

Abstract
Reprogrammed metabolism supports tumor growth and provides a potential source of therapeutic targets and disease biomarkers. Mass spectrometry-based metabolomics has emerged as a broadly informative technique for profiling metabolic features associated with specific oncogenotypes, disease progression, therapeutic liabilities and other clinically relevant aspects of tumor biology. In this review, we introduce the applications of metabolomics to study deregulated metabolism and metabolic vulnerabilities in cancer. We provide examples of studies that used metabolomics to discover novel metabolic regulatory mechanisms, including processes that link metabolic alterations with gene expression, protein function, and other aspects of systems biology. Finally, we discuss emerging applications of metabolomics for in vivo isotope tracing and metabolite imaging, both of which hold promise to advance our understanding of the role of metabolic reprogramming in cancer.
Keywords: Metabolomics, Cancer Metabolism, Metabolic Subtypes, Systems Biology, Isotope Tracing, Metabolite Imaging
Introduction
Metabolism supports various aspects of normal cell biology, including breakdown of fuels such as carbohydrates, fats, and amino acids to generate energy and biosynthetic precursors for growth[1]. These fundamental features of cellular metabolism are reprogrammed in cancer cells to support their pathological levels of growth and proliferation. Metabolic reprogramming in malignant cells is likely the result of the multifactorial effects of genomic alterations (i.e. mutations of oncogenes and tumor suppressors), the tumor microenvironment (which imposes metabolic stress caused by compromised nutrients and oxygen availability), and other influences[1–3]. We need to understand the complete breadth of metabolic abnormalities in cancer because some metabolic changes provide opportunities to develop novel therapeutic targets and predictive biomarkers.
Generations of studies reaching back to the 1920s have analyzed metabolic alterations in cancer, with enhanced glucose utilization being the most frequently and broadly observed. The clinical relevance of metabolic reprogramming in tumors is supported by routine use of the glucose analog fluorodeoxyglucose as a radiolabeled tracer for positron emission tomography-based imaging (FDG-PET)[4]. As newer technologies have become available to characterize tumor metabolism more broadly and specifically than ever before, many other examples of potentially clinically-actionable metabolic perturbations have become apparent, indicating that the propensity for enhanced glucose uptake is merely the tip of the iceberg[5].
Understanding cancer metabolism requires systematic application of analytical techniques to assess metabolite levels in biological samples from healthy and diseased tissues. Metabolomics has emerged as the most powerful platform to recognize metabolic anomalies in urine, serum or tissue samples[6, 7]. In general, metabolomics techniques provide semi-quantitative or quantitative information about the steady-state abundance of intermediates from many metabolic pathways simultaneously, providing the user with an overview of the metabolic network and its perturbation in disease[8, 9]. This review discusses metabolomics methods and presents examples where metabolomics has been used to uncover new concepts in cancer biology or to identify novel targets for diagnostic imaging and therapy.
Metabolomics: An informative platform to study cancer metabolism
Metabolomics requires analytical techniques such as nuclear magnetic resonance spectroscopy (NMR) and mass-spectrometry (MS) to measure metabolites in biological samples. NMR detects the magnetic spin of molecular nuclei under a defined magnetic frequency and is effective at identifying metabolites from complex mixtures, quantifying metabolite abundance, and assessing the position of specific nuclei (e.g. 13C) within a metabolite of interest, all with excellent reproducibility[10, 11]. NMR has the advantage of providing non-destructive analysis and the potential for in vivo metabolite detection in humans. Proton magnetic resonance spectroscopy (MRS) provides non-invasive detection of D-2-hydroxyglutarate (D-2HG) in gliomas with IDH1/2 mutations, and confirmed the previously observed profile of elevated choline and downregulated creatine and N-acetyl aspartate in gliomas compared to normal human brain[12, 13]. Similarly, 13C magnetic resonance spectroscopy (13C MRS) analysis of healthy individuals infused with [U-13C] glucose identified glucose flux into the TCA cycle via pyruvate dehydrogenase in healthy brain[14]. Limitations of NMR for comprehensive metabolomic assessment include its relatively low sensitivity and selectivity[11].
MS-based techniques rely on the mass/charge (m/z) ratio of a metabolite or its fragments. These techniques have extremely high sensitivity, with commercial instruments enabling the detection of metabolites in tiny samples of a few thousand cells or less and achieving femtomolar sensitivity[11, 15–17]. MS analysis can require parallel extraction procedures to recover polar, non-polar and volatile compounds for analysis, and extensive processing can lead to sample disintegration with loss of the most labile compounds. However, rapid advancements in mass spectrometry hardware, ionization techniques, and data-analysis software have steadily increased the scope of MS-based metabolomics experiments in both targeted and untargeted applications, making MS the most prominent technology in modern metabolomics[11, 17].
A common application of metabolomics has been to discover biomarkers for diagnosis or to predict therapeutic sensitivity and prognosis[18]. For example, relatively early metabolomics experiments in breast cancer identified positive associations between levels of choline, glycine, and lactate, and histopathological grade and tumor size[19, 20]. Similar work in tissue samples from ovarian[21], prostate[22, 23], brain[24, 25], and kidney[26, 27] cancers and breath samples from lung cancer patients[28] identified metabolic perturbations within tumor grades and sizes. Specifically, choline and related metabolites were elevated in prostate and pediatric brain tumors, while lipids were elevated in kidney tumors. These studies employed various forms of statistical modelling to determine metabolic signatures that differentiated tumor from noncancerous tissues, and distinguished tumor stages and grades from each other.
Metabolic disturbances associated with genomic alterations in metabolic enzymes
Many early metabolomic studies identified metabolic differences between tumors and non-cancerous tissues, but lacked understanding of the molecular basis for these differences. More recently, the simultaneous implementation of molecular biology techniques and other integrative strategies together with metabolomics has played an essential role in deciphering the molecular underpinnings of metabolic reprogramming in cancer. We now appreciate that genomic or gene expression alterations in key enzymes of metabolic pathways support oncogenic transformation and/or enable tumor growth and progression. A current challenge is to understand how these changes contribute to tumor biology and which might be amenable to therapeutic targeting. In a few important cases discussed in this section, mutations in metabolic enzymes result in the accumulation of metabolites that directly contribute to malignant transformation. These metabolites are commonly referred to as oncometabolites, and although they account for a small subset of reprogrammed metabolism, they are highly instructive because they provide insight into mechanisms of tumorigenesis and the impact of metabolic perturbation on tissue function.
A. Oncometabolites generated by gain of neomorphic enzyme activity
Isocitrate-dehydrogenases (IDH1, IDH2 and IDH3) catalyze the NAD+/NADP+-dependent decarboxylation of isocitrate to α-ketoglutarate (α-KG)[29]. While IDH1 is localized to cytosol and peroxisomes, IDH2 and IDH3 are mitochondrial enzymes. About 10 years ago, IDH1 and IDH2 mutations were identified in patients with low- and intermediate-grade gliomas and in glioblastomas arising from these initially less aggressive lesions[30, 31]. These mutations result in suppression of the canonical NADP+-dependent oxidative decarboxylation of α-KG. However, the mutations are monoallelic and essentially always located in the same residues in the IDH1/IDH2 active site, suggesting a gain-of-function mechanism relevant to tumor initiation. Metabolomics identified millimolar levels of D-2-hydroxyglutarate (D-2HG), a metabolite normally present at trace levels, in gliomas and cell lines expressing mutant IDH1[32]. The R132 mutation in IDH1 confers a neomorphic activity resulting in NADPH-dependent generation of D-2HG from α-KG[32] (Figure 1). Soon after this observation, mutations in IDH1 and IDH2 were identified in acute myeloid leukemia (AML)[33], thyroid cancer[34], and in other tumor types[35]. Invariably, R132 mutations in IDH1 result in D-2HG accumulation, suggesting a critical role for D-2HG in driving tumorigenesis. Orthogonal studies demonstrated that D-2HG functionally impairs α-KG dependent dioxygenases, such as histone and DNA demethylases and prolyl hydroxylases[35–37] (Figure 1). Small-molecules inhibiting mutant IDH1/2 were demonstrated to suppress D-2HG production, reduce tumor growth and/or induce differentiation of experimental models of glioma[38] and leukemia[39, 40], indicating the therapeutic potential of targeting these mutations. These remarkable discoveries ultimately led to the US Food and Drug Administration (FDA) approval of the mutant IDH2 inhibitor enasidenib (IDHIFA, Celgene Corp.) for the treatment of AML patients[41], with ongoing clinical trials in other forms of IDH-mutant cancer.
Figure 1. Oncometabolites inhibit α-KG-dependent dioxygenases.
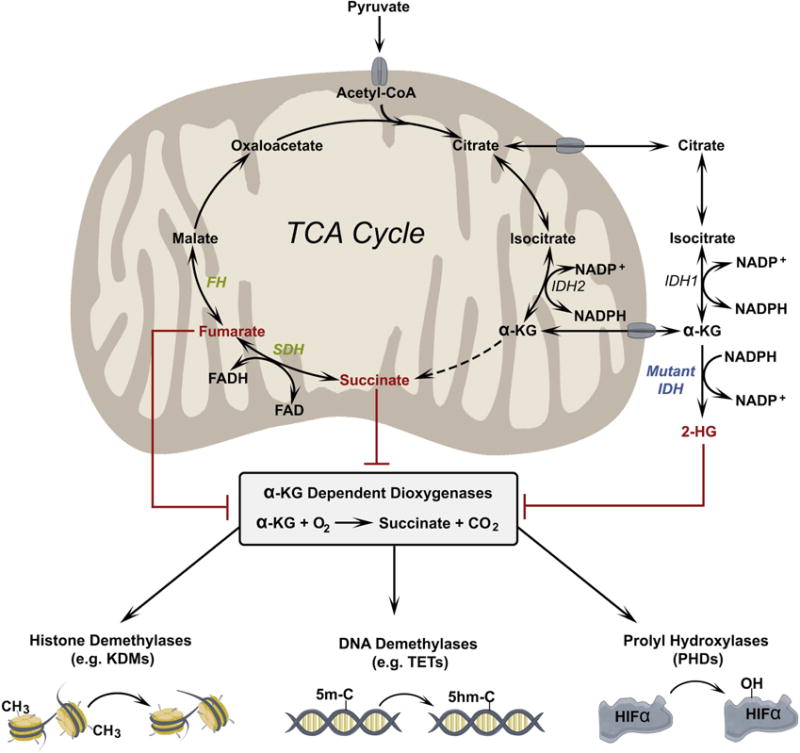
α-KG is required for the function of a family of dioxygenase enzymes including histone demethylases, which remove methyl groups from lysine residues in histone proteins; 5-methylcytosine hydroxylases, which initiate demethylation of cytosine bases; and prolyl hydroxylases, which hydroxylate proline residues in proteins such as the α subunits of hypoxia inducible factors (HIFs). These dioxygenases can be inhibited by high levels of other dicarboxylic acids, which compete with α-KG. Dicarboxylic acids demonstrated to inhibit dioxygenases include D-HG (a product of mutant IDH1/2) and fumarate and succinate, which accumulate due to loss-of-function mutations in FH and SDH, respectively.
B. Oncometabolites generated by loss of enzyme activity
Mutations in the TCA cycle enzymes succinate dehydrogenase (SDH)[42, 43] and fumarate hydratase (FH)[44] lead to familial cancer syndromes such as paraganglioma, pheochromocytoma and papillary renal cell carcinoma, indicating that these enzymes function as tumor suppressors[45]. Unlike IDH mutations, FH and SDH mutations result in the loss of enzymatic function, usually through the inheritance of one germline loss-of-function mutation followed by loss of the second allele in the tumor. This results in the accumulation of the substrates fumarate and succinate, both of which inhibit α-KG dependent dioxygenases (Figure 1). The mechanisms by which fumarate and succinate contribute to tumorigenesis in sensitive tissues are an area of active investigation, with evidence indicating some overlap with D-2HG effects. Mechanistically, with varying IC50 values, fumarate and succinate inhibit α-KG dependent prolyl hydroxylases[46, 47], TET enzyme-regulated hydroxylation of methyl-cytosine[48] including in the promoters of HIF-regulated genes[49], and histone demethylases[48], conferring genome and proteome modifications that support oncogenic transformation. Studies suggest that both metabolites can modulate the epithelial-mesenchymal transition (EMT). The fumarate driven EMT gene-signature is independent of HIF signaling but requires inhibition of TET enzyme-dependent demethylation of microRNA clusters that negatively regulate metastasis in papillary and renal cell carcinoma[50]. In paraganglioma and kidney cells lacking functional SDH, succinate inhibits α-KG dependent dioxygenases, resulting in histone hypermethylation and acquisition of EMT-like migratory phenotypes[50, 51]. Fumarate also covalently modifies reactive sulfhydryl groups on cysteine residues through a process called succination. Elevated levels of fumarate result in the conversion of glutathione (GSH) to succinated GSH, reducing NADPH abundance and enhancing ROS and HIF1α activation[52]. Fumarate also succinates cysteine residues of Kelch Like ECH Associated Protein 1 (KEAP1), abrogating its ability to repress nuclear factor erythroid 2–related factor 2 (NRF2), which regulates expression of antioxidant genes and promotes tumorigenesis[53].
Metabolomics identifies subtype-selective therapeutic liabilities in cancer
Metabolomics can be used to reveal metabolic differences among different tumor subclasses from the same anatomic location. Some of these differences reflect the cell-autonomous effects of specific oncogenotypes, while others reflect the complex effects of evolving tumor biology during cancer progression. Regardless of the cause, such metabolic changes might present new opportunities for diagnostic imaging or therapy. In this section, we discuss applications of metabolomics to identify metabolic features that stratify tumors into molecular or biological subclasses.
A. Metabolic abnormalities governed by cancer genotypes
In many non-transformed epithelial and hematopoietic cells, nutrient uptake is tightly regulated by growth factor signaling, allowing cells to engage growth-promoting metabolic pathways precisely when they receive exogenous signals to proliferate. The PI3K-AKT-mTOR signaling pathway is a major effector of growth factors and induces numerous bioenergetic and biosynthetic pathways through post-translational protein modifications, activating transcriptional networks, and other mechanisms[54–58]. This pathway frequently becomes constitutively activated in cancer due to mutations/amplifications of key regulatory subunits and/or deletion of tumor suppressors. For example, phosphatase and tensin homolog (PTEN), the key negative regulator of PI3K, is the most frequently deleted tumor suppressor[59–61]. PTEN transgenic mice with additional genomic copies of PTEN demonstrate increased energy expenditure and reduced body mass[62, 63], and embryonic fibroblasts from these mice display elevated oxidative phosphorylation and reduced glycolysis as a result of repressed PI3K/AKT signaling[63]. Chronic engagement of PI3K-AKT-mTOR signaling through a variety of molecular mechanisms enhances glycolysis and anabolic pathways to supply de novo biosynthesis of nucleotides, fatty acids, and amino acids for tumor growth. For instance, Epidermal Growth Factor Receptor (EGFR)-mutant lung adenocarcinoma cell lines require PI3K-AKT-mTOR signaling to maintain a growth-promoting metabolic program, and inhibition of the signaling pathway suppresses glycolysis[64]. These tumors do not respond to RAS/MEK/MAPK pathway inhibitors, but NRAS-mutant melanoma tumors do require concomitant MEK and PI3K signaling[65]. These examples highlight the orchestration of oncogenic signals to modulate cellular metabolism in cancer.
Several recent studies have used metabolomic profiling to identify metabolic liabilities within specific molecular subsets of cancer. In these cases, signaling or transcriptional networks imposed by a mutation or combination of mutations can render cells exquisitely dependent on an activity that is dispensable in cells with different oncogenotypes. For example, in non-small cell lung cancer (NSCLC), metabolomics revealed marked differences between cells and tumors with concomitant mutations in the oncogene KRAS and tumor suppressor protein LKB1 compared to cells/tumors with mutant KRAS and wild-type LKB1[66]. Specifically, the co-mutants had broad alterations in pathways of nitrogen metabolism and expressed high levels of carbamoyl phosphate synthetase-1 (CPS1), the rate-limiting enzyme of urea cycle. CPS1’s physiological role in the liver is to initiate nitrogen disposal by condensing ammonia and bicarbonate in the mitochondria. Surprisingly, in KRAS/LKB1 co-mutant NSCLC cells, carbamoyl phosphate from CPS1 instead stimulates an unconventional pathway of pyrimidine biosynthesis[66] (Figure 2a, Left). Suppressing CPS1 depletes the pyrimidine pool in co-mutant cells, resulting in DNA polymerase stalling, DNA damage, reduced tumor growth and increased sensitivity to cisplatin, whereas cells with wild-type LKB1 are resistant to CPS1 loss[66]. Similarly, high-grade KRASG12D/G12D-mutant lung cancer with TP53 null background exhibits elevated glycolysis and glucose-derived carbon flux into the TCA cycle and glutathione biosynthesis[67] (Figure 2a, Right). These tumors are strikingly more sensitive to combined treatment with the glycolytic inhibitor 2-deoxy-D-glucose (2DG) and the glutathione biosynthesis inhibitor buthionine sulfoximine (BSO), whereas both KRAS wild-type tumors and KRASG12D/+ tumors with wild-type TP53 are relatively resistant to these treatments.
Figure 2. Examples of genotype-driven metabolic reprogramming in cancer.
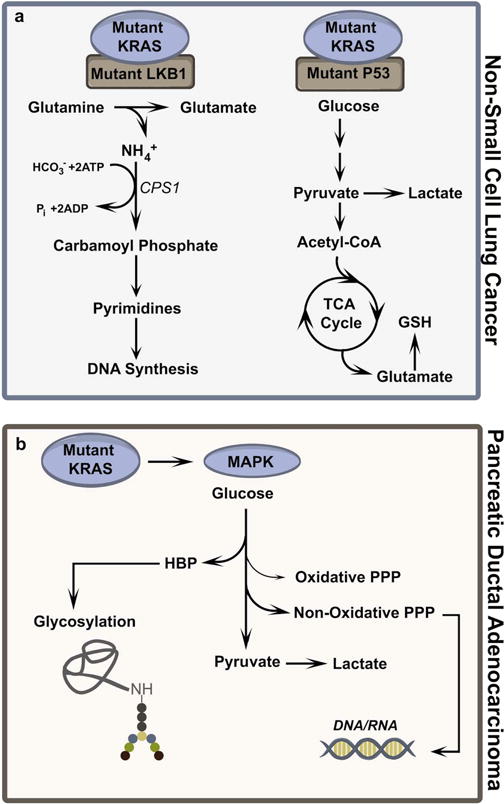
a. Non-small cell lung cancer (NSCLC) with concomitant mutations in KRAS and LKB1 use an unusual form of pyrimidine biosynthesis initiated by carbamoylphosphate synthetase-1 (CPS1). NSCLC with mutations in KRAS and p53 display glucose-dependent glutathione (GSH) biosynthesis. b. KRAS-mutant pancreatic ductal adenocarcinoma (PDAC) requires MAPK signaling to regulate glucose flux into the hexosamine biosynthesis pathway (HBP) and non-oxidative pentose phosphate pathway (Non-Oxidative PPP). These pathways contribute to protein glycosylation and nucleic acid synthesis, respectively.
More than 90% of pancreatic ductal adenocarcinoma (PDAC) contain the KRASG12D mutation[68]. Combined transcriptomics and metabolomics in KRASG12D-mutant PDAC revealed elevated glycolysis and increased fluxes into the hexosamine biosynthetic pathway (HBP) to maintain protein glycosylation and the non-oxidative pentose phosphate pathway (non-oxidative PPP) to generate DNA/RNA[68] (Figure 2b). These KRASG12D tumors require MAPK signaling, but not the PI3K-AKT pathway, to maintain glucose flux through the HBP and the non-oxidative PPP. Oncogenic KRAS also diverts glutamine-derived aspartate to malate in the cytoplasm through increased expression of glutamic-oxaloacetic transaminase 1 (GOT1) in PDAC cells[69]. Subsequently, malate-derived oxaloacetate generates pyruvate and NADPH via malic enzyme, and the NADPH is used to maintain a reduced GSH pool for ROS homeostasis[69]. Such rewiring of glutamine metabolism confers independence of the oxidative pentose phosphate pathway for NADPH production. KRAS-mutant colorectal cancer (CRC) cells convert glutamine-derived aspartate to asparagine and show sensitivity to inhibitors of asparagine synthetase (ASNS)[70]. Interestingly, KRAS-mutant CRC cells maintain asparagine levels through PI3K-AKT-mTOR pathway mediated ASNS expression[70]. Thus, oncogenic KRAS has pleiotropic metabolic effects that result in different metabolic liabilities in different cancer types.
BRAF, another member of the RAS family of oncogenes, is also frequently mutated in cancer. Numerous studies have illustrated the impact of the BRAF V600E on metabolic phenotypes. Genome wide shRNA screening identified 3-hydroxy-3-methylglutaryl-CoA lyase (HMGCL) as a metabolic vulnerability associated with BRAF V600E melanoma[71]. HMGCL generates the ketone body acetoacetate, which physically binds to mutant BRAF protein to stabilize its interaction with Mitogen-Activated Protein Kinase Kinase 1 (MEK1), thus potentiating BRAF-dependent signals[71]. These BRAF and MAPK dependent melanomas show reduced mitochondrial metabolism and decreased expression of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PCG1α)[72]. In patient-derived melanoma xenografts, the BRAF V600E genotype is associated with abundant glycolytic metabolites and enhanced activity of the glycolytic pathway in vivo[73].
MYC family members are the most frequently amplified or otherwise activated oncogenic transcription factors across human tumor types[74]. MYC stimulates glucose and glutamine metabolism by regulating expression of genes related to these pathways, including lactate dehydrogenase-A (LDHA)[75], glucose transporter (GLUT1)[76] and glutaminase (GLS1)[77]. Metabolomic studies have broadened our understanding of the diverse functions of MYC in regulating many other metabolic pathways in cancer. In triple negative breast cancer, MYC regulates metabolites and genes involved in fatty acid oxidation, resulting in enhanced levels of acylcarnitine intermediates and rendering tumors sensitive to inhibitors of carnitine palmitoyltransferase 1 (CPT1), the rate-limiting enzyme in this pathway[78]. MYC also regulates 2-hydroxyglutarate (2-HG)-dependent DNA hypermethylation in tripe negative breast cancer[79]. Global metabolic profiling of MYC-driven colorectal tumors identified significant metabolic dysregulation at the advanced adenoma stage[80]. In this model, MYC regulates genes involved in purine/pyrimidine metabolism, glycolysis, and the pentose phosphate pathway as well as fatty acid synthesis. However, in contrast to its role in triple negative breast cancer, MYC overexpression downregulates fatty acid oxidation genes in colorectal cancer[80], further emphasizing that the metabolic effects of oncogene activation may manifest differently in different tumors.
B. Metabolic dysregulation associated with tumor progression or aggressiveness
Cellular metabolism is thought to evolve during cancer progression, and several studies demonstrate how metabolomics can be used to nominate evolving metabolic features as biomarkers or therapeutic targets. To identify non-invasive biomarkers for diagnosis and prognosis of prostate cancer, Sreekumar et. al used metabolomics to assess global metabolic alterations in urine, serum, and prostate cancer tissues[81]. Sarcosine, a naturally-occurring N-methylated form of glycine, was progressively elevated in localized and metastatic tissues (Figure 3) and urine samples. Treating benign prostate epithelial cells with sarcosine or silencing the sarcosine-degrading enzyme sarcosine dehydrogenase (SARDH) enhanced invasive properties, while knockdown of the sarcosine-synthesizing enzyme glycine N-methyltransferase (GNMT) reduced invasion of prostate cancer cells[81]. Several[82, 83] but not all[84, 85] subsequent studies have validated elevated sarcosine level in tissues and urine samples from prostate cancer patients.
Figure 3. Metabolic rewiring during cancer progression.
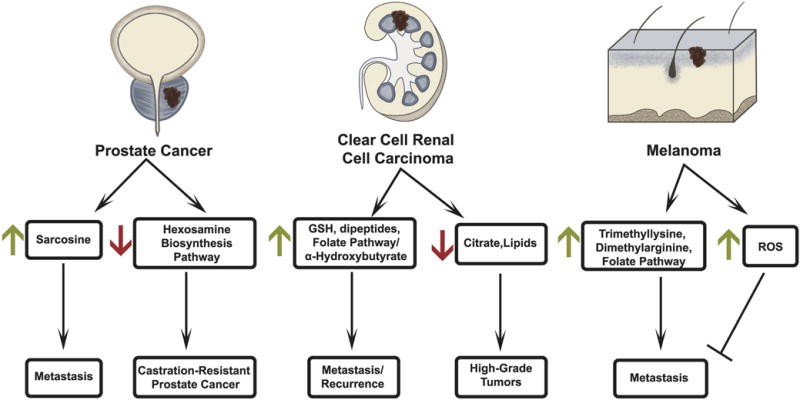
In prostate cancer, elevated sarcosine is associated with metastasis while downregulation of the hexosamine biosynthesis pathway (HBP) is associated with castration-resistance. In clear cell renal cell carcinoma (ccRCC), elevated glutathione (GSH), dipeptide metabolites, and metabolites from the 1-carbon/folate pathway are associated with metastasis while α-hydroxybutyrate is associated with disease recurrence. Decreased levels of lipids and citrate are observed as lower-grade tumors progress to high-grade ccRCC. In melanoma, trimethyllysine, dimethylarginine, and induction of the 1-carbon/folate pathway are associated with metastasis, while elevated ROS is associated with inhibition of metastasis.
The metabolomics data generated by Sreekumar et al[81] have subsequently been used to identify other metabolic networks contributing to prostate cancer progression. We observed that inhibition of the hexosamine biosynthesis pathway (HBP) promotes castration-resistant prostate cancer (CRPC)[86] (Figure 3). While the HBP is essential for localized prostate cancer growth[86], its downregulation promotes CRPC, making the HBP an example of metabolic rewiring associated with cancer progression. Additionally, metabolomics of androgen-dependent and castration-resistant prostate cancer cell lines identified correlations between UDP-glucuronosyltransferase expression and disease progression[87]. In CRPC, constitutive activation of the androgen receptor (AR) is associated with resistance to 2nd generation anti-androgens. Interestingly, AR-V7, a spliced form of full-length AR that is expressed in anti-androgen enzalutamide and abiraterone-acetate resistant CRPC patients[88], was found to decrease the abundance of citrate[89] and increase utilization of glutamine-derived reductive carboxylation of α-KG in prostate cancer LNCaP cells expressing AR-V7[89]. Similarly, steroid receptor coactivator-2 (SRC-2), a nuclear receptor interacting protein with histone acetyltransferase activity, is elevated in metastatic prostate cancer and regulates glutamine-derived reductive carboxylation of α-KG in CRPC and metastatic cell lines[90].
Many similar studies have focused on breast cancer, another hormone-sensitive cancer. However, breast cancer frequently becomes independent of hormone signaling in the aggressive/metastatic stages. Triple negative breast cancer (TNBC) is aggressive and therapeutically intractable. It is characterized by loss of estrogen and progesterone receptors (ER and PR) and lacks expression of the epidermal growth factor receptor HER-2. Cao et al. used high-resolution magic angle spinning magnetic resonance spectroscopy (HR MAS MRS) to differentiate metabolic profiles between TNBC and tumors positive for ER, PR, and HER-2[91]. TNBC tumors displayed elevations of choline and glutamate, while HER-2 positivity was associated with elevated glycine and glutamine[91]. Interestingly, in a separate study, choline and glutamic acid were identified as components of an 11-metabolite panel associated with breast cancer recurrence[92].
Clear cell renal cell carcinoma (ccRCC) is the most commonly diagnosed kidney cancer. These tumors typically display constitutive expression of hypoxia inducible factors (HIF1α and/or HIF2α) due to frequent loss of the tumor suppressor Von-Hippel Lindau (VHL), which normally facilitates the oxygen-dependent degradation of HIF-α subunits[93–95]. ccRCC displays hallmarks of perturbed metabolism including dramatic accumulation of lipids and glycogen. A large metabolomics analysis of more than 130 matched ccRCC tumors and adjacent kidney samples revealed decreased citrate and increased glutathione, dipeptides, and α-hydroxybutrate in stage 4 tumors[96] (Figure 3). The study also found positive associations between α-hydroxybutyrate and disease recurrence, and between metabolites of the one-carbon and cysteine/methionine cycles and disease progression. These latter correlations are consistent with several other studies in suggesting that ccRCC tumors acquire robust anti-oxidant and methylation capacity[97–100]. We used metabolomics, Dixon-MR imaging of fat content, and lipidomics to assess regional heterogeneity of fat deposition and its relationship with metabolite abundance and tumor grade in ccRCC. This study revealed marked heterogeneity of lipids and aqueous metabolites, with relative depletion of lipid content and accumulation of several amino acids in higher-grade tumors[100].
A comprehensive, integrative analysis incorporating gene expression and metabolomics data observed elevated glycolytic metabolites and decreased expression of the gluconeogenic enzyme fructose-1,6-bisphosphatase 1 (FBP1) in ccRCC, with FBP1 expression declining as the disease progressed[99]. FBP1 expression suppressed glycolysis, thereby interrupting the pseudohypoxic metabolic state of cells with chronic HIF-α expression. Surprisingly, this study identified a second, non-catalytic function of FBP1 in ccRCC. Nuclear-localized FBP1 directly associated with HIF-α subunits, interfering with the expression of HIF target genes and resulting in reduced cell proliferation.
A key question in cancer metabolism is how metabolic reprogramming influences metastasis, because metastasis is the primary determinant of mortality in many forms of cancer. Melanoma provides an excellent opportunity to study the metabolic basis of metastasis, because these tumors metastasize frequently in patients and because mouse models of melanoma exhibiting frequent metastasis are available for study. We used metabolomics to profile a panel of patient-derived melanoma xenografts (PDXs) in which the metastatic efficiency of the tumors in mice correlated with progression to stage IV melanoma (i.e. distant macrometastases) in the donor patients[101]. In this panel of PDXs, subcutaneous tumors that frequently gave rise to distant macrometastases had increased levels of trimethyllysine (TML) and dimethylarginine (DMA), two metabolites related to histone methylation[73] (Figure 3). TML abundance correlated with two distinct trimethylation marks on histone H3, H3K9me3 and H3K27me3. Erasing these marks by silencing or inhibiting the methyltransferases SET Domain Bifurcated 1 (SETDB1) and Enhancer Of Zeste Homolog 2 (EZH2) reduced free TML levels and decreased in vitro invasion and in vivo metastasis without impacting subcutaneous tumor growth[73]. These findings indicate that metabolomics can be sensitive enough to detect changes in the epigenetic state and can identify activities that enable metastasis in vivo.
Another study using these same melanoma PDXs focused on metabolic differences between tumor cells at the subcutaneous site, in the circulation and in metastases in visceral organs [102]. Tumor cells in the circulation and in visceral metastases had evidence of oxidative stress and enhanced activity of the folate pathway to generate NADPH for ROS homeostasis, indicating that ROS imposed a bottleneck on metastasis in these models (Figure 3). Treating tumor-bearing mice with the antioxidant N-acetyl cysteine (NAC) increased the number of tumor cells in circulation and enhanced metastatic burden, whereas imposing modest oxidative stress with low-dose methotrexate suppressed metastasis[102]. Additional mechanistic studies in cell culture further support a role for ROS mitigation in anchorage-independent survival and growth, key determinants of the metastatic cascade. Loss of NADPH production from the pentose phosphate pathway was observed to limit the survival of non-transformed cells during loss of anchorage, and this could be overcome by expressing an oncogene[103]. In cancer cells, loss of attachment to a 2-dimensional matrix was associated with enhanced levels of mitochondrial ROS which limited the growth of detached tumor spheroids[104]. In this system, mitigating mitochondrial ROS required transfer of NADPH from the pentose phosphate pathway in the cytosol into the mitochondria. The transfer mechanism involved NADPH-dependent reductive carboxylation of cytosolic α-KG by IDH1, followed by entry of the resulting isocitrate/citrate into the mitochondria where it supplied IDH2-depenent NADPH production[104]. Altogether, these studies demonstrate how metabolism is rewired to mitigate elevated ROS associated with disease progression and metastasis.
C. Metabolic cross-talk between cancer cells and immune cells
Cancer cells compete for nutrient availability with non-malignant cells in the tumor microenvironment, including macrophages, dendritic cells, lymphocytes, natural killer cells, fibroblasts, adipocytes, pericytes and others[105–107]. Metabolism of these cells may also be perturbed during tumorigenesis, and therefore, understanding their metabolism could help develop better therapeutic strategies. Tumor-infiltrating T cells (TILs) and tumor-associated macrophages (TAMs) exhibit marked metabolic adaptations in the tumor microenvironment. Activated CD8+ T cells demonstrate elevated HIF1α/VEGFA signaling which corresponds positively with their anti-tumor acivities[108]. Tumor-promoting TAMs demonstrate elevated oxidative phosphorylation and reliance on glutamine metabolism and fatty acid oxidation, while tumor-inhibiting TAMs demonstrate elevated glycolysis and pentose phosphate pathway activity[109]. Several excellent reviews describe metabolic adaptations in immune cells in various diseases, including cancer[106, 110].
With the approval of anti-PD1 and anti-PDL1 immunotherapy such as nivolumab for advanced cancer, there is an emerging interest in deciphering metabolic alterations associated with immunotherapy. Giannakis et al. used LC/MS-based metabolomics to identify changes in the serum of melanoma and RCC patients treated with nivolumab[111]. Melanoma patients that responded to therapy showed increased levels of kynurenine[111], an intermediate of tryptophan degradation with inflammatory and immunomodulatory properties[112]. In RCC, nivolumab non-responders showed increased adenosine and poor progression free survival[111]. Frankel et al. conducted a similar but prospective study to determine both gut microbiome and metabolite alterations in melanoma patients treated with ipilimumab (anti-CLA4 antibody); nivolumab (anti-PD1 antibody); ipilimumab with nivolumab; or pembrolizumab (anti-PD1 antibody). Metabolomics on stool samples revealed several altered pathways among the treatment groups, and metagenomics of stool revealed a significant increase in the expression of bacterial enzymes related to fatty acid synthesis in patients responding to therapy[113]. Many ongoing clinical trials will further explore the interplay between immunotherapy and cancer metabolism, with the goal of identifying metabolic inhibitors to enhance the efficacy of immunotherapy.
Systems biology to integrate metabolomics with genomics, transcriptomics, and proteomics
Cancer originates from genomic alterations that rewire the landscape of transcriptome, proteome, and metabolome. While changes in gene and protein expression have pleiotropic effects on the cell, changes in the metabolome often occur in the closest proximity to changes in cell biology; in other words, many changes in the cellular phenotype are most closely related to changes in metabolic activity downstream of altered transcription and protein function (Figure 4). This principle emphasizes the benefits of including metabolomics in the overall assessment of tumor phenotypes. Moreover, recent work has revealed many processes that place metabolic alterations “upstream” of changes in the genome, transcriptome, and proteome. Examples include the impact of 2-HG on epigenetic reprogramming, the ability of short-chain acyl-CoA esters to modify protein function, and the impact of ROS and xenobiotics on mutagenesis (Figure 4). Thus, metabolism should be considered as a dynamic network with the potential both to respond to and influence other networks. Systems biology provides a powerful approach to integrate high-content data generated from genomics, transcriptomics, proteomics, and metabolomics. For years, concordance-based analyses of high-content data sets have recognized roles of transcription factors and signaling pathways in regulating metabolism. Massie et al. identified increased AR binding to regulatory regions of the metabolic genes including glucose transporter (GLUT1), hexokinases (HK1/2), and glutathione-disulfide reductase (GSR), involved in glycolysis and amino acid metabolism in prostate cancer cells treated with androgen[114]. Subsequently, metabolomics analysis confirmed alterations in these pathways in androgen treated cells[114]. Hakimi et al used pathway-based enrichment analysis to identify alterations in several metabolic pathways in ccRCC, but also demonstrated lack of uniform concordance between metabolite levels and corresponding enzyme-coding gene expression in ccRCC[96]. This observation likely reflects the myriad regulatory mechanisms in addition to gene expression that regulate pathway activity, and argues that advanced applications of systems biology are needed to improve our understanding of interactions between genes, proteins, and metabolites.
Figure 4. Systems biology approaches to understand biological interactions between the metabolome and other regulatory networks.
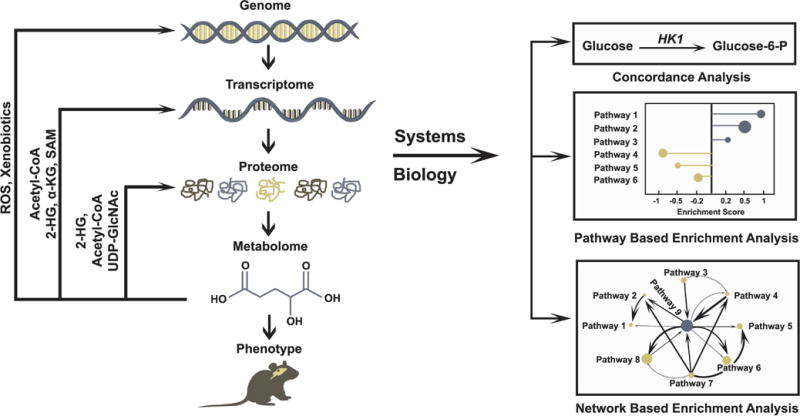
Metabolic changes define many phenotypic aspects of genetically-determined diseases. These diseases generally originate with genomic mutations and are executed through changes in the transcriptome, proteome and metabolome. Recent work has emphasized the importance of signaling effects caused by perturbed metabolic states, resulting in changes in protein function, transcription, and other effects. Examples include post-translational protein modification or regulation of these modifications by 2-HG, Acetyl-CoA, and UDP-GlcNac, all of which can impact cell signaling. Other metabolites regulate epigenetic control of the transcriptome or promote further genomic alterations. Systems biology provides systematic techniques to interrogate the complex interaction of genes and proteins with metabolites. Broadly, high throughput data generated from multiple compartments can be integrated with metabolomics using three different approaches. Concordance analysis uses direct information from the transcript/protein expression of enzymes and levels of product and substrate of the reaction. As an example, high levels of glucose and glucose 6-phosphate (Glucose-6-P) correlate with elevated hexokinase 1 (HK1) expression. Pathway based enrichment analysis uses statistical tests, such as Fisher’s exact test, to determine the likelihood of observing alterations in groups of metabolites/genes associated with specific metabolic pathways. In the corresponding figure, node size represents the number of metabolites in a pathway, and enrichment score represents directionality of enriched pathways based on composite score of differential metabolites. Network based integration uses interaction information about genes, proteins and metabolites as well as stoichiometry information of reactomes to design networks to test enrichment of metabolic pathways using several mathematical models. In the figure, node size corresponds to number of metabolites in a pathway, and interaction between pathways and directionality of flux are represented by arrows of varying width.
In recent years, novel pathway and network-based integrative approaches have been developed to combine data from multi-omics studies, especially metabolomics and transcriptomics. We used the Oncomine concept map (OCM)[87] and network based gene set analysis (NetGSA)[86] to combine data from transcriptomics and metabolomics in prostate cancer. OCM is a pathway-based analysis platform that requires a list of differential enzyme-coding genes or metabolites to test the enrichment of associated biological processes, also termed as molecular concepts, using Fisher’s exact test[115]. Advanced network-centric framework utilizes information on gene-gene/protein-protein interactions and reactome based information with associated stoichiometry. We applied network-based integrative analysis in prostate cancer to first derive pathway scores from gene-expression data using gene set analysis (GSA) and from metabolomics data using network-based gene set analysis (NetGSA)[86]. Unlike gene-set enrichment analysis, NetGSA utilizes reactome-based metabolite-interactions and stochiometric information, which increases the power of this statistical model[116]. Finally, the pathway scores were combined using a bootstrap resampling procedure to nominate significant enrichment of the HBP and pathways of riboflavin, biotin, cysteine, and valine-isoleucine metabolism in prostate cancer[86].
Others have applied similar network-based approaches. Zhang et al. used weighted co-expression network analysis (WGCNA) to identify highly interconnected nodes of metabolic pathways associated with fatty acid metabolism in pancreatic ductal adenocarcinoma (PDAC)[117]. First, the authors generated a matrix of pairwise Pearson correlation coefficients for each metabolite in all tumors, then defined an adjacent matrix using a power function. The resulted weighted network was assessed using WGCNA to enrich for highly interconnected modules based on network topology. Next, genes corresponding to top altered metabolites were analyzed in tumor samples to determine their association with pathways derived using WGCNA[117].
Several other studies have used various forms of integrative approaches to classify novel interactions between metabolomics and other dimensions of systems biology. Su et al. interrogated metabolomics and gene-expression from the NCI-60 cell lines to study relationships between metabolite and transcripts[118]. They observed that the metabolome can distinguish cancer subtypes and that metabolite levels correlate well with gene expression under strong correlation models[118]. Yang et al. performed a comprehensive assessment of metabolomics and gene expression from cervical cancer patients and identified potential diagnostic biomarkers[119]. Using a similar approach that also incorporated pathway over-representation analysis, Fahrmann et al. combined metabolomics and proteomics to identify altered nicotinamide and polyamine metabolism in lung adenocarcinoma[120]. Using parameters of biochemical reactions, enzyme expression and metabolite levels, Auslander et al. combined gene-expression and metabolomics data from breast cancer patients to identify significant metabolite-gene correlations in both malignant and non-malignant tissues, with more abundant correlations in cancer tissues[121]. Interestingly, the authors developed a support vector machine model that predicted metabolite levels based on gene-expression data and performed quite well in breast cancer and hepatocellular carcinoma[121]. Altogether, these studies highlight the potential of systems-based approaches, and many online tools are available to perform such integrative analyses[122–124].
A current challenge in integrative analysis is to identify common metabolomic alterations across multiple cancer types; such alterations could shed light on common mechanisms of transformation and might uncover opportunities for generalizable therapies. These analyses have proven difficult in part because of the lack of standard methods to combine datasets generated in different tumor types, from different institutions, and/or incorporating different metabolomics methods. These hurdles were highlighted in a meta-analysis of clinical metabolomics studies from diverse tumor types[125]. The authors combined data using binary vote-counting methods, which uses a voting function of +1 for elevation and −1 for downregulation for each metabolite across all cohorts to derive a composite voting score. This approach identified high levels of lactate and glutamic acid in cancer tissues from multiple cohorts. It also noted the dearth of complete and raw datasets in many of the published studies, perhaps because of the lack of universally-accepted metabolomics practices, limiting the scope of meta-analyses[125]. Hopefully the dissemination of standard metabolomics protocols via the Metabolomics Workbench[126] and other efforts will improve the implementation of integrative approaches.
A related challenge in integrative analysis is the incomplete coverage of the metabolome provided by different methods. To address this challenge, multiple groups are developing improved extraction and chromatography techniques and enhancing mass-spectrometry sensitivity to increase the breadth of metabolites that can be quantified in a single experiment[127, 128]. In recent years, techniques of untargeted LC/MS metabolomics have been successfully applied to improve metabolome coverage from biological samples. Novel bioinformatics software, such as XCMS[129–131], can perform non-linear integration of raw spectral peaks and identify thousands of novel metabolic features across multiple biological samples. Thus, the next few years should bring improved implementation of targeted and untargeted metabolomics techniques to map global metabolic changes in cancer and other diseases.
Advanced applications of metabolomics
In vivo isotope tracing and metabolite imaging have emerged as advanced techniques to assess metabolism. In this section, we briefly discuss applications of these techniques to generate insights about cancer metabolism in intact tissues.
A. Tracing of isotope labeled metabolites
Unlike metabolomics, isotope tracing (i.e. monitoring distribution of an isotope label originating on a nutrient of interest) provides information about metabolic pathway activity. Isotope tracing has been used extensively to characterize altered metabolic fluxes arising from mutations in tumor suppressors and oncogenes, or resulting from various metabolic stressors in cancer cells. The reader is referred to several reviews discussing principles and experimental techniques in isotope tracing[132–134]. Here we discuss a few original papers that helped shape current concepts in cancer metabolism. Isotope tracing with 13C-glucose and 13C-glutamine using NMR-compatible bioreactors that enabled long-term, steady-state labeling highlighted the prominence of anaplerotic fluxes in proliferating cancer cells; these fluxes, which can be provided by either glucose or glutamine, allow the TCA cycle to provide biosynthetic precursors for macromolecular synthesis[135, 136]. Subsequent isotope tracing in tumor-bearing mice and cancer patients also revealed extensive glucose oxidation and anaplerosis in vivo, with pyruvate carboxylation providing an anaplerotic flux in lung and brain tumors[137–140]. Several in vivo analyses of tumor metabolic flux have highlighted the importance of both cell-intrinsic (e.g. genetic) and cell-extrinsic (e.g. impact of the tissue/culture environment) determinants on metabolic phenotypes[137, 138, 141, 142].
The fact that cancer metabolism is influenced by such a complex set of factors makes metabolic analysis in primary human tumors essential. Neither the full complement of genetic diversity in human tumors nor the precise composition of human tumor microenvironments are recapitulated in mouse models of cancer. Progression of low-grade neoplastic lesions to disseminated macrometastatic disease can take years in humans, and it is not feasible to model this key aspect of cancer in mice. A number of studies have used isotope tracing in patients as a primary means to describe tumor metabolism in vivo, then turned to mouse models to test hypotheses arising from the human studies. Most published studies to date have focused on brain and lung tumors. Human gliomas and brain metastases tumors are metabolically active and display substantial glucose oxidation in vivo[139]. Non-small cell lung tumors are reported to a) oxidize glucose at rates exceeding the adjacent lung[143], a departure from the classical view of suppressed glucose oxidation in cancer cell lines (Figure 5, Left); b) display substantial inter- and intratumor metabolic heterogeneity[144]; and c) exhibit nutrient preferences associated with the degree of tissue perfusion, with the best-perfused areas complementing glucose oxidation with oxidation of additional fuels[144]. An interesting outcome of in vivo human infusion studies has been to identify some of these alternative fuels. Tumors in the human brain were observed to use the short chain fatty acid acetate as a carbon source for the TCA cycle[145]. In lung tumors in both humans and mice, lactate from the circulation provided carbon for the TCA cycle, a surprising finding given the long-standing expectation that lactate is primarily a waste product in cancer[146, 147].
Figure 5. Advanced in vivo applications of metabolomics.
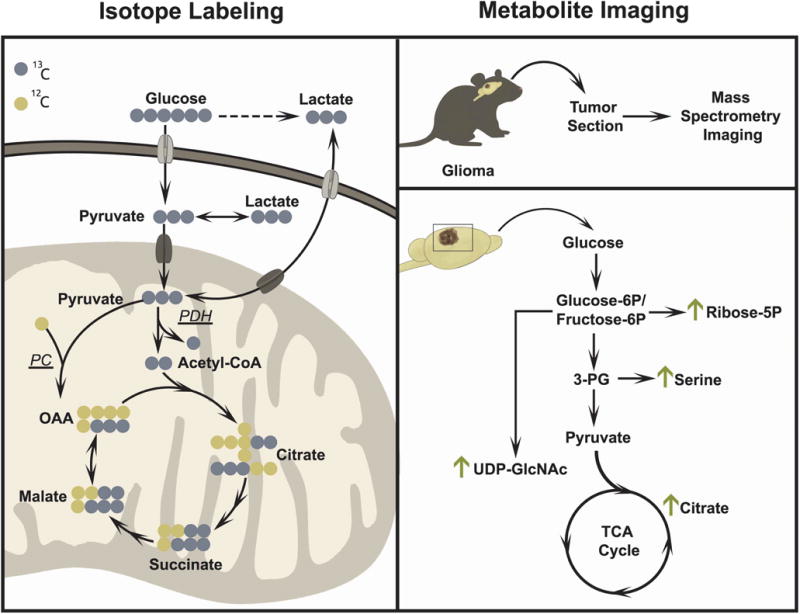
Isotope tracing (left) and metabolite imaging (right) are two examples of advanced applications of metabolomics. Isotope tracing studies in lung cancer patients have established that glucose and lactate are oxidized in the TCA cycle in vivo. These studies have also revealed the activity of both pyruvate dehydrogenase and pyruvate carboxylase (PDH and PC) in vivo. In the illustration, PDH activity results in TCA cycle intermediates with two 13C nuclei and PC activity results in TCA cycle intermediates with three 13C nuclei. Metabolite imaging (right) using matrix assisted laser desorption/ionization (MALDI) provides temporal and spatial resolution of metabolite abundance to observe metabolic differences across tissue sections. Metabolite imaging has been used in murine glioma models to assess changes in glycolytic and TCA cycle intermediates.
Conventional methods in isotope tracing usually capitalize on a priori knowledge of which downstream metabolites should carry label from the precursor. A number of newer approaches allow the user to perform unbiased detection of isotope-labeled molecules, providing the opportunity to detect truly novel pathways. These techniques, which include nontargeted tracer fate detection (NTFD)[148] and X13CMS[149], require extensive computation and statistical modelling and pose challenges in data interpretation[150]. Nevertheless, a recent study applied few of these concepts in a targeted analysis in mice fed with liquid diet containing [U-13C] glucose for up to 48 hours. This study demonstrated significant enrichment of 13C in wide range of metabolic pathways, including HBP, PPP, lipids, as well as in proteins[151], documenting the feasibility of global metabolite labeling in vivo.
B. Metabolite Imaging
The tumor metabolomics and isotope tracing experiments described above assessed metabolites extracted from tissue fragments containing mixtures of distinct cell types. In addition to cancer cells, the tumor microenvironment contains a number of non-malignant cell populations, including fibroblasts, endothelial cells, immune cells and others. A major ongoing challenge is to deconvolve the contributions of different cell types to understand how they interact metabolically with each other. High resolution matrix-assisted laser desorption ionization (MALDI)-based mass spectrometry imaging (MSI) has the potential to address this challenge by assessing localized metabolite distribution across a plane of tissue. This technique has proven to be particularly useful in assessing the spatial resolution of larger metabolites like lipids[152, 153]. Kawashima et al. used MALDI-MSI to identify spatial distribution of phosphatidylinositol species in malignant cells in breast cancer and to correlate the abundance of these lipids with invasive phenotypes[154]. Dilliol et al[155] used MALDI-MSI to study proteins and metabolites in mouse models of glioblastoma (Figure 5, Right). They employed Fourier Transform Ion Cyclotron Resonance (FTICR)-MALDI-MSI to demonstrate changes in protein compositions in high-grade gliomas, then validated their findings using microproteomics in laser-capture microdissected tumor tissue sections. MALDI-MSI also revealed accumulation of glucose-6-phosphate, ribose-5-phosphate, glycine, UDP-N acetyl glucosamine, and TCA cycle intermediates in tumors compared to adjacent tissue[155]. MALDI-MSI and the related technique desorption electrospray ionization MSI (DESI-MSI) have been used to assess regional metabolite distribution in tissues from human and mouse models of renal cell carcinoma[156, 157], prostate cancer[158], gastric cancer[159], and sarcoma[160], in some cases identifying specific metabolic effects of oncogenic drivers in the tumor tissue. MSI approaches have also been used to detect conversion of precursor to product molecules, thereby providing proof of principle that the technique has the capability to monitor some metabolic activities[161]. Altogether, these studies illustrate the emerging role of MSI in studying cancer metabolism and understanding the role of the native microenvironment in dictating metabolic phenotypes in vivo[162–164].
Future Perspective
Recent years have seen the expanded use of metabolomics to study cancer. These studies have been propelled by rapid improvements both in our understanding of the molecular basis of metabolic reprogramming, and in the analytical systems with which cancer metabolism can be studied. The availability of isotope labeling methods, metabolite imaging, and tools to integrate metabolic data with genomics, transcriptomics, and proteomics have the potential to accelerate research in cancer metabolism even further, particularly in the context of intact tumors in mice and humans. We anticipate further advancements in global metabolite profiling, hopefully with an increasing emphasis on methodological consistency to facilitate durable data sharing and reproducibility across centers.
Acknowledgments
We regret that due to limited space we were unable to include additional excellent work from many authors who contributed to our current understanding of cancer metabolism. We thank members of the DeBerardinis Lab for helpful suggestions and critique of the manuscript, and Katie Regan for assistance with the Figures. R.J.D. is supported by grants from the NCI (R35 CA220449 and P50 CA175754) and Cancer Prevention and Research Institute of Texas (RP160089).
Abbreviations
- MS
Mass-Spectrometry
- NMR
Nuclear Magnetic Resonance Spectroscopy
- MRS
Magnetic Resonance Spectroscopy
- D-2-HG
D-2-Hydroxyglutarate
- α-KG
α-Ketoglutarate
- OAA
Oxaloacetic acid
- TET
Ten-Eleven Translocation
- GSH
Glutathione
- ROS
Reactive Oxygen Species
- LKB1
Liver Kinase B1
- PPP
Pentose Phosphate Pathway
- HBP
Hexosamine Biosynthesis Pathway
- SAM
S-Adenosylmethionine
- UDP-GlcNAc
Uridine Diphosphate N-Acetylglucosamine
- NSCLC
Non-Small Cell Lung Cancer
- PDAC
Pancreatic Ductal Adenocarcinoma
- CRPC
Castration-Resistant Prostate Cancer
- CRC
Colorectal Cancer
- TNBC
Triple Negative Breast Cancer
- ccRCC
Clear Cell Renal Cell Carcinoma
- PDX
Patient-Derived Xenograft
- LC/MS
Liquid Chromatography-Mass Spectrometry
- OCM
Oncomine Concept Map
- NetGSA
Network-Based Gene Set Analysis
- WGCNA
Weighted Co-Expression Network Analysis
- MALDI
Matrix-Assisted Laser Desorption Ionization
- MSI
Mass Spectrometry Imaging
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2:e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell. 2017;168:657–669. doi: 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17:351–359. doi: 10.1038/ncb3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Almuhaideb A, Papathanasiou N, Bomanji J. 18F-FDG PET/CT imaging in oncology. Ann Saudi Med. 2011;31:3–13. doi: 10.4103/0256-4947.75771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armitage EG, Barbas C. Metabolomics in cancer biomarker discovery: current trends and future perspectives. J Pharm Biomed Anal. 2014;87:1–11. doi: 10.1016/j.jpba.2013.08.041. [DOI] [PubMed] [Google Scholar]
- 7.Serkova NJ, Glunde K. Metabolomics of cancer. Methods Mol Biol. 2009;520:273–295. doi: 10.1007/978-1-60327-811-9_20. [DOI] [PubMed] [Google Scholar]
- 8.Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vazquez-Fresno R, Sajed T, Johnson D, Li C, Karu N, Sayeeda Z, Lo E, Assempour N, Berjanskii M, Singhal S, Arndt D, Liang Y, Badran H, Grant J, Serra-Cayuela A, Liu Y, Mandal R, Neveu V, Pon A, Knox C, Wilson M, Manach C, Scalbert A. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 2018;46:D608–D617. doi: 10.1093/nar/gkx1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wishart DS, Mandal R, Stanislaus A, Ramirez-Gaona M. Cancer Metabolomics and the Human Metabolome Database. Metabolites. 2016;6 doi: 10.3390/metabo6010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Markley JL, Bruschweiler R, Edison AS, Eghbalnia HR, Powers R, Raftery D, Wishart DS. The future of NMR-based metabolomics. Curr Opin Biotechnol. 2017;43:34–40. doi: 10.1016/j.copbio.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emwas AH. The strengths and weaknesses of NMR spectroscopy and mass spectrometry with particular focus on metabolomics research. Methods Mol Biol. 2015;1277:161–193. doi: 10.1007/978-1-4939-2377-9_13. [DOI] [PubMed] [Google Scholar]
- 12.Andronesi OC, Kim GS, Gerstner E, Batchelor T, Tzika AA, Fantin VR, Vander Heiden MG, Sorensen AG. Detection of 2-hydroxyglutarate in IDH-mutated glioma patients by in vivo spectral-editing and 2D correlation magnetic resonance spectroscopy. Sci Transl Med. 2012;4:116ra114. doi: 10.1126/scitranslmed.3002693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi C, Ganji SK, DeBerardinis RJ, Hatanpaa KJ, Rakheja D, Kovacs Z, Yang XL, Mashimo T, Raisanen JM, Marin-Valencia I, Pascual JM, Madden CJ, Mickey BE, Malloy CR, Bachoo RM, Maher EA. 2-hydroxyglutarate detection by magnetic resonance spectroscopy in IDH-mutated patients with gliomas. Nat Med. 2012;18:624–629. doi: 10.1038/nm.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheshkov S, Dimitrov IE, Jakkamsetti V, Good L, Kelly D, Rajasekaran K, DeBerardinis RJ, Pascual JM, Sherry AD, Malloy CR. Oxidation of [U-(13) C]glucose in the human brain at 7T under steady state conditions. Magn Reson Med. 2017;78:2065–2071. doi: 10.1002/mrm.26603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dieterle F, Riefke B, Schlotterbeck G, Ross A, Senn H, Amberg A. NMR and MS methods for metabonomics. Methods Mol Biol. 2011;691:385–415. doi: 10.1007/978-1-60761-849-2_24. [DOI] [PubMed] [Google Scholar]
- 16.Agathocleous M, Meacham CE, Burgess RJ, Piskounova E, Zhao Z, Crane GM, Cowin BL, Bruner E, Murphy MM, Chen W, Spangrude GJ, Hu Z, DeBerardinis RJ, Morrison SJ. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature. 2017;549:476–481. doi: 10.1038/nature23876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bedair M, Sumner LW. Current and emerging mass-spectrometry technologies for metabolomics. TrAC Trends in Analytical Chemistry. 2008;27:238–250. [Google Scholar]
- 18.Gowda GA, Zhang S, Gu H, Asiago V, Shanaiah N, Raftery D. Metabolomics-based methods for early disease diagnostics. Expert Rev Mol Diagn. 2008;8:617–633. doi: 10.1586/14737159.8.5.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sitter B, Lundgren S, Bathen TF, Halgunset J, Fjosne HE, Gribbestad IS. Comparison of HR MAS MR spectroscopic profiles of breast cancer tissue with clinical parameters. NMR Biomed. 2006;19:30–40. doi: 10.1002/nbm.992. [DOI] [PubMed] [Google Scholar]
- 20.Cheng LL, Chang IW, Smith BL, Gonzalez RG. Evaluating human breast ductal carcinomas with high-resolution magic-angle spinning proton magnetic resonance spectroscopy. J Magn Reson. 1998;135:194–202. doi: 10.1006/jmre.1998.1578. [DOI] [PubMed] [Google Scholar]
- 21.Denkert C, Budczies J, Kind T, Weichert W, Tablack P, Sehouli J, Niesporek S, Konsgen D, Dietel M, Fiehn O. Mass spectrometry-based metabolic profiling reveals different metabolite patterns in invasive ovarian carcinomas and ovarian borderline tumors. Cancer Res. 2006;66:10795–10804. doi: 10.1158/0008-5472.CAN-06-0755. [DOI] [PubMed] [Google Scholar]
- 22.Cheng LL, Wu C, Smith MR, Gonzalez RG. Non-destructive quantitation of spermine in human prostate tissue samples using HRMAS 1H NMR spectroscopy at 9.4 T. FEBS Lett. 2001;494:112–116. doi: 10.1016/s0014-5793(01)02329-8. [DOI] [PubMed] [Google Scholar]
- 23.Swanson MG, Vigneron DB, Tabatabai ZL, Males RG, Schmitt L, Carroll PR, James JK, Hurd RE, Kurhanewicz J. Proton HR-MAS spectroscopy and quantitative pathologic analysis of MRI/3D-MRSI-targeted postsurgical prostate tissues. Magn Reson Med. 2003;50:944–954. doi: 10.1002/mrm.10614. [DOI] [PubMed] [Google Scholar]
- 24.Tzika AA, Cheng LL, Goumnerova L, Madsen JR, Zurakowski D, Astrakas LG, Zarifi MK, Scott RM, Anthony DC, Gonzalez RG, Black PM. Biochemical characterization of pediatric brain tumors by using in vivo and ex vivo magnetic resonance spectroscopy. J Neurosurg. 2002;96:1023–1031. doi: 10.3171/jns.2002.96.6.1023. [DOI] [PubMed] [Google Scholar]
- 25.Martinez-Bisbal MC, Marti-Bonmati L, Piquer J, Revert A, Ferrer P, Llacer JL, Piotto M, Assemat O, Celda B. 1H and 13C HR-MAS spectroscopy of intact biopsy samples ex vivo and in vivo 1H MRS study of human high grade gliomas. NMR Biomed. 2004;17:191–205. doi: 10.1002/nbm.888. [DOI] [PubMed] [Google Scholar]
- 26.Tate AR, Foxall PJ, Holmes E, Moka D, Spraul M, Nicholson JK, Lindon JC. Distinction between normal and renal cell carcinoma kidney cortical biopsy samples using pattern recognition of (1)H magic angle spinning (MAS) NMR spectra. NMR Biomed. 2000;13:64–71. doi: 10.1002/(sici)1099-1492(200004)13:2<64::aid-nbm612>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 27.Moka D, Vorreuther R, Schicha H, Spraul M, Humpfer E, Lipinski M, Foxall PJ, Nicholson JK, Lindon JC. Biochemical classification of kidney carcinoma biopsy samples using magic-angle-spinning 1H nuclear magnetic resonance spectroscopy. J Pharm Biomed Anal. 1998;17:125–132. doi: 10.1016/s0731-7085(97)00176-3. [DOI] [PubMed] [Google Scholar]
- 28.Machado RF, Laskowski D, Deffenderfer O, Burch T, Zheng S, Mazzone PJ, Mekhail T, Jennings C, Stoller JK, Pyle J, Duncan J, Dweik RA, Erzurum SC. Detection of lung cancer by sensor array analyses of exhaled breath. Am J Respir Crit Care Med. 2005;171:1286–1291. doi: 10.1164/rccm.200409-1184OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cohen AL, Holmen SL, Colman H. IDH1 and IDH2 mutations in gliomas. Curr Neurol Neurosci Rep. 2013;13:345. doi: 10.1007/s11910-013-0345-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, Keir S, Nikolskaya T, Nikolsky Y, Busam DA, Tekleab H, Diaz LA, Jr, Hartigan J, Smith DR, Strausberg RL, Marie SK, Shinjo SM, Yan H, Riggins GJ, Bigner DD, Karchin R, Papadopoulos N, Parmigiani G, Vogelstein B, Velculescu VE, Kinzler KW. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, Su SM. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, Fulton LA, Locke DP, Magrini VJ, Abbott RM, Vickery TL, Reed JS, Robinson JS, Wylie T, Smith SM, Carmichael L, Eldred JM, Harris CC, Walker J, Peck JB, Du F, Dukes AF, Sanderson GE, Brummett AM, Clark E, McMichael JF, Meyer RJ, Schindler JK, Pohl CS, Wallis JW, Shi X, Lin L, Schmidt H, Tang Y, Haipek C, Wiechert ME, Ivy JV, Kalicki J, Elliott G, Ries RE, Payton JE, Westervelt P, Tomasson MH, Watson MA, Baty J, Heath S, Shannon WD, Nagarajan R, Link DC, Walter MJ, Graubert TA, DiPersio JF, Wilson RK, Ley TJ. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hemerly JP, Bastos AU, Cerutti JM. Identification of several novel non-p.R132 IDH1 variants in thyroid carcinomas. Eur J Endocrinol. 2010;163:747–755. doi: 10.1530/EJE-10-0473. [DOI] [PubMed] [Google Scholar]
- 35.Yang H, Ye D, Guan KL, Xiong Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res. 2012;18:5562–5571. doi: 10.1158/1078-0432.CCR-12-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IK, Li XS, Woon EC, Yang M, McDonough MA, King ON, Clifton IJ, Klose RJ, Claridge TD, Ratcliffe PJ, Schofield CJ, Kawamura A. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, Kunii K, Pedraza A, Schalm S, Silverman L, Miller A, Wang F, Yang H, Chen Y, Kernytsky A, Rosenblum MK, Liu W, Biller SA, Su SM, Brennan CW, Chan TA, Graeber TG, Yen KE, Mellinghoff IK. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Losman JA, Looper RE, Koivunen P, Lee S, Schneider RK, McMahon C, Cowley GS, Root DE, Ebert BL, Kaelin WG., Jr (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339:1621–1625. doi: 10.1126/science.1231677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, Yang H, Gross S, Artin E, Saada V, Mylonas E, Quivoron C, Popovici-Muller J, Saunders JO, Salituro FG, Yan S, Murray S, Wei W, Gao Y, Dang L, Dorsch M, Agresta S, Schenkein DP, Biller SA, Su SM, de Botton S, Yen KE. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- 41.Mullard A. FDA approves first-in-class cancer metabolism drug. Nat Rev Drug Discov. 2017;16:593. doi: 10.1038/nrd.2017.174. [DOI] [PubMed] [Google Scholar]
- 42.Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW, 3rd, Cornelisse CJ, Devilee P, Devlin B. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 43.Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Skoldberg F, Husebye ES, Eng C, Maher ER. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. 2001;69:49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, Roylance RR, Olpin S, Bevan S, Barker K, Hearle N, Houlston RS, Kiuru M, Lehtonen R, Karhu A, Vilkki S, Laiho P, Eklund C, Vierimaa O, Aittomaki K, Hietala M, Sistonen P, Paetau A, Salovaara R, Herva R, Launonen V, Aaltonen LA, Multiple Leiomyoma C. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–410. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 45.Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, Griffiths JR, Dalgleish A, McGrath JA, Gleeson MJ, Hodgson SV, Poulsom R, Rustin P, Tomlinson IP. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet. 2005;14:2231–2239. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 46.Koivunen P, Hirsila M, Remes AM, Hassinen IE, Kivirikko KI, Myllyharju J. Inhibition of hypoxia-inducible factor (HIF) hydroxylases by citric acid cycle intermediates: possible links between cell metabolism and stabilization of HIF. J Biol Chem. 2007;282:4524–4532. doi: 10.1074/jbc.M610415200. [DOI] [PubMed] [Google Scholar]
- 47.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 48.Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, Guan KL. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Laukka T, Mariani CJ, Ihantola T, Cao JZ, Hokkanen J, Kaelin WG, Jr, Godley LA, Koivunen P. Fumarate and Succinate Regulate Expression of Hypoxia-inducible Genes via TET Enzymes. J Biol Chem. 2016;291:4256–4265. doi: 10.1074/jbc.M115.688762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sciacovelli M, Goncalves E, Johnson TI, Zecchini VR, da Costa AS, Gaude E, Drubbel AV, Theobald SJ, Abbo SR, Tran MG, Rajeeve V, Cardaci S, Foster S, Yun H, Cutillas P, Warren A, Gnanapragasam V, Gottlieb E, Franze K, Huntly B, Maher ER, Maxwell PH, Saez-Rodriguez J, Frezza C. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature. 2016;537:544–547. doi: 10.1038/nature19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, Buffet A, Marcaillou C, Bertherat J, Amar L, Rustin P, De Reynies A, Gimenez-Roqueplo AP, Favier J. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 52.Sullivan LB, Martinez-Garcia E, Nguyen H, Mullen AR, Dufour E, Sudarshan S, Licht JD, Deberardinis RJ, Chandel NS. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell. 2013;51:236–248. doi: 10.1016/j.molcel.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp GW, Wolhuter K, Stevens M, Fischer R, Carmeliet P, Maxwell PH, Pugh CW, Frizzell N, Soga T, Kessler BM, El-Bahrawy M, Ratcliffe PJ, Pollard PJ. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524–537. doi: 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol. 2013;15:555–564. doi: 10.1038/ncb2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB. Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev. 2008;18:54–61. doi: 10.1016/j.gde.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J, Schulze A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene. 2005;24:6465–6481. doi: 10.1038/sj.onc.1208802. [DOI] [PubMed] [Google Scholar]
- 57.Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell. 2007;18:1437–1446. doi: 10.1091/mbc.E06-07-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deberardinis RJ, Lum JJ, Thompson CB. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem. 2006;281:37372–37380. doi: 10.1074/jbc.M608372200. [DOI] [PubMed] [Google Scholar]
- 59.Georgescu MM. PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control. Genes Cancer. 2010;1:1170–1177. doi: 10.1177/1947601911407325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ortega-Molina A, Serrano M. PTEN in cancer, metabolism, and aging. Trends Endocrinol Metab. 2013;24:184–189. doi: 10.1016/j.tem.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X, Wu H. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell. 2003;4:209–221. doi: 10.1016/s1535-6108(03)00215-0. [DOI] [PubMed] [Google Scholar]
- 62.Ortega-Molina A, Efeyan A, Lopez-Guadamillas E, Munoz-Martin M, Gomez-Lopez G, Canamero M, Mulero F, Pastor J, Martinez S, Romanos E, Mar Gonzalez-Barroso M, Rial E, Valverde AM, Bischoff JR, Serrano M. Pten positively regulates brown adipose function, energy expenditure, and longevity. Cell Metab. 2012;15:382–394. doi: 10.1016/j.cmet.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 63.Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, de Boer VC, Anastasiou D, Ito K, Sasaki AT, Rameh L, Carracedo A, Vander Heiden MG, Cantley LC, Pinton P, Haigis MC, Pandolfi PP. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell. 2012;149:49–62. doi: 10.1016/j.cell.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Makinoshima H, Takita M, Saruwatari K, Umemura S, Obata Y, Ishii G, Matsumoto S, Sugiyama E, Ochiai A, Abe R, Goto K, Esumi H, Tsuchihara K. Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) Axis Is Responsible for Aerobic Glycolysis mediated by Glucose Transporter in Epidermal Growth Factor Receptor (EGFR)-mutated Lung Adenocarcinoma. J Biol Chem. 2015;290:17495–17504. doi: 10.1074/jbc.M115.660498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Posch C, Moslehi H, Feeney L, Green GA, Ebaee A, Feichtenschlager V, Chong K, Peng L, Dimon MT, Phillips T, Daud AI, McCalmont TH, LeBoit PE, Ortiz-Urda S. Combined targeting of MEK and PI3K/mTOR effector pathways is necessary to effectively inhibit NRAS mutant melanoma in vitro and in vivo. Proc Natl Acad Sci U S A. 2013;110:4015–4020. doi: 10.1073/pnas.1216013110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim J, Hu Z, Cai L, Li K, Choi E, Faubert B, Bezwada D, Rodriguez-Canales J, Villalobos P, Lin YF, Ni M, Huffman KE, Girard L, Byers LA, Unsal-Kacmaz K, Pena CG, Heymach JV, Wauters E, Vansteenkiste J, Castrillon DH, Chen BPC, Wistuba I, Lambrechts D, Xu J, Minna JD, DeBerardinis RJ. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature. 2017;546:168–172. doi: 10.1038/nature22359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature. 2016;531:110–113. doi: 10.1038/nature16967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, Yan H, Wang W, Chen S, Viale A, Zheng H, Paik JH, Lim C, Guimaraes AR, Martin ES, Chang J, Hezel AF, Perry SR, Hu J, Gan B, Xiao Y, Asara JM, Weissleder R, Wang YA, Chin L, Cantley LC, DePinho RA. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, DePinho RA, Cantley LC, Kimmelman AC. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Toda K, Kawada K, Iwamoto M, Inamoto S, Sasazuki T, Shirasawa S, Hasegawa S, Sakai Y. Metabolic Alterations Caused by KRAS Mutations in Colorectal Cancer Contribute to Cell Adaptation to Glutamine Depletion by Upregulation of Asparagine Synthetase. Neoplasia. 2016;18:654–665. doi: 10.1016/j.neo.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kang HB, Fan J, Lin R, Elf S, Ji Q, Zhao L, Jin L, Seo JH, Shan C, Arbiser JL, Cohen C, Brat D, Miziorko HM, Kim E, Abdel-Wahab O, Merghoub T, Frohling S, Scholl C, Tamayo P, Barbie DA, Zhou L, Pollack BP, Fisher K, Kudchadkar RR, Lawson DH, Sica G, Rossi M, Lonial S, Khoury HJ, Khuri FR, Lee BH, Boggon TJ, He C, Kang S, Chen J. Metabolic Rewiring by Oncogenic BRAF V600E Links Ketogenesis Pathway to BRAF-MEK1 Signaling. Mol Cell. 2015;59:345–358. doi: 10.1016/j.molcel.2015.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, Arany Z, Widlund HR. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell. 2013;23:302–315. doi: 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi X, Tasdogan A, Huang F, Hu Z, Morrison SJ, DeBerardinis RJ. The abundance of metabolites related to protein methylation correlates with the metastatic capacity of human melanoma xenografts. Sci Adv. 2017;3:eaao5268. doi: 10.1126/sciadv.aao5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, Metabolism, and Cancer. Cancer Discov. 2015;5:1024–1039. doi: 10.1158/2159-8290.CD-15-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A. 1997;94:6658–6663. doi: 10.1073/pnas.94.13.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275:21797–21800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- 77.Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, Dang CV. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, Eyob H, Kajimura S, Tward A, Krings G, Nomura DK, Goga A. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med. 2016;22:427–432. doi: 10.1038/nm.4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Terunuma A, Putluri N, Mishra P, Mathe EA, Dorsey TH, Yi M, Wallace TA, Issaq HJ, Zhou M, Killian JK, Stevenson HS, Karoly ED, Chan K, Samanta S, Prieto D, Hsu TY, Kurley SJ, Putluri V, Sonavane R, Edelman DC, Wulff J, Starks AM, Yang Y, Kittles RA, Yfantis HG, Lee DH, Ioffe OB, Schiff R, Stephens RM, Meltzer PS, Veenstra TD, Westbrook TF, Sreekumar A, Ambs S. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J Clin Invest. 2014;124:398–412. doi: 10.1172/JCI71180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Satoh K, Yachida S, Sugimoto M, Oshima M, Nakagawa T, Akamoto S, Tabata S, Saitoh K, Kato K, Sato S, Igarashi K, Aizawa Y, Kajino-Sakamoto R, Kojima Y, Fujishita T, Enomoto A, Hirayama A, Ishikawa T, Taketo MM, Kushida Y, Haba R, Okano K, Tomita M, Suzuki Y, Fukuda S, Aoki M, Soga T. Global metabolic reprogramming of colorectal cancer occurs at adenoma stage and is induced by MYC. Proc Natl Acad Sci U S A. 2017;114:E7697–E7706. doi: 10.1073/pnas.1710366114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, Laxman B, Mehra R, Lonigro RJ, Li Y, Nyati MK, Ahsan A, Kalyana-Sundaram S, Han B, Cao X, Byun J, Omenn GS, Ghosh D, Pennathur S, Alexander DC, Berger A, Shuster JR, Wei JT, Varambally S, Beecher C, Chinnaiyan AM. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457:910–914. doi: 10.1038/nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 82.Lucarelli G, Fanelli M, Larocca AM, Germinario CA, Rutigliano M, Vavallo A, Selvaggi FP, Bettocchi C, Battaglia M, Ditonno P. Serum sarcosine increases the accuracy of prostate cancer detection in patients with total serum PSA less than 4.0 ng/ml. Prostate. 2012;72:1611–1621. doi: 10.1002/pros.22514. [DOI] [PubMed] [Google Scholar]
- 83.Khan AP, Rajendiran TM, Ateeq B, Asangani IA, Athanikar JN, Yocum AK, Mehra R, Siddiqui J, Palapattu G, Wei JT, Michailidis G, Sreekumar A, Chinnaiyan AM. The role of sarcosine metabolism in prostate cancer progression. Neoplasia. 2013;15:491–501. doi: 10.1593/neo.13314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jentzmik F, Stephan C, Lein M, Miller K, Kamlage B, Bethan B, Kristiansen G, Jung K. Sarcosine in prostate cancer tissue is not a differential metabolite for prostate cancer aggressiveness and biochemical progression. J Urol. 2011;185:706–711. doi: 10.1016/j.juro.2010.09.077. [DOI] [PubMed] [Google Scholar]
- 85.Ankerst DP, Liss M, Zapata D, Hoefler J, Thompson IM, Leach RJ. A case control study of sarcosine as an early prostate cancer detection biomarker. BMC Urol. 2015;15:99. doi: 10.1186/s12894-015-0095-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kaushik AK, Shojaie A, Panzitt K, Sonavane R, Venghatakrishnan H, Manikkam M, Zaslavsky A, Putluri V, Vasu VT, Zhang Y, Khan AS, Lloyd S, Szafran AT, Dasgupta S, Bader DA, Stossi F, Li H, Samanta S, Cao X, Tsouko E, Huang S, Frigo DE, Chan L, Edwards DP, Kaipparettu BA, Mitsiades N, Weigel NL, Mancini M, McGuire SE, Mehra R, Ittmann MM, Chinnaiyan AM, Putluri N, Palapattu GS, Michailidis G, Sreekumar A. Inhibition of the hexosamine biosynthetic pathway promotes castration-resistant prostate cancer. Nat Commun. 2016;7:11612. doi: 10.1038/ncomms11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kaushik AK, Vareed SK, Basu S, Putluri V, Putluri N, Panzitt K, Brennan CA, Chinnaiyan AM, Vergara IA, Erho N, Weigel NL, Mitsiades N, Shojaie A, Palapattu G, Michailidis G, Sreekumar A. Metabolomic profiling identifies biochemical pathways associated with castration-resistant prostate cancer. J Proteome Res. 2014;13:1088–1100. doi: 10.1021/pr401106h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, Lotan TL, Zheng Q, De Marzo AM, Isaacs JT, Isaacs WB, Nadal R, Paller CJ, Denmeade SR, Carducci MA, Eisenberger MA, Luo J. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shafi AA, Putluri V, Arnold JM, Tsouko E, Maity S, Roberts JM, Coarfa C, Frigo DE, Putluri N, Sreekumar A, Weigel NL. Differential regulation of metabolic pathways by androgen receptor (AR) and its constitutively active splice variant, AR-V7, in prostate cancer cells. Oncotarget. 2015;6:31997–32012. doi: 10.18632/oncotarget.5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dasgupta S, Putluri N, Long W, Zhang B, Wang J, Kaushik AK, Arnold JM, Bhowmik SK, Stashi E, Brennan CA, Rajapakshe K, Coarfa C, Mitsiades N, Ittmann MM, Chinnaiyan AM, Sreekumar A, O’Malley BW. Coactivator SRC-2-dependent metabolic reprogramming mediates prostate cancer survival and metastasis. J Clin Invest. 2015;125:1174–1188. doi: 10.1172/JCI76029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cao MD, Lamichhane S, Lundgren S, Bofin A, Fjosne H, Giskeodegard GF, Bathen TF. Metabolic characterization of triple negative breast cancer. BMC Cancer. 2014;14:941. doi: 10.1186/1471-2407-14-941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Asiago VM, Alvarado LZ, Shanaiah N, Gowda GA, Owusu-Sarfo K, Ballas RA, Raftery D. Early detection of recurrent breast cancer using metabolite profiling. Cancer Res. 2010;70:8309–8318. doi: 10.1158/0008-5472.CAN-10-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wettersten HI, Aboud OA, Lara PN, Jr, Weiss RH. Metabolic reprogramming in clear cell renal cell carcinoma. Nat Rev Nephrol. 2017;13:410–419. doi: 10.1038/nrneph.2017.59. [DOI] [PubMed] [Google Scholar]
- 94.N. Cancer Genome Atlas Research. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–49. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Brauch H, Weirich G, Brieger J, Glavac D, Rodl H, Eichinger M, Feurer M, Weidt E, Puranakanitstha C, Neuhaus C, Pomer S, Brenner W, Schirmacher P, Storkel S, Rotter M, Masera A, Gugeler N, Decker HJ. VHL alterations in human clear cell renal cell carcinoma: association with advanced tumor stage and a novel hot spot mutation. Cancer Res. 2000;60:1942–1948. [PubMed] [Google Scholar]
- 96.Hakimi AA, Reznik E, Lee CH, Creighton CJ, Brannon AR, Luna A, Aksoy BA, Liu EM, Shen R, Lee W, Chen Y, Stirdivant SM, Russo P, Chen YB, Tickoo SK, Reuter VE, Cheng EH, Sander C, Hsieh JJ. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell. 2016;29:104–116. doi: 10.1016/j.ccell.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Abu Aboud O, Habib SL, Trott J, Stewart B, Liang S, Chaudhari AJ, Sutcliffe J, Weiss RH. Glutamine Addiction in Kidney Cancer Suppresses Oxidative Stress and Can Be Exploited for Real-Time Imaging. Cancer Res. 2017 doi: 10.1158/0008-5472.CAN-17-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wettersten HI, Hakimi AA, Morin D, Bianchi C, Johnstone ME, Donohoe DR, Trott JF, Aboud OA, Stirdivant S, Neri B, Wolfert R, Stewart B, Perego R, Hsieh JJ, Weiss RH. Grade-Dependent Metabolic Reprogramming in Kidney Cancer Revealed by Combined Proteomics and Metabolomics Analysis. Cancer Res. 2015;75:2541–2552. doi: 10.1158/0008-5472.CAN-14-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li B, Qiu B, Lee DS, Walton ZE, Ochocki JD, Mathew LK, Mancuso A, Gade TP, Keith B, Nissim I, Simon MC. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature. 2014;513:251–255. doi: 10.1038/nature13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang Y, Udayakumar D, Cai L, Hu Z, Kapur P, Kho EY, Pavia-Jimenez A, Fulkerson M, de Leon AD, Yuan Q, Dimitrov IE, Yokoo T, Ye J, Mitsche MA, Kim H, McDonald JG, Xi Y, Madhuranthakam AJ, Dwivedi DK, Lenkinski RE, Cadeddu JA, Margulis V, Brugarolas J, DeBerardinis RJ, Pedrosa I. Addressing metabolic heterogeneity in clear cell renal cell carcinoma with quantitative Dixon MRI. JCI Insight. 2017;2 doi: 10.1172/jci.insight.94278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Quintana E, Piskounova E, Shackleton M, Weinberg D, Eskiocak U, Fullen DR, Johnson TM, Morrison SJ. Human melanoma metastasis in NSG mice correlates with clinical outcome in patients. Sci Transl Med. 2012;4:159ra149. doi: 10.1126/scitranslmed.3004599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, Morrison SJ. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–191. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, Brugge JS. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, Terada LS, Adams ND, McCabe MT, Pietrak B, Schmidt S, Metallo CM, Dranka BP, Schwartz B, DeBerardinis RJ. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature. 2016;532:255–258. doi: 10.1038/nature17393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. Journal of Cell Science. 2012;125:5591–5596. doi: 10.1242/jcs.116392. [DOI] [PubMed] [Google Scholar]
- 106.Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, Kreutz M. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front Immunol. 2017;8:248. doi: 10.3389/fimmu.2017.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Palazon A, Tyrakis PA, Macias D, Velica P, Rundqvist H, Fitzpatrick S, Vojnovic N, Phan AT, Loman N, Hedenfalk I, Hatschek T, Lovrot J, Foukakis T, Goldrath AW, Bergh J, Johnson RS. An HIF-1alpha/VEGF-A Axis in Cytotoxic T Cells Regulates Tumor Progression. Cancer Cell. 2017;32:669–683 e665. doi: 10.1016/j.ccell.2017.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA. Macrophage Metabolism As Therapeutic Target for Cancer, Atherosclerosis, and Obesity. Front Immunol. 2017;8:289. doi: 10.3389/fimmu.2017.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell. 2017;169:570–586. doi: 10.1016/j.cell.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Giannakis M, Li H, Jin C, Gopal S, Desai K, Horak C, Wind-Rotolo M, Allen EMV, Clish C, Hodi FS, Garraway LA, Choueiri TK. Metabolomic correlates of response in nivolumab-treated renal cell carcinoma and melanoma patients. Journal of Clinical Oncology. 2017;35:3036–3036. [Google Scholar]
- 112.Wang Q, Liu D, Song P, Zou MH. Tryptophan-kynurenine pathway is dysregulated in inflammation, and immune activation. Front Biosci (Landmark Ed) 2015;20:1116–1143. doi: 10.2741/4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Frankel AE, Coughlin LA, Kim J, Froehlich TW, Xie Y, Frenkel EP, Koh AY. Metagenomic Shotgun Sequencing and Unbiased Metabolomic Profiling Identify Specific Human Gut Microbiota and Metabolites Associated with Immune Checkpoint Therapy Efficacy in Melanoma Patients. Neoplasia. 2017;19:848–855. doi: 10.1016/j.neo.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Massie CE, Lynch A, Ramos-Montoya A, Boren J, Stark R, Fazli L, Warren A, Scott H, Madhu B, Sharma N, Bon H, Zecchini V, Smith DM, Denicola GM, Mathews N, Osborne M, Hadfield J, Macarthur S, Adryan B, Lyons SK, Brindle KM, Griffiths J, Gleave ME, Rennie PS, Neal DE, Mills IG. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011;30:2719–2733. doi: 10.1038/emboj.2011.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, Varambally S, Ghosh D, Chinnaiyan AM. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–180. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Shojaie A, Michailidis G. Network enrichment analysis in complex experiments, Stat. Appl Genet Mol Biol. 2010;9 doi: 10.2202/1544-6115.1483. Article22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang G, He P, Tan H, Budhu A, Gaedcke J, Ghadimi BM, Ried T, Yfantis HG, Lee DH, Maitra A, Hanna N, Alexander HR, Hussain SP. Integration of metabolomics and transcriptomics revealed a fatty acid network exerting growth inhibitory effects in human pancreatic cancer. Clin Cancer Res. 2013;19:4983–4993. doi: 10.1158/1078-0432.CCR-13-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Su G, Burant CF, Beecher CW, Athey BD, Meng F. Integrated metabolome and transcriptome analysis of the NCI60 dataset. BMC Bioinformatics. 2011;12(Suppl 1):S36. doi: 10.1186/1471-2105-12-S1-S36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yang K, Xia B, Wang W, Cheng J, Yin M, Xie H, Li J, Ma L, Yang C, Li A, Fan X, Dhillon HS, Hou Y, Lou G, Li K. A Comprehensive Analysis of Metabolomics and Transcriptomics in Cervical Cancer. Sci Rep. 2017;7:43353. doi: 10.1038/srep43353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fahrmann JF, Grapov DD, Wanichthanarak K, DeFelice BC, Salemi MR, Rom WN, Gandara DR, Phinney BS, Fiehn O, Pass H, Miyamoto S. Integrated Metabolomics and Proteomics Highlight Altered Nicotinamide- and Polyamine Pathways in Lung Adenocarcinoma. Carcinogenesis. 2017 doi: 10.1093/carcin/bgw205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Auslander N, Yizhak K, Weinstock A, Budhu A, Tang W, Wang XW, Ambs S, Ruppin E. A joint analysis of transcriptomic and metabolomic data uncovers enhanced enzyme-metabolite coupling in breast cancer. Sci Rep. 2016;6:29662. doi: 10.1038/srep29662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kuo TC, Tian TF, Tseng YJ. 3Omics: a web-based systems biology tool for analysis, integration and visualization of human transcriptomic, proteomic and metabolomic data. BMC Syst Biol. 2013;7:64. doi: 10.1186/1752-0509-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cavill R, Jennen D, Kleinjans J, Briede JJ. Transcriptomic and metabolomic data integration. Brief Bioinform. 2016;17:891–901. doi: 10.1093/bib/bbv090. [DOI] [PubMed] [Google Scholar]
- 124.Wanichthanarak K, Fahrmann JF, Grapov D. Genomic, Proteomic, and Metabolomic Data Integration Strategies. Biomark Insights. 2015;10:1–6. doi: 10.4137/BMI.S29511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Goveia J, Pircher A, Conradi LC, Kalucka J, Lagani V, Dewerchin M, Eelen G, DeBerardinis RJ, Wilson ID, Carmeliet P. Meta-analysis of clinical metabolic profiling studies in cancer: challenges and opportunities. EMBO Mol Med. 2016;8:1134–1142. doi: 10.15252/emmm.201606798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sud M, Fahy E, Cotter D, Azam K, Vadivelu I, Burant C, Edison A, Fiehn O, Higashi R, Nair KS, Sumner S, Subramaniam S. Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 2016;44:D463–470. doi: 10.1093/nar/gkv1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sitnikov DG, Monnin CS, Vuckovic D. Systematic Assessment of Seven Solvent and Solid-Phase Extraction Methods for Metabolomics Analysis of Human Plasma by LC-MS. Sci Rep. 2016;6:38885. doi: 10.1038/srep38885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lindahl A, Saaf S, Lehtio J, Nordstrom A. Tuning Metabolome Coverage in Reversed Phase LC-MS Metabolomics of MeOH Extracted Samples Using the Reconstitution Solvent Composition. Anal Chem. 2017;89:7356–7364. doi: 10.1021/acs.analchem.7b00475. [DOI] [PubMed] [Google Scholar]
- 129.Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem. 2006;78:779–787. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 130.Tautenhahn R, Patti GJ, Rinehart D, Siuzdak G. XCMS Online: a web-based platform to process untargeted metabolomic data. Anal Chem. 2012;84:5035–5039. doi: 10.1021/ac300698c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Huan T, Forsberg EM, Rinehart D, Johnson CH, Ivanisevic J, Benton HP, Fang M, Aisporna A, Hilmers B, Poole FL, Thorgersen MP, Adams MWW, Krantz G, Fields MW, Robbins PD, Niedernhofer LJ, Ideker T, Majumder EL, Wall JD, Rattray NJW, Goodacre R, Lairson LL, Siuzdak G. Systems biology guided by XCMS Online metabolomics. Nat Methods. 2017;14:461–462. doi: 10.1038/nmeth.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Metallo CM, Walther JL, Stephanopoulos G. Evaluation of 13C isotopic tracers for metabolic flux analysis in mammalian cells. J Biotechnol. 2009;144:167–174. doi: 10.1016/j.jbiotec.2009.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Buescher JM, Antoniewicz MR, Boros LG, Burgess SC, Brunengraber H, Clish CB, DeBerardinis RJ, Feron O, Frezza C, Ghesquiere B, Gottlieb E, Hiller K, Jones RG, Kamphorst JJ, Kibbey RG, Kimmelman AC, Locasale JW, Lunt SY, Maddocks OD, Malloy C, Metallo CM, Meuillet EJ, Munger J, Noh K, Rabinowitz JD, Ralser M, Sauer U, Stephanopoulos G, St-Pierre J, Tennant DA, Wittmann C, Vander Heiden MG, Vazquez A, Vousden K, Young JD, Zamboni N, Fendt SM. A roadmap for interpreting (13)C metabolite labeling patterns from cells. Curr Opin Biotechnol. 2015;34:189–201. doi: 10.1016/j.copbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Faubert B, DeBerardinis RJ. Analyzing Tumor Metabolism In Vivo. Annual Review of Cancer Biology. 2017;1:99–117. [Google Scholar]
- 135.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM, DeBerardinis RJ. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A. 2011;108:8674–8679. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, Gui DY, Sullivan LB, Wasylenko TM, Subbaraj L, Chin CR, Stephanopolous G, Mott BT, Jacks T, Clish CB, Vander Heiden MG. Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab. 2016;23:517–528. doi: 10.1016/j.cmet.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Marin-Valencia I, Yang C, Mashimo T, Cho S, Baek H, Yang XL, Rajagopalan KN, Maddie M, Vemireddy V, Zhao Z, Cai L, Good L, Tu BP, Hatanpaa KJ, Mickey BE, Mates JM, Pascual JM, Maher EA, Malloy CR, Deberardinis RJ, Bachoo RM. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012;15:827–837. doi: 10.1016/j.cmet.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Maher EA, Marin-Valencia I, Bachoo RM, Mashimo T, Raisanen J, Hatanpaa KJ, Jindal A, Jeffrey FM, Choi C, Madden C, Mathews D, Pascual JM, Mickey BE, Malloy CR, DeBerardinis RJ. Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed. 2012;25:1234–1244. doi: 10.1002/nbm.2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sellers K, Fox MP, Bousamra M, 2nd, Slone SP, Higashi RM, Miller DM, Wang Y, Yan J, Yuneva MO, Deshpande R, Lane AN, Fan TW. Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest. 2015;125:687–698. doi: 10.1172/JCI72873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yuneva MO, Fan TW, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, Mates JM, Alonso FJ, Wang C, Seo Y, Chen X, Bishop JM. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 2012;15:157–170. doi: 10.1016/j.cmet.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, Lau AN, Ji BW, Dixit PD, Hosios AM, Muir A, Chin CR, Freinkman E, Jacks T, Wolpin BM, Vitkup D, Vander Heiden MG. Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science. 2016;353:1161–1165. doi: 10.1126/science.aaf5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fan TW, Lane AN, Higashi RM, Farag MA, Gao H, Bousamra M, Miller DM. Altered regulation of metabolic pathways in human lung cancer discerned by (13)C stable isotope-resolved metabolomics (SIRM) Mol Cancer. 2009;8:41. doi: 10.1186/1476-4598-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, Wodzak M, Klimko C, McMillan E, Butt Y, Ni M, Oliver D, Torrealba J, Malloy CR, Kernstine K, Lenkinski RE, DeBerardinis RJ. Metabolic Heterogeneity in Human Lung Tumors. Cell. 2016;164:681–694. doi: 10.1016/j.cell.2015.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, Huang Z, Barnett S, Mickey BE, DeBerardinis RJ, Tu BP, Maher EA, Bachoo RM. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. 2014;159:1603–1614. doi: 10.1016/j.cell.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Z, Yanxiang Guo J, White E, Rabinowitz JD. Glucose feeds the TCA cycle via circulating lactate. Nature. 2017;551:115–118. doi: 10.1038/nature24057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, Li H, Huet G, Yuan Q, Wigal T, Butt Y, Ni M, Torrealba J, Oliver D, Lenkinski RE, Malloy CR, Wachsmann JW, Young JD, Kernstine K, DeBerardinis RJ. Lactate Metabolism in Human Lung Tumors. Cell. 2017;171:358–371 e359. doi: 10.1016/j.cell.2017.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hiller K, Metallo CM, Kelleher JK, Stephanopoulos G. Nontargeted elucidation of metabolic pathways using stable-isotope tracers and mass spectrometry. Anal Chem. 2010;82:6621–6628. doi: 10.1021/ac1011574. [DOI] [PubMed] [Google Scholar]
- 149.Huang X, Chen YJ, Cho K, Nikolskiy I, Crawford PA, Patti GJ. X13CMS: global tracking of isotopic labels in untargeted metabolomics. Anal Chem. 2014;86:1632–1639. doi: 10.1021/ac403384n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Weindl D, Wegner A, Hiller K. Metabolome-Wide Analysis of Stable Isotope Labeling-Is It Worth the Effort? Front Physiol. 2015;6:344. doi: 10.3389/fphys.2015.00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sun RC, Fan TW, Deng P, Higashi RM, Lane AN, Le AT, Scott TL, Sun Q, Warmoes MO, Yang Y. Noninvasive liquid diet delivery of stable isotopes into mouse models for deep metabolic network tracing. Nat Commun. 2017;8:1646. doi: 10.1038/s41467-017-01518-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sparvero LJ, Amoscato AA, Dixon CE, Long JB, Kochanek PM, Pitt BR, Bayir H, Kagan VE. Mapping of phospholipids by MALDI imaging (MALDI-MSI): realities and expectations. Chem Phys Lipids. 2012;165:545–562. doi: 10.1016/j.chemphyslip.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Landgraf RR, Garrett TJ, Conaway MC, Calcutt NA, Stacpoole PW, Yost RA. Considerations for quantification of lipids in nerve tissue using matrix-assisted laser desorption/ionization mass spectrometric imaging. Rapid Commun Mass Spectrom. 2011;25:3178–3184. doi: 10.1002/rcm.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kawashima M, Iwamoto N, Kawaguchi-Sakita N, Sugimoto M, Ueno T, Mikami Y, Terasawa K, Sato TA, Tanaka K, Shimizu K, Toi M. High-resolution imaging mass spectrometry reveals detailed spatial distribution of phosphatidylinositols in human breast cancer. Cancer Sci. 2013;104:1372–1379. doi: 10.1111/cas.12229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dilillo M, Ait-Belkacem R, Esteve C, Pellegrini D, Nicolardi S, Costa M, Vannini E, Graaf EL, Caleo M, McDonnell LA. Ultra-High Mass Resolution MALDI Imaging Mass Spectrometry of Proteins and Metabolites in a Mouse Model of Glioblastoma. Sci Rep. 2017;7:603. doi: 10.1038/s41598-017-00703-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jones EE, Powers TW, Neely BA, Cazares LH, Troyer DA, Parker AS, Drake RR. MALDI imaging mass spectrometry profiling of proteins and lipids in clear cell renal cell carcinoma. Proteomics. 2014;14:924–935. doi: 10.1002/pmic.201300434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Shroff EH, Eberlin LS, Dang VM, Gouw AM, Gabay M, Adam SJ, Bellovin DI, Tran PT, Philbrick WM, Garcia-Ocana A, Casey SC, Li Y, Dang CV, Zare RN, Felsher DW. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc Natl Acad Sci U S A. 2015;112:6539–6544. doi: 10.1073/pnas.1507228112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Banerjee S, Zare RN, Tibshirani RJ, Kunder CA, Nolley R, Fan R, Brooks JD, Sonn GA. Diagnosis of prostate cancer by desorption electrospray ionization mass spectrometric imaging of small metabolites and lipids. Proc Natl Acad Sci U S A. 2017;114:3334–3339. doi: 10.1073/pnas.1700677114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kunzke T, Balluff B, Feuchtinger A, Buck A, Langer R, Luber B, Lordick F, Zitzelsberger H, Aichler M, Walch A. Native glycan fragments detected by MALDI-FT-ICR mass spectrometry imaging impact gastric cancer biology and patient outcome. Oncotarget. 2017;8:68012–68025. doi: 10.18632/oncotarget.19137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lou S, Balluff B, Cleven AH, Bovee JV, McDonnell LA. Prognostic Metabolite Biomarkers for Soft Tissue Sarcomas Discovered by Mass Spectrometry Imaging. J Am Soc Mass Spectrom. 2017;28:376–383. doi: 10.1007/s13361-016-1544-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.O’Brien PJ, Lee M, Spilker ME, Zhang CC, Yan Z, Nichols TC, Li W, Johnson CH, Patti GJ, Siuzdak G. Monitoring metabolic responses to chemotherapy in single cells and tumors using nanostructure-initiator mass spectrometry (NIMS) imaging. Cancer Metab. 2013;1:4. doi: 10.1186/2049-3002-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Quanico J, Franck J, Wisztorski M, Salzet M, Fournier I. Progress and Potential of Imaging Mass Spectrometry Applied to Biomarker Discovery. Methods Mol Biol. 2017;1598:21–43. doi: 10.1007/978-1-4939-6952-4_2. [DOI] [PubMed] [Google Scholar]
- 163.McDonnell LA, Angel PM, Lou S, Drake RR. Mass Spectrometry Imaging in Cancer Research: Future Perspectives. Adv Cancer Res. 2017;134:283–290. doi: 10.1016/bs.acr.2016.11.010. [DOI] [PubMed] [Google Scholar]
- 164.Baker TC, Han J, Borchers CH. Recent advancements in matrix-assisted laser desorption/ionization mass spectrometry imaging. Curr Opin Biotechnol. 2017;43:62–69. doi: 10.1016/j.copbio.2016.09.003. [DOI] [PubMed] [Google Scholar]