

Abstract
A series of trisubstituted thiazoles have been identified as potent inhibitors of Plasmodium falciparum (Pf) cGMP-dependent protein kinase (PfPKG) through template hopping from known Eimeria PKG (EtPKG) inhibitors. The thiazole series has yielded compounds with improved potency, kinase selectivity and good in vitro ADME properties. These compounds could be useful tools in the development of new anti-malarial drugs in the fight against drug resistant malaria.
Keywords: Plasmodium falciparum, Malaria, PfPKG, Thiazole
Malaria causes up to 450 000 deaths every year, most of which occur in sub-Saharan Africa. 1 Most of the malaria mortality is caused by Plasmodium falciparum (Pf), one of the four species known to infect humans. Transmitted through the bites of Anopheles mosquitoes, the symptoms include fever, headache, chills and vomiting and if not treated quickly, anaemia, respiratory distress and multi organ failure. The population groups who are at highest risk are young children and pregnant women. Chloroquine was the first line of treatment for malaria for many years; however due to the development of resistance, its use has been substituted by drugs, the current standard of care being artemisinin-based combination therapy (ACT). However, parasite resistance to ACTs in South East Asia has been detected and is predicted to grow; therefore development of other treatments is highly desirable.2
One promising antimalarial drug target that is being investigated is Pf cGMP-dependent protein kinase (PfPKG), an enzyme which is conserved to a high degree of homology in other species of therapeutic relevance such as P. vivax and P. berghei. This protein kinase is essential in all the key phases of the Pf parasite life cycle and in the blood stage, inhibiting PfPKG stops replication by preventing merozoite egress3 and invasion.4 First generation PfPKG inhibitors have also been shown to block gametogenesis5 and ookinete motility6 - two key developmental events required for mosquito-borne transmission. It has also been shown that PKG is essential for sporozoite motility and liver cell invasion7 as well as late liver stage development,8 making PfPKG an attractive target for new classes of anti-malarial drugs.
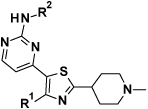
Herein, we will discuss PfPKG inhibitors derived from PKG inhibitors (compound 1 and 2) developed to treat Eimeria infections in poultry9 (Figure 1). Both of these compounds showed low nanomolar potencies in a biochemical assay against PfPKG10 and moderate potency in an in vitro cell viability assay, the hypoxanthine incorporation assay (HXI).10
Figure 1.

Structure and in vitro data of compounds of 1 (data unpublished) and 210.
This paper is focused on the monocyclic compound 1, containing a pyrrole and an unflanked 4-pyridyl, both considered undesirable motifs for further SAR development. Furthermore, poor kinase selectivity was seen with 1, as it also showed potent activity against several other human kinases. Due to these unfavourable properties of 1, an alternative core was sought for further analogue development with the aim of enhancing anti-parasitical activity against PfPKG.
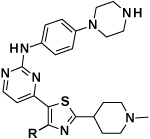
Literature searches on similar structures to compound 2 had showed examples where the imidazopyridine core could be swapped for a thiazole core, while still maintaining the interactions formed by the appended groups.11 In addition, homology modelling (data not shown) indicated a good fit for our proposed compounds. With this knowledge, we swapped the monocyclic pyrrole core to a thiazole core, and the pyridine for the 2-aminopyrimidine from compound 2, to give thiazole 3 (Figure 2).
Figure 2.

Structure and in vitro data of thiazole 3.
Compound 3, when tested, showed similar biochemical potency and a slight drop in cellular potency when compared to compounds (1) and (2) (Figure 2), which was seen as a positive result for the changed thiazole core.
To optimize the potency, we first examined the pendent 2-aminopyrimidine (Scheme 1).
Scheme 1.

Reagents and conditions (a) LiHMDS (1M in THF), THF, 0 °C to r.t., 25%; (b) (i) (Me)3SiCl, (nBu)4NBr, DMSO, THF, 0 °C to r.t., (ii) tert-butyl 4-carbamothioyl-piperidine-1-carboxylate, EtOH, reflux, 79%; (c) (i) 4M HCl/dioxane (ii) HCHO, Na(OAc)3BH, AcOH, CH2Cl2, 47%; (d) (i) H2O2, Na2WO4.2H2O, AcOH; (ii) NH4OAc, 130 °C or AlkNH2, THF, 70 °C or ArNH2, TFA, sBuOH, 130 °C, 10-45%.
Alkylation of 4 with benzoate 5 was achieved using LiHMDS to give ketone 6. This was then reacted with (Me)3SiCl and (nBu)4NBr to yield the α-chloro ketone in situ, which was cyclized with the Boc-protected thioamide in refluxing ethanol to give 7. Deprotection of 7, and subsequent reductive amination gave 8, and on oxidation, the resulting sulfone could be displaced by the appropriate amine to yield 9a-d. Two alternatives to the N-methylpiperidine ring were also made. 9e was synthesized following sulfone oxidation of 7, with Boc deprotection occurring in the displacement step by the amine. 9f was synthesized from tert-butyl 3-carbamothioylpyrrolidine-1-carboxylate and 6 using similar chemistry.
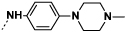
Introduction of a methyl group on the amino pyrimidine (9a) had little effect on potency but extending to the phenylpiperazines (9b-c), gave a 10-fold improvement in both biochemical and cellular potencies. The more basic phenylpiperidine (9d) gave a further boost in potency with an IC50 of 300 pM in the biochemical assay and a cellular potency of 25 nM. Not converting the piperidine N-Boc to methyl allowed access to 9e, while swapping the piperidine group of the thiazole 2-position for the pyrrolidine (9f) gave an equipotent compound to 9b. With the generation of an apo X-ray structure we were able to model the compounds into the ATP binding site of PfPKG. This revealed the key interactions made by compound 3 and the rationale for improved IC50 values of compounds 9b-f. As shown in Figure 3, the 4-fluorophenyl is deeply buried in a hydrophobic pocket between the catalytic lysine (K570) and the small gatekeeper residue (T618).12 The piperidine attached to the thiazole core projects out towards solvent, engaging in charge interactions with D682. The 2-aminopyrimidine forms a pair of H-bonds with the backbone of V621 on the hinge and offers a vector for growth from that amine. The pendent phenyl ring forms edge-face aromatic interactions with Y822 and positions the terminal amine to form charge interactions with D628 at the entrance to the pocket.
Figure 3.

Compounds 3 (left) and 9d (right) docked into an apo crystal structure of PfPKG (PDB:5DYK)13 using Schrodinger GlideSP. The protein surface is coloured by electrostatic potential (red negative, blue positive) and ligand H-bonding interactions are shown in magenta (distance in angstroms), with aromatic interactions in cyan.
The kinase selectivity of 9c when measured against a panel of human kinases at a concentration of 100 nM (Figure 5) showed extensive inhibition of several kinases across the panel. It was thought that selectivity could be achieved via the gatekeeper or DGF loop. Therefore, with the phenyl piperazine in place, variations were then explored around the 4-fluorophenyl portion of the molecule. It was postulated that due to the proximity of this group to the threonine gatekeeper residue,12 which is much smaller than the corresponding residues in most human serine/threonine kinases,14 suitable changes here might enhance the selectivity of the compounds (Table 3).
Figure 5.

Selectivity of compounds 9c, 16a and 23 against a panel of human kinases at a concentration of 100 nM.16 Red is >90% inhibition; yellow between 90% and 70% inhibition, green <70% inhibition, white—not tested
Table 3. Sulfone and sulfonamide analogues.
 | ||||
|---|---|---|---|---|
| Entry | R1 | R2 |
PfPKG IC50 (nM) |
HXI EC50 (nM) |
| 16b | 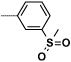 |
- H | 33 | 4570 |
| 16c | 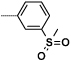 |
 |
9 | 1560 |
| 16d | 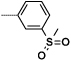 |
 |
17 | NT |
| 16e | 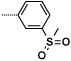 |
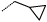 |
6 | 1330 |
| 23 | 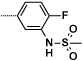 |
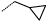 |
2 | 113 |
Methyl derivative (10) and cyclopropyl, (11) were synthesized using the same route as shown in Scheme 1, from the appropriate ester starting material. The syntheses of 16a-e are shown in Scheme 2, starting with the methyl m-(methylthio)benzoate 12, which was elaborated via similar chemistry to intermediate 15. Compound 15 then underwent a double SMe oxidation to the bis-sulfone with hydrogen peroxide and catalytic sodium tungstate, followed by displacement of the (methylsulfonyl)pyrimidine by the requisite amine.
Scheme 2.

Reagents and conditions (a) LiHMDS (1M in THF), THF, 0 °C to r.t., 80%; (b) (i) (Me)3SiCl, (nBu)4NBr, DMSO, THF, 0 °C to r.t., (ii) tert-butyl 4-carbamothioylpiperidine-1-carboxylate, EtOH, reflux, 25%; (c) (i) 4M HCl/dioxane (ii) HCHO, Na(OAc)3BH, AcOH, CH2Cl2; (d) (i) H2O2, Na2WO4.2H2O, AcOH; (ii) NH4OAc, 130 °C or AlkNH2, THF, 70 °C or ArNH2, TFA, sBuOH, 130°C,12-35% from (14)
Replacement of the 4-fluoropenyl moiety with alkyl substituents gave rise to weakly active analogues (10, 11) which both showed a significant drop in biochemical potency when compared to 9c. The lower activity seen with the alkyl substituents could be attributed to their inability to sufficiently fill the hydrophobic pocket between the catalytic lysine (K570) and the small gatekeeper residue (T618) (Figure 3). Despite the binding potency of 11, it showed similar cellular potency to 9c, possibly resulting from poor kinase selectivity as 11 is capable of binding to kinases in the cell with larger gatekeepers.14 Introduction of the sulfone (16a) gave a compound with comparable IC50 values to 9c, but with a much improved kinase selectivity profile (Figure 5).
To further enhance the kinase selectivity of the compounds, additional analogues were made with groups of greater polarity in an attempt to capitalize on additional interactions with the PfPKG enzyme. The new analogues were synthesized with smaller alkyl groups on the pyrimidine ring instead of the large phenyl piperazine group (Table 3).
The synthesis of sulfonamide 23 is highlighted in Scheme 3.
Scheme 3.

Reagents and conditions (a) (i) H-cube, 10% Pd/C, 1 atm, r.t. (ii) methyl chloroformate, pyridine, CH2Cl2, 0 °C to r.t., 94%; (b) LiHMDS (1M in THF), THF, 0 °C to r.t., 67%; (c) (i) (Me)3SiCl, (nBu)4NBr, DMSO, THF, 0 °C to r.t., (ii) tert-butyl 4-carbamothioylpiperidine-1-carboxylate, EtOH, reflux; (d) (i) 4M HCl/dioxane (ii) HCHO, Na(OAc)3BH, AcOH, CH2Cl2; (e) MeSO2Cl, Et3N, CH2Cl2, 0 °C to r.t. 13% from (19); (f) 2M NaOH (aq), dioxane, 80 °C, 49%; (g) (i) H2O2, Na2WO4.2H2O, AcOH; (ii) cyclopropylmethylamine, THF, 70 °C, 11% from (21)
The nitroester (17) was hydrogenated to the aniline, and protected as the methyl carbamate to yield 18 which was subjected to subsequent ketone formation, then chlorination and cyclization to give thiazole 20. Boc deprotection and reductive amination, followed by hydrolysis with sodium hydroxide gave the unprotected aniline, 21. Sulfonylation of 21 was carried out with two equivalents of methanesulfonyl chloride to give firstly the bis sulfonamide, which on exposure to excess sodium hydroxide, yielded the mono sulfonamide 22. Oxidation to the sulfone and displacement with cyclopropylmethylamine yielded 23.
Replacement of the phenyl piperazine by a hydrogen to give the unsubstituted amino pyrimidine (16b) showed a drop in potency, due to the loss of the aforementioned charge interaction with D682. Small alkyl substituents on the amino pyrimidine with the methyl sulphone (16c-e) all showed good potency in the biochemical assay but this did not translate into good potency in the cell assay.
The fluorosulfonamide (23) was the most potent analogue in the biochemical assay and docking of this compound (as shown in Figure 4) revealed the additional polar interactions responsible. The cyclopropylmethyl efficiently fills the small hydrophobic pocket between V621 and Y822, whilst the sulphonamide forms H-bonds with the DFG loop and a charge interaction with K570.
Figure 4.

Compounds 3 (left) and 23 (right) docked into an apo crystal structure of PfPKG (PDB:5DYK)13 using Schrodinger GlideSP. The protein surface is coloured by electrostatic potential (red negative, blue positive) and ligand H-bonding interactions are shown in magenta (distance in angstroms).
Additionally 23 showed very good selectivity against human kinases, likely arising from the small threonine gatekeeper residue as described earlier. Compound 23 and two additional key examples were then profiled in a number of in vitro ADME assays (Table 4). Data for 9c showed a very good overall profile, good logD and stability along with good PAMPA and kinetic solubility. Despite an otherwise excellent profile, the LogD of 16a was low when measured, potentially contributing to the poor permeability seen. Compound 23 was found to be metabolically stable in human and mouse liver microsomes, and also showed good permeability (Table 4). In transforming 9c (cLogP 3.76 pIC50 9.16 LLE15 5.40) to 23 (clogP 2.83 pIC50 8.70 LLE 5.87), we were able to reduce reliance on potency gained from the large, lipophilic phenylpiperazine, and instead improve alternate polar interactions.
Table 4. In vitro ADME data for compounds 9c, 16a and 23.
| Entry | MLMa (% rem) |
HLMa (% rem) |
mLogD (@ pH 7.4) |
PAMPA (nm/s) |
Kinetic Solubility (µM) |
|---|---|---|---|---|---|
| 9c | 97 | 91 | 1.8 | 75 | 209 |
| 16a | 89 | 84 | 0.3 | 0 | 208 |
| 23 | 89 | 95 | 1.9 | 57 | 190 |
% remaining at 40 mins
In summary, several PfPKG inhibitors have been developed and synthesized around the new thiazole core with several possessing low nanomolar biochemical potencies and cellular potencies around 100 nM. Excellent selectivity was achieved over human kinases through modification of the 4-fluorophenyl group to more polar substituents such as 23, removing previous inhibition seen against p38a MAPK and PKA kinases. Furthermore, the anti-parasitical activity of the compounds has been enhanced greatly, as 23 shows a 4-fold improvement in cellular potency over compound 1. Presently, the thiazole series is being further developed to improve anti-parasite activity.
Table 1. Biochemical and cell potencies of thiazole analogues.
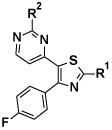 | ||||
|---|---|---|---|---|
| Entry | R1 | R2 | PfPKG IC50 (nM) |
HXI EC50 (nM) |
| 3 | 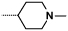 |
- NH2 | 17 | 1810 |
| 9a | 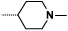 |
- NHMe | 15 | 1490 |
| 9b | 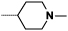 |
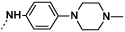 |
1 | 155 |
| 9c | 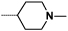 |
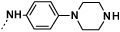 |
0.7 | 92 |
| 9d | 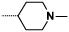 |
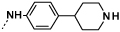 |
0.3 | 25 |
| 9e | 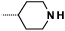 |
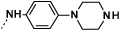 |
0.5 | 73 |
| 9f | 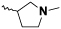 |
 |
6 | 144 |
Table 2. Variations around the 4-fluorophenyl.
 | |||
|---|---|---|---|
| Entry | R |
PfPKG IC50 (nM) |
HXI EC50 (nM) |
| 9c | 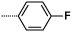 |
0.7 | 92 |
| 10 | - Me | 149 | NT |
| 11 | 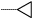 |
47 | 90 |
| 16a | 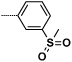 |
0.9 | 105 |
NT – not tested
Acknowledgements
This work was funded by MRC DPFS grant no G1000779. We thank David Tickle, Sadhia Khan and Katie Barnes (LifeArc) for compound purification and in vitro ADME data, Win Gutteridge and Simon Croft for many helpful discussions, and Jeremy Burrows and Sir Simon Campbell (Medicines for Malaria Venture) for their support of this work
References and Notes
- 1.World Health Organisation. World Malaria Report 2017. [Google Scholar]
- 2.(a) World Health Organisation. World Malaria Report 2014. [Google Scholar]; (b) Dondorp AM, Fairhurst RM, Slutsker L, MacArthur JR, Breman JG, Guerin PJ, Wellems TE, Ringwald P, Newman RD, Plowe CV. New Engl J Med. 2011;365:1073. doi: 10.1056/NEJMp1108322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor HM, McRobert L, Grainger M, Sicard A, Dluzewski AR, Hopp CS, Holder AA, Baker DA. Euk Cell. 2010;9:37. doi: 10.1128/EC.00186-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alam MM, Solyakov L, Bottrill AR, Flueck C, Siddiqui FA, Singh S, Mistry S, Viskaduraki M, Lee K, Chitnis CE, Doerig C, et al. Nat Commun. 2015;6:7285. doi: 10.1038/ncomms8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McRobert L, Taylor CJ, Deng W, Fivelman QL, Cummings RM, Polley SD, Billker O, Baker DA. PLoS Biol. 2008;6:e139. doi: 10.1371/journal.pbio.0060139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moon RW, Taylor CJ, Bex C, Schepers R, Goulding D, Janse CJ, Waters AP, Baker DA, Billker O. PLoS Path. 2009;5:e1000599. doi: 10.1371/journal.ppat.1000599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Govindasamy K, Jebiwott S, Jaijyan DK, Davidow A, Ojo KK, Van Voorhis WC, Brochet M, Billker O, Bhanot P. Mol Microbiol. 2016;102:349. doi: 10.1111/mmi.13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Falae A, Combe A, Amaladoss A, Carvalho T, Menard R, Bhanot P. J Biol Chem. 2010;285:3282. doi: 10.1074/jbc.M109.070367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biftu T, Feng D, Fisher M, Liang G, Qian X, Scribner A, Dennis R, Lee S, Liberator PA, Brown C, Gurnett A, et al. Bioorg Med Chem Lett. 2006;16:2479. doi: 10.1016/j.bmcl.2006.01.092. [DOI] [PubMed] [Google Scholar]
- 10.Full experimental details can be found in Baker DA, Stewart LB, Large JM, Bowyer PW, Ansell KH, Jiménez-Díaz MB, El Bakkouri M, Birchall K, Dechering KJ, Bouloc NS, Coombs PJ, et al. Nat Commun. 2017;8:430. doi: 10.1038/s41467-017-00572-x.
- 11.Stellwagen JC, Adjabeng GM, Arnone MR, Dickerson SH, Han C, Hornberger KR, King AJ, Mook RA, Petrov KG, Rheault TR, Rominger CM, et al. Bioorg Med Chem Lett. 2011;15:4436. doi: 10.1016/j.bmcl.2011.06.021. [DOI] [PubMed] [Google Scholar]
- 12.(a) Donald RG, Zhong T, Wiersma H, Nare B, Yao D, Lee A, Allocco J, Liberator PA. Mol Biochem Parasitol. 2006;149:86. doi: 10.1016/j.molbiopara.2006.05.003. [DOI] [PubMed] [Google Scholar]; (b) Gurnett AM, Liberator PA, Dulski PM, Salowe SP, Donald RG, Anderson JW, Wiltsie J, Diaz CA, Harris G, Chang B, Darkin-Rattray SJ, et al. J Biol Chem. 2002;277:15913. doi: 10.1074/jbc.M108393200. [DOI] [PubMed] [Google Scholar]
- 13.Wernimont AK, Tempel W, He H, Seitova A, Hills T, Neculai AM, Baker DA, Flueck C, Kettleborough CA, Arrowsmith CH, Edwards AM, et al. Crystal structure of PF3D7_1436600. Structural Genomics Consortium; to be published. [Google Scholar]
- 14.Huang D, Zhou T, Lafleur K, Nevado C, Caflisch A. Bioinformatics. 2010;26:198. doi: 10.1093/bioinformatics/btp650. [DOI] [PubMed] [Google Scholar]
- 15.LLE = pIC50 - cLogP
- 16.Kinase selectivity profiling was carried out at the MRC Protein Phosphorylation Unit at the University of Dundee.Kinase list (reading left to right): MKK1, ERK1, ERK2, JNK1, JNK2, p38a, MAPK, RSK1, RSK2, PDK1, PKBa, PKBb, SGK1, S6K1, PKA, ROCK 2, PRK2, PKCa, PKCz, PKD1, MSK1, MNK1, MNK2, PRAK, CAMKKb, CAMK1, SmMLCK, PHK, CHK1, CHK2, GSK3b, CDK2-Cyclin A, PLK1, Aurora A, Aurora B, AMPK, MARK3, BRSK2, MELK, CK1, CK2, DYRK1A, NEK2a, NEK6, IKKb, PIM1, SRPK1, MST2, EF2K, HIPK2 PAK4, Src, Lck, CSK, FGF-R1, IRR, EPHA2, MST4, SYK, YES1, IGF-1R, VEG-FR, BTK, EPH-B3, TBK1, IKKe, GCK, IRAK4, NUAK1, MLK1, MINK1, MLK3, LKB1, HER4, TTK, IR, RIPK2, TAK1 MEKK1, TrkA, JAK2, DAPK1, IRAK1.