

Abstract
The concerted metabolic reprogramming across cancer and normal cellular compartments of the tumor microenvironment can favor tumorigenesis by increasing the survival and proliferating capacities of transformed cells. p62 has emerged as a critical signaling adaptor, beyond its role in autophagy, by playing an intricate context-dependent role in metabolic reprogramming of the cell types of the tumor and stroma, which shapes the tumor microenvironment to control tumor progression. Focusing on metabolic adaptations, we review the cellular processes upstream and downstream of p62 that regulate how distinct cell types adapt to the challenging and evolving environmental conditions during tumor initiation and progression. In addition, we describe partners of p62 that, in a collaborative or independent manner, can also rewire cell metabolism. Finally, we discuss the potential therapeutic implications of targeting p62 in cancer, considering its multifaceted roles in diverse cell types of the tumor microenvironment.
Keywords: p62, tumor microenvironment, metabolic reprogramming, inflammation, cancer
1. Introduction
Two critical characteristics of cancer cells are their ability to sustain proliferative signaling and evade death [1]. By adapting to their environment, tumor cells are able to survive and grow even when their bioenergetic demands outpace nutrient availability. One way that they do this is by reprograming their cellular metabolism [2]. Solid tumors are embedded in a complex heterocellular system of resident and recruited normal cells, termed the “tumor microenvironment” (TME). In parallel to tumor cell metabolic reprogramming, surrounding normal cells also undergo metabolic changes that might have profound impact on tumor behavior [3]. Emerging evidence suggests that a key factor mediating these metabolic alterations throughout the TME is p62 (also known as sequestosome 1; SQSTM1).
p62 has been most studied as an autophagy adaptor that recruits polyubiquitinated cargo into the autophagy machinery where it also undergoes degradation. Many of these studies suggest that p62 directly promotes autophagy-dependent cell survival under metabolic stress [4]. However, it is now known that the function of p62 extends well beyond its canonical role in autophagy. For example, although p62 lacks intrinsic enzymatic activity, it acts as a multifunctional signaling hub by utilizing its different conserved structural elements. This allows p62 to directly interact with protein adaptors at signaling nodes of pathways controlling inflammation, cell death, survival, and metabolic reprogramming [4]. With respect to metabolic reprogramming, p62’s impact on cellular metabolism in the TME is cell-type dependent [3]. Specifically, p62 has pro-tumorigenic functions in the tumor epithelium, yet plays a tumor-suppressor role in fibroblasts, hepatic stellate cells (HSC) and macrophages [5–8]. Accordingly, p62 is often over-expressed in malignant epithelial cells, but lost in the surrounding stroma [6, 9–11]. Consequently, understanding the role of p62 in metabolic reprogramming and its implications for tumorigenesis requires focused consideration of the specific cell types within the TME.
In this review, we will describe how p62 regulates cellular metabolism in the tumor epithelia, as well as in several cellular types of the stroma. In addition, we will highlight how these changes affect tumor-to-stroma crosstalk and shaping of the TME. Finally, we will discuss some of the ways in which interactors of p62 exert independent functions that also impact cell metabolism.
2. p62 and the tumor epithelia
The malignant transformation of a normal cell is the consequence of intrinsic and extrinsic events, such as genetic aberrations and chronic inflammation, respectively [1, 12]. To counteract these stressors, affected cells must balance regulatory pathways that either promote survival by repairing damage or eliminate irreversibly injured cells by executing cell death programs. When this response system fails, damaged cells can survive and accumulate genetic aberrations, which can lead to transformation. High intracellular levels of p62 can tip the balance in favor of tumorigenesis, by promoting cell survival and resistance to stress, and by exerting a potent anabolic signaling that further drives tumor growth [4].
Not surprisingly, several intrinsic pathways control the homeostatic levels of p62 to prevent tumorigenesis. At the transcriptional level, p62 mRNA expression has been reported to be regulated by four main signaling nodes that integrate inflammatory, oncogenic, oxidative, and endoplasmic reticulum (ER) stress signals [10, 13–15] (Figure 1). Post-transcriptionally, p62 is an extremely short-lived protein, which is constantly degraded by autophagy and the proteasome [16, 17] (Figure 1). Despite these inherent regulatory mechanisms, p62 accumulation has been documented in most, if not all, tumors of epithelial origin [4]. This is often the result of a self-amplifying auto-regulatory loop that starts with impairment of autophagy, and the resulting accumulated p62 sequesters inhibitors of its own transcriptional activators, which further drives p62 expression (Figure 1). Another mechanism of p62 accumulation involves amplification of its gene (SQSTM1) within the commonly found amplicon of chromosome 5q [18, 19] (Figure 1). Exemplifying its potent tumor-promoting role, p62 was found to be the driver gene in ccRCC, among more than 61 recurrently amplified genes within the 5q amplicon [9].
Figure 1. p62 in the tumor epithelia.
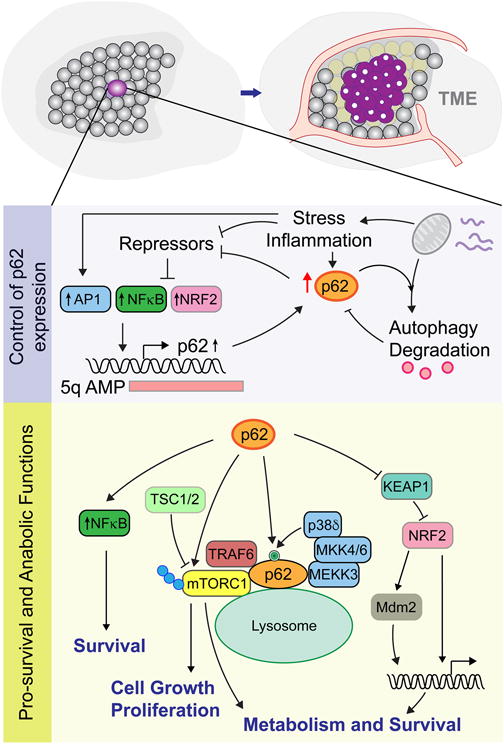
p62 is transcriptionally regulated by inflammation and stress acting via upstream activators (eg, AP1, NFκB and NRF2) that are repressed under homeostatic conditions. Autophagy impairment promotes p62 accumulation and de-repression of the p62 activators, leading to further p62 expression. p62 is also frequently found amplified within the 5q amplicon in ccCRC. p62 downstream functions include activation of NFκB to activate pro-survival pathways, mTORC1 to activate cell growth and proliferation and NRF2 to drive genes of redox detoxification and metabolic pathways.
Central to its housekeeping function, p62 plays important roles in both “bulk” and “selective” autophagy. In selective autophagy, it is believed that p62 and other adaptor proteins (eg, NBR1, TAX1BP1, NDP52, and OPTN) specifically recognize and bind K63-polyubiquitinated cargos through their UBA domains, and tethers them to the LC3-coated phagophore – thanks to the ability of the LIR domain to bind LC3. This biological redundancy suggests that selective autophagy requires a high degree of specificity to initiate waste removal programs that guarantee optimal cellular quality control. Thus, selective autophagy, acting through p62 and other adaptor proteins, plays an important housekeeping function in non-stressed cells by removing damaged or overabundant poly-ubiquitinated intracellular structures that are too big to be degraded by the proteasome [20]. In contrast, bulk autophagy non-selectively engulfs macromolecules and damaged organelles under conditions of stress, such as nutrient shortage or hypoxia, to guarantee energy production and availability of anabolic intermediates so as to sustain cell homeostasis [21]. Since cargo specificity is no longer needed, it is a process thought to be independent of autophagy adaptor function, yet p62 degradation under “bulk” autophagy is required to prevent tumorigenesis. Counterintuitively, the genetic inactivation of p62, rather than suppressing the autophagic flux, actually promotes “bulk” autophagy indirectly by inhibiting mTORC1 function [22]. Conversely, impairment of the autophagic flux promotes p62 accumulation to activate mTORC1 that further inhibits autophagy and results in increased p62 accumulation and tumorigenesis [10, 11] (Figure 1). This exemplifies the delicate balance and interplay between selective and bulk autophagy in regulating cell survival, and suggests that one of the main tumor-suppressor functions of autophagy is to prevent p62 accumulation [23] (Figure 1). Therefore, while autophagy is a clear positive regulator of cancer, it also shows a tumor suppressive role by keeping p62 levels at homeostatically tolerable levels.
Tumor initiation requires the stepwise acquisition of several driver mutations [24]. These oncogenic events can also induce cellular stress, which triggers cell-intrinsic safety mechanisms to induce senescence and cell-death [25]. Thus, for cells to become fully transformed, they must overcome the oncogene-induced stress response. In this context, p62 upregulation cooperates with acquired oncogenic drivers to promote pro-survival signaling pathways, which allows cells to withstand stress and accelerate cancer-driving mutations [10, 11] (Figure 1). One of the first examples of such cooperation came from studies done in a model of lung cancer, in which constitutively active KRas mutant (KRasG12D) led to p62 overexpression that, in turn, promoted an NFκB-dependent inflammatory response that stimulated cell survival [10] (Figure 1). Importantly, this study further showed that p62 was fundamental for KRas-transformed cells to survive oncogene-associated stress, as p62-deleted cells were resistant to Ras activation [10]. In keeping with this, other studies found that accumulation of p62, due to defects in autophagy, caused similar hyperactivation of NFκB-controlled survival programs that promoted tumorigenesis [23] (Figure 1). These pioneering works were the first indications that p62’s main function during tumorigenesis stems from its ability to promote specific pro-survival pathways that cooperate with driver oncogenic mutations.
As a more comprehensive view of p62 function emerges, it is now clear that, in addition to its role in autophagy, p62 exerts its tumor-promoting role through its activity as a multifunctional signaling hub [4]. Indeed, besides the LIR and UBA domains, p62 contains several other discrete substructures that bind important regulators of cellular stress and metabolism. For instance, the KEAP1 Interacting Region (KIR) mediates interaction with KEAP1, a substrate adaptor of the E3 ubiquitin ligase Cullin-3 (CUL3), which is responsible for ubiquitination and subsequent proteasomal degradation of transcription factor NRF2 [26–28] (Figure 1). Thus, p62 accumulation and binding to KEAP1 prevents KEAP1-dependent NRF2 inhibition, leading to stabilization of the transcription factor, which drives expression of a battery of antioxidant and cellular protective genes responsible for glutathione synthesis, drug efflux transport, detoxification of xenobiotics, NADPH synthesis, and elimination of reactive oxygen species (ROS) (Figure 1). While the role of NRF2 in cancer depends on timing and context [29], pan-cancer genomic studies have revealed several genomic mutations that give rise to hyperactivation of the NRF2 pathway, either by loss of function of KEAP1 or gain of function in NRF2. In addition, activation of NRF2 has been directly associated with promotion of survival in oncogene-induced senescent cells by its ROS-detoxification mechanism [30]. Similarly, it has been reported that glucose deprivation leads to glycosylation of KEAP1, which leads to activation of NRF2 [31]. Importantly, activation of the KEAP1/NRF2 cascade has been implicated in the control of cancer cell metabolism [32, 33]. That is, by controlling expression of PHGDH, PSAT1 and SHMT2 – all via ATF4, NRF2 has been reported to regulate the serine biosynthesis pathway [32], which supports glutathione and nucleotide production. Interestingly, NRF2 shifts glucose metabolism toward purine nucleotide synthesis, which is necessary to sustain increased cell proliferation [33]. Thus, beyond promoting an antioxidant response, NRF2 is likely to be instrumental in the control of key enzymes in the oxidative and non-oxidative arms of the pentose phosphate pathway that promote metabolic alterations that impact cell proliferation [33].
Besides increased survival capacity, tumor-initiating cells also need potent anabolic drivers of cell growth. In this regard, p62 is an important contributor to the activation of mTORC1, a major anabolic driver and regulator of cell growth and proliferation [34, 35] (Figure 1). In both tumor and normal cells, mTORC1 plays a fundamental role in promoting metabolic reprogramming, by acting as a metabolic switch that senses energy and nutrient availability and integrates them into the appropriate anabolic response [6, 36–38]. In normal cells, under conditions of nutrient shortage and absence of growth signals, mTORC1 remains inactive, due to the TSC1-TSC2 complex that maintains mTORC1-activating GTPase (Rheb1) in the inactive, GDP conformation [39]. Conversely, nutrient abundance is sensed by upstream activators of mTORC1 and, thus, promotes mTORC1 activation [38]. Mechanistically, upon amino acid sensing, mTORC1 is rapidly repositioned to the lysosomal membrane thanks to the actions of Rag GTPases and the v-ATPase multimeric complex [40, 41]. Recent work has unveiled additional p62-driven mechanisms that fine-tune amino-acid-driven mTORC1 activation. By binding MEKK3, p62 initiates a MKK3/6 signaling cascade, leading to activation of p38δ and, ultimately, direct phosphorylation of p62 at residues T269 and S272, which can be important for full activation of mTORC1 [35] (Figure 1). In fact, p62 phosphorylation-deficient mutants defectively activate mTORC1 in response to amino acids [35]. Taken together, these studies underscore the importance of p62 in the activation of mTORC1 to sustain anabolic pathways conducive to tumorigenesis.
Our understanding of the role of p62 in mTORC1 activation has been greatly advanced by liver cancer studies. Hepatocyte-specific deletion of TSC1 promoted hepatocellular carcinoma (HCC) by allowing constitutive mTORC1 activation [42]. And deletion of p62 in TSC1-KO hepatocyte suppressed HCC incidence by preventing mTORC1 activation [11]. Conversely, HCC was triggered in hepatocytes by both the overexpression of wild-type p62 or that of an autophagy-deficient p62 mutant (p62-ΔUBA) [11]. On the other hand, KIR-deficient p62 did not induce HCC [11]. These important experiments strongly suggest that overexpression of p62 is oncogenic in itself, and that this oncogenicity is mediated by an autophagy-independent mechanism. Combined with the fact that p62 accumulates in preneoplastic hepatocytes, these data also suggest that hepatocyte stress-driven p62 accumulation triggers sustained activation of mTORC1 to support cell growth, and, through stimulation of NRF2, maintains pro-survival pathways – all of which promote cell transformation [11] (Figure 1).
This paradigm also seems to hold true in pancreatic cancer [43]. That is, in pancreatic ductal adenocarcinoma (PDAC) development, acquisition of KRas mutations is an early event [44, 45]. Fortunately for the hosts, partially transformed pancreatic cells tend to succumb to oncogenic-mediated stress, and PanIN1 lesions, the early form of PDAC, rarely progress to full malignancies [46, 47]. Accumulation of p62 has been observed in mouse and human pancreatitis [48], which is thought to be the result of impaired autophagy. This negative regulation of autophagy can occur under conditions of obesity, hypernutrition, alcohol consumption and tobacco smoking, all well-established risk factors for pancreatic cancer [43]. In fact, during pancreatitis, quiescent differentiated pancreatic ductal cells acquire tumor progenitor characteristics, when p62 accumulation induces NRF2-dependent activation of Mdm2, which mediates p53-dependent and -independent abrogation of the regulatory blockade of cell transformation [43] (Figure 1).
In summary, these data suggest that p62 functions as a critical regulator of tumorigenesis by impinging on two key events: cell detoxification through selective autophagy and activation of NRF2 and NFκB [10, 11, 23], and cell anabolism through regulation of mTORC1 [11, 22] (Figure 1). Therefore, by supporting pro-survival and anabolic pathways p62 helps to fuel tumor cell growth and proliferation.
3. p62 and the tumor microenvironment
The TME contains a large variety of normal cells, especially stromal fibroblasts, and these contribute to the acquisition of additional tumor hallmarks [1]. For their part, tumor cells create a hostile environment by, for example, depleting nutrients, disrupting organ architecture and flooding the area with byproducts. While the initial host response is to fend off the outgrowth of transformed cells, tumors eventually gain the upper hand [49]. Further, cells surrounding the tumor epithelium exhibit altered biological properties, compared to their tumor-naïve counterparts [50]. Importantly, p62 has emerged as a critical regulator of metabolic reprogramming in normal cells of the TME, with working mechanisms that greatly differ from its pro-tumorigenic role in the tumor epithelia [5–7].
While tumor cells exhibit p62 accumulation that plays an instrumental role in tumorigenesis, the surrounding stromal cells exhibit depressed levels of p62 that, nevertheless, also play a pivotal role in tumor progression. Downregulation of p62 in stromal cells is particularly well documented in prostate and liver [5–7]. In stromal fibroblasts in prostate, loss of p62 induced a cancer-associated fibroblast (CAF) phenotype by impairing the mTORC1-c-Myc axis – and, thereby, downregulating the capacity of these cells to efficiently drive metabolic detoxification [6] (Figure 2). More specifically, reduced p62 levels inhibit glutamine and glucose consumption, which impaired the synthesis of key intermediaries required for glutathione production and, thereby, glutathione-mediated cellular detoxification. This metabolic reprogramming led to accumulation of reactive oxygen species (ROS) in the stromal fibroblasts and, as a result, release of the pro-survival inflammatory cytokine IL-6 – serving as a key autocrine signal for stromal fibroblasts to become CAFs, and as a paracrine signal to sustain invasiveness and growth of prostate cancer cells. This study by Valencia et al. (2014) was the first to demonstrate the important contribution of p62-driven metabolic reprogramming to the tumor-promoting role of activated fibroblasts.
Figure 2. p62 and the tumor microenvironment.
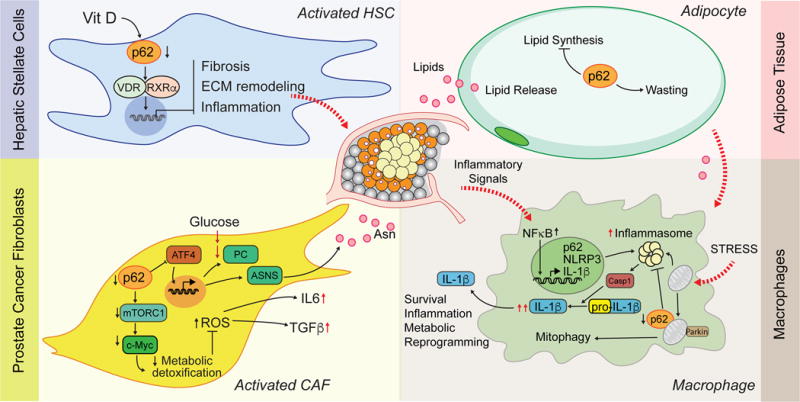
Role of p62 in four compartments of the TME. HSC activation is repressed when p62 activates VDR/RXR function, which controls transcription of genes mediating ECM remodeling, fibrosis and inflammation. Decreased p62 in prostate CAFs downregulates mTORC1 and c-Myc and, thus, cellular detoxification capacity; as a result, ROS accumulation is elevated, leading to increased secretion of IL-6 and TGFβ. In addition, downregulation of p62 promotes ATF4 upregulation, which drives PC and ASNS to increase fibroblast fitness and survival conditions of glutamine deprivation and causes release of Asparagine (Asn), which supports tumor growth. p62 regulates lipid biosynthesis and mobilization in adipocytes. Inflammatory cues originating in the TME, increase p62 levels in macrophages, which then mediates recycling of damaged mitochondria through parkin-dependent mitophagy to ultimately limit inflammasome-dependent production of IL-1β.
Consistent with this finding, a more recent study revealed that downregulation of p62 in prostate fibroblasts endowed them with an increased ability to survive glutamine deprivation [5], which is common in the TME of highly aggressive cancers [51]. This is important because glutamine is the source for key anaplerotic intermediates in the TCA, and the obligate nitrogen donor for many cellular biosynthetic processes [52]. Interestingly, under conditions of glutamine abundance, p62 facilitates proteasome-mediated degradation of ATF4 by the SCF (βTrCP) ubiquitin ligase [5] (Figure 2). Thus, downregulation of p62 allows ATF4 to accumulate and drive a transcriptional program that includes two fundamental metabolic enzymes, pyruvate carboxylase (PC) and asparagine synthetase (ASNS) [5] (Figure 2). Nutrient tracing experiments in p62-deficient fibroblasts under glutamine deprivation revealed increased glucose consumption and shuttling of glucose-derived carbons into the TCA cycle for biosynthesis of amino acids (eg, glutamate and aspartate/asparagine). In addition, upregulation of PC partially compensated for the lack of glutamine-derived α-ketoglutarate by providing oxaloacetate and acetyl-CoA, which can generate α-ketoglutarate when combined with citrate [5].
Of note, the ATF4 upregulation driven by p62 loss while noted in stromal fibroblasts, was not seen in tumor epithelial cell lines – supporting the notion that p62’s mechanisms of action are cell-type specific. Importantly, in vivo genetic ablation of p62 in the fibroblast compartment alone was sufficient to induce prostate epithelial hyperplasia, which usually precedes malignant lesions, and also promoted a strong stromal reaction that included upregulation of ATF4, ASNS and PC [5] (Figure 2). Of great relevance for the tumor-to-stroma crosstalk and the tumor-promoting role of CAFs, the authors showed that under glutamine-free conditions, p62-deficient stromal cells sustained prostate cancer cell growth by secreting asparagine [5] (Figure 2). Interestingly, a recent study showed that increased asparagine in tumor cells favored growth in low-glutamine conditions; this could be to be due to asparagine’s ability to maintain protein synthesis, rather than being a product of catabolism [53]. Similarly, a parallel study showed that KRas-dependent upregulation of ASNS elevated intracellular asparagine to sustain mTORC1 and proliferation, and block apoptosis [54]. Asparagine might activate mTORC1 through its exchange for other amino acids in the TME (eg, serine, arginine and histidine) [55]. These studies demonstrate that asparagine from p62-depleted CAFs, is a key nutrient for sustaining anabolism in tumor cells. Finally, it is highly significant that asparaginase, which hydrolyzes asparagine to aspartate, is routinely used to treat leukemia [56], and is showing promising preclinical results as a treatment for solid tumors [53, 54, 57]. Targeting asparagine secretion from metabolically reprogrammed CAFs could be an alternative or complementary approach to treat some solid tumors. Taken together, these studies show that p62 loss in TME fibroblasts leads to increased fibroblast cell fitness and metabolic reprogramming to overcome glutamine deprivation. Therefore, the emerging model is that tumor-to-stroma cross-talk is also impacted by p62, with reciprocal tumor feeding by asparagine secretion and paracrine inflammatory feedback through IL-6 synthesis, which is sufficient to drive hyperproliferation of normal cells and promote conditions conducive to prostate cancer, even in the absence of oncogenic drivers [5, 6].
Like fibroblasts, stellate cells of the pancreas (PSC) and liver (HSC) are of mesenchymal origin and have a fundamental role in providing the structural framework of their tissue parenchymas [58]. While stellate cells are normally quiescent, they can be activated by tissue injury and metabolic stress. Once activated, stellate cells lose their characteristic lipid droplets, proliferate, mobilize to damaged areas, and undergo morphological and gene expression changes – that all promote tissue remodeling to help restore homeostasis [59]. A key modulator of stellate cell activation and fibrosis is the vitamin D receptor (VDR) [60, 61]. In that context, total body loss of p62 in mice fed a high fat diet and treated with DEN carcinogen promoted HCC development [7] – rather than inhibiting tumorigenesis, as it would have been initially expected, given the tumor-promoting activities of p62 in hepatocytes [11] (Figure 2). Further analysis in cell-type-specific p62 KO mice revealed that this was at least partly the result of HSC activation [7]. This is important because it suggests that loss of p62 pro-survival signaling in the hepatocytes was compensated by the inflammation and fibrosis in the p62-deficient activated stroma [7] (Figure 2). Importantly, p62’s tumor-suppressive effect in HSC was clearly dependent on its ability to mediate ligand-dependent VDR activation [7], although still to-be-determined additional metabolic mechanisms could also come into play, such as release or recycling of products of the lipid droplets lost during stellate cell activation.
In addition to these resident cells, a repertoire of normal recruited cells also forms part of the tumor niche. Among them, macrophages patrol the TME – playing a prominent generally protumoral role in the tumor-to-stroma crosstalk [62]. Because macrophages detect and engulf extracellular pathogens and damaged cells in highly inflamed tissues, they need a well-balanced intrinsic regulatory mechanism to control their own level of inflammation. PAMP or DAMP activation of these innate immune cells is mediated by major signaling pathways, including NFκB, which promotes the synthesis of pro-IL1β that is processed into mature IL-1β by the inflammasome [63]. The mechanism that opposes excessive activation of the inflammasome, also depends on NFκB and, as it turns out, p62 (but no other autophagy adaptors). Specifically, macrophage-activating signals (eg, LPS) induce p62, which is required to limit inflammasome complex formation and production of IL-1β downstream of NFκB, and to recycle mitochondria damaged by signals that culminate in activation of the NLRP3-dependent inflammasome [8] (Figure 2). Thus, when p62 was genetically ablated in macrophages in two different mouse models of NLRP3-dependent inflammation, and the macrophages were activated, damaged mitochondria accumulated, and this induced excessive IL-1β-dependent inflammation and increased tissue damage [8] (Figure 2). Based on these results, loss of p62 in tumor-associated macrophages (TAM) would be expected to promote tumorigenesis, at least in part through release of cytokine IL-1β, which has been suggested to contribute to reprogramming the TME [64, 65] (Figure 2).
In addition to p62’s role in cancer, we have also learned a great deal about its role in systemic metabolic disease. For example, p62 regulates heterocellular crosstalk in the context of adipose tissue inflammation, which promotes insulin resistance and so is conducive to type-2 diabetes [66]. In this regard, adipocyte conditioned media activates adipose tissue macrophages (ATM), a process termed “metabolic activation”, which results in an increase of p62 and PPARγ levels [67]. In agreement with the proposed role of p62 in limiting excessive inflammation [8], genetic ablation of p62 in ATM markedly increase their production of TNFα and IL-1β [67]. Similarly, chemical inhibition of PPARγ increased the metabolic activation of ATM [67], consistent with PPARγ’s anti-proliferative role. Of note, increased p62 was caused by a deficiency in autophagy, induced by the metabolic intermediate palmitate secreted by adipocytes. This adipocyte-macrophage crosstalk through palmitate exemplifies how metabolic functions in neighboring cell compartments can impact one another’s behavior. Taken together, these studies suggest that p62 and PPARγ restrict macrophage-induced inflammation during metabolic activation in adipose tissue.
In addition to its function in macrophages, p62 also controls metabolic functions in adipocytes [66, 68]. Total body ablation of p62 [66] and adipocyte-specific p62 KO [68] both induced mature-onset obesity, due to increased adiposity and reduced energy expenditure (Figure 2). Interestingly, the actions of adipocyte-specific p62 deletion were cell-autonomous – originating by impairment of mitochondrial biogenesis, and were independent of development or differentiation [68]. This obesity model provides an opportunity to study tumor-adipocyte crosstalk independent of caloric intake – that is, without relying on administration of hyper-nutritious diets, or genetic ablation of leptin [69]. This is important, given that epidemiological studies have found significant associations between obesity and increased risk of cancer, both in its incidence and in its progression [70]. While adipocytes form fat tissue where tumors do not usually originate, they are also part of the TME – especially in organs surrounded by visceral fat [71]. In fact, cancer cells can mobilize adipocyte-stored lipids to induce a wasting program leading to release of these lipids and a consequent loss of fat mass [72, 73]. This process, known as “fat cachexia”, has been shown to represent a key mechanism for promoting tumor progression, by providing both an accessible stash of nutrients and increased levels of pro-inflammatory cytokines [72]. While the adipocyte-specific function of p62 has not been investigated in the context of cancer, its role as a multifunctional adaptor in adipocyte homeostasis (eg, sensing beta-adrenergic inputs) [68], suggests that it might suppress the tumorigenic role of the adipose tissue.
A broader view of the metabolic actions of p62 in non-cancer contexts was gained by studies of one of p62’s closest homologs, the PB1-containing autophagy adaptor NBR1. NBR1 promotes an inflammatory circuit in ATM, by mediating formation of a signaling complex required for JNK activation [74]. In contrast to p62’s anti-inflammatory effect in macrophages [8, 67], in vivo deletion of NBR1 in the myeloid compartment inhibited ATM inflammatory pathways – resulting in impaired macrophage function, M1 polarization and chemotactic activity, which prevented adipose tissue inflammation and improved glucose tolerance in obese mice [74]. Opposing actions of NBR1 and p62 were also found in osteoblasts, where p62 limited macrophage-dependent NFκB activation through autophagy, which prevented excessive inflammation and release of chemokines that retain hematopoietic progenitors (HPs) in the bone marrow [75]. Consequently, deletion of p62 in osteoblasts promoted excessive NFκB-driven inflammation, loss of chemoattractant production and HPs egress from the bone marrow. In contrast, concomitant deletion of p62 and NBR1 rescued the release of HP-attractant chemokines, thereby rescuing the HP-retention capacity [75]. These findings suggest opposite roles for p62 and NBR1 in mediating inflammation, and that autophagy, which has been suggested to degrade inflammasomes and limit IL-1β [76], can also contribute to p62-dependent mitophagy to limit inflammation [8].
In summary, p62 has emerged as a multifaceted protein that functions as a tumor suppressor in fibroblasts, stellate cells and macrophages by regulating activation of pro-survival pathways and promoting cellular adaptation to nutrient deprivation. In adipocytes, p62 might also have a metabolic reprogramming effect, which likely impacts tumor progression.
4. p62 partners as independent regulators of cancer metabolism
It now seems clear that p62 plays an important role as an adaptor protein in regulating tumor metabolism. In addition, many of the proteins that interact with p62 (eg, PKCζ, NBR1 and TRAF6) have also been reported to directly regulate metabolic reprogramming of cancer cells, although in an apparently p62-independent manner (Figure 3). Interestingly, p62 was discovered as an atypical protein kinase C (PKC)-interacting protein, binding via the Phox/Brem 1p (PB1) domain [77, 78] (Figure 3). PKCζ plays an essential role in colorectal cancer (CRC) by reprogramming the metabolism of cancer cells under glucose-deprivation conditions [79]. More specifically, in the absence of glucose, loss of PKCζ allows cancer cells to utilize glutamine through 3-phosphoglycerate dehydrogenase (PHGDH) and phosphoserine aminotransferase (PSAT1), key enzymes in the serine biosynthesis pathway [79]. Since CRC cells utilize high amounts of glucose to survive and proliferate, PKCζ deficiency enables these cells to adjust their metabolism to keep up with their bioenergetics demands, even when glucose is low.
Figure 3. p62 partners as independent regulators of cancer metabolism.
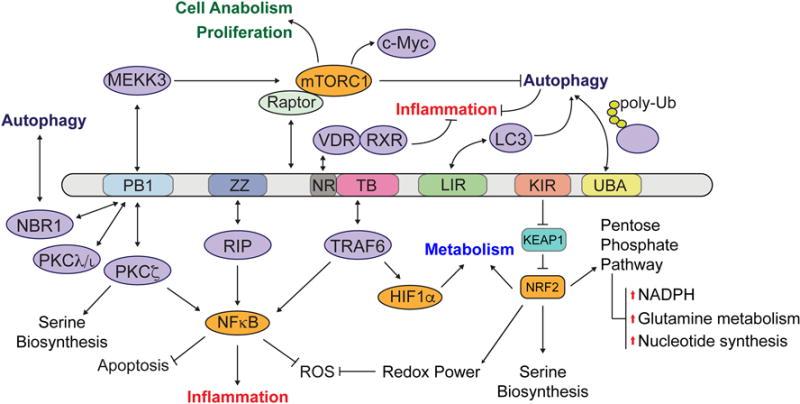
Partners of p62 interact through specific p62 domains: PB1 domain binds other PB1-containing proteins to control autophagy, inflammation and serine biosynthesis; ZZ domain binds RIP and stimulates NFκB pathway; NR box binds VDR to limit inflammatory responses; TB domain binds TRAF6 to promote NFκB activation; LIR domain binds LC3 protein to promote autophagy degradation; KIR domain binds NRF2 inhibitor KEAP1, which activates several downstream transcriptional programs (eg, cellular detoxification, survival, pentose phosphate pathway, serine biosynthesis and lipid metabolism); UBA domain binds polyubiquitinated proteins that, together with the LC3 domain, serve as a bridge to exert its function as an autophagy adaptor. Raptor also binds p62 and is involved in regulating mTORC1 activation.
Much like p62, NBR1 functions as an autophagy adaptor, accumulates upon autophagy blockade, and can be found within cytosolic aggregates of misfolded and ubiquitinated proteins [80, 81]. In addition, NBR1 interacts with p62 through its PB1 domain [82], which has been suggested to have a cooperative effect in mediating selective autophagy of ubiquitinated proteins [81] (Figure 3). However, NBR1 also works independently of p62. For instance, NBR1-dependent autophagy controls local remodeling of focal adhesions to control cell migration [83]. Furthermore, NBR1 has been shown to localize to peroxisomes, where it mediates their selective degradation through a form of autophagy termed “pexophagy” [84]. This molecular process is especially relevant in ccCRC, which is characterized by high levels of hypoxia and activation of Hif2α to promote NBR1-dependent pexophagy [85]. In turn, increased pexophagy can lead to metabolic reprogramming of the lipid metabolism [85], which has been suggested to play a role in the initiation and progression of ccRCC, a cancer characterized by accumulation of neutral lipids and glycogen [86]. Interestingly, while NBR1 is necessary and sufficient for pexophagy, p62 is not required in the process, but does increase its efficiency [84]. Thus, in addition to its impact on metabolism through p62-dependent autophagy, NBR1 functions as an independent adaptor to regulate additional biological processes, such as migration and pexophagy, that also impact cell metabolism of tumor cells [83, 85]. These, and additional future studies, will hopefully help clarify the apparent redundancy of having multiple autophagy adaptors and the relation between them.
TRAF6 interacts with p62’s TB domain [87]. Importantly, this interaction makes p62 a key component for TRAF6 to function as a mediator of inflammatory cues, such as those triggered by TNFα or IL-1β, which ultimately activate NFκB signaling [87]. Beyond inflammation, TRAF6 and p62 interaction is also necessary to promote mTORC1 activation in the lysosomal membrane in response to amino acids [34]. However, TRAF6 has other important functions in regulating tumor cell metabolism to which the contribution of p62 is unknown. A recent example of that is the ability of TRAF6 to counteract the tumor suppressive function of p53 by restricting its mitochondrial translocation [88]. Given the pleiotropic functions of p53, which include the control of cellular metabolism [89], this newly uncovered TRAF6-dependent regulation of p53 could have larger implications than just blocking spontaneous apoptosis. Furthermore, TRAF6 also regulates HIF-1α by both oxygen-independent and -dependent mechanisms [90, 91]. In cancer cells, under normoxic conditions, TRAF6 mediates ubiquitination of HIF-1α, which enhances HIF-1α protein levels that, in turn, lead to increased HIF-1α transcriptional activity and tumor angiogenesis and growth [90] (Figure 3). In breast cancer, TRAF6 also regulates HIF-1α signaling, in this case through monoubiquitination of H2AX, which might modulate progression of the malignancy [91]. This is important because HIF-1α has a critical role in reprogramming cancer cell metabolism – specifically in shifting glucose metabolism from mitochondrial oxidative phosphorylation to anaerobic glycolysis [92], which is a hallmark of cancer cell metabolism. Thus, PKCζ, NBR1 and TRAF6 all function in p62-independent pathways but also interact with p62. It is therefore tempting to speculate that p62 might work as a buffer essentially sequestering these proteins as a means to regulate metabolic cascades. Alternatively, p62 might fine-tune the actions of its partners adding another layer of regulation.
5. Concluding remarks
The role of p62 in cancer is growing in complexity and relevance. Well beyond its role in autophagy, p62 has emerged as a signaling adaptor protein that is capable of orchestrating metabolic reprogramming of tumor epithelial cells, but also normal cells in the tumor niche – modulating their ability to support tumor growth and proliferation. While p62 might appear to be an appealing target in cancer, important limitations arise due to its potent tumor suppressor role in certain normal cellular compartments within the TME. On the other hand, in cases where p62 has already been lost in the tumor stroma, patients could still benefit from p62 inhibition therapies since ablation of p62 in the tumor epithelium dramatically inhibits tumorigenesis in vitro and in vivo. However, therapeutic applications targeting p62 need to consider both cellular contexts.
Acknowledgments
Studies in the author’s labs were supported by grants from NIH (R01CA172025, R01CA207177, R01DK108743, R01CA211794 to J.M.; R01CA192642, R01CA218254 to M.T.D.-M.). M.R.C is supported by “La Caixa” fellowship for studies in North America. We thank Rebecca Tuttle for help in editing the manuscript.
Abbreviations
- TME
tumor microenvironment
- SQSTM1
Sequestosome 1
- ccRCC
clear cell renal cell carcinoma
- NBR1
neighbor of BRCA1 Gene 1
- TAX1BP1
Tax1 binding protein 1
- NDP52
nuclear domain 10 protein 52
- OPTN
Optineurin
- LC3
light chain 3
- LIR
LC3 interacting region
- UBA
ubiquitin-associated
- p62-ΔUBA
truncated p62 with deletion of UBA domain
- mTORC1
mammalian target of rapamycin complex 1
- NRF2
nuclear receptor factor 2
- KEAP1
Kelch Like ECH Associated Protein 1
- HCC
hepatocellular carcinoma
- PDAC
pancreatic ductal adenocarcinoma
- KRas
Kirsten Ras
- Mdm2
mouse double minute 2
- TSC1/2
tuberous sclerosis complex 1/2
- Rheb1
Ras homolog enriched In brain 1
- Raptor
regulatory associated protein of mTOR complex 1
- Rag
Ras related GTP binding
- MEKK3
mitogen-activated protein kinase kinase kinase 3
- p38δ, alias for
mitogen-activated protein kinase 13 (MAPK13)
- NFκB
nuclear factor kappa B
- c-myc
proto-oncogene c-Myc
- ROS
reactive oxygen species
- IL-6
interleukin 6
- ATF4
activating transcription factor 4
- SCF
Skp, Cullin, F-box containing complex
- β-TRcP
beta-transducin repeat containing E3 ubiquitin protein ligase
- PC
pyruvate carboxylase
- ASNS
asparagine synthethase
- TCA
tricarboxylic acid cycle
- VDR
Vitamin-D receptor
- RXR
retinoic X receptor
- NRB
nuclear receptor box
- TAM
tumor-associated macrophages
- PAMP(s)
pathogen-associated molecular pattern(s)
- DAMP(s)
damage-associated molecular pattern(s)
- IL-1β
interleukin 1 beta
- LPS
lipopolysaccharide
- NLRP3
NLR family pyrin domain containing 3
- TNFα
tumor necrosis factor alpha
- ATM(s)
adipose tissue macrophage(s)
- PPARγ
peroxisome proliferator activated receptor gamma
- PB1
Phox/Bem 1
- PKC
protein kinase C
- PHGDH
phosphoglycerated dehydrogenase
- PSAT1
phosphoserine aminotransferase 1
- Vhl
von hippel lindeau
- Hif
hypoxia inducible factor
- SHMT2
serine hydroxymethyltransferase 2
- G6PD
glucose 6 phosphate dehydrogenase
- PGD
phosphogluconate dehydrogenase
- TKT
transketolase
- TALDO1
transaldolase 1
- PPP
pentose phosphate pathway
- NADPH
nicotinamide adenine dinucleotide phosphate
- ZZ
zinc finger domain
- TRAF6
TNF receptor associated factor 6
- mUb
mono Ubiquitin
- H2AX
H2A histone family member X
- CK1
casein kinase 1 alpha 1
- NASH
non-alcoholic steatohepatitis
- NAFLD
non-alcoholic fatty liver disease
- HSC
hepatic stellate cells
- PSC
pancreatic stellate cells
- DEN
diethylnitrosamine
- HP
hematopoietic progenitor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reina-Campos M, Moscat J, Diaz-Meco M. Metabolism shapes the tumor microenvironment. Curr Opin Cell Biol. 2017;48:47–53. doi: 10.1016/j.ceb.2017.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moscat J, Karin M, Diaz-Meco MT. p62 in Cancer: Signaling Adaptor Beyond Autophagy. Cell. 2016;167:606–609. doi: 10.1016/j.cell.2016.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linares JF, Cordes T, Duran A, Reina-Campos M, Valencia T, Ahn CS, Castilla EA, Moscat J, Metallo CM, Diaz-Meco MT. ATF4-Induced Metabolic Reprograming Is a Synthetic Vulnerability of the p62-Deficient Tumor Stroma. Cell Metab. 2017;26:817–829 e816. doi: 10.1016/j.cmet.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valencia T, Kim JY, Abu-Baker S, Moscat-Pardos J, Ahn CS, Reina-Campos M, Duran A, Castilla EA, Metallo CM, Diaz-Meco MT, Moscat J. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell. 2014;26:121–135. doi: 10.1016/j.ccr.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duran A, Hernandez ED, Reina-Campos M, Castilla EA, Subramaniam S, Raghunandan S, Roberts LR, Kisseleva T, Karin M, Diaz-Meco MT, Moscat J. p62/SQSTM1 by Binding to Vitamin D Receptor Inhibits Hepatic Stellate Cell Activity, Fibrosis, and Liver Cancer. Cancer Cell. 2016;30:595–609. doi: 10.1016/j.ccell.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, McGeough MD, Ellisman MH, Seki E, Gustafsson AB, Hoffman HM, Diaz-Meco MT, Moscat J, Karin M. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell. 2016;164:896–910. doi: 10.1016/j.cell.2015.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li L, Shen C, Nakamura E, Ando K, Signoretti S, Beroukhim R, Cowley GS, Lizotte P, Liberzon E, Bair S, Root DE, Tamayo P, Tsherniak A, Cheng SC, Tabak B, Jacobsen A, Hakimi AA, Schultz N, Ciriello G, Sander C, Hsieh JJ, Kaelin WG., Jr SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell. 2013;24:738–750. doi: 10.1016/j.ccr.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–354. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Umemura A, He F, Taniguchi K, Nakagawa H, Yamachika S, Font-Burgada J, Zhong Z, Subramaniam S, Raghunandan S, Duran A, Linares JF, Reina-Campos M, Umemura S, Valasek MA, Seki E, Yamaguchi K, Koike K, Itoh Y, Diaz-Meco MT, Moscat J, Karin M. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell. 2016;29:935–948. doi: 10.1016/j.ccell.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ling J, Kang Y, Zhao R, Xia Q, Lee DF, Chang Z, Li J, Peng B, Fleming JB, Wang H, Liu J, Lemischka IR, Hung MC, Chiao PJ. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:105–120. doi: 10.1016/j.ccr.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.B’Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41:7683–7699. doi: 10.1093/nar/gkt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24:8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fang J, Barker B, Bolanos L, Liu X, Jerez A, Makishima H, Christie S, Chen X, Rao DS, Grimes HL, Komurov K, Weirauch MT, Cancelas JA, Maciejewski JP, Starczynowski DT. Myeloid malignancies with chromosome 5q deletions acquire a dependency on an intrachromosomal NF-kappaB gene network. Cell Rep. 2014;8:1328–1338. doi: 10.1016/j.celrep.2014.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beroukhim R, Brunet JP, Di Napoli A, Mertz KD, Seeley A, Pires MM, Linhart D, Worrell RA, Moch H, Rubin MA, Sellers WR, Meyerson M, Linehan WM, Kaelin WG, Jr, Signoretti S. Patterns of gene expression and copy-number alterations in von-hippel lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009;69:4674–4681. doi: 10.1158/0008-5472.CAN-09-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moscat J, Diaz-Meco MT. To aggregate or not to aggregate? A new role for p62. EMBO Rep. 2009;10:804. doi: 10.1038/embor.2009.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461–472. doi: 10.1038/nrm4024. [DOI] [PubMed] [Google Scholar]
- 22.Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44:134–146. doi: 10.1016/j.molcel.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, Dipaola RS, Karantza-Wadsworth V, White E. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–315. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 26.Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, Hoshii T, Hirao A, Takagi K, Mizushima T, Motohashi H, Lee MS, Yoshimori T, Tanaka K, Yamamoto M, Komatsu M. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–631. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 27.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 28.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564–571. doi: 10.1038/nrc3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA, Tuveson DA. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA, Corvalan AH, Biswal S, Swisher SG, Bekele BN, Minna JD, Stewart DJ. Wistuba, II, Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res. 2010;16:3743–3753. doi: 10.1158/1078-0432.CCR-09-3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeNicola GM, Chen PH, Mullarky E, Sudderth JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, Wistuba II, Minna JD, DeBerardinis RJ, Cantley LC. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015;47:1475–1481. doi: 10.1038/ng.3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitsuishi Y, Motohashi H, Yamamoto M. The Keap1-Nrf2 system in cancers: stress response and anabolic metabolism. Front Oncol. 2012;2:200. doi: 10.3389/fonc.2012.00200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell. 2013;51:283–296. doi: 10.1016/j.molcel.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Linares JF, Duran A, Reina-Campos M, Aza-Blanc P, Campos A, Moscat J, Diaz-Meco MT. Amino Acid Activation of mTORC1 by a PB1-Domain-Driven Kinase Complex Cascade. Cell Rep. 2015;12:1339–1352. doi: 10.1016/j.celrep.2015.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pusapati RV, Daemen A, Wilson C, Sandoval W, Gao M, Haley B, Baudy AR, Hatzivassiliou G, Evangelista M, Settleman J. mTORC1-Dependent Metabolic Reprogramming Underlies Escape from Glycolysis Addiction in Cancer Cells. Cancer Cell. 2016;29:548–562. doi: 10.1016/j.ccell.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 37.Angela M, Endo Y, Asou HK, Yamamoto T, Tumes DJ, Tokuyama H, Yokote K, Nakayama T. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARgamma directs early activation of T cells. Nat Commun. 2016;7:13683. doi: 10.1038/ncomms13683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;169:361–371. doi: 10.1016/j.cell.2017.03.035. [DOI] [PubMed] [Google Scholar]
- 39.Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, Freinkman E, Sabatini DM. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science. 2017;358:807–813. doi: 10.1126/science.aan6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kenerson HL, Yeh MM, Kazami M, Jiang X, Riehle KJ, McIntyre RL, Park JO, Kwon S, Campbell JS, Yeung RS. Akt and mTORC1 have different roles during liver tumorigenesis in mice. Gastroenterology. 2013;144:1055–1065. doi: 10.1053/j.gastro.2013.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Todoric J, Antonucci L, Di Caro G, Li N, Wu X, Lytle NK, Dhar D, Banerjee S, Fagman JB, Browne CD, Umemura A, Valasek MA, Kessler H, Tarin D, Goggins M, Reya T, Diaz-Meco M, Moscat J, Karin M. Stress-Activated NRF2-MDM2 Cascade Controls Neoplastic Progression in Pancreas. Cancer Cell. 2017;32:824–839 e828. doi: 10.1016/j.ccell.2017.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988;16:7773–7782. doi: 10.1093/nar/16.16.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 46.Hruban RH, Maitra A, Goggins M. Update on pancreatic intraepithelial neoplasia. Int J Clin Exp Pathol. 2008;1:306–316. [PMC free article] [PubMed] [Google Scholar]
- 47.Collins MA, Pasca di Magliano M. Kras as a key oncogene and therapeutic target in pancreatic cancer. Front Physiol. 2013;4:407. doi: 10.3389/fphys.2013.00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li N, Wu X, Holzer RG, Lee JH, Todoric J, Park EJ, Ogata H, Gukovskaya AS, Gukovsky I, Pizzo DP, VandenBerg S, Tarin D, Atay C, Arkan MC, Deerinck TJ, Moscat J, Diaz-Meco M, Dawson D, Erkan M, Kleeff J, Karin M. Loss of acinar cell IKKalpha triggers spontaneous pancreatitis in mice. J Clin Invest. 2013;123:2231–2243. doi: 10.1172/JCI64498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tape CJ, Ling S, Dimitriadi M, McMahon KM, Worboys JD, Leong HS, Norrie IC, Miller CJ, Poulogiannis G, Lauffenburger DA, Jorgensen C. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell. 2016;165:1818. doi: 10.1016/j.cell.2016.05.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lyssiotis CA, Kimmelman AC. Metabolic Interactions in the Tumor Microenvironment. Trends Cell Biol. 2017;27:863–875. doi: 10.1016/j.tcb.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu X, Yang Y, Hernandez-Davies JE, Rosales KK, Li H, Hugo W, Song C, Xu X, Schones DE, Ann DK, Gradinaru V, Lo RS, Locasale JW, Kong M. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol. 2016;18:1090–1101. doi: 10.1038/ncb3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahn CS, Metallo CM. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015;3:1. doi: 10.1186/s40170-015-0128-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pavlova NN, Hui S, Ghergurovich JM, Fan J, Intlekofer AM, White RM, Rabinowitz JD, Thompson CB, Zhang J. As Extracellular Glutamine Levels Decline, Asparagine Becomes an Essential Amino Acid. Cell Metab. 2018 doi: 10.1016/j.cmet.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gwinn DM, Lee AG, Briones-Martin-Del-Campo M, Conn CS, Simpson DR, Scott AI, Le A, Cowan TM, Ruggero D, Sweet-Cordero EA. Oncogenic KRAS Regulates Amino Acid Homeostasis and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to L-Asparaginase. Cancer Cell. 2018;33:91–107 e106. doi: 10.1016/j.ccell.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krall AS, Xu S, Graeber TG, Braas D, Christofk HR. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat Commun. 2016;7:11457. doi: 10.1038/ncomms11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pieters R, Hunger SP, Boos J, Rizzari C, Silverman L, Baruchel A, Goekbuget N, Schrappe M, Pui CH. L-asparaginase treatment in acute lymphoblastic leukemia: a focus on Erwinia asparaginase. Cancer. 2011;117:238–249. doi: 10.1002/cncr.25489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim K, Jeong JH, Lim D, Hong Y, Lim HJ, Kim GJ, Shin SR, Lee JJ, Yun M, Harris RA, Min JJ, Choy HE. L-Asparaginase delivered by Salmonella typhimurium suppresses solid tumors. Mol Ther Oncolytics. 2015;2:15007. doi: 10.1038/mto.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest. 2007;117:50–59. doi: 10.1172/JCI30082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397–411. doi: 10.1038/nrgastro.2017.38. [DOI] [PubMed] [Google Scholar]
- 60.Ding N, Yu RT, Subramaniam N, Sherman MH, Wilson C, Rao R, Leblanc M, Coulter S, He M, Scott C, Lau SL, Atkins AR, Barish GD, Gunton JE, Liddle C, Downes M, Evans RM. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell. 2013;153:601–613. doi: 10.1016/j.cell.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, Martin P, Tseng TW, Dawson DW, Donahue TR, Masamune A, Shimosegawa T, Apte MV, Wilson JS, Ng B, Lau SL, Gunton JE, Wahl GM, Hunter T, Drebin JA, O’Dwyer PJ, Liddle C, Tuveson DA, Downes M, Evans RM. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80–93. doi: 10.1016/j.cell.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Agostini L, Martinon F, Burns K, McDermott MF, Hawkins PN, Tschopp J. NALP3 forms an IL-1beta-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity. 2004;20:319–325. doi: 10.1016/s1074-7613(04)00046-9. [DOI] [PubMed] [Google Scholar]
- 64.Wu T, Hong Y, Jia L, Wu J, Xia J, Wang J, Hu Q, Cheng B. Modulation of IL-1beta reprogrammes the tumor microenvironment to interrupt oral carcinogenesis. Sci Rep. 2016;6:20208. doi: 10.1038/srep20208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Apte RN, Dotan S, Elkabets M, White MR, Reich E, Carmi Y, Song X, Dvozkin T, Krelin Y, Voronov E. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006;25:387–408. doi: 10.1007/s10555-006-9004-4. [DOI] [PubMed] [Google Scholar]
- 66.Rodriguez A, Duran A, Selloum M, Champy MF, Diez-Guerra FJ, Flores JM, Serrano M, Auwerx J, Diaz-Meco MT, Moscat J. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 2006;3:211–222. doi: 10.1016/j.cmet.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 67.Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, Landerholm RW, Crouthamel M, Gozal D, Hwang S, Singh PK, Becker L. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014;20:614–625. doi: 10.1016/j.cmet.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Muller TD, Lee SJ, Jastroch M, Kabra D, Stemmer K, Aichler M, Abplanalp B, Ananthakrishnan G, Bhardwaj N, Collins S, Divanovic S, Endele M, Finan B, Gao Y, Habegger KM, Hembree J, Heppner KM, Hofmann S, Holland J, Kuchler D, Kutschke M, Krishna R, Lehti M, Oelkrug R, Ottaway N, Perez-Tilve D, Raver C, Walch AK, Schriever SC, Speakman J, Tseng YH, Diaz-Meco M, Pfluger PT, Moscat J, Tschop MH. p62 links beta-adrenergic input to mitochondrial function and thermogenesis. J Clin Invest. 2013;123:469–478. doi: 10.1172/JCI64209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Calle EE, Kaaks R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nature Reviews Cancer. 2004;4:579–591. doi: 10.1038/nrc1408. [DOI] [PubMed] [Google Scholar]
- 71.Iyengar NM, Gucalp A, Dannenberg AJ, Hudis CA. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J Clin Oncol. 2016;34:4270–4276. doi: 10.1200/JCO.2016.67.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Porporato PE. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis. 2016;5:e200. doi: 10.1038/oncsis.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hoy AJ, Balaban S, Saunders DN. Adipocyte-Tumor Cell Metabolic Crosstalk in Breast Cancer. Trends Mol Med. 2017;23:381–392. doi: 10.1016/j.molmed.2017.02.009. [DOI] [PubMed] [Google Scholar]
- 74.Hernandez ED, Lee SJ, Kim JY, Duran A, Linares JF, Yajima T, Muller TD, Tschop MH, Smith SR, Diaz-Meco MT, Moscat J. A macrophage NBR1-MEKK3 complex triggers JNK-mediated adipose tissue inflammation in obesity. Cell Metab. 2014;20:499–511. doi: 10.1016/j.cmet.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chang KH, Sengupta A, Nayak RC, Duran A, Lee SJ, Pratt RG, Wellendorf AM, Hill SE, Watkins M, Gonzalez-Nieto D, Aronow BJ, Starczynowski DT, Civitelli R, Diaz-Meco MT, Moscat J, Cancelas JA. p62 is required for stem cell/progenitor retention through inhibition of IKK/NF-kappaB/Ccl4 signaling at the bone marrow macrophage-osteoblast niche. Cell Rep. 2014;9:2084–2097. doi: 10.1016/j.celrep.2014.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. doi: 10.1038/ni.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Puls A, Schmidt S, Grawe F, Stabel S. Interaction of protein kinase C zeta with ZIP, a novel protein kinase C-binding protein. Proc Natl Acad Sci U S A. 1997;94:6191–6196. doi: 10.1073/pnas.94.12.6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sanchez P, De Carcer G, Sandoval IV, Moscat J, Diaz-Meco MT. Localization of atypical protein kinase C isoforms into lysosome-targeted endosomes through interaction with p62. Mol Cell Biol. 1998;18:3069–3080. doi: 10.1128/mcb.18.5.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ma L, Tao Y, Duran A, Llado V, Galvez A, Barger JF, Castilla EA, Chen J, Yajima T, Porollo A, Medvedovic M, Brill LM, Plas DR, Riedl SJ, Leitges M, Diaz-Meco MT, Richardson AD, Moscat J. Control of nutrient stress-induced metabolic reprogramming by PKCzeta in tumorigenesis. Cell. 2013;152:599–611. doi: 10.1016/j.cell.2012.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kirkin V, Lamark T, Sou YS, Bjorkoy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Overvatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–516. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 81.Kirkin V, Lamark T, Johansen T, Dikic I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy. 2009;5:732–733. doi: 10.4161/auto.5.5.8566. [DOI] [PubMed] [Google Scholar]
- 82.Lamark T, Perander M, Outzen H, Kristiansen K, Overvatn A, Michaelsen E, Bjorkoy G, Johansen T. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J Biol Chem. 2003;278:34568–34581. doi: 10.1074/jbc.M303221200. [DOI] [PubMed] [Google Scholar]
- 83.Kenific CM, Stehbens SJ, Goldsmith J, Leidal AM, Faure N, Ye J, Wittmann T, Debnath J. NBR1 enables autophagy-dependent focal adhesion turnover. J Cell Biol. 2016;212:577–590. doi: 10.1083/jcb.201503075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Deosaran E, Larsen KB, Hua R, Sargent G, Wang Y, Kim S, Lamark T, Jauregui M, Law K, Lippincott-Schwartz J, Brech A, Johansen T, Kim PK. NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci. 2013;126:939–952. doi: 10.1242/jcs.114819. [DOI] [PubMed] [Google Scholar]
- 85.Walter KM, Schonenberger MJ, Trotzmuller M, Horn M, Elsasser HP, Moser AB, Lucas MS, Schwarz T, Gerber PA, Faust PL, Moch H, Kofeler HC, Krek W, Kovacs WJ. Hif-2alpha promotes degradation of mammalian peroxisomes by selective autophagy. Cell Metab. 2014;20:882–897. doi: 10.1016/j.cmet.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 86.Drabkin HA, Gemmill RM. Cholesterol and the development of clear-cell renal carcinoma. Curr Opin Pharmacol. 2012;12:742–750. doi: 10.1016/j.coph.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 87.Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19:1576–1586. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang X, Li CF, Zhang L, Wu CY, Han L, Jin G, Rezaeian AH, Han F, Liu C, Xu C, Xu X, Huang CY, Tsai FJ, Tsai CH, Watabe K, Lin HK. TRAF6 Restricts p53 Mitochondrial Translocation, Apoptosis, and Tumor Suppression. Mol Cell. 2016;64:803–814. doi: 10.1016/j.molcel.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14:359–370. doi: 10.1038/nrc3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sun H, Li XB, Meng Y, Fan L, Li M, Fang J. TRAF6 upregulates expression of HIF-1alpha and promotes tumor angiogenesis. Cancer Res. 2013;73:4950–4959. doi: 10.1158/0008-5472.CAN-13-0370. [DOI] [PubMed] [Google Scholar]
- 91.Rezaeian AH, Li CF, Wu CY, Zhang X, Delacerda J, You MJ, Han F, Cai Z, Jeong YS, Jin G, Phan L, Chou PC, Lee MH, Hung MC, Sarbassov D, Lin HK. A hypoxia-responsive TRAF6-ATM-H2AX signalling axis promotes HIF1alpha activation, tumorigenesis and metastasis. Nat Cell Biol. 2017;19:38–51. doi: 10.1038/ncb3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Semenza GL. Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin Cancer Biol. 2009;19:12–16. doi: 10.1016/j.semcancer.2008.11.009. [DOI] [PubMed] [Google Scholar]