

Abstract
Well-defined heparin like oligosaccharides up to decasaccharides were synthesized. It was discovered for the first time that heparin oligosaccharides, as short as a tetrasaccharide, can bind with the most toxic tau species, i.e., tau oligomers with nM KD. The binding significantly reduced cellular uptake of the toxic tau oligomers and protected cells from tau oligomer induced cytotoxicity.
Graphical Abstract
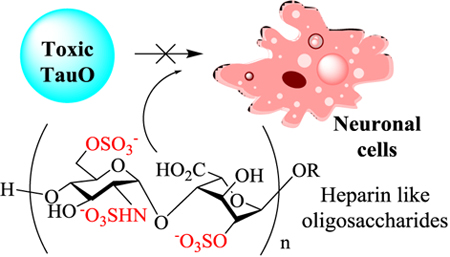
Heparin oligosaccharides can mitigate the cytotoxic effects of tau oligomers towards neuronal cells.
Alzheimer’s disease (AD) is a progressive degenerative brain disease, which is estimated to affect 5.5 million Americans in 2017.1 Although the causes for most AD cases have not been firmly established, the pathology of tau protein is believed to play a pivotal role in AD and other tauopathies.2 While large molecular weight tau aggregates, known as neurofibrillary tangles (NFTs), are abundant in the brains of late stage AD patients,3 it is generally believed that these large insoluble aggregates may not be the key toxic species in AD.4 There are strong evidences supporting that the soluble, oligomeric tau (TauO) rather than the NFTs are likely the most toxic species causing disease and efficiently seeding the propagation of the pathology.5, 6 TauO have been found in the extracellular space around neurons in human AD patients.7 They can propagate through different brain regions, spreading neuronal death and associated learning and memory deficits.8, 9 Therefore, TauO are potential new therapeutic targets, and strategies that can reduce TauO-associated neurotoxicity are highly desired.
Heparan sulfate (HS) and its more sulfated analog heparin are a class of highly negatively charged polysaccharides present on mammalian cells including neuronal cells. HS and heparin are composed of repeating disaccharide subunits with D-glucosamine (GlcN) α−1,4 linked with a uronic acid (either L-uronic acid (IdoA) or D-glucuronic acid (GlcA)).10, 11 The amine moiety, 3-OH and 6-OH of GlcN and 2-OH of the uronic acid of heparin can be sulfated. With the positively charged proline-rich domain and the C-terminal tail in Tau protein,12 heparin can bind with high molecular tau aggregates.13–15 However, it is not known whether heparin can interact with TauO. Herein, using structurally well-defined synthetic oligosaccharides, we report for the first time that heparin like oligosaccharides, as small as tetrasaccharides, can bind with the toxic TauO. Furthermore, treatment of cells with heparin like oligosaccharides protects them from TauO-induced toxicity providing an exciting new direction in targeting AD and related tauopathies.
In order to obtain heparin like oligosaccharides, we based our synthetic design on disaccharide modules 1, 2 and 3. Disaccharides 1 and 2 were synthesized starting from disaccharide 4.16 For the non-reducing end disaccharide module, while TBS bearing disaccharide 4 could be used, we found it was impossible to remove the TBS group from sulfated oligosaccharides during deprotection.16 This consideration prompted us to prepare the 4’-O-benzyl (Bn) protected disaccharide 3. Pre-activation of Bn protected glucosamine donor 5 with p-TolSCl and AgOTf17 followed by the addition of acceptor 6, gave the α-linked disaccharide 7 in 87% yield (Scheme 1a). The 1,2-cis linkage in the newly formed glycosidic bond was confirmed by NMR analysis (JH1’-H2’ = 3.5 Hz, JH1’-C1’ = 171.0 Hz). Protective group manipulation of 7 yielded the disaccharide module 3. Reaction of the donor 3 with acceptor 1 generated tetrasaccharide 8 in 85% yield (Scheme 1b). Alternatively, 3 glycosylated the reducing end disaccharide module 2 giving tetrasaccharide 9 (Scheme 1b). In a similar manner, TBS bearing tetrasaccharide donor 10 was formed (Scheme 1c).
Scheme 1.
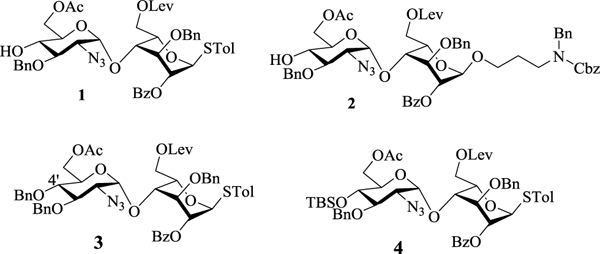
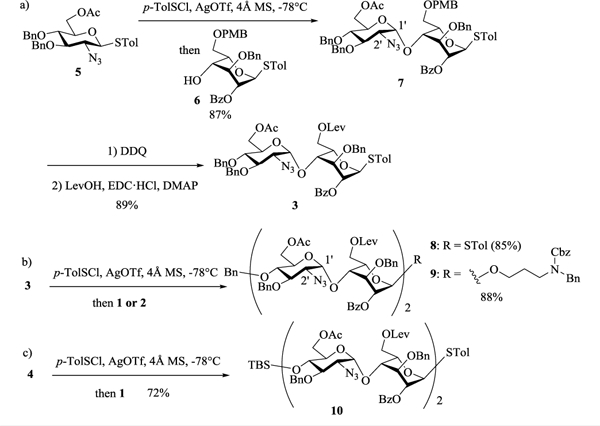
Synthesis of tetrasaccharide backbones 8–10
To produce the hexasaccharide backbone, a 4+2 glycosylation was carried out between the tetrasaccharide donor 8 and acceptor 2 leading to hexasaccharide 11 (Scheme 2a). The 4-O-TBS protected tetrasaccharide donor 10 also reacted well with disaccharide 2. Removal of the TBS group from the glycosylation product produced the hexasaccharide acceptor 12 (Scheme 2b), which was subsequently glycosylated by tetrasaccharide donor 8, forming decasaccharide 13 in 84% yield (Scheme 2c).
Scheme 2.
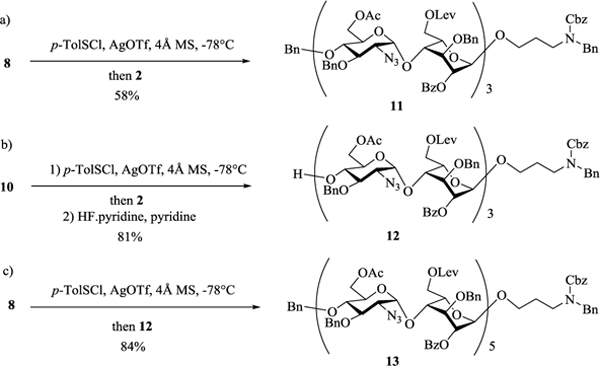
Constructions of heparin hexa- and deca-saccharide backbones.
The deprotections and modifications of the backbones were carried out first by removal of 6-O-Lev from fully protected tetra-, hexa- and deca-saccharide 9, 11 and 13 respectively with hydrazine acetate exposing the 6-OH (Scheme 3a). The conversion of these primary hydroxyl groups to carboxylic acids was mediated by bis(acetoxy)iodobenzene (BAIB) assisted 2, 2, 6, 6-tetramethyl-1-piperidinyloxyl (TEMPO) oxidation.18 Since free carboxylic acids were found to lead to low yields in subsequent sulfation reactions,16 they were protected as either methyl (83% for tetrasaccharide 17 in 2 steps) or benzyl (77% for hexasaccharide 18 and 81% for decasaccharide 19 in 2 steps) esters. Removal of the acyl protecting groups was accomplished by treating oligosaccharides 17-19 with sodium methoxide, which gave 20, 21 and 22 respectively (Scheme 3a).
Scheme 3.
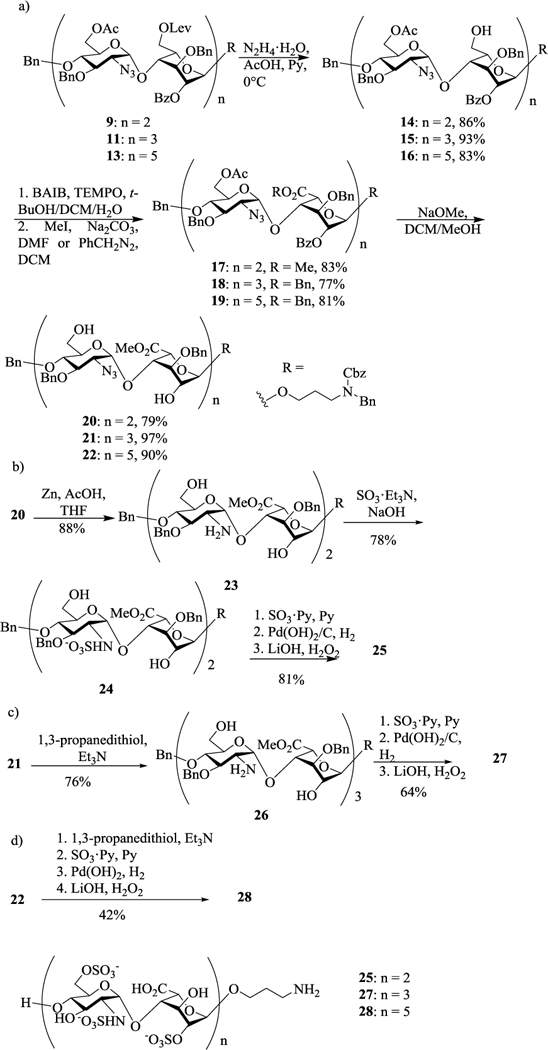
Deprotection and sulfation of heparin oligosaccharides.
The two azido groups in tetrasaccharide 20 were reduced by zinc powder to provide 23 with two free amine groups (Scheme 3b). Sulfations of free hydroxyls and amines of 23 were performed stepwise. Firstly, 23 was dissolved in methanol with aqueous NaOH solution adjusting the pH to 9.5 in order to deprotonate amine groups. N-sulfation was then performed by adding excess SO3·Et3N complex to the mixture to give 24 in 78% yield, which was then subjected to O-sulfation with SO3·pyridine complex in pyridine overnight at 55 °C. Subsequent hydrogenolysis and saponification produced sulfated tetrasaccharide 25.
For heparin hexasaccharide synthesis, the azido groups in 21 were first reduced to yield amine 26 (Scheme 3c). Similar stepwise sulfation as in synthesis of tetrasaccharide 25 was attempted on hexasaccharide 26, which only led to decomposition of the starting materials. Analysis of the reaction mixture showed the formation of side products due to β-elimination with the oligosaccharide backbone cleaved. Instead, treatment of hexasaccharide 26 with 600 mM SO3·py in pyridine at 55 °C successfully installed both N- and O-sulfation in one step. Interestingly, the free amine 26 obtained through 1, 3-propanedithiol reduction16 of 21 gave higher yields during sulfation compared to the corresponding compound produced by zinc power reduction. Subsequent catalytic hydrogenation and methyl ester hydrolysis gave the final heparin like hexasaccharide 27 at 64% yield over 3 steps (Scheme 3c). Analogously, the heparin like decasaccharide 28 was synthesized with an overall yield of 42% from 22 (Scheme 3d).
With the synthetic oligosaccharides in hand, their binding with TauO was analysed. 25, 27, and 28 were biotinylated, immobilized on streptavidin coated biolayer interferometry (BLI) sensors and incubated with various concentrations of TauO. The sensorgrams showed that tetrasaccharide 25 could bind to TauO with a KD value of 2.79 × 10−7 M. Increasing the backbone length of the oligosaccharide to hexa- and deca-saccharides led to enhancements in TauO binding, with KD values of 1.41 × 10−7 M and 3.49 × 10−8 M for oligosaccharides 27 and 28, respectively. These results suggest that glycans, as short as tetrasaccharide, can already exhibit significant interactions with TauO and longer oligosaccharide backbones enhance the binding.
With the establishment of binding, we evaluated the abilities of heparin like oligosaccharides to modulate the aggregation state and toxicity of TauO. Oligomeric tau species were incubated with and without oligosaccharides at room temperature for 16 hours under oligomerization conditions.19 Atomic force microscopy (AFM) was performed to visualize and characterize the morphology and aggregation state, which reflects the distinct effects of the compounds on TauO. Classically, TauO exist as homogeneous spherical structures (Fig. 2a), while in the presence of heparin like oligosaccharides (Fig. 2b-d), TauO were converted into larger aggregates.
Fig. 2. Morphology characterization of TauO alone or in the presence of heparin like oligosaccharides.

Atomic Force Microscopy images of TauO without (a) and with oligosaccharides b) 25, c) 27, and d) 28 (5X). AFM images show the ability of the glycans to modulate TauO aggregation states converting TauO into much larger aggregates. Scale bars = 100 nm.
Next, we evaluated the ability of the heparin like oligosaccharides to prevent TauO-induced cytotoxicity using human neuroblastoma SH-SY5Y purchased from American Type Culture Collection. Cells were incubated with tau alone or in the presence of heparin like oligosaccharides 25, 27 and 28, respectively. The cytotoxicity to SH-SY5Y cells was determined in a lactate dehydrogenase (LDH)-based assay, with the amount of LDH released into the culture media from damaged cells indicative of cellular cytotoxicity and cytolysis. Consistent with the idea that TauO being the more toxic tau species, TauO treatment of SH-SY5Y cells led to significantly higher LDH release, while cells incubated with the higher molecular aggregate tau fibrils released much less LDH (Fig. 3a). Excitingly, in the presence of oligosaccharides (10 μM), TauO induced significantly lower levels of LDH release, compared to TauO alone (Fig. 3a). Moreover, cells exposed to each condition were evaluated for morphological differences. In the presence of TauO alone, there was significant cell shrinkage compared to either the control cells (Ctrl) or cells treated with TauO in the presence of heparin like oligosaccharides (Fig. 3b). Taken together, these results suggest that heparin like oligosaccharides interact with and subsequently convert the toxic TauO into larger aggregates modulating their toxicity.
Fig. 3.
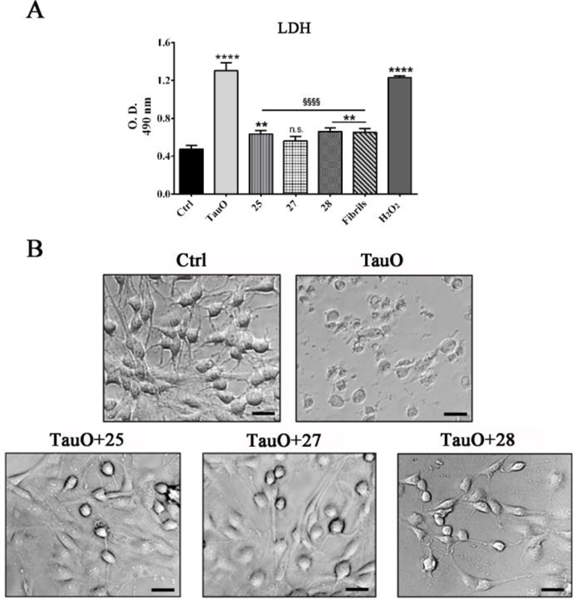
(A) SH-SY5Y cells cytotoxicity after exposure to 2μM TauO, or 2μM TauO with 10μM of oligosaccharide (25, 27, 28) and untreated control (Ctrl). Treatment of SH-SY5Y cells with TauO had significantly higher LDH release compared to the untreated control, cells exposed to TauO in the presence of oligosaccharides or cells exposed to high molecular weight tau fibrils. Each experiment was performed in triplicate (n = 3). Data were compared by one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparison test: Ctrl vs TauO, 25, 27, and 28, Fibrils: ****p<0.0001, **p<0.01; TauO vs 25, 27, and 28: §§§§p<0.0001. Bars and error bars represent means and standard deviations performed. (B) Untreated SH-SY5Y cells were compared to cells treated for 24 hours with TauO alone or TauO in the presence of oligosaccharides (25, 27, and 28) and evaluated for morphological changes. Scale bar =20 μm.
To further confirm our findings and gain a better understanding of the protective effects of the oligosaccharides, SH-SY5Y cells were incubated with sub-lethal concentration of TauO and imaged by confocal microscopy. TauO were observed in large areas of the cytoplasm of cells, indicating extensive cellular internalization of TauO (Fig. S1a). Intracellular TauO can instigate mitochondrial damage, induce cytochrome c release and stimulate reactive oxygen species production, which are potential mechanisms for their cytotoxicity.20, 21 SH-SY5Y cells treated with TauO, co-incubated with oligosaccharide 25, 27 and 28, showed a significant reduction in the percentage of area positive of TauO staining (Fig. S1b). These results indicate that the glycan-induced aggregates are less prone to be uptaken by the cells explaining their reduced cytotoxicity. Altogether, our findings suggest that the glycans interact with TauO leading to the formation of larger tau aggregates and inhibiting them from cellular internalization.
In conclusion, with the increasing appreciation of the high toxicity of TauO, finding an effective prevention and treatment strategy available for tauopathies, has becoming important. In this work, using fully synthetic well-defined glycans, we report the new finding that heparin like oligosaccharide bearing 2-O, 6-O and N-sulfation can bind strongly with the most toxic tau species, i.e., TauO. Heparin oligosaccharides can convert TauO into less toxic high molecular weight species, and mitigate TauO-associated cytotoxicity. In addition, the glycans significantly reduce TauO cellular internalization, which is critical for the progression of the pathology. While heparin is known to bind with high molecular tau aggregates,13–15 this is the first time that heparin oligosaccharide is shown to interact with TauO. Importantly, the newly synthesized heparin oligosaccharides could aid in the development of novel therapeutic approaches for AD and related tauopathies. Further studies are ongoing to establish the impacts of heparin structures such as sulfation patterns and backbone sequences on TauO interactions.
We are grateful for financial support from the National Institutes of Health (R01GM72667 and U01GM116262 to XH and R01AG054025, R01NS094557 to RK), Mitchell Center for Neurodegenerative Diseases, and the Gillson Longenbaugh Foundation. FLC was also supported by UTMB-University of Palermo joint PhD program. We thank Dr. Mauro Montalbano for his technical assistance with confocal microscopy.
Supplementary Material
Fig. 1. Sensograms of heparin like oligosaccharide binding with TauO.
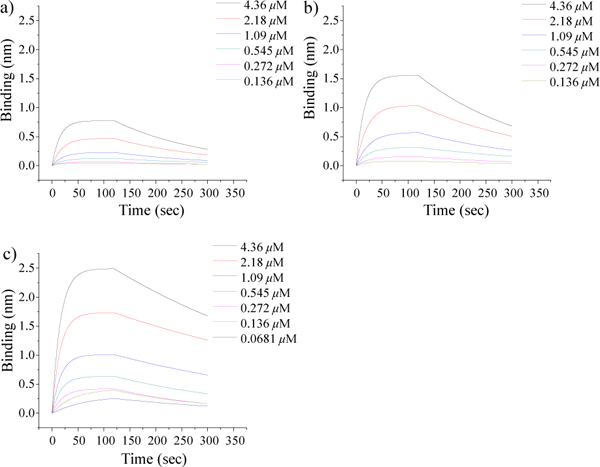
Fit curves of interactions between BLI sensors loaded with a) 25, b) 27 and c) 28 and TauO at various concentrations were obtained using models from Octet Data Analysis 9.0.0.12. Tetrasaccharide 25 exhibited significant binding to TauO, with longer backbone length leading to stronger binding.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.2017 Alzheimer’s Disease facts and figures, https://www.alz.org/documents_custom/2017-facts-and-figures.pdf, 2017.
- 2.Šimić G, Babić Leko M, Wray S, Harrington C, Delalle I, Jovanov-Milošević N, Bažadona D, Buée L, de Silva R, Di Giovanni G, Wischik C and Hof PR, Biomolecules, 2016, 6, 6 doi: 10.3390/biom6010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arriagada PV, Growdon JH, Hedley-Whyte ET and Hyman BT, Neurology, 1992, 42, 631–639. [DOI] [PubMed] [Google Scholar]
- 4.Cowan CM, Quraishe S and Mudher A, Biochem. Soc. Trans, 2012, 40, 693–697. [DOI] [PubMed] [Google Scholar]
- 5.Gerson JE, Mudher A and Kayed R, Crit. Rev. Biochem. Mol. Biol, 2016, 51, 482–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guerrero-Munoz MJ, Gerson JE and Castillo-Carranza DL, Front. Cell. Neurosci, 2015, 9, 464 https://doi.org/410.3389/fncel.2015.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Medina M and Avila J, Front. Cell. Neurosci, 2014, 8, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Kiritoshi T, Neugebauer V, Jackson GR and Kayed R, Sci. Rep, 2012, 2, 700 doi: 710.1038/srep00700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuster-Matanzo A, Hernandez F and Avila J, Int. J. Mol. Sci, 2018, 19, E645, doi: 610.3390/ijms19030645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linhardt RJ, Dordick JS, Deangelis PL and Liu J, Semin. Thromb. Hemost, 2007, 33, 453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dulaney SB and Huang X, Adv. Carbohydr. Chem. Biochem, 2012, 67, 95–136 and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kolarova M, García-Sierra F, Bartos A, Ricny J and Ripova D, Int. J. Alzheimer’s Dis, 2012, 2012, 731526 http://dx.doi.org/731510.731155/732012/731526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu HL, Fernández C, Fan JB, Shewmaker F, Chen J, Minton AP and Liang Y, J. Biol. Chem, 2010, 285, 3592–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, Kotzbauer PT, Miller TM, Papy-Garcia D and Diamond MI, Proc. Natl. Acad. Sci., USA, 2013, 110, E3138–E3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao J, Huvent I, Lippens G, Eliezer D, Zhang A, Li Q, Tessier P, Linhardt RJ, Zhang F and Wang C, Biophysical J, 2017, 112, 921–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dulaney SB, Xu Y, Wang P, Tiruchinapally G, Wang Z, Kathawa J, El-Dakdouki MH, Yang B, Liu J and Huang X, J. Org. Chem, 2015, 80, 12265–12279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang X, Huang L, Wang H and Ye X-S, Angew. Chem. Int. Ed, 2004, 43, 5221–5224. [DOI] [PubMed] [Google Scholar]
- 18.van den Bos LJ, Codee JD, van der Toorn JC, Boltje TJ, van Boom JH, Overkleeft HS and van der Marel GA, Org. Lett, 2004, 6, 2165–2168. [DOI] [PubMed] [Google Scholar]
- 19.Lo Cascio F and Kayed R, ACS Chem. NeuroSci, 2018, in press, DOI: 10.1021/acschemneuro.1027b00501. [DOI] [PubMed] [Google Scholar]
- 20.Shafiei SS, Guerrero-Munoz MJ and Castillo-Carranza DL, Front. Aging Neurosci, 2017, 9, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Usenovic M, Niroomand S, Drolet RE, Yao L, Gaspar RC, Hatcher NG, Schachter J, Renger JJ and Parmentier-Batteur S, J. Neurosci, 2015, 21, 14234–14250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.