

Abstract
Background:
The mouse mammary tumor virus (MMTV) encodes a functional signal peptide (SP), a cleavage product of envelope and Rem proteins. SP interacts with a 3’ cis-acting RNA element, the Rem-responsive element (RmRE), to facilitate expression of both unspliced genomic (gRNA) and spliced mRNAs. An additional RmRE, has been proposed at the 5’ end of the genome, facilitating nuclear export of the unspliced gRNA, whereas the 3’ RmRE could facilitate translation of all other mRNAs, including gRNA.
Results:
To address this hypothesis, a series of mutations were introduced into a 24-nt region found exclusively in the unspliced gRNA. Mutant clones using MMTV or hCMV promoters were tested in both transient and stable transfections to determine their effect on gRNA nuclear export, stability, and translation. Nuclear export of the gRNA was affected only in a small mutant subset in stably-transfected Jurkat T cells. Quantitative real-time RT-PCR of Actinomycin D-treated cells expressing MMTV revealed that multiple mutants were severely compromised for RNA expression and stability. Both genomic and spliced nuclear RNAs were reduced, leading to abrogation of Gag and Env protein expressed from unspliced and spliced mRNAs, respectively. RT-PCRs with multiple primer pairs indicated failure to elongate genomic MMTV transcripts beyond ~500 nt compared to the wild-type in a cell line dependent manner.
Conclusions:
MMTV contains a novel cis-acting element downstream of the major splice donor critical for facilitating MMTV gRNA elongation and stability. Presence of a mirror repeat within the element may represent important viral/host factor binding site(s) within MMTV gRNA.
Keywords: MMTV, Rem, 5’ RmRE, Transcript elongation, RNA nuclear export, RNA stability element.
Graphical Abstract
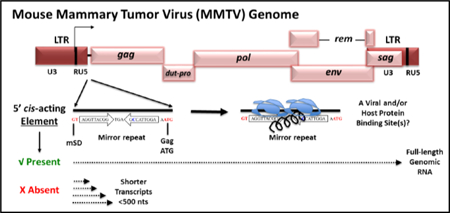
Introduction
The mouse mammary tumor virus (MMTV) is the prototypic oncogenic member of the genus Betaretrovirus of the Retroviridae family that causes breast cancer and lymphomas in mice [1]. The single-stranded RNA genome encodes for four canonical genes found in all retroviruses: gag, pro, pol, and env. Additionally, MMTV specifies three regulatory/accessory genes, rem, dut, and superantigen (sag), which encode proteins that regulate viral gene expression, facilitate successful infection of quiescent cells, and transmission in mice (reviewed by [2]). The rem gene encodes a precursor, which is cleaved into a signal peptide (SP), which interacts with cis-acting sequences, the Rem-responsive element (RmRE), to increase Gag and Pol expression from MMTV RNA [3, 4]. SP is a homolog of the Rev/Rex/Rec/Rej proteins encoded by lentiviruses such as human, simian, and feline immunodeficiency viruses (HIV/SIV/FIV) as well as other retroviruses such as human T cell leukemia virus (HTLV), human endogenous retrovirus K (HERV-K), and Jaagsiekte sheep retrovirus (JSRV) [5–11].
Expression of the MMTV structural and accessory genes is further controlled by cis- acting regulatory elements within the unique 3’ region (U3) of the viral long terminal repeats (LTRs). Elements within U3 include promoters, enhancers, and transcription factor binding sites, hormone response elements (HREs), as well as negative regulatory elements (NREs) important for basal, hormone-inducible, and tissue-specific gene expression, a hallmark of MMTV gene expression [12–17]. MMTV differentially regulates its own gene expression in a tissue-specific manner, inhibiting virus expression in lymphocytes, but responding to the differentiated state of the mammary epithelial cells, the cell type responsible for MMTV transmission from mothers to offspring through milk. Together, these and many other features make MMTV a powerful vector system for human gene therapy vector development[18].
We have studied MMTV gene expression to understand how virus replication leads to tumor induction, and how a modified genome might be exploited as gene therapy vectors.Binding of SP protein to its cognate cis-acting element, RmRE, located at the 3’ end of the viral genome [19, 20] facilitates the nuclear export and expression of full length RNA [3, 4]. However, SP also has post-nuclear export functions, such as translation [21]. Further, we expressed MMTV Gag/Pol proteins by substitution of the RmRE with the MPMV CTE in expression constructs [22]. Although this strategy has been used successfully for efficiently expressing HIV-1, SIV, and FIV Gag/Pol proteins [23–25], the absence or presence of the CTE in our system did not affect nuclear export of MMTV gRNA used for Gag/Pol expression. Instead, the inclusion of the CTE in these MMTV expression plasmids restored the translation of the Gag/Pol proteins [22]. This observation suggested that these Gag/Pol expression plasmids contained a cis-acting element required for the nuclear export of MMTV gRNA, but lacked the 3’ RmRE needed for translation, which could be compensated by the MPMV CTE. A similar observation was made when MMTV SP and other Rev-like proteins were tested for their ability to facilitate nuclear export of RNAs containing the luciferase gene within the 3’ end of the MMTV genome [21]. Protein expression, but not nuclear export, of the unspliced luciferase reporter RNA was affected by the presence of the 3’ RmRE [21]. Another study indicated that MMTV Gag production requires the 3’ RmRE and splice sites flanking gag in vector constructs together with Rem expression [26]. These observations strongly suggested that the Rem/RmRE regulatory system has multiple functions, including both nuclear export and post-nuclear export activities.
In a complex retrovirus such as HIV-1, an additional 5’ Rev-responsive element (5’ RRE) that binds Rev has been postulated [27, 28]. Our previous experiments suggested that MMTV has two RmREs: a 5’ RmRE present at the 5’ end of gRNA that is required for nuclear export of unspliced RNA and a 3’RmRE present in all MMTV mRNAs that is needed for efficient translation [4, 21]. To explore this hypothesis, we introduced deletion and substitution mutations at the 5’ end of the MMTV genome (between SD1 and the Gag start codon) present only in the full-length gRNA. Transient and stable transfections of mutant clones revealed a major defect in overall expression of spliced and unspliced MMTV mRNAs; however, contrary to our expectations, the primary defect occurred through reduced transcript elongation as well as poor RNA stability. These results reveal that the 5’ end of the MMTV genome harbors a cis-acting element(s) containing viral and/or cellular protein binding site(s) that control the transcription and elongation of viral transcripts, reminiscent of the Tat/TAR system of HIV-1 [29–31].
Results
Experimental Design.
To test for a potential 5’ RmRE present within the 5’ UTR of MMTV genome, a series of deletion and substitution mutations were introduced into the small 24 nt region from the 5’ splice donor site (SD1) to the gag start codon, a sequence found exclusively in the gRNA synthesized from a replication-competent molecular clone of MMTV, HYB-MTV [32]. This region is highly conserved among various MMTV strains with only the GR strain showing a two-nucleotide variation (Fig. S1). Since the MMTV promoter has a low basal activity in most cell types without hormone induction, the U3 region of HYB-MTV was substituted with the constitutively active hCMV promoter-enhancer (Fig. 1). This design has been used successfully to express MMTV sub-genomic RNAs for packaging studies in a single-round replication assay [33–35]. The molecular clones SQ5 to SQ9 all contained deletions in the test region from SD1 to the gag gene ATG. SQ5 had a complete deletion of the entire 24 bp region, whereas SQ6 deletion removed the first 12 bp of the region. The SQ7 mutation removed 12 bp from the 3’ half of the region, the SQ8 mutation removed the first 6 bp, and the SQ9 mutation removed 9 bp in the middle of the region. SQ10-SQ14 were substitution mutants that contained heterologous sequences in place of the deletions described in SQ5-SQ9 (Fig. 1). The introduced mutations were confirmed by sequencing, and the molecular clones were used in transient transfections to analyze their effect on nuclear export of full-length gRNA and its translation into Gag/Pol proteins.
Figure 1. Illustration of the mutations introduced into the putative 5’ RmRE along with the MMTV transcripts expressed using specific splice sites.
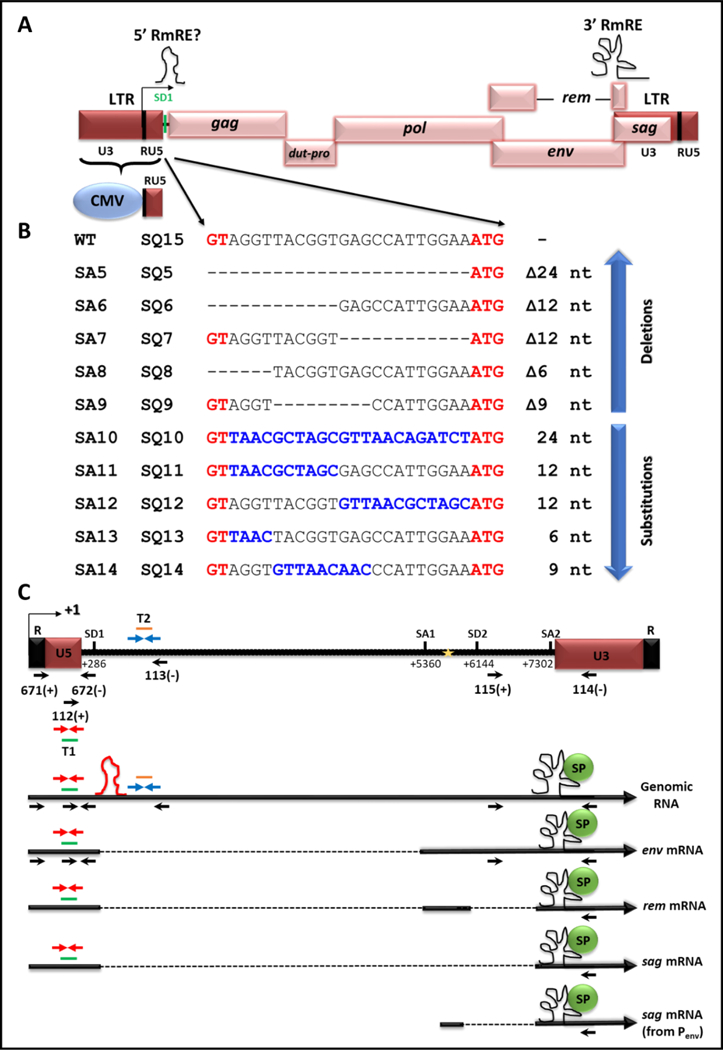
(A) Schematic representation of the MMTV genome with and without the hybrid (CMV/R/U5) LTR. Locations of the 3’ RmRE as well as that of the putative 5’ RmRE are demarcated on the genome with schematics of higher order structures. (B) Depiction of mutations introduced between SD1 and gag start codon (ATG) and cloned into the full length wild type (WT) clone, HYB-MTV (SA series of clones), or cloned into MMTV with the hybrid hCMV/R/U5 5’ LTR (SQ series of clones). The sequences in black represent the WT sequences, whereas the sequences in red represent the SD1 (GT) and gag initiation codon (ATG). Dashes represent the deleted sequences, whereas sequences in blue denote substituted sequences. (C) Illustration of MMTV gRNA followed by the major transcripts produced from the use of the indicated splice sites. The mapped primers show the location of common primers used in this study. For the sake of clarity, the words OTR and OFM have been omitted from the primer names, showing only primer numbers (see Table 1 for primer sequences). SD1, major splice donor; SD2, splice donor in the env gene. Gold star; the internal promoter in the env gene. The red stem loop depicts the location of the putative 5’ cis-acting element. The black structural element depicts the 3’ Rem responsive element (RmRE). T1, Taqman Assay 1 used to quantitate all MMTV mRNAs; T2, Taqman Assay 2 used to quantitate the full-length gRNA.
The wild-type (WT) and mutant constructs containing the desired mutations (SQ5- SQ14) were co-transfected into HEK293T cells with the luciferase reporter vector, pGL3C, to monitor transfection efficiencies. Three-days post transfection, the transfected cells were harvested, and RNAs from nuclear (NUC) and cytoplasmic (CYT) fractions were prepared for RT-PCR analysis. Lysates were also prepared for protein expression studies. Prior to cDNA preparation, the RNA samples were treated with DNase I to prevent amplification of genomic or plasmid DNA contaminating the RNA fractions and tested by PCR amplification using GAPDH-specific and MMTV-specific primers (Table 1). PCR amplification confirmed that all RNA samples were devoid of any contaminating DNA (Fig. 2A; panels I-IV). Next, cDNAs were prepared and analyzed for the quality of the subcellular fractionation by using β-actin primers specific for unspliced and spliced RNA (Table 1). As expected, both spliced and unspliced β -actin were observed in NUC fractions, whereas CYT fractions revealed low levels of unspliced products (Fig. 2B; panels I-IV). These controls revealed slight leakage between the two compartments during fractionation, but consistency among samples indicated that slight contamination was insufficient to affect the interpretation of our results.
Table 1.
Description of primer sets used for the selective amplification of different MMTV and host transcripts.
| Target mRNAs | Primer Set Used and its Description |
|---|---|
| All MMTV mRNAs from the LTR promoter, including gRNA. | OTR671(+)/OTR672(−). Binds within 5’ R/PBS. |
| All MMTV spliced mRNAs except gRNA due to long length. | OFM112(+)/OFM114(−). Binds within 5’ U5 and 3’ U3 flanking the genome. |
| MMTV gRNA only. | OFM112(+)/OFM113(−). Binds a region flanking SD1 (5’ U5 and Gag ORF) |
| MMTV spliced mRNAs from the LTR promoter. | OFM112(+)/OFM114(−). Binds within 5’ U5 and 3’ U3 flanking the genome. |
| MMTV spliced mRNAs from the Env promoter. | OFM115(+)/OFM114(−). Binds downstream of SD2 and 3’ U3 region. |
| All MMTV mRNAs, both spliced and unspliced. |
 Taqman assay 1 (T1). Binds within 5’ U5 (nt 1192- _ 1259) and can detect a minimum transcript size of 86 nt.
Taqman assay 1 (T1). Binds within 5’ U5 (nt 1192- _ 1259) and can detect a minimum transcript size of 86 nt. |
| Unspliced MMTV gRNA only. |
 Taqman assay 2 (T2). Binds within gag, 270 nt 3’ from SD1 (nt 1729–1791) and can detect a minimum transcript size of 618 nt.
Taqman assay 2 (T2). Binds within gag, 270 nt 3’ from SD1 (nt 1729–1791) and can detect a minimum transcript size of 618 nt. |
| GAPDH host mRNA. | ORT581(+)/OTR582(−). Detects host gapdh gene or mRNA. |
| Spliced β-actin mRNA. | OTR580(+)/OTR581(−). Detects spliced β-actin mRNA. |
| Unspliced β-actin mRNA. | OTR582(+)/OTR581(−). Detects unspliced β-actin gene or mRNA. |
Figure 2: A region between SD1 and the gag ATG is important for MMTV mRNA expression.
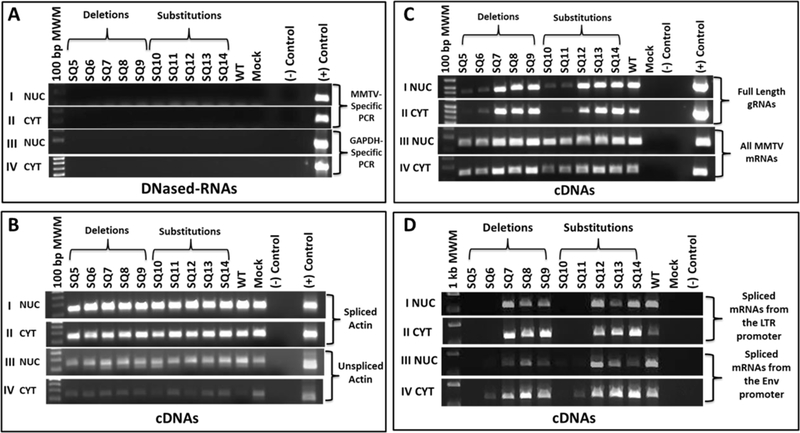
(A) DNase-treatment of nuclear (NUC) and cytoplasmic (CYT) RNAs were tested by PCR. Panels I & II: MMTV-specific primers (OTR671/OTR672) for plasmid DNA contamination; Panels III & IV: Cellular GAPDH primers for genomic DNA contamination. (B) RT-PCR amplification of spliced and unspliced ß-actin mRNA as a subcellular-fractionation control. Panels I & II: Spliced mRNA amplification of ß-actin in NUC and CYT fractions, respectively. Panels III & IV: Unspliced mRNA amplification of ß-actin in NuC and CYT cDNAs. (C) RT-PCR analysis of: Panels I & II, full-length (OFM112/OFM113) and Panels III & IV, all MMTV mRNAs (OTR671/OTR672) in NUC and CYT cDNA fractions. (D) RT-PCR analysis of other MMTV mRNAs using: Panels I & II, OFM112/OFM114 primers that detect spliced mRNAs from the LTR promoter. Expected sizes: env 2485 bp/rem 1324 bp/sag 542 bp. The band corresponding to sag mRNA is shown since the other bands could not be detected consistently; Panels III & IV: OFM115/OFM114 primers that detect spliced mRNAs from the env promoter. Expected sizes: env 1728 bp/rem and sag 567 bp. Once again, only the 567 bp band could be detected under the PCR conditions used.
Mutations within the 5’ UTR Reduce Full-Length gRNA Expression.
RT-PCR analysis of gRNA expression using primers both upstream and downstream of SD1 (OFM112/OFM113; Fig. 1 & Table 1) revealed compromised expression of full-length RNA from the SQ5 and SQ6 deletions and corresponding substitutions (SQ10 and SQ11), both in NUC and CYT cellular fractions (Fig. 2C; panels I & II). Next, expression of all MMTV mRNAs was analyzed using primers OTR671/OTR672 that bind to a region common to all MMTV mRNAs (within R and PBS of the 5’ end of the viral genome; Fig. 1 & Table 1). A slight decrease in all MMTV mRNAs expression was observed using the SQ5, SQ6, SQ10, and SQ11 mutants, as mentioned above compared to the WT after 30 cycles of PCR amplification (Fig. 2C; panels III & IV). Further, we investigated the expression of MMTV spliced mRNAs, including transcripts for env, sag, and rem using OFM112/OFM114 or OFM115/OFM114 (Fig. 1 & Table 1). Amplification with LTR-promoter-specific primers, which should only detect the spliced sag and rem mRNAs under the conditions used, revealed that the same subset of mutants (SQ5/SQ6 and SQ10/SQ11) showed a complete lack of MMTV-specific spliced gene expression compared to WT in both the NUC and CYT fractions (Fig. 2D; panels I & II). Amplification with Env-promoter-specific primers showed a similar result except SQ6 and SQ11 showed a weak signal in the CYT fraction (Fig. 2D; panel IV). As expected, splicing patterns were altered. These observations suggested that SQ5/SQ6 and SQ10/SQ11 were defective for both the full-length and spliced mRNA production.
To quantify the defect in gRNA expression being observed, we established a custom- made Taqman quantitative PCR (qPCR) assay. This assay employed primers and a FAM- MGB-labeled probe in the gag gene downstream of SD1 and, therefore, present only in MMTV gRNA but outside of introduced mutations (Table 1 & Fig. 1). A commercially available human β-actin VIC-MGB assay served as the endogenous control. Mutants SQ5, SQ6, SQ10, and SQ11 showed a delayed amplification of gRNA (appearing at cycle threshold (Ct) of ~23) compared to that observed for the WT or other mutants (appearing at Ct range of ~15) in both the NUC and CYT fractions compared to the WT or other mutants (Fig. 3A; panel I & 3B; panel IV). All constructs showed similar amplification of human β- actin RNA in both fractions (Fig. 3A; panel II & 3B; panel V). Relative quantification (RQ) of the gRNA expression between the mutants and WT was conducted by normalizing the MMTV expression with the endogenous β-actin control. Further normalization was performed to eliminate differences in transfection efficiencies of the various mutants. Compromised expression of gRNA was observed after transfection of SQ5, SQ6, SQ10, and SQ11 mutants in both the NUC and CYT compartments compared to the WT or other mutants that was statistically significant (p<0.001; Fig. 3A; panel III & 3B; panel VI). Reduced expression in the nuclear compartment suggested that the mutations in SQ5, SQ6, SQ10, and SQ11 affected the transcription or stability of the gRNAs (Fig. 3A; panel III).
Figure 3. RT-qPCR analysis of MMTV full-length gRNA reveals reduced mRNA levels expressed from mutants SQ5/SQ6 and SQ10/SQ11.
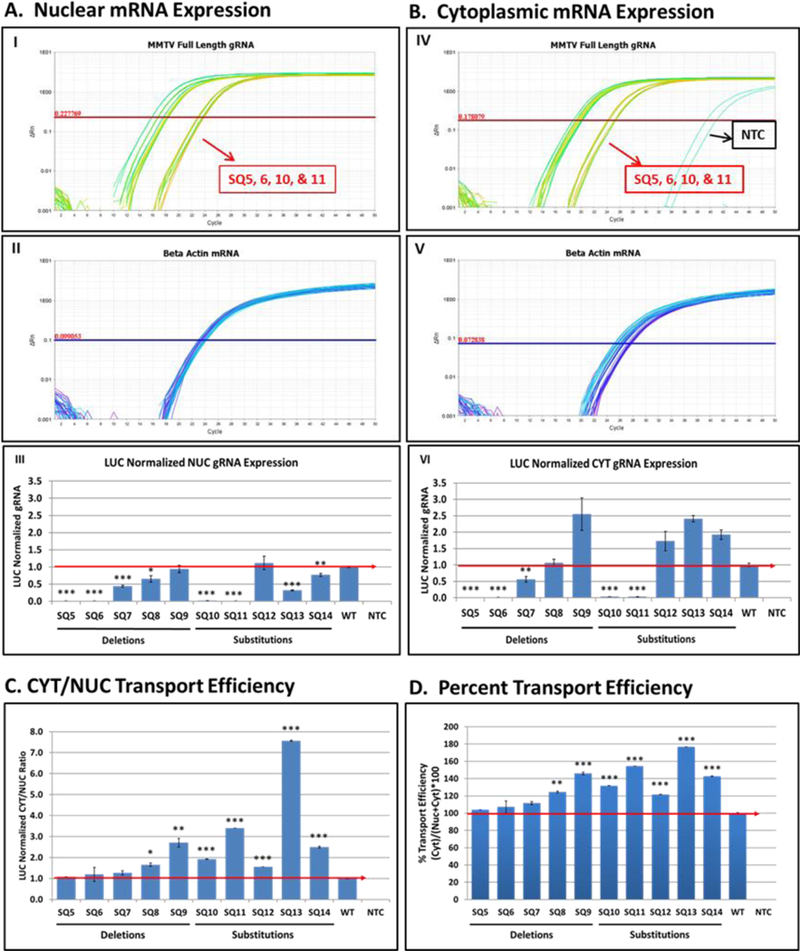
Relative quantification of MMTV full- length gRNA and ß-actin mRNA levels in HEK293T cells transfected with wild type (WT) and MMTV mutant clones using Taqman Assay 2 (see Fig. 1 and Table 1 for details). (A) Nuclear (NUC) and (B) cytoplasmic (CYT) levels of full-length gRNA (panels I and IV) and endogenous ß-actin mRNA expression (panels II and V). Panels III and VI are the LUC-normalized gRNA expression. Nucleocytoplasmic transport efficiency of gRNA using (C) CYT/NUC or (D) percentage method. The red line indicates the wild type levels of RNA. NTC, no template control. * indicates statistically significant differences between the WT and mutants (* = p <0.01; ** = p <0.01 but > 0.001; *** = p <0.001).
The 5’ UTR Mutations Did Not Affect Nuclear Export of MMTV gRNA in HEK293T Cells.
Next, we analyzed whether the nuclear export of full-length gRNA was affected in these mutants by calculating the CYT/NUC ratio of full-length gRNA levels using normalized RQ values relative to the wild type. The relative nuclear export efficiency of the deletion as well as substitution mutants appeared to be equivalent or better when compared to those from WT (Fig. 3C). The relative export efficiency was also verified by the percentages (Fig. 3D). Similar to the export ratio of full-length gRNA, the percentage export efficiency of mutants appeared similar or greater than that calculated for the WT. Both methods supported our conclusion that the deletion or substitution of this 5’ UTR region did not affect the nuclear export of full-length gRNAs in HEK293T cells (Fig. 3C & D).
The 5’ UTR Mutants are Defective for Gag/Pol and Env Expression.
Next, we determined the effect of introduced mutations on the translation of the Gag structural proteins expressed from the full-length gRNA as well as Env expressed from singly spliced mRNA. Total cellular protein lysates were prepared from transiently transfected HEK293T cells with the WT (SQ15) and mutant (SQ5-SQ14) clones. As expected, due to the reduced RNA expression, western blot analysis of these lysates using antibodies for either MMTV Gag (α-CA) or α-Env revealed a complete abrogation of protein expression in these mutants (SQ5, SQ6, SQ10, and SQ11) compared to the WT (Fig. 4A & B). The SQ7-SQ9 deletions and SQ12–14 substitutions showed little difference in Gag or Env expression. Although SQ8 had normal Gag expression, the Env precursor was decreased (Fig. 4B), consistent with loss of SD1 and spliced env mRNA. Equal protein loading was confirmed by β-actin expression. These results confirmed the defect in full-length and/or spliced mRNA expression of selected mutants within the MMTV 5’ UTR.
Figure 4. The 5’ UTR mutants are defective for Gag/Pol and Env protein expression.
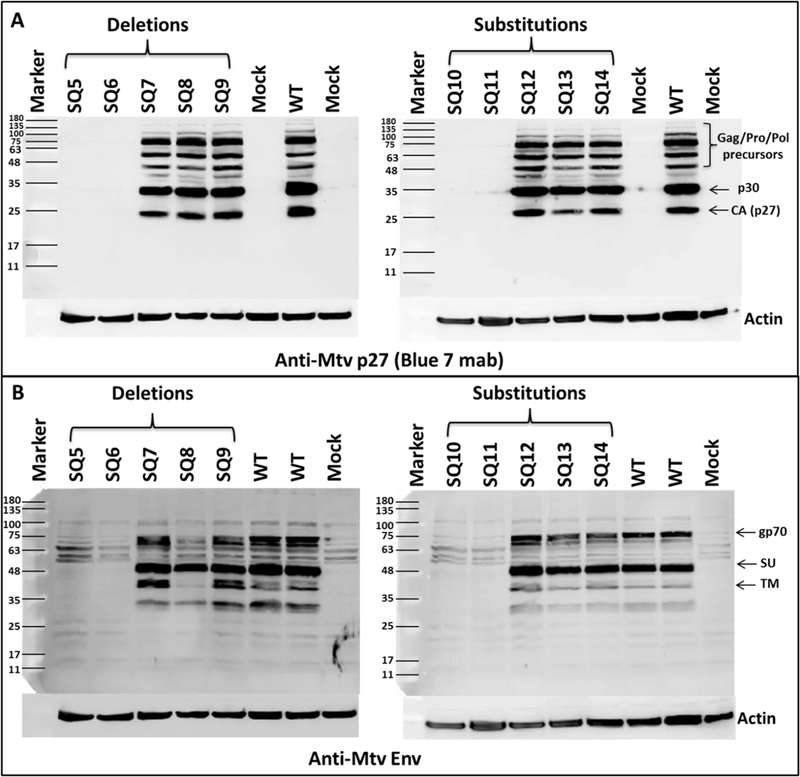
Western blot analysis of HEK293T cells transfected with the wild type (WT) and indicated mutant clones. (A) α- MMTV CA (Blue 7) antibody. (B) Env-specific antibody (α-MTV Env) with human β-actin serving as a loading control in both gels. Mock, HEK293T cells transfected with pcDNA3 plasmid alone.
MMTV Full-Length gRNA is Stable in Mutants with Reduced mRNA Levels.
We then determined whether the low expression of spliced and unspliced MMTV mRNAs in the nuclear fractions of SQ5, SQ6, SQ10, and SQ11 mutants was due to mRNA instability. Two mutants that exhibited the most pronounced effect on MMTV mRNA expression (SQ5 and SQ6) were transiently transfected into HEK293T cells for stability analysis. The cultures were treated with Actinomycin D (a classical transcription inhibitor) 48 hours post transfection. These cells were then processed for subcellular fractionation at 0, 8, 16, 24, and 48 hours post treatment. Total, NUC, and CYT RNA fractions were isolated and confirmed to lack contaminating DNA (Fig. 5A). Nuclear cDNAs were synthesized and analyzed for the stability of spliced and unspliced β-actin as well as all MMTV mRNAs as controls. As reported [36, 37], unspliced β -actin mRNA was unstable within 8 hours of treatment, whereas spliced β-actin mRNA was stable for 48 hours after Actinomycin D treatment (Fig. 5B, panels I & II). Similar to the spliced β-actin mRNA, we detected all MMTV mRNAs at similar levels at each time point, revealing the overall stability of MMTV mRNAs (Fig. 5B, panel III). The cDNAs were then analyzed for stability of the full-length unspliced gRNAs. The mutants SQ5 and SQ6 expressed much lower initial levels of full- length gRNAs in the nucleus compared to the WT (Fig. 5C, panel I), but once expressed, the mRNAs were stable for up to 48 hours and exported efficiently to the cytoplasm (Fig. 5C, panels I & II). Analysis of the total RNA fraction gave a similar result, with a greater decline of SQ5 and SQ6 gRNAs at 48 hours (Fig. 5C, panel III).
Figure 5: RNA stability analysis of 5’ UTR mutants.
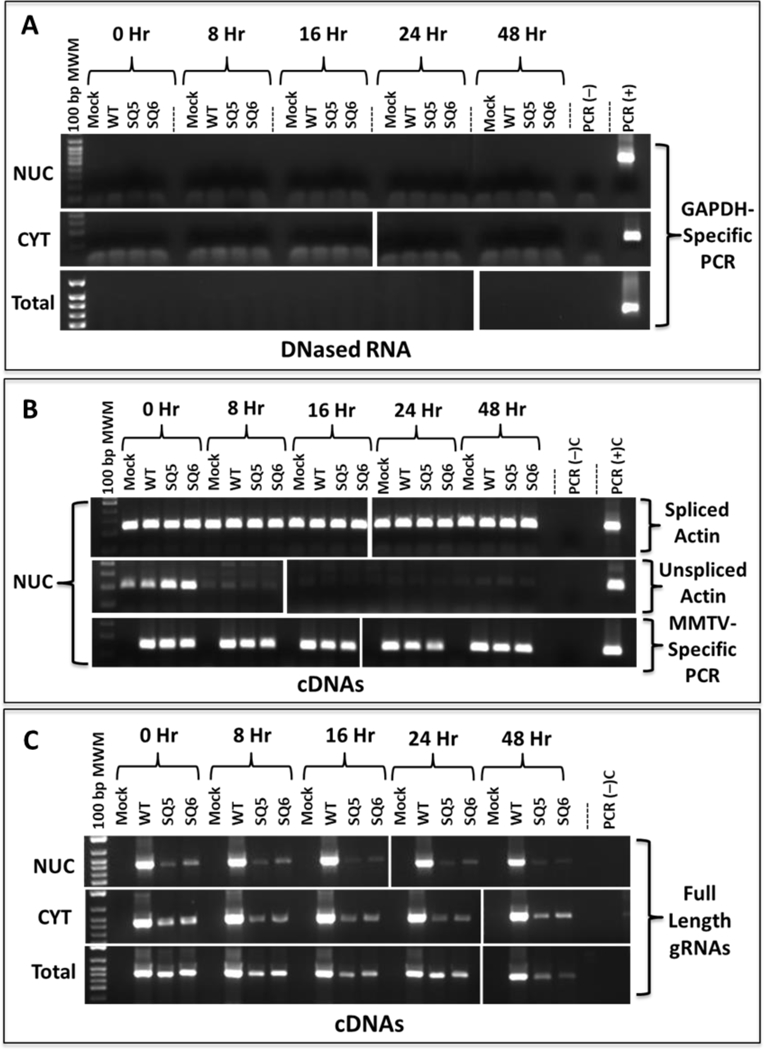
HEK293T cells transfected with mutant and wild type (WT) MMTV constructs and treated with Actinomycin D for various time points. Nuclear (NUC), cytoplasmic (CYT), and total extracted RNAs were DNase-treated and analyzed by RT-PCR for: (A) Lack of DNA contamination in Panels I, NUC, II, CYT, and III, Total RNA samples using GAPDH-specific primers. (B) Panel I, spliced B-actin (247 bp), panel II, unspliced ß-actin (203 bp), as well as panel III, all MMTV mRNAs (142 bp) in the NUC fractions using primers OTR671 and OTR672. (C) MMTV full-length genomic RNAs (723 bp) in panel I, NUC, panel II, CYT, and panel III, total RNA using primer pair OFM112/OFM113. Mock, HEK293T cells transfected with pcDNA3 plasmid alone.
To quantify the full-length gRNA levels with time, we tested the NUC, CYT, and total cDNA samples using our RT-qPCR Taqman assay designed for gRNA (Fig. 6). Analysis of the full-length gRNA levels after treatment with Actinomycin D revealed that SQ5 and SQ6 were highly deficient (by >two logs) in mRNA expression from the start at time zero prior to drug addition, compared to the WT in the nucleus (Fig. 6A). This difference remained essentially the same throughout the 24-hour time course of drug treatment for both the wild type and mutant RNAs in both the nucleus and the cytoplasm except for a small increase in SQ6 mRNA at 8 hours that went down again at 16 hours (Fig. 6B). The same could be observed in the analysis of the total RNA as well (Fig. 6C). Given the low level of expression being detected in the two mutants, the slight fluctuations being observed in their expression with time are probably insignificant. Based on these observations, we conclude that mutants SQ5 and SQ6 had defective steady-state expression of full-length gRNA in the nucleus of transfected cells, reflecting lower levels in the cytosol.
Figure 6: Lack of MMTV full length gRNA in the nucleus suggests defects in transcription.
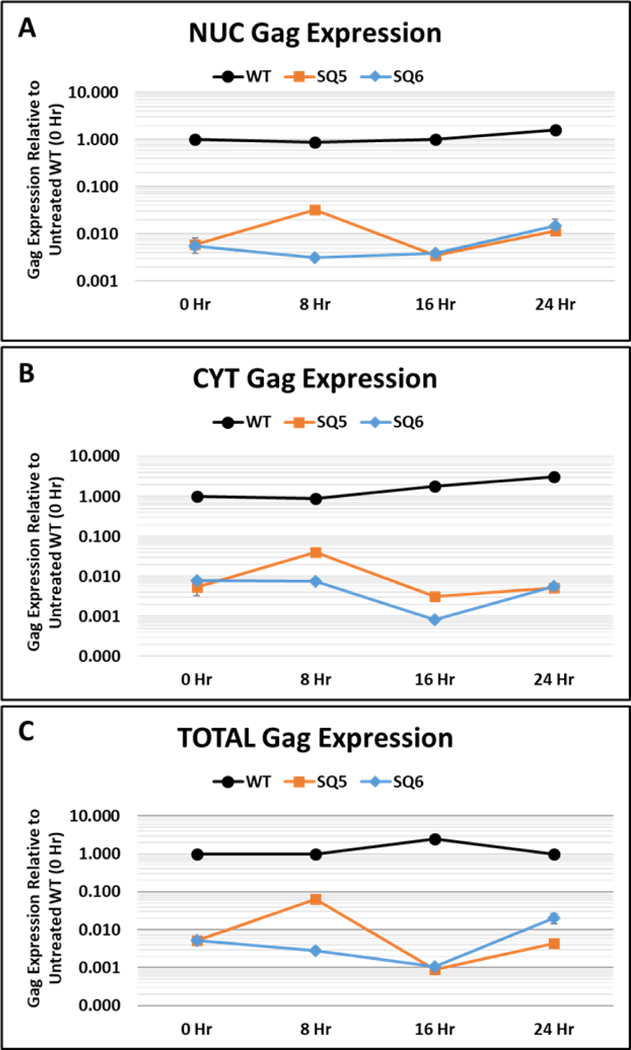
qPCR analysis of wild type (WT) full-length gRNA up to 48 hours after Actinomycin D treatment in: (A) nuclear (NUC), (B) cytoplasmic (CYT), and (C) total cDNAs prepared from HEK293T cells transiently transfected with the indicated clones. Full-length gRNA expression was quantified using the custom-made Taqman Assay 2 that binds between nt 1729–1791 of HYB-MTV genome, located at the 5’ end of the gag gene (see Fig. 1 and Table 1 for details).
The 5’ UTR Mutants Show Reduced RNA Levels in Stably-transfected Jurkat T Cells.
Since we had substituted the MMTV 5’ U3 region of the proviral LTR with the hCMV promoter, we also tested the same mutations after cloning into the intact 5’ MMTV LTR.These mutants, labelled SA5-SA14, correspond to previously tested mutants SQ5-SQ14 (Fig. 1B). To further ensure that the results were not due to the transient nature of the transfections, stable cell lines were prepared using Jurkat T cells, which are permissive for MMTV production and transmission to mice [38]. PCR on genomic DNA confirmed the presence of MMTV sequences in the gDNA prepared from all stably transfected clones (Fig. S2A; panel I). The stable cell lines were also analyzed for all MMTV mRNA expression by RT-PCR. Appearance of MMTV-specific PCR-amplified bands confirmed the expression of the WT and all mutant viruses in these stably transfected cell lines (Fig. S2A; panel II).
To determine whether the 5’ UTR mutations affected viral mRNA in Jurkat cells, nuclear and cytoplasmic RNA samples from the stable WT and mutant MMTV-expressing cells were confirmed to lack detectable DNA (Fig. S2B). After conversion into cDNAs, the integrity of the RNA fractionation was verified using primers for spliced and unspliced β-actin primers (Fig. S2C; panels I-IV). Subsequently, the NUC and CYT cDNAs were analyzed for MMTV gene expression using RT-PCR with MMTV-specific primers upstream of the SD1 to detect all MMTV mRNAs from the LTR promoter, OTR671/OTR672 (Fig. 1 and Table 1). Most mutant and WT NUC cDNAs gave the expected product, although weak bands were observed for SA5, SA6, and SA14 (Fig. S2D; panel I). Interestingly, in contrast to what was observed in 293T cells (Fig. 2C; panels III & IV), defective nuclear export of these RNAs was observed in the Jurkat stables; while some of the deletion mutants (SA7-SA9) could inefficiently transport these RNAs to the cytosol, none of the substitution mutants could, except weakly by the SA12 mutant (Fig. S2D; panel II). These NUC and CYT cDNAs were then analyzed for full-length gRNA using primers that should detect only the unspliced gRNA (Fig. 1 and Table 1). Consistent with our earlier observations, RT-PCR analysis revealed defective expression of gRNA in SA5 and SA6. However, additionally, most of the substitution mutants were also defective for gRNA expression within the nucleus, unlike in the HEK293T cells (Fig. S2D; panel III). Furthermore, no mutant RNA expression was observed in cytosolic fractions, although SA8 had nuclear RNA levels comparable to WT (Fig. S2D; panel III & IV). Based on these observations, we conclude that an export defect could be observed in Jurkat cells relative to that observed in HEK293T cells.
We also used real-time RT-qPCR analysis to confirm that most constructs expressed MMTV gDNA within 2-fold of the WT levels except for SA7, SA11, and SA13 (Fig. S3A). Using the same assay, the RNA expression (all MMTV mRNAs) of the clones was quantitated using total cellular RNA (Fig. S3B) and then normalized to the integrations observed (Fig. S3C). This analysis revealed that SA6 and its substitution mutant, SA11, had the lowest levels of viral RNA as observed for the hCMV-based clones (Figs. 2–6).
The 5’ UTR Mutant gRNAs Have Reduced Transcription Elongation.
Our previous results indicated a discrepancy in MMTV RNA expression depending on the primers used for RT-PCR (Fig. 2C; panel I vs panel III), suggesting a potential problem with mRNA elongation. To characterize this defect more systematically, we re-analyzed nuclear mRNAs expressed by the wild-type and mutant constructs (SQ5 and SQ6 from the transiently- transfected HEK293T cells) as well as SA5 and SA6 from the Jurkat stable transfectants following Actinomycin D treatment after 0, 8, 16, & 24 hours. The length of the mRNAs expressed by these constructs was tested by RT-PCR by maintaining the 5’ end primer (OTR671) while incrementally increasing the distance of the 3’ primer on the genomic RNA (Fig. 7A). Since these primer sets flanked SD1, the PCR product had the potential to detect and discriminate between the spliced and unspliced mRNAs, depending upon the transcript size. This experimental design allowed us to analyze MMTV nuclear transcripts for size and stability irrespective of the MMTV or CMV promoter. Control amplifications using primers for unspliced β-actin RNA confirmed that the amount of cDNA used for each PCR was equivalent (Fig. 7A).
Figure 7: Inability of 5’ UTR mutants to successfully elongate genomic transcripts.
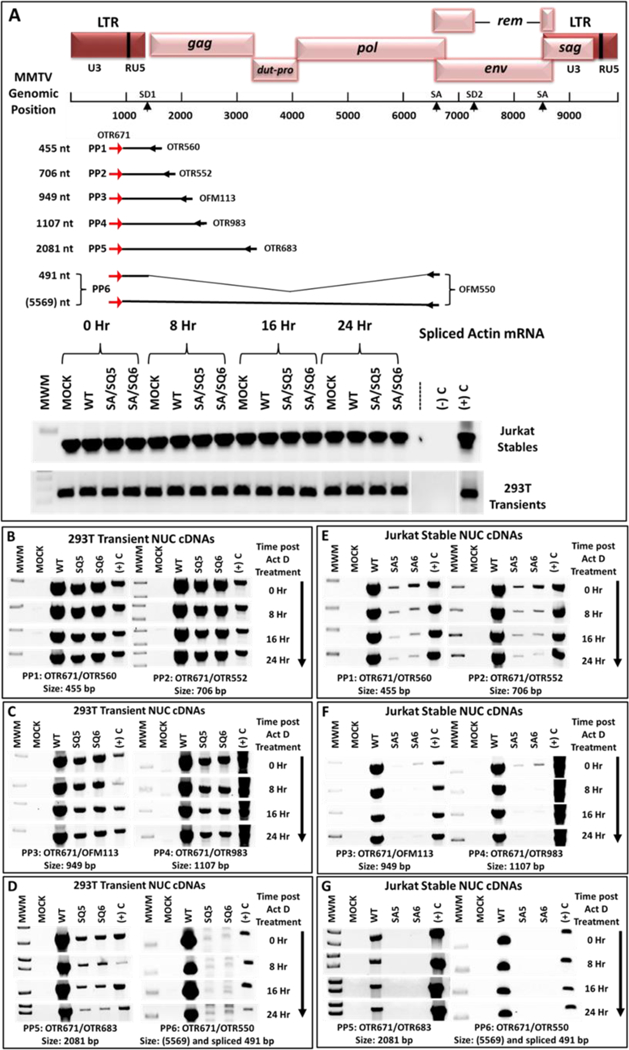
(A) Map of the MMTV genome showing the location and coordinates of the primers used for the transcript elongation analysis. Length of the gRNA transcripts expressed was analyzed using the six primer pairs (PP1-PP6) shown with their product size. The forward primer (OTR671) was common to all the primer pairs with a different reverse primer located progressively downstream from the transcriptional start site. PP6 could amplify both the genomic as well as the spliced mRNAs; however, due to the long length of the gRNA (5569 nt), this mRNA could not be amplified by PP6 under the conditions used. The detected band is the spliced mRNA only. Below the map are the control splice actin mRNA amplifications, showing that the amount of input cDNAs in each lane was equivalent. (B-D) RT-PCR analysis of cDNAs from HEK293T cell lines transiently transfected with MMTV WT or mutant SQ5 and SQ6 clones containing the CMV promoter. (E-G) RT-PCR analysis of cDNAs prepared from stable Jurkat cell lines expressing either the wild type (WT) or mutant SA5 and SA6 clones. Note that the results for length of transcript elongation are different in HEK293T and Jurkat cells. For ease of observation, the inverted images of the gels are presented.
Primer pairs PP1 and PP2 binding to the 5’ end of the gRNA efficiently detected the full-length gRNA from both the WT and mutant constructs SQ5 and SQ6 in HEK293T cells, revealing that mRNAs of at least 706 nt length were efficiently expressed and stable up to 24 hours in the nucleus of both the WT and mutant clones, with mutant levels slightly less than those of the WT (Fig. 7B). However, the 949 and 1107 nt products were expressed much less efficiently by the mutants compared to the WT, although these transcripts were stable up to 24 hours (Fig. 7C). Amplification of 2081 nt transcripts using PP5 revealed even lower efficiency of initial expression by the mutants at time zero in the nucleus; furthermore, the mutant mRNAs were unstable, as observed by the incremental decrease in the PCR product with time (Fig. 7D). Amplification of the 5569 nt product was not possible under the test conditions used, yet this primer set could detect the spliced mRNA (491 nt long) efficiently in the WT (Fig. 7D). In sharp contrast, this 491 nt mRNA was absent in both mutants, suggesting that spliced mRNA was absent due to failure of genomic RNA to be extended (Fig. 7A).
Analysis of 5’ UTR mutants in Jurkat cells revealed a similar result, although PP1 and PP2 could only amplify very low levels of an unstable mRNA in the nucleus of the mutantexpressing cells (Fig. 7E). Amplification with PP3 and PP4 further reduced the PCR product observed with mutant RNAs, and the products with PP5 and PP6 were undetectable at the initial time period (Fig. 7F and 7G). Although the results are not quantitative, 50 cycles of PCR on total cDNAs from both cell lines gave very similar results (data not shown). Thus, transcriptional elongation beyond ~700 nt in HEK293T cells and <450 nt in Jurkat cells was severely affected due to mutations within the 5’ UTR downstream of SD1.
Discussion
The study presented here investigated the hypothesis that the MMTV genome may have two RmREs: the previously described element at the 3’ end present in all the mRNAs and regulating their translation [21] and a putative element at the 5’ end present only in the genomic RNA and regulating its nuclear export. This idea stemmed from our earlier observation that the MPMV CTE allowed expression of MMTV Gag/Pol independently of the Rem/RmRE system [22]. Characterization of these constructs revealed that the CTE was critical for the translation of Gag/Pol proteins, but not for the nucleocytoplasmic transport of MMTV gag-pol mRNAs. In addition, MMTV luciferase plasmids that expressed the reporter in a Rem/RmRE-dependent manner did not affect nuclear export of the reporter RNA [21]. Our current results reveal that the MMTV genome harbors a novel 12 bp cis-acting element immediately downstream of the SD1 within the 5’ UTR. The introduced mutations affected transcriptional elongation and stability of full-length genomic transcripts. We speculate that this region acts as a binding site for either a cellular or virally-encoded protein that assists with transcription and elongation of viral transcripts. This observation is further substantiated by previous reports of an element within the MMTV 5’ UTR that enhances env gene expression [39]. Using subgenomic Env- and luciferase expression constructs, their results showed that incorporation of the 5’ UTR enhanced nuclear export as well as stability of env and luciferase mRNAs in an orientation-dependent manner. Unlike these previous experiments, our study used genomic constructs and mapped an essential element to within 12 nt of SD1 with clear effects on mRNA elongation and RNA stability.
Transcription factor-binding sites, such as the NF1, Oct1, AP-2, SATB1, Cux1 and TFIID, are observed in the MMTV U3 region (reviewed in [13, 16, 17, 40–44]. TFIID activates gene expression from RNA polymerase II-transcribed genes, whereas NF1, Oct1, and AP-2 are specific transcription factors that regulate expression from certain cellular and viral genes that contain their binding sites. In the case of MMTV, NF1, Oct1, and AP-2 induce not only basal transcription from the viral promoter, but further enhance hormone-dependent transcription, including chromatin remodeling (reviewed in [14, 45, 46]). The MMTV U3 region also contains multiple binding sites for SATB1 and Cux1 to repress transcription in T cells and undifferentiated mammary cells [17, 43, 44]. Therefore, the position of the 5’ UTR element identified here suggests a novel element outside of the U3 region and the previously identified initiator within the R region of the LTR [47]. The possibility remains that the 5’ UTR mutations affect more than one cis-acting element in this region. Transcription factor binding at the DNA level may disrupt basal transcription and elongation, whereas other factors may bind to a cis-acting stability element on viral RNA (reviewed in [48]).
Stability and elongation of gRNA is affected by the 5’ cis-acting element.
We tested the effect of the mutations on mRNA stability using the transcription inhibitor Actinomycin D. Our analysis revealed some effects on gRNA stability post Actinomycin D treatment in the nucleus of HEK293T cells transfected with clones with the hybrid CMV/RU5 promoter (Fig 5C; panel 1 & Fig. 7D). This effect was more pronounced in the Jurkat stable transfectants (Fig. 7E-G), due perhaps to the lower efficiency of MMTV LTR promoter. The primary effect of the 5’ UTR mutations was on MMTV gRNA levels and not mRNA nuclear export, leading to abrogation of Gag/Pol and Env protein expression (Fig. 4). Nevertheless, we observed some effects on RNA export to the cytoplasm in Jurkat cells stably transfected with some mutants, but not in 293T cells. Thus, some tissue specific factor(s) may be involved in this phenotype. Furthermore, it has been shown previously that MMTV env mRNA is not Crm1-dependent despite the fact that all MMTV mRNAs contain the 3’ RmRE [18, 20]. Therefore, Env expression should not have been affected by mutants outside the SD1. MMTV and HIV-1 differ in this respect since the partially spliced HIV-1 env mRNA is Rev/RRE-dependent [49, 50]. Since the 5’ UTR mutations severely reduced gRNA levels, expression of all partially spliced and multiply-spliced RNAs was also affected (Fig. 2C & D).
In mutants with decreased nuclear gRNA (SQ5, SQ6, SQ10, and SQ11), we amplified the MMTV RNA transcripts using primers that bound within R/PBS and hence to all MMTV mRNAs (Fig. 2C; panels III & IV), but not with primers that bound upstream of SD1, as well as the 3’ ends of the mRNAs (Fig. 2D; panels I & II). This observation suggested failure to elongate transcription. Although elongation of HIV-1 transcripts is ensured by the binding of the virally-encoded Tat protein to a TAR element present in the 5’ UTR [30], no such mechanism has been reported for MMTV.
Potential factor-binding sites in the MMTV 5’ UTR.
Our observation that only a 12 bp deletion compromised transcription in the SA6/SQ6 mutants also suggests that an RNA and/or DNA-binding site was disrupted. The ZASC1 cellular transcription factor, which belongs to the zinc finger family, has DNA-binding sites in the U3 region of murine leukemia virus (MLV) and HIV-1 LTRs to regulate transcription [51, 52]. ZASC1 binds to nearly three identical sites (a 12-bp DNA element) in the MLV LTR promoter to activate gene expression by interaction and stabilization of the pre-initiation complex within the promoter in both mouse and human cells [51]. In HIV-1, four possible ZASC1 binding sites have been verified, but the two almost palindromic sites downstream of the TATA box are more important for viral gene expression. Furthermore, in contrast to MLV, ZASC1 primarily affects HIV-1 transcriptional elongation by a TAR-independent recruitment of Tat on the HIV-1 core promoter in both tissue culture and primary T-cell lines [52]. The MMTV 5’ UTR element contains a mirror repeat that potentially regulates MMTV gene expression. This region has an imperfect repeat: gt AGGTTACgG tga GcCATTGGA aatg (shown in capital letters) separated by three nucleotides. The repeat sequence was in the reverse orientation on the same strand, unlike the well-known “inverted repeats” found as factor binding sites in the reverse orientation on the opposite strand. Our mutational analysis suggests that the first “repeat” sequence immediately downstream of the SD1 “gt” is the most important since its deletion or substitution in SQ6/SA6 & SQ11/SA11 was sufficient to create a phenotype comparable to deletion of the whole 24-bp region (in SQ5/SA5 and SQ10/SA10). Interestingly, deletion or substitution of the distal imperfect repeat had no effect on MMTV gene expression, suggesting lack of functional redundancy.
Named “mirror repeat”, the presence of the inverted repeat sequences on the same strand, prevents them from forming hairpins or cruciform (double hairpin) structures [53]. Instead, mirror repeats are part of a special type of triple DNA helix formation called H-DNA, which is found in polypyrimidine or polypurine-rich sequences, implicated in transcriptional regulation [54]. Recently, conserved triple-helix forming RNA stability elements have also been observed in the genomes of diverse DNA and RNA viruses involving polyadenylation signals that stabilize long noncoding RNAs by preventing their nuclear decay via deadenylation [55]. Thus, such a sequence upstream of MMTV genes may also be involved in the stability of MMTV transcripts by creating induced-platforms for the recruitment of cellular or viral factors or inhibiting transcript nuclear decay. Further studies are needed to clarify the nature of this region and to identify the factor(s) that regulate MMTV gene expression.
Material and Methods
Genome Nucleotide Numbering System.
The MMTV nucleotide numbering system refers to the nucleotide positions to HYBMTV, a molecular clone created by Shackleford and Varmus [32].
MMTV Full-length Genomic Clones.
A series of deletion and substitution mutations were introduced into the region from SD1 and Gag start codon in the context of the full-length genome of MMTV, HYB-MTV [32], using splice overlap extension (SOE) PCR [56]. The resulting amplified products containing the designed mutations were then cloned through a series of steps and the final clones were named SA5-SA14 (Fig. 1). Details of the steps of the various steps of cloning are available from the authors upon request.
In another set of clones, the U3 sequences in the 5’ LTR of the clones SA5-SA14 were substituted with the human cytomegalovirus (hCMV) promoter. Briefly, a ~891 nt fragment containing hCMV promoter sequence along with ‘R’ and ‘U5’ sequences of MMTV was removed from DA018 MMTV transfer vector [22] using SalI and BstEIII restriction endonucleases and replaced with a 1276-nt long fragment in the wild type as well as the mutant clones mentioned above to obtain the wild type (SQ15) and mutant clones (SQ5- SQ14), respectively.
Cell Lines and Culture Conditions.
The human embryonic kidney (HEK) 293T cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) (Hyclone, USA) supplemented with 10% fetal bovine serum (FBS; Hyclone). Jurkat human T lymphoma cells were grown in RPMI) 1640 (Hyclone), supplemented with 10% FBS. Culture media for these cell lines were further supplemented with 1% penicillin (10,000U/ml) and streptomycin (10,000ug/ml; Life Technologies, USA), and 0.1% gentamicin (50 mg/ml w/v solution) (Life Technologies).All cell lines were maintained in a water-jacketed incubator (Forma series II, Thermo Scientific) at 37°C and 5% CO2.
Transient and Stable Transfections.
The HEK 293T cells were transiently transfected using the Calcium Phosphate Transfection Kit (Invitrogen Life Technologies, USA) as described by the manufacturer. Briefly, 5 × 105 cells were plated per well of a 6-well plate the day before transfection. The test plasmid DNA (6 μg/well) and reporter plasmid DNA (pGL3C, 250 ng/well) were combined with the DNA cocktail buffer (525 μl/6 wells) and added dropwise to 525 μl of 2X HEPES-buffered saline (HBS) while bubbling the mixture. After a 30-minute room temperature incubation, the mixture was added to cells dropwise while swirling the plates, which were returned to the incubator subsequently. After ~24 hours post transfection, cells were washed with 1X phosphate buffer saline (PBS) (Hyclone) to remove DNA precipitates and fresh media was added. The transfected cells were harvested ~72 hours post transfection for isolation of RNA and proteins.
The Jurkat T cells were stably transfected by electroporation using Gene Pulser II Electroporator (BioRad, USA). The cells were cultured at 5 × 105 cells/ml of growth media ~24 hours before transfection. On the day of the transfection, 1.2 × 107 cells were resuspended in 500 ^l of serum-free RPMI medium and mixed gently with a DNA cocktail consisting of 40 μg of test plasmids and 250 ng of reporter plasmid (pGL3C) on ice. The mixture was then electroporated at 260 V, 950 μF and R10 (720 Ω) using 0.4 cm electroporation cuvettes (BioRad). The electroporated cells were allowed to recover by incubating on ice for 5–10 minutes and then re-plated in T-25 flasks in 5 ml of complete RPMI media containing 20% FBS. After 2–3 days, the stably-expressing cells were selected with media containing 0.8 mg/ml hygromycin antibiotic for 2–3 weeks. Periodically, dead cells were removed by slow speed centrifugation prior to stable transfectant outgrowth.
Subcellular Fractionation.
Subcellular fractionation was performed by spinning 2/3rd of the harvested transfected cells from a 6-well plate at low speed (2400 rpm) in a microcentrifuge for 2 minutes at 4oC. The resulting pellets were gently lysed in 800 μl of diethyl pyrocarbonate-treated RLN buffer (50 mM Tris-HCl, pH 8.0, 140 mM NaCl, 1.5 mM MgCl2) supplemented with 0.5% NP40. After a 2-minute incubation on ice, the lysed cells were subjected to centrifugation at 2400 rpm for 2 minutes at 4oC and 700 μl of the supernatant (cytoplasmic fraction) was carefully transferred to fresh microcentrifuge tubes, whereas the pellets (nuclear fraction) were rinsed with 1 ml of fresh RLN buffer and lysed in 1 ml TRIzol reagent (Invitrogen Life Technologies). The cytoplasmic fraction was spun again at 5000 rpm for 5 minutes at 4oC to remove residual nuclear components. The upper 600 μl of the supernatant was transferred to a fresh tube and spun again at 12,000 rpm for 5 minutes at 4oC. Finally, 500 μl of the clarified supernatant was mixed with 600 μl of TRIzol LS reagent (Invitrogen Life Technologies). Both the nuclear and cytoplasmic fractions in TRIzol were frozen at −80oC until processed for RNA isolation, as described by the manufacturer.
RT-PCR Assays.
After isolation, RNA was quantified using a Nanodrop spectrophotometer and 2 μg RNA from transfected 293T cells or 6 μg from Jurkat stables was treated with 2 units (U) of Turbo DNase I (Invitrogen Life Technologies) in the presence of 40 U of Recombinant RNasin (Promega) for 30 minutes at 37oC to ensure DNA-free preparations. The DNase I enzyme was inactivated using 1/10th volume of Turbo DNase Inactivation Reagent, and 1 μl of the reaction mix was subjected to 30 cycles of PCR to test for the presence of contaminating genomic or plasmid DNA using GAPDH (OFM24/OFM25) and/or MMTV-specific primers (OTR671/OTR672) (Tables 1 and S1). The DNase-treated RNA was then used to synthesize cDNA in a reverse transcription reaction. First strand cDNA synthesis was achieved by the addition of 1.5 μg of poly(dT17) or random dodecamers and 0.5 mM deoxynucleotide triphosphates (dNTPs). Primer annealing was performed at 70oC for 5 minutes, followed by immediate incubation on ice for 5 minutes. Reverse transcription was performed by the addition of 200 U of Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLV RT) (Invitrogen Life Technologies) and 40 U of RNase inhibitor at 37oC for 1 hour. Freshly prepared cDNAs were aliquoted and stored at −80oC, and 1–2 μl was used to check for the quality of cDNA using primers specific for spliced β-actin (OTR580/OTR581), unspliced β-actin (OTR581/OTR582) as well as for MMTV expression (OTR671/OTR672). All primers used for gene expression analysis or cloning in this study are described in Tables 1 and S1 and were commercially synthesized by Macrogen, South Korea.
PCRs were performed using the following general conditions: initial denaturation at 94oC for 2 minutes, 30 cycles of denaturation at 90oC for 45 seconds, primer annealing at 50–60oC for 45 seconds depending upon the primer melting temperature, and a final extension at 72oC for 45 seconds. The amplified PCR products were analyzed by electrophoresis at 100 volts on 0.8–2% agarose gels (Invitrogen Life Technologies), stained with ethidium bromide, and viewed using a gel documentation system (UVP Biospectrum 610 Imaging System).
Real-time Quantitative PCR (qPCR) Assays.
Quantitative PCR was used to calculate the level of expression and transport efficiency of all MMTV mRNAs as well as the unspliced genomic RNAs from the nucleus to cytoplasm using two different custom-made Taqman assays shown in Fig. 1 and described in Table 1. Expression of all MMTV mRNAs was quantitated using the previously validated and published Taqman Assay 1 that bound within HYB-MTV 5’ U5 region, (nt 1192–1259; [34,35], while expression of the gRNA was quantified using Taqman Assay 2 that bound within a region in MMTV Gag (nts1729–1791) present in both the wild type and mutant clones, but outside the region of introduced mutations. Specifically, it consisted of a FAM-labelled probe (nt 1752–1769), a forward primer (nt 1729– 1750F), and a reverse primer (nt 1791–1771). The human β-actin Endogenous Control Assay (VIC/MGB probe, Cat# 4326315E, Applied Biosystems (ABI), USA) was used as the control in assays where cellular cDNA (2 μl/sample) served as the template, whereas the human β-actin Endogenous Control Assay (FAM/MGB probe, Cat# 401846, ABI,) was used as the endogenous control with 50 μg gDNA/sample as the template. Standard PCR conditions were used for amplification in triplicates using QuantStudio™ 7 Flex Real-Time PCR System: a 2-minute initial incubation at 50oC and 94oC for 10 minutes to inactivate uracil N-glycosylase and to denature the template, followed by 50 cycles of denaturation at 94oC for 15 seconds and annealing/extension at 60oC for 1 minute for amplification. The relative quantification (RQ) values were obtained by normalization to the endogenous β-actin control using the -ΔΔCt method. These values were further normalized to the relative firefly luciferase expression per ^g of protein (RLUC/μg) to account for differences in transfection efficiencies and reported relative to the expression of wild type levels. To calculate the percentage of cytoplasmic RNA, the CYT expression of gRNA was divided by the total of CYT and NUC full-length gRNA levels and multiplied by 100.
mRNA Stability Assays.
The mRNA stability analysis was performed by treating transfected cells with 5 μg/ml of Actinomycin D (Sigma-Aldrich, USA) transcription inhibitor 48 hours post transfection. Cells were harvested at 0, 8, 16, 24, and 48 hours post treatment and resuspended in 1 ml of 1X PBS before dividing into three parts: 50 μl of cell suspension was used for firefly luciferase and protein quantification assay, 100 μl of cells were used for total RNA extraction, and 850 μl of cells were used for subcellular fractionation, as described earlier.
Western Blot Analysis.
Whole cell extracts were prepared in RIPA buffer (10 mM Tris-Cl [pH 8.0], 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, and 140 mM NaCl) supplemented per ml with 50 μl of β-mercaptoethanol and 1 mM PMSF (Sigma). Proteins were clarified from nuclear debris through centrifugation and quantified using the Bradford reagent, as described by the manufacturer. Each lysate (50 μg) was separated on 8–10% SDS polyacrylamide gels and transferred overnight at 30 volts at 4oC onto nitrocellulose membranes (GE Healthcare, USA). Membranes were blocked in 5% non-fat milk and incubated with primary antibodies (1:100 dilution of anti-MMTV CA (p27) Blue 7 [57] or 1:500 dilution of goat anti-Env (National Cancer Institute/Biological Carcinogenesis Branch [NCI/BCB] Repository, National Institutes of Health, USA) in 2% non-fat milk, followed by incubation in their respective horseradish peroxidase (HRP)-conjugated secondary antibodies. Chemiluminescent signal was detected using the ECL Plus western blotting substrate (Thermo Scientific, USA) as recommended by the manufacturer and captured using Typhoon FLA 9500 (GE Healthcare).
Statistical Analysis.
For determination of statistically significant differences between the wild type and mutant clones, the standard paired, two-tailed Students t-test was performed. A threshold p value of 0.01 was considered to be significant. The extent of significance was shown as either one, two, or three stars, depending upon the values obtained (* = p <0.01; ** = p <0.01 but >.001; *** = p <0.001).
Supplementary Material
Highlights:
A 12 nt cis-acting element upstream of Gag ATG is critical for MMTV gene expression
This element does not significantly affect nuclear export of gRNA in HEK293T cells
Mutations in element primarily affect gRNA transcription, elongation, & stability
The 5’ UTR element contains a mirror repeat with potential for protein binding
Acknowledgements
This study was supported by United Arab Emirates University (UAEU) Zayed Center for Health Sciences and Terry Fox Funds for Cancer Research grants (31R020 and 21M095, respectively) and College of Medicine & Health Sciences grant (NP 14–34) to TAR and in part by UAEU Start-up and Zayed Center for Health Sciences grants (31M122 and 31R122, respectively) and College of Medicine & Health Sciences grant (31M331) to FM. JPD was funded by the National Institutes of Health grant R01 CA167053. Funding for open access charge: UAE University.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication.As a service to our customers we are providing this early version of the manuscript.The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Dudley JP, Golovkina TV, Ross SR. Lessons Learned from Mouse Mammary Tumor Virus in Animal Models. ILAR J. 2016;57:12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ross SR. Mouse mammary tumor virus molecular biology and oncogenesis. Viruses. 2010;2:2000–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Indik S, Gunzburg WH, Salmons B, Rouault F. Mouse mammary tumor virus infects human cells. Cancer Res. 2005;65:6651–9. [DOI] [PubMed] [Google Scholar]
- [4].Mertz JA, Simper MS, Lozano MM, Payne SM, Dudley JP. Mouse mammary tumor virus encodes a self-regulatory RNA export protein and is a complex retrovirus. J Virol. 2005;79:14737–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cheng SM, Blume M, Lee SG, Hung PP, Hirsch VM, Johnson PR. The simian immunodeficiency virus (SIV) rev gene regulates env expression. J Med Primatol. 1990;19:167–76. [PubMed] [Google Scholar]
- [6].Hidaka M, Inoue J, Yoshida M, Seiki M. Post-transcriptional regulator (rex) of HTLV-1 initiates expression of viral structural proteins but suppresses expression of regulatory proteins. EMBO J. 1988;7:519–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Malim MH, Hauber J, Fenrick R, Cullen BR. Immunodeficiency virus rev trans-activator modulates the expression of the viral regulatory genes. Nature. 1988;335:181–3. [DOI] [PubMed] [Google Scholar]
- [8].Malim MH, Bohnlein S, Fenrick R, Le SY, Maizel JV, Cullen BR. Functional comparison of the Rev trans-activators encoded by different primate immunodeficiency virus species. Proc Natl Acad Sci U S A. 1989;86:8222–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Phillips TR, Lamont C, Konings DA, Shacklett BL, Hamson CA, Luciw PA, et al. Identification of the Rev transactivation and Rev-responsive elements of feline immunodeficiency virus. J Virol. 1992;66:5464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Palmarini M, Fan H. Molecular biology of jaagsiekte sheep retrovirus. Curr Top Microbiol Immunol. 2003;275:81–115. [DOI] [PubMed] [Google Scholar]
- [11].Lower R, Tonjes RR, Korbmacher C, Kurth R, Lower J. Identification of a Rev-related protein by analysis of spliced transcripts of the human endogenous retroviruses HTDV/HERV-K. J Virol. 1995;69:141–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bramblett D, Hsu CL, Lozano M, Earnest K, Fabritius C, Dudley J. A redundant nuclear protein binding site contributes to negative regulation of the mouse mammary tumor virus long terminal repeat. J Virol. 1995;69:7868–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gunzburg WH, Salmons B. Factors controlling the expression of mouse mammary tumour virus. Biochem J. 1992;283 ( Pt 3):625–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kusk P, John S, Fragoso G, Michelotti J, Hager GL. Characterization of an NF-1/CTF family member as a functional activator of the mouse mammary tumor virus long terminal repeat 5’ enhancer. J Biol Chem. 1996;271:31269–76. [DOI] [PubMed] [Google Scholar]
- [15].Liu J, Bramblett D, Zhu Q, Lozano M, Kobayashi R, Ross SR, et al. The matrix attachment region-binding protein SATB1 participates in negative regulation of tissue- specific gene expression. Mol Cell Biol. 1997;17:5275–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Grimm SL, Nordeen SK. Mouse mammary tumor virus sequences responsible for activating cellular oncogenes. J Virol. 1998;72:9428–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhu Q, Dudley JP. CDP binding to multiple sites in the mouse mammary tumor virus long terminal repeat suppresses basal and glucocorticoid-induced transcription. J Virol. 2002;76:2168–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Indik S Mouse mammary tumor virus-based vector for efficient and safe transgene delivery into mitotic and non-mitotic cells‥ CELL & GENE THERAPY 2016;2:589–98. [Google Scholar]
- [19].Mertz JA, Chadee AB, Byun H, Russell R, Dudley JP. Mapping of the functional boundaries and secondary structure of the mouse mammary tumor virus Rem-responsive element. J Biol Chem. 2009;284:25642–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mullner M, Salmons B, Gunzburg WH, Indik S. Identification of the Rem-responsive element of mouse mammary tumor virus. Nucleic Acids Res. 2008;36:6284–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mertz JA, Lozano MM, Dudley JP. Rev and Rex proteins of human complex retroviruses function with the MMTV Rem-responsive element. Retrovirology. 2009;6:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rizvi TA, Ali J, Phillip PS, Ghazawi A, Jayanth P, Mustafa F. Role of a heterologous retroviral transport element in the development of genetic complementation assay for mouse mammary tumor virus (MMTV) replication. Virology. 2009;385:464–72. [DOI] [PubMed] [Google Scholar]
- [23].Rizvi TA, Panganiban AT. Simian immunodeficiency virus RNA is efficiently encapsidated by human immunodeficiency virus type 1 particles. J Virol. 1993;67:2681–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rizvi TA, Schmidt RD, Lew KA, Keeling ME. Rev/RRE-independent Mason-Pfizer monkey virus constitutive transport element-dependent propagation of SIVmac239 vectors using a single round of replication assay. Virology. 1996;222:457–63. [DOI] [PubMed] [Google Scholar]
- [25].Browning MT, Schmidt RD, Lew KA, Rizvi TA. Primate and feline lentivirus vector RNA packaging and propagation by heterologous lentivirus virions. J Virol. 2001;75:5129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Boeras I, Sakalian M, West JT. Translation of MMTV Gag requires nuclear events involving splicing motifs in addition to the viral Rem protein and RmRE. Retrovirology. 2012;9:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gallego J, Greatorex J, Zhang H, Yang B, Arunachalam S, Fang J, et al. Rev binds specifically to a purine loop in the SL1 region of the HIV-1 leader RNA. J Biol Chem. 2003;278:40385–91. [DOI] [PubMed] [Google Scholar]
- [28].Greatorex JS, Palmer EA, Pomerantz RJ, Dangerfield JA, Lever AM. Mutation of the Rev-binding loop in the human immunodeficiency virus 1 leader causes a replication defect characterized by altered RNA trafficking and packaging. J Gen Virol. 2006;87:3039–44. [DOI] [PubMed] [Google Scholar]
- [29].Bannwarth S, Gatignol A. HIV-1 TAR RNA: the target of molecular interactions between the virus and its host. Curr HIV Res. 2005;3:61–71. [DOI] [PubMed] [Google Scholar]
- [30].Brady J, Kashanchi F. Tat gets the “green” light on transcription initiation.Retrovirology. 2005;2:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Sharp PA, Marciniak RA. HIV TAR: an RNA enhancer? Cell. 1989;59:229–30. [DOI] [PubMed] [Google Scholar]
- [32].Shackleford GM, Varmus HE. Construction of a clonable, infectious, and tumorigenic mouse mammary tumor virus provirus and a derivative genetic vector. Proc Natl Acad Sci U S A. 1988;85:9655–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rizvi TA, Kenyon JC, Ali J, Aktar SJ, Phillip PS, Ghazawi A, et al. Optimal packaging of FIV genomic RNA depends upon a conserved long-range interaction and a palindromic sequence within gag. J Mol Biol. 2010;403:103–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mustafa F, Al Amri D, Al Ali F, Al Sari N, Al Suwaidi S, Jayanth P, et al. Sequences within both the 5’ UTR and Gag are required for optimal in vivo packaging and propagation of mouse mammary tumor virus (MMTV) genomic RNA. PLoS One. 2012;7:e47088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Aktar SJ, Vivet-Boudou V, Ali LM, Jabeen A, Kalloush RM, Richer D, et al. Structural basis of genomic RNA (gRNA) dimerization and packaging determinants of mouse mammary tumor virus (MMTV). Retrovirology. 2014;11:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Johnson TR, Trojan J, Rudin SD, Blossey BK, Ilan J, Ilan J. Effects of actinomycin D and cycloheximide on transcript levels of IGF-I, actin, and albumin in hepatocyte primary cultures treated with growth hormone and insulin. Mol Reprod Dev. 1991;30:95–9. [DOI] [PubMed] [Google Scholar]
- [37].Leclerc GJ, Leclerc GM, Barredo JC. Real-time RT-PCR analysis of mRNA decay: half-life of Beta-actin mRNA in human leukemia CCRF-CEM and Nalm-6 cell lines. Cancer Cell Int. 2002;2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Mustafa F, Bhadra S, Johnston D, Lozano M, Dudley JP. The type B leukemogenic virus truncated superantigen is dispensable for T-cell lymphomagenesis. J Virol. 2003;77:3866–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hohenadl C, Gunzburg WH, Salmons B, Indik S. The 5’ leader sequence of mouse mammary tumor virus enhances expression of the envelope and reporter genes. J Gen Virol. 2012;93:308–18. [DOI] [PubMed] [Google Scholar]
- [40].Cordingley MG, Riegel AT, Hager GL. Steroid-dependent interaction of transcription factors with the inducible promoter of mouse mammary tumor virus in vivo. Cell. 1987;48:261–70. [DOI] [PubMed] [Google Scholar]
- [41].Toohey MG, Lee JW, Huang M, Peterson DO. Functional elements of the steroid hormone-responsive promoter of mouse mammary tumor virus. J Virol. 1990;64:4477–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mink S, Hartig E, Jennewein P, Doppler W, Cato AC. A mammary cell-specific enhancer in mouse mammary tumor virus DNA is composed of multiple regulatory elements including binding sites for CTF/NFI and a novel transcription factor, mammary cell-activating factor. Mol Cell Biol. 1992;12:4906–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zhu Q, Maitra U, Johnston D, Lozano M, Dudley JP. The homeodomain protein CDP regulates mammary-specific gene transcription and tumorigenesis. Mol Cell Biol. 2004;24:4810–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhu Q, Gregg K, Lozano M, Liu J, Dudley JP. CDP is a repressor of mouse mammary tumor virus expression in the mammary gland. J Virol. 2000;74:6348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zhao FQ. Octamer-binding transcription factors: genomics and functions. Front Biosci (Landmark Ed). 2013;18:1051–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chikhirzhina GI, Al’-Shekhadat RI, Chikhirzhina EV. [Transcription factors of the nuclear factor 1 (NF1) family. Role in chromatin remodelation]. Mol Biol (Mosk). 2008;42:388–404. [PubMed] [Google Scholar]
- [47].Fee BE, Steinke JW, Pierce J, Peterson DO. Initiation site binding protein and the initiator-like promoter element of mouse mammary tumor virus. Virology. 2002;302:185–94. [DOI] [PubMed] [Google Scholar]
- [48].Cheng J, Maier KC, Avsec Z, Rus P, Gagneur J. Cis-regulatory elements explain most of the mRNA stability variation across genes in yeast. RNA. 2017;23:1648–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hope TJ. The ins and outs of HIV Rev. Arch Biochem Biophys. 1999;365:186–91. [DOI] [PubMed] [Google Scholar]
- [50].Pollard VW, Malim MH. The HIV-1 Rev protein. Annu Rev Microbiol. 1998;52:491–532. [DOI] [PubMed] [Google Scholar]
- [51].Bruce JW, Hierl M, Young JA, Ahlquist P. Cellular transcription factor ZASC1 regulates murine leukemia virus transcription. J Virol. 2010;84:7473–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bruce JW, Reddington R, Mathieu E, Bracken M, Young JA, Ahlquist P. ZASC1 stimulates HIV-1 transcription elongation by recruiting P-TEFb and TAT to the LTR promoter. PLoS Pathog. 2013;9:e1003712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bikard D, Loot C, Baharoglu Z, Mazel D. Folded DNA in action: hairpin formation and biological functions in prokaryotes. Microbiol Mol Biol Rev. 2010;74:570–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mirkin SM, Frank-Kamenetskii MD. H-DNA and related structures. Annu Rev Biophys Biomol Struct. 1994;23:541–76. [DOI] [PubMed] [Google Scholar]
- [55].Tycowski KT, Shu MD, Borah S, Shi M, Steitz JA. Conservation of a triple-helix-forming RNA stability element in noncoding and genomic RNAs of diverse viruses. Cell Rep. 2012;2:26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gibbs JS, Regier DA, Desrosiers RC. Construction and in vitro properties of SIVmac mutants with deletions in “nonessential” genes. AIDS Res Hum Retroviruses. 1994;10:607–16. [DOI] [PubMed] [Google Scholar]
- [57].Purdy A, Case L, Duvall M, Overstrom-Coleman M, Monnier N, Chervonsky A, et al. Unique resistance of I/LnJ mice to a retrovirus is due to sustained interferon gamma- dependent production of virus-neutralizing antibodies. J Exp Med. 2003;197:233–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.