (Figure S2C).
Effect of mcm2-3A on parental histone transfer using the recombination-induced tag switch system
To validate the impact of mcm2-3A on histone distribution using an independent approach, we used the published Recombination-Induced Tag Exchange (RITE) system (Verzijlbergen et al., 2010), which makes it possible to mark parental and new histone H3 with two different tags. Briefly, the engineered H3 gene initially expresses the HA-tagged H3 (H3-HA), but then undergoes homologous recombination during induction of the Cre recombinase in G0-arrested cells, thus replacing the HA tag with the T7 tag. Therefore, H3-HA and H3-T7 represent parental and newly synthesized H3 (Figure 2A), respectively, when cells are subsequently released into S phase from G0. The H3-T7 and H3K56ac eSPAN peaks in mcm2-3A cells at early S phase showed a strong lagging-strand bias (Figure 2 C-F and Figure S2D), whereas the H3-HA and H3K4me3 eSPAN peaks exhibited a strong bias towards leading strands (Figure 2G-J and Figure S2D). Thus, using different antibodies and two different yeast backgrounds (W303 and S288C; Table S1), we could confirm that Mcm2 regulates the transfer of parental (H3-H4)2 to lagging strands.
Figure 2. Analysis of new and parental histone H3 at leading and lagging strands in mcm2-3A mutant cells using the Recombination-Induced Tag Exchange (RITE) system.

(A) Outline of the experimental conditions employed for tagging parental and new histone H3 with the HA epitope (H3-HA) and T7 (H3-T7), respectively. (B) Snapshot of BrdU IP-ssSeq, H3-HA (parental), and H3-T7 (new) eSPAN peaks at individual nucleosomes surrounding ARS1309 in mcm2-3A mutant cells. (C-F) Analysis of the bias ratio and pattern of new H3 eSPAN peaks, representing by H3-T7 (C-D) and H3K56ac (E-F) in mcm2-3A mutant cells. (G-J) Parental H3 eSPAN peaks in mcm2-3A mutant cells as detected by both H3-HA (G-H) and H3K4me3 (I-J) show a strong leading-strand bias. See also Figure S2.
Ctf4 associates with both sides of the replication fork
The Mcm2 histone-binding domain is attached to the N-tier ring of the MCM2-7 complex within the CMG helicase, which moves at the heart of the replisome along the leading-strand template (Douglas et al., 2018; Georgescu et al., 2017; Noguchi et al., 2017). The CMG helicase is connected to the Pol α primase on lagging strands via Ctf4, which interacts with the Sld5 subunit of GINS in the CMG helicase and with Pol1, the catalytic subunit of Pol α primase (Simon et al., 2014; Villa et al., 2016; Zhu et al., 2007). Therefore, we hypothesized that Mcm2 facilitates the transfer of parental (H3-H4)2 onto lagging strands via the CMG-Ctf4-Pol1 complex. To test this hypothesis, we first analyzed the location of Ctf4 at DNA replication forks using Ctf4 ChIP-ssSeq and eSPAN. Like Pol1 (Yu et al., 2017), Ctf4 ChIP-ssSeq peaks showed (+) strand bias, suggesting that Ctf4 is enriched on lagging-strand template (Figure 3A and 3B, see Fig. 3D for explanation). However, in contrast to Pol1, the Ctf4 eSPAN peaks showed a bias towards the leading strand side of the fork (Figure 3A and 3C), a bias pattern similar to the Mcm6 subunit of the CMG helicase (Yu et al., 2014). These results indicated that Ctf4 is not only cross-linked to lagging-strand templates, possibly via Pol1 and/or other proteins on lagging strands, but also to replicating leading strand, possibly via the CMG helicase (Figure 3D). Consistent with this interpretation, the ctf4-4E mutant, which contains four amino acid substitutions in the Ctf4 domain that disrupts the binding to its partners including Sld5 and Pol1 (Villa et al., 2016), showed no apparent binding to DNA replication origins (Figure 3A and Figure 3E). These results indicate that the unique binding pattern of Ctf4 with DNA replication forks is likely mediated through its interactions with proteins on both leading and lagging strand sides of the fork. The association of Ctf4 with DNA replication origins (Figure 3E) and with lagging-strand templates (Figure 3F) was not affected to a detectable degree in the pol1-4A mutant that is defective in Ctf4 binding (Villa et al., 2016). These results suggested that in addition to Pol1, Ctf4 might also interact with another protein(s) on lagging-strand template. Thus, Ctf4 connects the CMG helicase on the leading strand to Polα on the lagging strand side of replication forks, possible also interacting with another protein on lagging strands (Figure 3D), or else simply projecting from CMG towards lagging-strand template.
Figure 3. Ctf4 is cross-linked to both lagging-strand template and replicating leading strand.

(A) Snapshot of Ctf4 ChIP-ssSeq peaks in different strains with the indicated genotype (top) and of Ctf4, Pol1 (catalytic subunit of Polα), and Mcm6 ChIP-ssSeq and eSPAN peaks in wild type cells (bottom) at the region surrounding replication origin ARS1309. (B) Average bias of Ctf4, Pol1, and Mcm6 ChIP-ssSeq peaks at 134 early replication origins in wild type cells, which shows that Ctf4 and Pol1, but not Mcm6, bind to lagging-strand template. (C) Average bias of Ctf4, Pol1, and Mcm6 eSPAN peaks showing that Ctf4 and Mcm6, but not Pol1, bind nascent leading strands. (D) A model for the homotrimer Ctf4 interacting with replicating leading strand and lagging-strand template indirectly via protein-protein interaction. (E) Analysis of the average Ctf4 ChIP-ssSeq read density in wild type, ctf4-4E, pol1-4A, and ctf4Δ cells at 134 early replication origins. (F) Average bias of Ctf4 ChIP-ssSeq peaks in wild type, ctf4-4E, pol1-4A, and ctf4Δ cells at 134 early replication origins.
Deletion of CTF4 impairs the transfer of parental H3-H4 to lagging strands
Next, we analyzed how ctf4Δ mutants affected the distribution of H3K56ac and H3K4me3 on replicating DNA. The overall nucleosome occupancy surrounding DNA replication origins detected by MNase-ssSeq and H3K4me3 ChIP-ssSeq in ctf4Δ cells was not affected to a detectable degree compared to wild type cells (Figure S3A-B). We noticed, however, that nucleosome occupancy as detected by H3K56ac ChIP-ssSeq and BrdU-IP-ssSeq in ctf4Δ cells was markedly reduced compared to wild type cells (Figure S3C-D). Moreover, DNA synthesis in ctf4Δ cells proceeded further than wild type cells (Figure S3E), which is likely due to increased dNTP concentrations (Poli et al., 2012). Therefore, when normalized against total sequence reads, the peak height of H3K56ac ChIP-ssSeq and BrdU-IP-ssSeq was reduced, which contributes, at least partly, to the apparently reduced nucleosome occupancy at replicated chromatin as detected by H3K56ac ChIP-ssSeq and BrdU-IP-ssSeq in ctf4Δ cells. The impact of ctf4Δ on DNA synthesis is distinct from mcm2-3A, consistent with the idea that Ctf4 likely has roles in other processes, such as sister chromatin cohesion (Samora et al., 2016), which are not shared with the Mcm2 histone-binding domain.
Like mcm2-3A cells, we observed that H3K56ac and H3K4me3 eSPAN peaks in ctf4Δ cells showed a strong lagging and leading strand bias, respectively (Figure 4A-E). We also analyzed the relative amount of H3K56ac and H3K4me3 at leading and lagging strands in ctf4Δ cells compared to wild type cells using eSPAN datasets and observed that H3K4me3 at lagging strands was markedly reduced, with a slight increase at leading strands in ctf4Δ cells (Figure 4F). Loss of Ctf4 had minimal impact on the relative amount of H3K56ac at both leading and lagging strands (Figure S3F). The effect of ctf4Δ on the H3K4me3 distribution was highly correlated with that caused by the mcm2-3A allele (Figure 4G). Taken together, these results indicate that Ctf4 also has a role in the transfer of parental (H3-H4)2 to lagging strands, most likely through same pathway as the Mcm2 histone-binding domain.
Figure 4. Deletion of CTF4 impairs the transfer of parental H3-H4 to lagging strands.

(A) Snapshot of BrdU-IP-ssSeq, H3K56ac and H3K4me3 eSPAN peaks at ARS1309 in ctf4Δ mutant cells. (B-E) Analysis of the average bias ratio and pattern of H3K56ac (B-C) and H3K4me3 (D-E) eSPAN peaks in ctf4Δ mutant cells. H3K4me3 eSPAN peaks in ctf4Δ mutant cells show a strong leading-strand bias. Data in C and E are represented as mean ± SEM from two independent experiments. (F) Analysis of the relative levels of H3K4me3 at leading and lagging strands of newly replicated chromatin in ctf4Δ mutant cells compared to wild type cells. The representation and calculation methods are the same as in Figure 1F. (G) The effect of mcm2-3A mutation on H3K4me3 eSPAN peaks is highly correlated to that of ctf4Δ cells. One dot represents the bias ratio of eSPAN peaks at individual nucleosomes (±10 nucleosomes) surrounding each of 134 early DNA replication origins. See also Figure S3.
Analysis of the impact of ctf4Δ on histone segregation onto replicating DNA during unperturbed S phase
All the experiments presented above utilized cells released from G1 into early S phase in the presence of HU. We have shown that HU has no apparent effect on the distribution of histones onto leading and lagging strand DNA in dpb3Δ mutant cells (Yu et al., 2018). To test whether HU affects histone distribution in ctf4Δ cells, we performed H3K56ac and H3K4me3 eSPAN experiments using ctf4Δ mutant cells released into S phase at 25 °C for 30 and 40 minutes in the absence of HU. We observed that H3K56ac and H3K4me3 eSPAN peaks in ctf4Δ in the absence of HU showed the same bias pattern as those in the presence of HU (Figure 5 and Figure S4). Moreover, the impact of ctf4Δ mutation on H3K4me3 eSPAN peaks in the absence of HU was highly correlated with that in the presence of HU (Figure S4F), indicating that the presence of HU does not have a major impact on the histone distribution in ctf4Δ cells, and possibly also in mcm2-3A cells.
Figure 5. The ctf4Δ mutation also impacts the transfer of parental H3-H4 to lagging strands during normal S phase without HU.

(A) Snapshot of BrdU IP-ssSeq, H3K56ac and H3K4me3 eSPAN peaks at ARS1309 in ctf4Δ mutant cells during normal S phase. (B-E) Analysis of the average bias pattern of H3K56ac (B-C) and H3K4me3 (D-E) eSPAN peaks in ctf4Δ mutant cells using early S phase cells 30 minutes after release from G1 block without HU. The same bias pattern is observed in ctf4Δ mutant cells 40 minutes after release from G1 (see also Figure S4).
Effect of ctf4-4E and pol1-4A mutants on parental histone transfer and epigenetic silencing
To gain additional insight into the impact of ctf4Δ on parental H3-H4 transfer, we tested nucleosome assembly of H3K56ac and H3K4me3 in the ctf4-4E mutant, defective in binding to both the CMG helicase and Pol1 (Simon et al., 2014; Villa et al., 2016) (Figure 6A). The ctf4-4E mutant cells showed almost identical effects as ctf4Δ on the distribution of H3K56ac and H3K4me4 based on MNase-ssSeq, H3K56ac and H3K4me3 ChIP-ssSeq and eSPAN (Figure 6B-C and Figure S5) at replicating chromatin. These results strongly supported the idea that the ability of Ctf4 to associate with the CMG helicase, and with other partners such as Pol α, is critical for Ctf4 to perform its function in histone transfer to lagging strands. To test this idea further, we analyzed the impact on nucleosome assembly at replicating chromatin in pol1-4A mutant cells where Pol1 is defective in binding to Ctf4. Like mcm2-3A cells, the pol1-4A mutation had little impact on the overall nucleosome occupancy in replicating chromatin as compared to wild type cells (Figure S6A-D). Moreover, the H3K4me3 eSPAN peaks showed a strong leading-strand bias in pol1-4A cells (Figure 6D-E), albeit a smaller bias ratio than that observed in ctf4Δ or mcm2-3A cells. Interestingly, H3K56ac eSPAN peaks in pol1-4A cells did not show a significant bias towards either leading or lagging strands (Figure S6E). These results indicate that the ability of Pol1 to interact with Ctf4 is important for its role in the transfer of parental (H3-H4)2 to lagging strands. Consistent with this interpretation, the impacts of ctf4-4E and pol1-4A on the H3K4me3 transfer were highly correlated with each other and with that of mcm2-3A mutant (Figure 6F-H). Finally, using an assay monitoring the transient loss of heterochromatin silencing during cell division (Dodson and Rine, 2015; Janke et al., 2018), mcm2-3A, ctf4Δ and pol1-4A cells also showed increased rates of loss of HML silencing (Figure S6F), with pol1-4A showing the smallest effect among these three mutants. The silencing defects reported here are consistent with the silencing defects previously observed in ctf4Δ and pol1-4A mutant cells (Evrin et al., 2018; Suter et al., 2004). These results support the idea that a defect in the transfer of parental (H3-H4)2 to lagging strands in these mutant cells compromises the inheritance of silent chromatin at the HML locus. Taken together, these results indicate that Mcm2, Ctf4 and Pol1 function together to transfer parental histones (H3-H4)2 to lagging strands (Figure 6I).
Figure 6. Disruption of the CMG-Ctf4-Pol1 interaction impairs the transfer of parental histone (H3-H4)2 to lagging strands.

(A) Mutation of the Ctf4-interacting-peptide of Pol1 (pol1-4A) or the Ctf4 mutant defective in binding to both CMG and Pol1 (ctf4-4E) uncouples the CMG helicase from the Polα primase at lagging strand. (B-E) Analysis of the average bias pattern of H3K4me3 eSPAN peaks in ctf4-4E (B-C) or pol1-4A (D-E) mutant cells. The leading strand bias ratio of H3K4me3 eSPAN in pol1-4A cells is smaller than that of mcm2-3A, ctf4Δ, or ctf4-4E cells. (F-H) The dot scatter plots show the H3K4me3 bias pattern in ctf4-4E vs mcm2-3A (F), pol1-4A vs mcm2-3A (G), andpol1-4A vs ctf4-4E (H). Each dot represents bias ratio of H3K4me3 eSPAN peaks at a single nucleosome among 134 early DNA replication origins (from −10 to +10, n=2680). (I) Model depicting how the MCM helicase and Pol1, connected by Ctf4, regulate the transfer of parental (H3-H4)2 to the lagging strand. See also Figures S5 and S6.
Discussion
The Mcm2-Ctf4-Pol α axis facilitates the transfer of parental H3-H4 to lagging strands
The Mcm2 histone-binding domain is located at the front of the replisome, facing the parental chromatin (Georgescu et al., 2017; Noguchi et al., 2017). Therefore, it is likely that parental (H3-H4)2 tetramers dissembled from nucleosomes, by chromatin remodeling complexes and/or by the action of the CMG helicase, will first encounter and bind to the Mcm2 histone-binding domain, together with other proteins such as FACT. Since the CMG helicase travels on the leading-strand template at replication forks, one might expect that Mcm2 facilitates the transfer of parental (H3-H4)2 to leading strands. Surprisingly, we observed that the histone-binding mcm2-3A mutant impairs the transfer of parental (H3-H4)2 to lagging strands in two different yeast backgrounds. We present several lines of evidence supporting the model that parental (H3-H4)2 tetramers, once bound to Mcm2, are transferred to lagging strands via Ctf4 and Pol α, possibly requiring contact with another protein on lagging strands as well (Figure 6I). First, deletion of CTF4, or mutation of its domain that binds to the CMG helicase and Pol1, produces the same defect in parental H3-H4 transfer as the mcm2-3A mutant. Second, pol1-4A mutant cells, which cannot bind to Ctf4, also show a defect in the transfer of parental (H3-H4)2 to lagging strands. Finally, we show that the effect of the ctf4Δ, ctf4-4E and pol1-4A mutants on parental histone (H3-H4)2 transfer to lagging strands is highly correlated with the phenotype of mcm2-3A cells. Thus, the Mcm2-Ctf4-Pol α axis participates in the transfer of parental histone (H3-H4)2 to lagging strands, revealing a previously undocumented role for this complex in this poorly understood but important process. Recently, it has been shown that the Pol α also contains a conserved histone-binding motif, which in the human orthologue is specific for H2A-H2B (Evrin et al., 2018). Mutations of conserved residues in this domain produce the same silencing defects as seen in mcm2-3A budding yeast cells (Evrin et al., 2018). In the future, it would be interesting to determine whether mutating the Pol1 histone-binding motif affects the transfer of parental (H3-H4)2. We note that human Mcm2 also contains the conserved histone-binding domain at its N-terminus (Huang et al., 2015; Richet et al., 2015). Moreover, human CTF4/And-1 interacts with both the MCM helicase and the catalytic subunit of Pol α primase (Zhu et al., 2007). Therefore, we suggest that the function of the Mcm2-Ctf4-Pol α axis in the transfer of parental (H3-H4)2 is likely conserved from yeast to human cells. Supporting this idea, while the current manuscript is under review, skewed histone deposition was detected in the Mcm2 mutant defective in binding to H3-H4 in mouse embryonic stem (ES) cells (Petryk et al., 2018). It would be interesting to determine whether the skewed histone deposition in ES cells is due to impaired transfer of parental H3-H4 to lagging strands and whether Ctf4 and Polα also have roles in this process in the future.
We observed that Ctf4 remains associated with lagging-strand template in pol1-4A mutant cells. This association is unlikely due to Ctf4’s interaction with the CMG helicase, which travels on the leading-strand template. Consistent with this idea, the impacts of the pol1-4A mutation in parental H3-H4 transfer, as well in HML silencing, is smaller than that observed in mcm2-3A, ctf4Δ or ctf4-4E cells. This suggests that in addition to Pol1, Ctf4 might interact with another protein(s) at lagging strands for the transfer of parental H3-H4 to lagging strands. In addition to Sld5 and Pol1, Ctf4 interacts with the Chl1 helicase for the establishment of sister-chromatid cohesion (Samora et al., 2016), and with the Dna2 and Tof2 proteins that help maintain the integrity of rDNA repeats (Villa et al., 2016). Moreover, RPA co-purified with Ctf4 (Luciano et al., 2015). Dna2 is best known for its role in lagging-strand maturation (Ayyagari et al., 2003). RPA is enriched at lagging-strand template as Pol1 (Yu et al., 2017). Therefore, it would be interesting to determine whether Dna2, RPA and/or another protein at lagging strands might also aid the transfer of parental (H3-H4)2 tetramers to lagging strands during chromatin replication.
How are parental (H3-H4)2 tetramers returned to their “original nucleosome positions” on replicating chromatin?
We observed that the average bias ratio of H3K4me3 eSPAN peaks in wild type cells (compare Figure 1F to 1D) (Yu et al., 2018), mcm2-3A (compare Figure 1F to 1D, compare Figure 2H to 2D), ctf4Δ (Figure 4E to 4C), and pol1-4A cells (Figure 6E to Figure S6E) is significantly larger than that of the H3K56ac eSPAN peaks in the corresponding mutant cells. We suggested previously that this could be due to the fact that nucleosomes formed with parental (H3-H4)2 tetramers are more stable and resistant to MNase digestion than nucleosomes formed with new (H3-H4)2 tetramers (Yu et al., 2018). Nucleosome stability in newly replicated chromatin is likely linked to nucleosome positions, because in order to maintain chromatin states, newly-formed nucleosomes irrespective of containing parental (H3-H4)2 or new (H3-H4)2 must assume the original/starting nucleosome positions. Therefore, the large bias ratio of H3K4me3 eSPAN peaks likely suggests that nucleosomes formed with parental H3K4me3-H4 are more likely to be transferred to their starting positions immediately following DNA replication, whereas nucleosomes formed with newly synthesized (H3-H4)2 need longer time to return to the original/starting position, possibly with the help of chromatin remodeling complexes. Consistent with this idea, it has been shown previously that in the Xenopus laevis extract DNA replication system, a high fraction of parental histones (H3-H4)2 are retained at their starting positions following DNA replication (Madamba et al., 2017). In contrast, in the SV40 DNA replication system, parental (H3-H4)2 are dispersed from the original location onto newly replicating chromatin. It was proposed that the CMG helicase, which is replaced by large T antigen in SV40 DNA replication system, is one of the factors involved in directing parental (H3-H4)2 to its starting position. We speculate that the CMG-Ctf4-Pol1 complex may serve as a “molecular ruler” that directs parental (H3-H4)2 tetramers to their original locations on lagging strands, whereas other proteins such as Pol ε help parental (H3-H4)2 assume the original nucleosome position at leading strands.
Functional implications of the utilization of distinct factors for the transfer of parental (H3-H4)2 to replicating DNA strands
In addition to Mcm2, we have recently shown that Dpb3 and Dpb4, two subunits of the leading strand DNA polymerase, Pol ε, facilitate the transfer of parental (H3-H4)2 to leading strands (Yu et al., 2018). Thus, replisomes may serve as the distribution center for parental histones to both replicating leading and lagging strands. The division of labor in the transfer of parental (H3-H4)2 to leading and lagging strands likely has two implications. First, this will ensure that parental (H3-H4)2 complexes are stably transferred to both leading and lagging strands. In this way, chromatin states marked by modifications on parental (H3-H4)2 will be maintained following DNA replication in both daughter cells. Second, we speculate that the utilization of different factors in parental histone transfer for leading and lagging strands will endow cells with the ability to switch chromatin states during development and/or during asymmetric cell division. Stem cells including cancer stem cells undergo asymmetric cell divisions (Morrison and Kimble, 2006). During the asymmetric division of Drosophila male germ cells, which is an extreme case, it has been shown that the daughter cell harboring ‘stemness’ properties will receive mostly parental (H3-H4)2 and the differentiated daughter cell contains mostly new (H3-H4)2 (Tran et al., 2012). If the Mcm2-Ctf4-Pol α pathway were somehow inactivated during replication of a particular domain, this would lead to high enrichment of parental histone (H3-H4)2 complexes onto leading strands, which would provide a mechanism for one daughter cell to receive more parental (H3-H4)2 than the other cell at that specific locus, thus establishing asymmetric histone distribution in the particular part of the genome.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zhiguo Zhang (zz2401@cumc.columbia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All yeast strains used in this study were of the W303 (leu2-3, 112 ura3-1 his3-11, trp1-1, ade2-1 can1-100) genetic background except the histone H3-tag switch strain (CYC662, which is S228C background, see Table S1). Mutagenesis was performed using PCR-based methods (Noguchi et al., 2008) or using the CRISPR/Cas9 plasmid pML104 (Laughery et al., 2015) along with the primers described in Table S2 for strains ASC118 and ASC131.
METHOD DETAILS
Yeast cell culture, chromatin immunoprecipitation (ChIP) and eSPAN
Yeast cells were synchronized and cultured following the standard protocol (Dunham et al., 2015). ChIP and eSPAN sample collection was performed following as described briefly below (Gan et al., 2017; Yu et al., 2014). Yeast cells were synchronized by adding alpha factor at 5μg/ml to an exponentially growing culture in YPD medium at OD600 = 0.4-0.5. Cells were arrested for 3 hours at 25°C and then released into fresh YPD medium containing 400 μg/ml BrdU with 200 mM HU for 45 minutes or without HU for 40 minutes. For the histone H3 Recombination-Induced Tag Exchange (RITE) experiments, overnight cultures grown in YPD in the presence of hygromycin B (200 μg/mL, Invitrogen) were diluted 1:10 into two fresh YPD flasks, with or without hygromycin B. After a 32h incubation to reach the G0 state, cells were collected by centrifugation at 3000 rpm for 10 minutes and the culture with hygromycin B was transferred to another flask containing depleted medium without hygromycin B. To induce recombination of the H3 tag, 1 μM β-estradiol (E-8875, Sigma-Aldrich) was added to the culture, and cells were grown for 16 additional hours. Cells were then diluted 1:25 in fresh YPD media containing 200 mM HU and 400 μg/ml BrdU at 30°C for 90 minutes.
Freshly-made formaldehyde was added to each culture at 1% at 25°C for 20 minutes and then quenched with 0.125M glycine for 5 minutes. After harvesting by centrifugation, cells were lysed with glass beads in ChIP lysis buffer (50 mM HEPES, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, and 0.1% sodium deoxycholate). For the H3K4me3 and H3K56ac ChIP and eSPAN experiments, chromatin was fragmented using MNase digestion to map nucleosomes (Wal and Pugh, 2012). Briefly, the pellet was washed twice with NP buffer (1.6 M sorbitol, 2 mM CaCl2, 5mM MgCl, 50 mM NaCl, 14 mM β-mercaptoethanol, 10 mM Tris-HCl (pH 7.4), 0.075% NP-40, 5 mM spermidine). A proper amount of MNase was added to each sample with incubation at 37°C for 20 minutes to ensure that the majority of chromatin was mono- and di-nucleosomes. The reaction was stopped with the addition of 5μl 0.5M EDTA. ¼ volume 5X ChIP lysis buffer was added to the digested chromatin and samples were incubated on ice for 30 minutes. For the Ctf4, Pol1, and Mcm6 ChIP and eSPAN experiments, chromatin was fragmented using sonication (12 cycles at 15 seconds on, 30 seconds off, Bioruptor Pico, Diagenode). In all cases, after clarification by centrifugation, soluble chromatin was immunoprecipitated with specific antibodies, including anti-H3K4me3 antibody (ab8580), anti-H3K56ac antibody (Han et al., 2007), anti-Ctf4 antibody (Karim Labib), and antibodies against epitopes fused with H3, MCM6, or Pol1 (anti-HA antibody: 12CA5, anti-T7 antibody: A190-117A BETHYL, and anti-Flag antibody: F1804). ChIP DNA was recovered using Chelex-100 (Nelson et al., 2006).
BrdU immunoprecipitation was performed as briefly described below (Gan et al., 2017; Yu et al., 2014). ChIP or total DNA was first incubated for 5 minutes at 100°C for denaturation and then immediately placed on an ice water bath for 5 minutes. DNA was diluted ten times with BrdU IP solution (PBS, 0.0625% Triton X-100(v/v), 6.7 μg/mL Escherichia coli tRNA, 0.17 μg/mL BrdU antibody (BD Bioscience)) and incubated for two hours at 4°C. Afterward, 20 μl of Protein G beads (GE Healthcare) were added to each sample and incubated for an additional hour at 4°C. After washing Protein G beads extensively, DNA was eluted with 100 μl of 1XTE buffer containing 1% SDS and incubation at 65°C for 15 minutes. Eluted DNA was purified using a Qiagen MinElute PCR Purification kit. Quantitative PCR method was used to test ChIP or eSPAN DNA using primers amplifying the early replication origin ARS607 and distal site ARS607+14kb primers (Han et al., 2010). The ssDNA libraries were prepared using the Accel-NGS 1S Plus (Cat no. 10096 Swift Bioscience) kit.
Sequence mapping and data analysis
The sequence mapping, nucleosome mapping, ChIP-ssSeq and eSPAN analysis was performed as described briefly below (Gan et al., 2017; Yu et al., 2018). The ssDNA libraries were sequenced using the paired-end method by Illumina sequencing platforms (Hi-seq 2000 and 2500 machine) at Mayo Clinic and Columbia University.
Sequence reads were first mapped to the yeast reference genome (sacCer3) with the Bowtie2 software (Langmead and Salzberg, 2012). Only paired-end reads with both ends mapped correctly were selected for continued analysis. Self-developed Perl programs were used to separate the forward and reverse reads following the reference genome. Genome-wide read coverage was calculated by BEDTools (Quinlan and Hall, 2010) and in-house Perl programs.
To obtain the nucleosome occupancy, 120-170bp DNA fragments calculated by the paired-reads were used. In order to increase sensitivity, DNA fragment length was then shortened to half of the nucleosome size (74bp) following the previous reference (Zhang et al., 2008). Finally, the count number of shortened DNA fragments across the whole genome was used to calculate nucleosome occupancy. Total sequence reads, including both Watson and Crick strands reads, were used to call BrdU-IP-ssSeq peaks using SICER software with a false discovery rate (FDR) cut-off of 0.01 (Zang et al., 2009). To calculate the average bias pattern of individual nucleosomes, total eSPAN sequence reads at each individual nucleosomes (±10 nucleosomes) surrounding the 134 early DNA replication origins were counted separately and were assigned to individual nucleosome positions determined previously (Brogaard et al., 2012). The log2 ratio of Watson strand reads over Crick strand reads at each nucleosome position was used to obtain the average bias pattern of eSPAN after normalization against the corresponding BrdU-IP-ssSeq. Similarly, to analyze the bias pattern of eSPAN peaks at individual origins, we calculated the log2 ratios of the Watson strand reads over Crick strand reads at each nucleosome position of each of the 134 early replication origins (±10 nucleosomes).
The calculation of relative amounts of parental or new histones at leading or lagging strands between mutant strains and WT determined was as follows: the corresponding BrdU-IP-ssSeq peaks were used to identify replication regions; the eSPAN reads were split into left and right halves on the basis of the location of the replication origin; each replication origin was separated into the following four quadrants: Watson strand at the left (WL) and the right (WR) of each origin; Crick strand at the left (CL) and the right (CR) of each origin; finally, the equation: log2 (number of sequencing reads in mutant strain divided by number of sequence reads in wild type) was used to calculate the relative amount of parental and new H3 in mutant cells compared to wild type cells.
Analysis of silencing-loss at the HML locus using the CRASH assay
The apparent silencing-loss rate at the HML loci was measured in wild-type (JRY9628), mcm2-3A (ASC118), ctf4Δ (ASC127), and pol1-4A (ASC131) strains using the CRASH (Cre-reported altered states of heterochromatin) assay (Dodson and Rine, 2015; Janke et al., 2018). Briefly, 10 colonies of each strain were grown separately in YPD medium to saturation, diluted to OD600=0.01 in YPD, and grown for 5 hours at 30°C. For each colony, 50,000 events were collected using a BD Fortessa cytometer. The apparent silencing-loss rate at the HML locus was calculated by dividing the number of RFP+ GFP+ cells (cells that have recently undergone the Cre-mediated recombination leading to GFP expression but still contain RFP) by the total number of cells with the potential to lose silencing (RFP+ GFP− and RFP+ GFP+).
QUANTIFICATION AND STATISTICAL ANALYSIS
For peak calling, the false discovery rate (FDR) was calculated by SICER software (Zang et al., 2009), and the cut-off value was set to 0.01. For the bias quantification, the binomial distribution was used to calculate the p value, and the cut-off value was set to 1×10−5. For the silencing-loss assay, p values were calculated using Welch’s t-test. The coefficient of correlation was calculated using Pearson's Correlation Coefficient method. Table S3 lists the number of repeats for each dataset including MNase-ssSeq, ChIP-ssSeq and eSPAN in wild type and different mutant yeast background.
Supplementary Material
Related to STAR methods. The number of repeats for each dataset
Highlights.
Histone binding by Mcm2 promotes parental H3-H4 transfer to lagging strands
The Mcm2-Ctf4-Polα axis mediates the parental histone transfer to lagging strands
Parental H3-H4 is likely transferred to the original location after DNA replication
Acknowledgments
We thank Dr. Richard Baer and Rebecca Burgess for editing and comments on this manuscript, Dr. Sontao Jia for discussion, and Dr. Jasper Rine for yeast strains. This study was supported by NIH grant (R35GM118015 to Z.Z.), the MRC (MC_UU_12016/13 to KL) and the Wellcome Trust (reference 102943/Z/13/Z to KL).
Footnotes
DATA AND SOFTWARE AVAILABILITY
All raw and analyzed sequencing data have been deposited in the NCBI with a GEO number (GSE118580).
Conflict of interests: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ayyagari R, Gomes XV, Gordenin DA, and Burgers PM (2003). Okazaki fragment maturation in yeast. I. Distribution of functions between FEN1 AND DNA2. J Biol Chem 278, 1618–1625. [DOI] [PubMed] [Google Scholar]
- Bell SP, and Dutta A (2002). DNA replication in eukaryotic cells. Annu Rev Biochem 71, 333–374. [DOI] [PubMed] [Google Scholar]
- Bell SP, and Labib K (2016). Chromosome Duplication in Saccharomyces cerevisiae. Genetics 203, 1027–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogaard K, Xi LQ, Wang JP, and Widom J (2012). A map of nucleosome positions in yeast at base-pair resolution. Nature 486, 496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgers PMJ, and Kunkel TA (2017). Eukaryotic DNA Replication Fork. Annu Rev Biochem 86, 417–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos EI, Stafford JM, and Reinberg D (2014). Epigenetic inheritance: histone bookmarks across generations. Trends Cell Biol 24, 664–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R, et al. (2013). The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 27, 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman RT, and Struhl G (2017). Causal role for inheritance of H3K27me3 in maintaining the OFF state of a Drosophila HOX gene. Science 356, 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devbhandari S, Jiang J, Kumar C, Whitehouse I, and Remus D (2017). Chromatin Constrains the Initiation and Elongation of DNA Replication. Mol Cell 65, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson AE, and Rine J (2015). Heritable capture of heterochromatin dynamics in Saccharomyces cerevisiae. Elife 4, e05007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas ME, Ali FA, Costa A, and Diffley JFX (2018). The mechanism of eukaryotic CMG helicase activation. Nature 555, 265–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham MJ, Gartenberg MR, and Brown GW (2015). Methods in yeast genetics and genomics : a Cold Spring Harbor Laboratory course manual / Maitreya J. Dunham, University of Washington, Marc R. Gartenberg, Robert Wood Johnson Medical School, Rutgers, The State University of New Jersey, Grant W. Brown, University of Toronto, 2015 edition / edn.
- Ehrenhofer-Murray AE, Kamakaka RT, and Rine J (1999). A role for the replication proteins PCNA, RF-C, polymerase epsilon and Cdc45 in transcriptional silencing in Saccharomyces cerevisiae. Genetics 153, 1171–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrin C, Maman JD, Diamante A, Pellegrini L, and Labib K (2018). Histone H2A-H2B binding by Pol alpha in the eukaryotic replisome contributes to the maintenance of repressive chromatin. EMBO J [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foltman M, Evrin C, De Piccoli G, Jones RC, Edmondson RD, Katou Y, Nakato R, Shirahige K, and Labib K (2013). Eukaryotic replisome components cooperate to process histones during chromosome replication. Cell Rep 3, 892–904. [DOI] [PubMed] [Google Scholar]
- Fu YV, Yardimci H, Long DT, Ho TV, Guainazzi A, Bermudez VP, Hurwitz J, van Oijen A, Scharer OD, and Walter JC (2011). Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell 146, 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambus A, van Deursen F, Polychronopoulos D, Foltman M, Jones RC, Edmondson RD, Calzada A, and Labib K (2009). A key role for Ctf4 in coupling the MCM2–7 helicase to DNA polymerase alpha within the eukaryotic replisome. EMBO J 28, 2992–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan H, Yu C, Devbhandari S, Sharma S, Han J, Chabes A, Remus D, and Zhang Z (2017). Checkpoint Kinase Rad53 Couples Leading- and Lagging-Strand DNA Synthesis under Replication Stress. Mol Cell 68, 446–455 e443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu R, Yuan Z, Bai L, de Luna Almeida Santos R, Sun J, Zhang D, Yurieva O, Li H, and O'Donnell ME (2017). Structure of eukaryotic CMG helicase at a replication fork and implications to replisome architecture and origin initiation. Proc Natl Acad Sci U S A 114, E697–E706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth A, Rocha W, Verreault A, and Almouzni G (2007). Chromatin challenges during DNA replication and repair. Cell 128, 721–733. [DOI] [PubMed] [Google Scholar]
- Han J, Li Q, McCullough L, Kettelkamp C, Formosa T, and Zhang Z (2010). Ubiquitylation of FACT by the cullin-E3 ligase Rtt101 connects FACT to DNA replication. Genes Dev 24, 1485–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Zhou H, Horazdovsky B, Zhang K, Xu RM, and Zhang Z (2007). Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Science 315, 653–655. [DOI] [PubMed] [Google Scholar]
- Huang H, Stromme CB, Saredi G, Hodl M, Strandsby A, Gonzalez-Aguilera C, Chen S, Groth A, and Patel DJ (2015). A unique binding mode enables MCM2 to chaperone histones H3-H4 at replication forks. Nat Struct Mol Biol 22, 618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke R, King GA, Kupiec M, and Rine J (2018). Pivotal roles of PCNA loading and unloading in heterochromatin function. Proc Natl Acad Sci U S A 115, E2030–E2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurat CF, Yeeles JT, Patel H, Early A, and Diffley JF (2017). Chromatin Controls DNA Replication Origin Selection, Lagging-Strand Synthesis, and Replication Fork Rates. Mol Cell 65, 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langston LD, Zhang D, Yurieva O, Georgescu RE, Finkelstein J, Yao NY, Indiani C, and O'Donnell ME (2014). CMG helicase and DNA polymerase epsilon form a functional 15-subunit holoenzyme for eukaryotic leading-strand DNA replication. Proc Natl Acad Sci U S A 111, 15390–15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprell F, Finkl K, and Muller J (2017). Propagation of Polycomb-repressed chromatin requires sequence-specific recruitment to DNA. Science 356, 85–88. [DOI] [PubMed] [Google Scholar]
- Laughery MF, Hunter T, Brown A, Hoopes J, Ostbye T, Shumaker T, and Wyrick JJ (2015). New vectors for simple and streamlined CRISPR-Cas9 genome editing in Saccharomyces cerevisiae. Yeast 32, 711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, and Allis CD (2013). Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 340, 857–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Xu Z, Leng H, Zheng P, Yang J, Chen K, Feng J, and Li Q (2017). RPA binds histone H3-H4 and functions in DNA replication-coupled nucleosome assembly. Science 355, 415–420. [DOI] [PubMed] [Google Scholar]
- Luciano P, Dehe PM, Audebert S, Geli V, and Corda Y (2015). Replisome function during replicative stress is modulated by histone h3 lysine 56 acetylation through Ctf4. Genetics 199, 1047–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, and Richmond TJ (1997). Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260. [DOI] [PubMed] [Google Scholar]
- Madamba EV, Berthet EB, and Francis NJ (2017). Inheritance of Histones H3 and H4 during DNA Replication In Vitro. Cell Rep 21, 1361–1374. [DOI] [PubMed] [Google Scholar]
- Martin C, and Zhang Y (2007). Mechanisms of epigenetic inheritance. Curr Opin Cell Biol 19, 266–272. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, and Kimble J (2006). Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 441, 1068–1074. [DOI] [PubMed] [Google Scholar]
- Nakayama J, Allshire RC, Klar AJ, and Grewal SI (2001). A role for DNA polymerase alpha in epigenetic control of transcriptional silencing in fission yeast. EMBO J 20, 2857–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JD, Denisenko O, and Bomsztyk K (2006). Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1, 179–185. [DOI] [PubMed] [Google Scholar]
- Noguchi C, Garabedian MV, Malik M, and Noguchi E (2008). A vector system for genomic FLAG epitope-tagging in Schizosaccharomyces pombe. Biotechnol J 3, 1280–1285. [DOI] [PubMed] [Google Scholar]
- Noguchi Y, Yuan Z, Bai L, Schneider S, Zhao G, Stillman B, Speck C, and Li H (2017). Cryo-EM structure of Mcm2–7 double hexamer on DNA suggests a lagging-strand DNA extrusion model. Proc Natl Acad Sci U S A 114, E9529–E9538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryk N, Dalby M, Wenger A, Stromme CB, Strandsby A, Andersson R, and Groth A (2018). MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science. [DOI] [PubMed] [Google Scholar]
- Poli J, Tsaponina O, Crabbe L, Keszthelyi A, Pantesco V, Chabes A, Lengronne A, and Pasero P (2012). dNTP pools determine fork progression and origin usage under replication stress. EMBO J 31, 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragunathan K, Jih G, and Moazed D (2015). Epigenetics. Epigenetic inheritance uncoupled from sequence-specific recruitment. Science 348, 1258699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom M, Dennehey BK, and Tyler JK (2010). Chaperoning histones during DNA replication and repair. Cell 140, 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richet N, Liu D, Legrand P, Velours C, Corpet A, Gaubert A, Bakail M, Moal-Raisin G, Guerois R, Compper C, et al. (2015). Structural insight into how the human helicase subunit MCM2 may act as a histone chaperone together with ASF1 at the replication fork. Nucleic Acids Res 43, 1905–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samora CP, Saksouk J, Goswami P, Wade BO, Singleton MR, Bates PA, Lengronne A, Costa A, and Uhlmann F (2016). Ctf4 Links DNA Replication with Sister Chromatid Cohesion Establishment by Recruiting the Chl1 Helicase to the Replisome. Mol Cell 63, 371–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra-Cardona A, and Zhang Z (2018). Replication-Coupled Nucleosome Assembly in the Passage of Epigenetic Information and Cell Identity. Trends Biochem Sci 43, 136–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon AC, Zhou JC, Perera RL, van Deursen F, Evrin C, Ivanova ME, Kilkenny ML, Renault L, Kjaer S, Matak-Vinkovic D, et al. (2014). A Ctf4 trimer couples the CMG helicase to DNA polymerase alpha in the eukaryotic replisome. Nature 510, 293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, and Whitehouse I (2012). Intrinsic coupling of lagging-strand synthesis to chromatin assembly. Nature 483, 434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter B, Tong A, Chang M, Yu L, Brown GW, Boone C, and Rine J (2004). The origin recognition complex links replication, sister chromatid cohesion and transcriptional silencing in Saccharomyces cerevisiae. Genetics 167, 579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran V, Lim C, Xie J, and Chen X (2012). Asymmetric division of Drosophila male germline stem cell shows asymmetric histone distribution. Science 338, 679–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verzijlbergen KF, Menendez-Benito V, van Welsem T, van Deventer SJ, Lindstrom DL, Ovaa H, Neefjes J, Gottschling DE, and van Leeuwen F (2010). Recombination-induced tag exchange to track old and new proteins. Proc Natl Acad Sci U S A 107, 64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa F, Simon AC, Ortiz Bazan MA, Kilkenny ML, Wirthensohn D, Wightman M, Matak-Vinkovic D, Pellegrini L, and Labib K (2016). Ctf4 Is a Hub in the Eukaryotic Replisome that Links Multiple CIP-Box Proteins to the CMG Helicase. Mol Cell 63, 385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wal M, and Pugh BF (2012). Genome-Wide Mapping of Nucleosome Positions in Yeast Using High-Resolution MNase ChIP-Seq. Nucleosomes, Histones & Chromatin, Pt B 513, 233–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, and Moazed D (2017). DNA sequence-dependent epigenetic inheritance of gene silencing and histone H3K9 methylation. Science 356, 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Long C, Chen X, Huang C, Chen S, and Zhu B (2010). Partitioning of histone H3-H4 tetramers during DNA replication-dependent chromatin assembly. Science 328, 94–98. [DOI] [PubMed] [Google Scholar]
- Yeeles JTP, Janska A, Early A, and Diffley JFX (2017). How the Eukaryotic Replisome Achieves Rapid and Efficient DNA Replication. Mol Cell 65, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Gan H, Han J, Zhou ZX, Jia S, Chabes A, Farrugia G, Ordog T, and Zhang Z (2014). Strand-Specific Analysis Shows Protein Binding at Replication Forks and PCNA Unloading from Lagging Strands when Forks Stall. Mol Cell 56, 551–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Gan H, Serra-Cardona A, Zhang L, Gan S, Sharma S, Jojansson E, Chabes A, Xu RM, and Zhang Z (2018). A mechanism for preventing asymmetric histone segregation onto replicating DNA strands Science, In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Gan H, and Zhang Z (2017). Both DNA Polymerases delta and epsilon Contact Active and Stalled Replication Forks Differently. Mol Cell Biol 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang C, Schones DE, Zeng C, Cui K, Zhao K, and Peng W (2009). A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25, 1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Shin H, Song JS, Lei Y, and Liu XS (2008). Identifying positioned nucleosomes with epigenetic marks in human from ChIP-Seq. BMC Genomics 9, 537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Shibahara K, and Stillman B (2000). PCNA connects DNA replication to epigenetic inheritance in yeast. Nature 408, 221–225. [DOI] [PubMed] [Google Scholar]
- Zhu W, Ukomadu C, Jha S, Senga T, Dhar SK, Wohlschlegel JA, Nutt LK, Kornbluth S, and Dutta A (2007). Mcm10 and And-1/CTF4 recruit DNA polymerase alpha to chromatin for initiation of DNA replication. Genes Dev 21, 2288–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Related to STAR methods. The number of repeats for each dataset