

Abstract
Thiopeptins are highly decorated thiopeptide antibiotics similar in structure to thiostrepton A and harbor two unusual features. All thiopeptins contain a thioamide, a rare moiety among natural products, and a subset of thiopeptins present with a piperidine in the core macrocycle rather than the more oxidated dehydropiperidine or pyridine rings typically observed in the thiopeptides. Here, we report the identification of the thiopeptin biosynthetic gene (tpn) cluster in Streptomyces tateyamensis and the gene product, TpnL, which shows sequence similarity to (deaza)flavin-dependent oxidoreductases. Heterologous expression of TpnL in the thiostrepton A producer Streptomyces laurentii led to the production of a piperidine-containing analogue. Binding studies revealed that TpnL preferentially binds the deazaflavin cofactor coenzyme F420, and in vitro reconstitution of TpnL activity confirmed that this enzyme is an F420H2-dependent dehydropiperidine reductase. The identification of TpnL and its activity establishes the basis for the piperidine-containing series a thiopeptides, one of the five main structural groups of this diverse family of antibiotics.
INTRODUCTION
Thiopeptides are ribosomally synthesized and post-translationally modified peptides with extensive chemical modifications that impart potent antiinfective properties, particularly against drug-resistant strains of Gram-positive pathogens.1 Despite the impressive antibacterial activity of thiopeptides, their clinical use has been stymied in large part by their low aqueous solubility. There is therefore interest in the discovery, design, and production of thiopeptide analogues with improved pharmacokinetic parameters.2,3 For example, semisynthetic derivatization of the thiopeptide GE2270A led to the development of an analogue that retained antibacterial efficacy and was improved in aqueous solubility.2 The elucidation of biosynthetic pathways of thiopeptide metabolites, especially those that include enzymes with additional biochemical activities and associated modifications, is essential to expand the biosynthetic toolkit available to produce derivatives that can combat the ever-growing threat of antibiotic-resistant bacterial pathogens.
To date, over 100 distinct thiopeptides have been identified and are classified as structural series a–e on the basis of the oxidation state of the central, six-membered nitrogenous ring in the core macrocycle (Figure 1A).1 Bearing a strikingly similar structure to the prototypical thiopeptide thiostrepton A (Figure 1B, 1), the thiopeptins (Figure 1B, 2–5) are a complex of thiopeptide antibiotics produced by Streptomyces tateyamensis ATCC 21389 (S. tateyamensis).4 Both 1 and 2–5 are 17 amino acids long, differing in sequence only in the first residue, and both include a second quinaldic acid-containing macro-cycle. The thiopeptins, however, are distinguished by a thioamide moiety and either a piperidine (series a) or dehydropiperidine (series b) ring in the core macrocycle.5 Thioamides are a rare feature in natural products, with only a handful of known examples, and only a few members of series a thiopeptides have been identified to date, which include 2, 3, and Sch 18640 (Figure 1B, 6).4–8 In addition to piperidine and dehydropiperidine rings, thiopeptides can also possess either a dihydroimidazopiperidine (Figure 1A, series c), pyridine (Figure 1A, series d), or hydroxypyridine (Figure 1A, series e).1 Recent studies established that the series d pyridine and core macrocycle are installed by a dedicated synthase that condenses two serine-derived dehydroalanine (Dha) residues of a linear precursor in a [4 + 2] cycloaddition, followed by the dehydration and elimination of the leader peptide to generate the aromatic nitrogenous heterocycle.9,10 The core macrocycle of series a and b thiopeptides also originates from the condensation of two Dha residues, although it is not yet understood how this macrocyclization process leads to retention of the N-terminal portion of the peptide and a more reduced piperidine or dehydropiperine ring.11 In this work, we identify the S. tateyamensis thiopeptin biosynthetic gene (tpn) cluster and establish the biosynthetic basis of a piperidine-containing series a thiopeptide.
Figure 1.

Thiopeptide structural series, example thiopeptide structures from series a and b, and the thiopeptin biosynthetic gene cluster. (A) Thiopeptide structural series a–e. R1−R4 represent the remaining thiopeptide structures. (B) Structures of thiostrepton A (1), the thiopeptins (2–5), and Sch18640 (6). The (dehydro)piperidine rings are shown in blue, and the (thio)amide moiety is shown in green, and the quinaldic acid moiety is shown in red. (C) Organization of the tpn and tsr biosynthetic gene clusters. Homologous genes are indicated by dashed lines. Abbreviations: dehydroalanine (Dha), dehydrobutyrine (Dhb), 4-(1-hydroxyethyl)quinoline-2-carboxylic acid (HEQ), dehydropiperidine (Pip), quinaldic acid moiety (Q), thiazole (Thz), and thiazoline (Tzn).
RESULTS
Identification of the Thiopeptin Biosynthetic Gene (tpn) Cluster.
To pinpoint the genes responsible for 2–5 biosyntheses, the S. tateyamensis genome was partially sequenced to 6.92 Mb on 2,838 contiguous fragments and scanned for a nucleotide sequence that could encode the core peptide (VASASCTTCICTCSCSS). The gene encoding the thiopeptin precursor peptide, tpnA, was identified, and the sequence of the tpn locus was fully assembled by sequencing the fosmid clones from an S. tateyamensis genomic library that were identified in a PCR screen for tpn genes. The tpn cluster encompasses 23 open reading frames very similar in composition and organization to the thiostrepton biosynthetic gene (tsr) cluster (Figure 1C and Table S1). On the basis of sequence similarity to proteins encoded in the tsr and other thiopeptide biosynthetic gene clusters (Table S1), TpnA, together with TpnBCDEFG, comprise the minimum set of proteins expected to be required to assemble the core thiopeptide scaffold. The thiazol(in)es are likely installed by TpnEFG. TpnF and TpnG are similar to leader peptide binding proteins and YcaO domain-containing cyclodehydratases, respectively, which operate in concert to form thiazoline rings from the amide backbone and Cys residues of the thiopeptide.10 The dehydrogenase TpnE is expected to oxidize the intermediate thiazoline to the thiazole.10,12 A split LanB-type dehydratase appears to be encoded by tpnBC and likely catalyzes the dehydrations to generate the Dha and dehydrobutyrine residues.10 Two Dha residues are likely coupled together by a putative [4 + 2] cyclase, TpnD, to form a dehydropiperidine and the thiopeptin core macrocycle.
The quinaldic acid moiety of 1, and likely of 2–5, is derived from L-tryptophan. TpnOPUVW are expected to convert the amino acid into 4-(1-hydroxyethyl)quinoline-2-carboxylic acid (HEQ) by a process that includes an oxidative ring expansion (Scheme S1).13–15 TpnO is similar to TsrM, a radical SAM and cobalamin-dependent methyltransferase that generates 2-methyl-L-tryptophan as the first step in this process (Scheme S1).16 The second step is catalyzed by an aminotransferase (Schemes S1 and S2).14 TpnW shares homology with histidinol-phosphate transaminases, demonstrating 40% identity (50% similarity) with the thiostrepton pathway transaminase TsrV.14 To determine if TpnW could be involved in 2–5 biosynthesis, we reconstituted TpnW activity in vitro. TpnW does convert 2-methyl-L-tryptophan to 3-(2-methylindolyl)pyruvate in the presence of α-keto acid acceptors 3-indolylpyruvate, p-hydroxyphenylpyruvate, and phenylpyruvate, but not in the presence of pyruvate, oxaloacetate, or α-ketoglutarate (Figures S1–S3). Similar preferences for α-keto acid acceptors were observed with TsrV.14,15 These results indicate that TpnW is a 2-methyl-L-tryptophan aminotransferase involved in HEQ biosynthesis for 2–5.
The quinaldic acid-containing macrocycle is likely installed in the thiopeptin scaffold by the actions of TpnHIQ.17–19 TpnI is similar to adenylyltransferases and is expected to attach HEQ to Thr12 of the peptide, likely followed by TpnH-catalyzed epoxygenation.14,17,19 TpnQ, similar to the macro-cyclase TsrB, is a candidate to mediate macrocyclization of the quinaldic acid containing loop through attack of the core peptide’s N-terminus upon the epoxide.18 The thiopeptins from S. tateyamensis possess either a methyl ester (2 and 4) or a carboxylic acid (3 and 5) at their C-termini; none containing the C-terminal amide observed in 1 have been reported. Accordingly, a homologue encoding the thiostrepton amidotransferase TsrT is absent.14 TpnT is similar to TsrP and may function as a carboxymethyltransferase to produce the C-terminal methyl ester.14,20 A homologue of the methylesterase TsrU is not present in the tpn cluster.20 Instead, TpnR, with similarity to other α/β-hydrolases, likely serves this role.
Three genes of the tpn cluster, tpnLMN, do not share homology with known thiopeptide biosynthetic genes and are therefore candidates for the thioamide and piperidine features unique to 2–5. TpnMN show sequence similarity to YcaO proteins and TfuA-like proteins, respectively, and are similar to the TvnHI pair encoded in the thioviridamide gene cluster, another thioamidated metabolite.7 The involvement of a YcaO–TfuA pair in the post-translational thioamidation of methyl-coenzyme A reductase in Methanosarcina acetivorans was recently established.21,22 The YcaO protein activates the peptide backbone in an ATP-dependent manner.22 Although the function of the TfuA protein is unclear, the YcaO and TfuA proteins are both required for the thioamidation of the peptide substrate.22 It is therefore likely that TpnM (YcaO protein) and TpnN (TfuA protein) work together to install the thiopeptin thioamide.
TpnL, on the other hand, shares homology to proteins annotated as FMN-dependent pyridoxine/pyridoxamine 5′-phosphate oxidases and other enzymes of the flavin/deazaflavin oxidoreductase subgroup B (FDOR-B).23 The only remaining thiopeptin modification unaccounted for is a reduction to produce the piperidine ring of the series a thiopeptins 2 and 3, and an oxidoreductase is a candidate enzyme for that step.
Heterologous Expression of TpnL in S. laurentii.
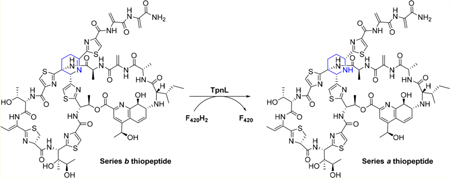
The structures of 1 and 2–5 are closely related, and we reasoned that TpnL may be able to intercept the appropriate thiostrepton biosynthetic intermediate as a substrate, generating a new metabolite and shedding light on the function of this protein. To this end, TpnL was heterologously expressed in Streptomyces laurentii ATCC 31255 (S. laurentii), a 1 producer.14 S. laurentii HI1 contains the integrative plasmid pSET1520-TpnL, expressing TpnL under control of the constitutive ermE* promoter, and S. laurentii HI0 houses an empty pSET1520 vector. S. laurentii HI1 still produced 1 (tR =23.8 min and [M + 2H]2+ m/z 833) but also produced the new thiopeptide metabolite 7 (tR = 23.7 min and [M + 2H]2+ m/z 834) with a mass increased by 2 Da relative to 1 (Figure 2A and Figure S4). 7 was isolated, and a molecular formula of C72H88N19O18S5 was determined by HR-ESI-MS (observed m/z 1666.5157 [M + H]+, calculated m/z 1666.5158). Additional MS/MS and NMR analyses indicated that 7, thiostrepton Aa, is a piperidine-containing analogue of 1 (Scheme 1 and Figures S5–S13 and Tables S2–S3). 1H−1H ROESY correlations of 7 between Pip-6-H and Pip-4-HB and between Pip-4-HB and Pip-2-H suggest that these atoms are all in axial positions of the sixmembered piperidine ring (Figures S7, S14, and S15 and Tables S3 and S4). On the basis of the previously determined structure of 1, we propose that the newly introduced stereocenter in 7 presents the same absolute configuration as is observed in the other series a thiopeptide metabolites 2 and 3.5,24 The piperidine ring in 7, characteristic of a series a thiopeptide, suggests that TpnL reduces a precursor dehydropiperidine.
Figure 2.

TpnL-mediated conversion of thiostrepton A (1) to thiostrepton Aa (7). (A) HPLC analyses of culture extracts from (a) S.laurentii, (b) S. laurentii HI0, (c) S. laurentii HI1, (d) coinjection of S. laurentii and HI1 extracts, and (e) coinjection of S. laurentii HI0 and HI1 extracts. (B) HPLC analyses of TpnL activity. Reactions omitting TpnL are shown at the top and reactions including TpnL are shown at the bottom. Reaction mixtures were incubated for (i) 0 min, (ii) 10 min, (iii) 60 min, and (iv) 6 h. Absorbance was monitored at 254 nm. 1 is represented by ○ and 7 by ●.
Scheme 1. TpnL-Catalyzed Dehydropiperidine Reduction.

The aqueous solubility of 7 (26.01 ± 0.05 μM) is slightly improved relative to the 21.88 ± 0.05 μM value for 1.25 The antibacterial activities of 7 are comparable to those of 1, demonstrating minimum inhibitory concentrations of 0.018–0.098 μg/mL (0.010–0.058 μM; Table S5). Since 1, and presumably 2–5, bind to the ribosome and impair its activity, inhibition of prokaryotic protein synthesis by 7 was determined using an in vitro transcription-translation assay.25,26 Under these conditions, the half-maximum inhibitory concentration of 7 was 0.26 ± 0.04 μM, similar to the 0.50 ± 0.04 μM value for 1 and consistent with the activities of the two metabolites against the indicator bacterial strains. The piperidine modification does not significantly alter the antibacterial properties of 7 under the conditions tested, consistent with prior examination of the semisynthetic piperidine-containing 1 analogue.27
TpnL Is an F420H2-Dependent Dehydropiperidine Reductase.
FDOR-Bs are a diverse protein family and can utilize cofactor F420, FAD, FMN, or heme, and the F420-dependent proteins catalyze a variety of reductions.28–31 TpnL contains a lysine residue (K55) conserved in most F420- dependent FDOR-Bs, part of a region including several positively charged residues, that facilitates binding the polyglutamate tail of the cofactor (Figures S16 and S17).23,32 To determine whether TpnL preferentially binds F420 or a flavin, fluorescence binding assays were performed. TpnL was heterologously expressed in E. coli with an N-terminal His6-tag. The purified protein (Figure S18) was colorless, suggesting that it does not co-purify with the flavins present in E. coli, a microorganism that does not produce F420. The intrinsic tryptophan fluorescence quenching in the presence of F420 or the flavins was used to indicate cofactor binding to TpnL (Figure S19).33 The KD value of F420 for TpnL (8.5 ± 1.4 μM) is 10 times lower than those for FAD and FMN (88.3 ± 24.4 and 105.9 ± 13.2 μM, respectively). Although the reduced form, F420H2, is most likely the native cofactor for TpnL, this protein does preferentially bind F420 over FAD or FMN.
To confirm that TpnL reduces the thiopeptide dehydropiperidine, we reconstituted its activity in vitro. Piperidine formation in series a thiopeptides is expected to be among the latter maturation steps, as the [4 + 2] condensation that assembles the core macrocycle occurs after many of the post-translational modifications on the precursor peptide.10,34 We therefore reasoned that 1 may be a substrate of TpnL (Scheme 1). To generate the reduced coenzyme F420H2 in situ, F420- dependent glucose-6-phosphate dehydrogenase (FGD) and glucose-6-phosphate were included in the assay mixture with TpnL and 1.33 TpnL catalyzed the reduction of 1 to 7, as detected by HPLC and LC-MS (Figure 2B and Figure S20). TpnL activity appears to be subject to substrate inhibition at concentrations greater than 2 μM 1 (Figure S21). To examine if the in situ generation of F420H2 could be limiting TpnL activity, F420 reduction by FGD was monitored during the time course of the TpnL assay and was found to be unaffected by TpnL and 1 (Figure S22). An additional assessment of TpnL activity at 2 and 10 μM 1 using increased concentrations of F420 (by 4.4-fold) and FGD (by 2-fold) did not demonstrate a significant alteration of TpnL activity (Figure S21). The assay conditions examined here prevented a determination of KM, but an apparent kcat/KM (2.80 × 104 M−1 s−1) was derived from the linear region of substrate dependence on rate. Our results clearly demonstrate that TpnL is an F420H2-dependent dehydropiperidine reductase.
DISCUSSION
A defining characteristic of thiopeptides is the core macrocycle linked through a nitrogenous heterocycle derived from two Dha residues via a formal [4 + 2] cycloaddition.10 Series d thiopeptides utilize a dedicated pyridine synthase to catalyze this cyclization, followed by dehydration and elimination of the leader peptide.9 A similar process might also lead to the core macrocycle of series a and b thiopeptides, although it is not yet understood how this permits retention of the N-terminal portion of the peptide and a more reduced ring system. There are, however, no obvious reductase candidates in either the 1 or siomycin (another series b thiopeptide) biosynthetic gene clusters, leaving the origin of the dehydropiperidine ring enigmatic.14,35 Identification of TpnL as a dehydropiperidine reductase demonstrates that the series a thiopeptide piperidine originates from the reduction of a series b thiopeptide intermediate. TpnL does accept 1, a mature metabolite bearing both the core macrocycle and the quinaldic acid loop, as a substrate. It is not yet known, however, if TpnL would act upon a dehydropiperidine-containing intermediate in which the second, quinaldic acid containing macrocycle has not yet been installed.18,36 The TpnL-dependent reduction shown here occurs independently of additional ancillary recognition elements such as the leader peptide, in contrast to many modifications in thiopeptide biosynthesis that do require the presence of the leader peptide.10
An in silico search for TpnL homologues in the NCBI database revealed potential thiopeptide biosynthetic gene clusters co-localized with a putative F420H2-dependent reductase (Figure 3). Two of these clusters appear to be capable of producing close structural homologues to 2–5, identified in Amycolatopsis saalfeldensis DSM 44993 (A. saalfeldensis) and Micromonospora carbonacea DSM 43168 (DSM 43168). The TpnL homologue identified in A. saalfeldensis (SEO90151.1) shares 70% sequence identity (78% similarity) to TpnL. The associated gene cluster is identical in composition and organization to the tpn cluster, including homologues of tpnMN that are proposed to be involved in thioamidation of the peptide backbone (Table S6). The core peptide sequence encoded in the A. saalfeldensis cluster (ASSSSCTTCICTCSCSS; SEO89922.1) is very similar to the sequence in TpnA and the thiostrepton precursor peptide TsrA.14 The putative thiopeptide biosynthetic gene cluster in DSM 43168 differs in organization from the tpn and A. saalfeldensis clusters but does contain a full set of tpn-/tsr-like genes (Figure 3 and Table S7) and also encodes homologues of TpnLMN, with the TpnL-like protein (SCF235261.1) bearing 52% identity (64% similarity) to TpnL. The core peptide sequence in DSM 43168 (SCF23644.1) differs from the thiopeptin sequence only at the first residue, is identical with the thiostrepton amino acid sequence, and is identical with that predicted for Sch 18640 6 (IASASCTTCICTCSCSS). Thus far, 6 has only been isolated from Micromonospora arborensis, and it is likely that DSM 43168 is another producer of this metabolite.6 The gene clusters identified in A. saalfeldensis and DSM 43168 may therefore be involved in the generation of thioamidated series a and b thiopeptides, similar to the complex of 2–5 produced by S. tateyamensis.
Figure 3.

Thiopeptin biosynthetic gene (tpn) cluster and possible thiopeptide biosynthetic gene clusters containing a homologue of tpnL. Two orfs in Micromonospora carbonacea var. africana ATCC 39149 based on incomplete nucleotide data in the NCBI database are shown with dashes and dotted lines. The accession numbers are indicated below each species, and the range of values are abbreviated by the “x” in the accession number. The individual orfs are labeled according to the abbreviated accession numbers.
To date, only one dihydroimidazopiperidine-containing series c thiopeptide has been identified, Sch 40832 (Figure S23) isolated from Micromonospora carbonacea var. Africana ATCC 39149 (ATCC 39149).37 The genome sequence of ATCC 39149 contains a candidate biosynthetic gene cluster for this unusual thiopeptide (Figure 3), and the precursor peptide found encoded in this locus (EEP73621.1) includes a match for a predicted Sch 40832 core peptide (TSSSSCTTCICTCSCSS). Like 1 and 2–5, Sch 40832 houses a second, quinaldic acid containing macrocycle, and the genes encoding HEQ biosynthetic enzyme homologues are present in the proposed cluster (Figure 3 and Table S8). The structure reported for Sch 40832 included a disaccharide tethered to the β-hydroxyl of Thr1, and a potential glycosyltransferase (EEP73625.1) is encoded in this cluster (Table S8). A homologue of TpnL (EEP73610.1) is present in the ATCC 39149 cluster, but there do not appear to be any homologues to TpnMN, the proposed thioamidation YcaO-TfuA-like proteins. Accordingly, the reported structure of Sch 40832 lacks a thioamide.37 The role of an F420H2-dependent reductase in the biosynthesis of a series c thiopeptide is not entirely clear. It could reduce a dehydropiperidine precursor, forming a nascent piperidine subjected to additional oxidative modifications elaborating the central nitrogenous ring into the dihydroimidazopiperidine scaffold. Alternatively, the TpnL homologue could mediate an entirely different reductive step in the maturation of a series c thiopeptide.
Actinoalloteichus hoggarensis DSM 45943 (A. hoggarensis) also identifies with a putative thiopeptide biosynthetic gene cluster harboring a TpnL-like protein and contains many similarities to the components of the ATCC 39149 biosynthetic gene cluster (Figure 3 and Tables S8 and S9). The A. hoggarensis core peptide (WP_093941958.1) is identical with the proposed Sch 40832 core peptide of ATCC 39149. The A. hoggarensis cluster, however, also includes homologues to the proposed TpnMN thioamidating system. It is therefore possible that A. hoggarensis may generate another series c thiopeptide bearing a thioamide.
This is the first report of involvement of the unusual coenzyme F420 in thiopeptide biosynthesis. F420 is widely produced by methanogenic archaea and Actinobacteria, including Streptomyces and Mycobacteria, and F420-related pathways have attracted attention as targets for the selective inhibition of pathogenic Mycobacteria.29,38 F0, a biosynthetic precursor of F420, is derived from the condensation of L-tyrosine and 5-amino-6-ribitylamino-2,4(1H,3H)-pyrimidinedione, both primary metabolites (Scheme S3).39 F0 synthase (FbiC) marks the first committed step in F420 biosynthesis, and at least one copy of an FbiC candidate is present in S. tateyamensis and the S. laurentii genomes (Figure S24).40 FbiA and FbiB are involved in the attachment of the phospho-L-lactyl and polyglutamyl tail on F420, respectively.41 The incomplete sequence of the S. tateyamensis genome prevented identification of encoded FbiA and FbiB homologues, but both are encoded in the complete genome sequence of S. laurentii (Figures S25 and S26).40 S. laurentii and, presumably, S. tateyamensis both appear to be capable of producing the mature cofactor F420, and the biosynthetic pathway for this cofactor is often present in actinomycetes.38 Indeed, FbiABC homologues are encoded in several thiopeptide-producing strains, including Planobispora rosea (series d; GE2270A), Streptomyces actuosus (series e; nosiheptide), and the putative series a–c producers identified above (Table S10).42,43
Several homologues of TpnL were identified in the NCBI database, only a few of which appear to be involved in thiopeptide biosynthesis. A phylogenetic analysis of TpnL was conducted alongside its homologues and established F420H2- dependent FDOR-Bs from Mycobacterium species and Streptomyces rimosus.23,28,32,44–46 The TpnL-like proteins affiliated with a known or predicted thiopeptide biosynthetic gene cluster are in a distinct clade from the other FDOR-B proteins (Figure 4). The two proteins that may be involved in biosynthesis of dihydroimidazopiperidine-containing (series c) thiopeptides form a separate subclade from the homologues identified from bacteria that are or may be involved in the biosynthesis of series a thiopeptides.
Figure 4.

Phylogenetic tree of TpnL and other F420-dependent FDOR-B homologues. TpnL homologues co-localized with predicted thiopeptide biosynthetic gene clusters are shown in blue, and other known and putative F420-dependent FDOR-Bs are shown in black. The F420-dependent FDOR-Bs from Actinobacteria that have been biochemically characterized are indicated with a bracket. The branch distances are indicated at the nodes and are based on the number of substitutions in the amino acid sequences between homologues, an indicator of genetic divergence. The scale bar represents 10 amino acid substitutions per 100 amino acid residues. Accession numbers for the sequences used are given in Table S11.
A growing number of F420H2-dependent enzymes have been proposed to reduce a wide variety of complex substrates in actinomycete secondary metabolism. In addition to the reduction of the dehydropiperidine ring in a heavily decorated thiopeptide macrocycle as shown here (Scheme 1), an F420H2- dependent FDOR-B family protein from Streptomyces rimosus reduces an alkene in the biosynthesis of the polyketide oxytetracycline.28 A separate subset of enzymes binding F420 belonging to the luciferase-like monooxygenase (LLM) family appear to be more broadly distributed and have been implicated in the biosynthesis of the 4-alkyl-L-proline substituent found in the nonribosomal peptide metabolites hormaomycin and the pyrrolo[1,4]benzodiazepines: e.g., lincomycin.38,47,48 LLM-family F420H2-dependent reductases are also suggested to be involved in the biosyntheses of the aminoglycoside kasugamycin and a coronafacoyl phytotoxin produced in a streptomycete.49,50 It is likely that the number and types of transformations attributable to F420H2-dependent enzymes will continue to expand as more metabolic pathways are characterized.
CONCLUSION
In summary, this work presents the initial biochemical characterization of an enzyme responsible for the formation of a series a thiopeptide and establishes the basis for biosynthesis of the thiopeptins. TpnL is an unusual F420H2- dependent enzyme that reduces the dehydropiperidine housed within the highly modified macrocycle of 1 to a piperidine and is expected to carry out a similar transformation for thiopeptin production. Further characterization of TpnL will be needed to determine whether this reduction reflects the terminal step in series a thiopeptide biosynthesis or if the true biological substrate is an earlier intermediate. Regardless, TpnL represents a biosynthetic tool that may be useful in the development of additional thiopeptide metabolites.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Georgia Institute of Technology and by the National Institutes of Health (R01GM090327). A GAANN predoctoral fellowship was provided to H.I., and G.B. is supported by a Sir Charles Hercus Fellowship through the Health Research Council of New Zealand. David Bostwick and Dr. Cameron Sullards (Georgia Institute of Technology) are thanked for their assistance with MS analyses, and Dr. Les Gelbaum (Georgia Institute of Technology) is thanked for his assistance with NMR analyses. Daniel Sircar is thanked for his assistance in the construction of plasmids.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b04238.
Detailed experimental procedures (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1). Bagley MC; Dale JW; Merritt EA; Xiong X Chem. Rev 2005, 105, 685–714. [DOI] [PubMed] [Google Scholar]
- (2). LaMarche MJ; Leeds JA; Amaral A; Brewer JT; Bushell SM; Deng G; Dewhurst JM; Ding J; Dzink-Fox J; Gamber G; Jain A; Lee K; Lee L; Lister T; McKenney D; Mullin S; Osborne C; Palestrant D; Patane MA; Rann EM; Sachdeva M; Shao J; Tiamfook S; Trzasko A; Whitehead L; Yifru A; Yu D; Yan W; Zhu QJ Med. Chem 2012, 55, 2376–2387. [DOI] [PubMed] [Google Scholar]
- (3). Xu L; Farthing AK; Dropinski JF; Meinke PT; McCallum C; Hickey E; Liu K Bioorg. Med. Chem. Lett 2013, 23, 366–369. [DOI] [PubMed] [Google Scholar]
- (4). Miyairi N; Miyoshi T; Aoki H; Kosaka M; Ikushima H Antimicrob. Agents Chemother 1972, 1, 192–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5). Hensens OD; Albers-Schonberg GJ Antibiot. 1983, 36, 814–831. [DOI] [PubMed] [Google Scholar]
- (6). Puar MS; Ganguly AK; Afonso A; Brambilla R; Mangiaracina P; Sarre O; MacFarlane RD J. Am. Chem. Soc 1981, 103, 5231–5233. [Google Scholar]
- (7). Izawa M; Kawasaki T; Hayakawa Y Appl. Environ. Microbiol 2013, 79, 7110–7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8). Lincke T; Behnken S; Ishida K; Roth M; Hertweck C Angew. Chem., Int. Ed 2010, 49, 2011–2013. [DOI] [PubMed] [Google Scholar]
- (9). Cogan DP; Hudson GA; Zhang Z; Pogorelov TV; van der Donk WA; Mitchell DA; Nair SK Proc. Natl. Acad. Sci. U. S.A 2017, 114, 12928–12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10). Hudson GA; Zhang Z; Tietz JI; Mitchell DA; van der Donk WA J. Am. Chem. Soc 2015, 137, 16012–16015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11). Mocek U; Zeng Z; O’Hagan D; Zhou P; Fan L-DG; Beale JM; Floss HG J. Am. Chem. Soc 1993, 115, 7992–8001 [Google Scholar]
- (12). Dunbar KL; Chekan JR; Cox CL; Burkhart BJ; Nair SK; Mitchell DA Nat. Chem. Biol 2014, 10, 823–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13). Duan L; Wang S; Liao R; Liu W Chem. Biol 2012, 19, 443–448. [DOI] [PubMed] [Google Scholar]
- (14). Kelly WL; Pan L; Li CJ Am. Chem. Soc 2009, 131, 4327–4334. [DOI] [PubMed] [Google Scholar]
- (15). Lin Z; Ji J; Zhou S; Zhang F; Wu J; Guo Y; Liu WJ Am. Chem. Soc 2017, 139, 12105–12108. [DOI] [PubMed] [Google Scholar]
- (16). Benjdia A; Pierre S; Gherasim C; Guillot A; Carmona M; Amara P; Banerjee R; Berteau O Nat. Commun 2015, 6, 8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17). Priestley ND; Smith TM; Shipley PR; Floss HG Bioorg. Med. Chem 1996, 4, 1135–1147. [DOI] [PubMed] [Google Scholar]
- (18). Zheng Q; Wang S; Duan P; Liao R; Chen D; Liu W Proc. Natl. Acad. Sci. U. S. A 2016, 113, 14318–14323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19). Zheng Q; Wang S; Liao R; Liu W ACS Chem. Biol 2016, 11, 2673–2678. [DOI] [PubMed] [Google Scholar]
- (20). Liao R; Liu WJ Am. Chem. Soc 2011, 133, 2852–2855. [DOI] [PubMed] [Google Scholar]
- (21). Nayak DD; Mahanta N; Mitchell DA; Metcalf WW eLife 2017, 6, e29218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22). Mahanta N; Liu A; Dong S; Nair SK; Mitchell DA Proc. Natl. Acad. Sci. U. S. A 2018, 115, 3030–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23). Ahmed FH; Carr PD; Lee BM; Afriat-Jurnou L; Mohamed AE; Hong NS; Flanagan J; Taylor MC; Greening C; Jackson CJ J. Mol. Biol 2015, 427, 3554–3571. [DOI] [PubMed] [Google Scholar]
- (24). Hensens OD; Albers-Schonberg G; Anderson BF J. Antibiot 1983, 36, 799–813. [DOI] [PubMed] [Google Scholar]
- (25). Zhang F; Kelly WL ACS Chem. Biol 2015, 10, 998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26). Harms JM; Wilson DN; Schluenzen F; Connell SR; Stachelhaus T; Zaborowska Z; Spahn CMT; Fucini P Mol. Cell 2008, 30, 26–38. [DOI] [PubMed] [Google Scholar]
- (27). Jonker HR; Baumann S; Wolf A; Schoof S; Hiller F; Schulte KW; Kirschner KN; Schwalbe H; Arndt HD Angew. Chem., Int. Ed 2011, 50, 3308–3312. [DOI] [PubMed] [Google Scholar]
- (28). Wang P; Bashiri G; Gao X; Sawaya MR; Tang YJ Am. Chem. Soc 2013, 135, 7138–7141. [DOI] [PubMed] [Google Scholar]
- (29). Hagemeier CH; Shima S; Thauer RK; Bourenkov G; Bartunik HD; Ermler UJ Mol. Biol 2003, 332, 1047–1057. [DOI] [PubMed] [Google Scholar]
- (30). Cellitti SE; Shaffer J; Jones DH; Mukherjee T; Gurumurthy M; Bursulaya B; Boshoff HI; Choi I; Nayyar A; Lee YS; Cherian J; Niyomrattanakit P; Dick T; Manjunatha UH; Barry CE 3rd; Spraggon G; Geierstanger BH Structure 2012, 20, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31). Johnson EF; Mukhopadhyay BJ Biol. Chem 2005, 280, 38776–38786. [DOI] [PubMed] [Google Scholar]
- (32). Mashalidis EH; Gittis AG; Tomczak A; Abell C; Barry CE 3rd; Garboczi DN Protein Sci. 2015, 24, 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33). Oyugi MA; Bashiri G; Baker EN; Johnson-Winters K Biochemistry 2016, 55, 5566–5577. [DOI] [PubMed] [Google Scholar]
- (34). Zhang Z; Hudson GA; Mahanta N; Tietz JI; van der Donk WA; Mitchell DA J. Am. Chem. Soc 2016, 138, 15511–15514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35). Liao R; Duan L; Lei C; Pan H; Ding Y; Zhang Q; Chen D; Shen B; Yu Y; Liu W Chem. Biol 2009, 16, 141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36). Li C; Zhang F; Kelly WL Chem. Commun 2012, 48, 558–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37). Puar MS; Chan TM; Hegde V; Patel M; Bartner P; Ng KJ; Pramanik BN; MacFarlane RD J. Antibiot 1998, 51, 221–224. [DOI] [PubMed] [Google Scholar]
- (38). Selengut JD; Haft DH J. Bacteriol 2010, 192, 5788–5798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39). Decamps L; Philmus B; Benjdia A; White R; Begley TP; Berteau OJ Am. Chem. Soc 2012, 134, 18173–18176. [DOI] [PubMed] [Google Scholar]
- (40). Doi K; Fujino Y; Nagayoshi Y; Ohshima T; Ogata S Genome Announc. 2016, 4, e00360–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41). Choi KP; Bair TB; Bae YM; Daniels LJ Bacteriol. 2001, 183, 7058–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42). Flinspach K; Kapitzke C; Tocchetti A; Sosio M; Apel AK PLoS One 2014, 9, e90499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43). Yu Y; Duan L; Zhang Q; Liao R; Ding Y; Pan H; Wendt-Pienkowski E; Tang G; Shen B; Liu W ACS Chem. Biol 2009, 4, 855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44). Ahmed FH; Mohamed AE; Carr PD; Lee BM; Condic-Jurkic K; O’Mara ML; Jackson CJ Protein Sci. 2016, 25, 1692–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45). Greening C; Jirapanjawat T; Afroze S; Ney B; Scott C; Pandey G; Lee BM; Russell RJ; Jackson CJ; Oakeshott JG; Taylor MC; Warden AC Front. Microbiol 2017, 8, 01000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46). Manjunatha UH; Boshoff H; Dowd CS; Zhang L; Albert TJ; Norton JE; Daniels L; Dick T; Pang SS; Barry CE 3rd Proc. Natl. Acad. Sci. U. S. A 2006, 103, 431–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47). Cai X; Teta R; Kohlhaas C; Crusemann M; Ueoka R; Mangoni A; Freeman MF; Piel J Chem. Biol 2013, 20, 839–846. [DOI] [PubMed] [Google Scholar]
- (48). Jiraskova P; Gazak R; Kamenik Z; Steiningerova L; Najmanova L; Kadlcik S; Novotna J; Kuzma M; Janata J Front. Microbiol 2016, 7, 00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49). Bown L; Altowairish MS; Fyans JK; Bignell DR Mol. Microbiol 2016, 101, 122–135. [DOI] [PubMed] [Google Scholar]
- (50). Ikeno S; Aoki D; Hamada M; Hori M; Tsuchiya KS J. Antibiot 2006, 59, 18–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.