

SUMMARY
In HIV vaccine studies, a major research objective is to identify immune response biomarkers measured longitudinally that may be associated with risk of HIV infection. This objective can be assessed via joint modeling of longitudinal and survival data. Joint models for HIV vaccine data are complicated by the following issues: (i) left truncations of some longitudinal data due to lower limits of quantification; (ii) mixed types of longitudinal variables; (iii) measurement errors and missing values in longitudinal measurements; (iv) computational challenges associated with likelihood inference. In this article, we propose a joint model of complex longitudinal and survival data and a computationally efficient method for approximate likelihood inference to address the foregoing issues simultaneously. In particular, our model does not make unverifiable distributional assumptions for truncated values, which is different from methods commonly used in the literature. The parameters are estimated based on the h-likelihood method, which is computationally efficient and offers approximate likelihood inference. Moreover, we propose a new approach to estimate the standard errors of the h-likelihood based parameter estimates by using an adaptive Gauss–Hermite method. Simulation studies show that our methods perform well and are computationally efficient. A comprehensive data analysis is also presented.
Keywords: Adaptive Gauss–Hermite, Cox model, h-Likelihood, Lower limit of quantification, Mixed-effect model, Shared-parameter model
1. Introduction
In preventive HIV vaccine efficacy trials, participants are randomized to receive a series of vaccinations or placebos and are followed until the day of being diagnosed with HIV infection or until the end of study follow-up. We are often interested in the times to HIV infection. Meanwhile, blood samples are repeatedly collected over time for each participant in order to measure immune responses induced by the vaccine, such as CD4 T cell responses. A major research interest in HIV vaccine studies is to identify potential immune response biomarkers for HIV infection. For example, for many infectious diseases antibodies induced by a vaccine can recognize and kill a pathogen before it establishes infection; therefore high antibody levels are often associated with a lower risk of pathogen infection. Since longitudinal trajectories of some immune responses are often associated with the risk of HIV infection, in statistical analysis it is useful to jointly model the longitudinal and survival data. Moreover, such joint models can be used to address measurement errors and non-ignorable missing data in the longitudinal data.
There has been active research on joint models of longitudinal and survival data in recent years. Lawrence Gould and others (2015) have given a comprehensive review in this field. Rizopoulos and others (2009) proposed a computational approach based on the Laplace approximation for joint models of continuous longitudinal response and time-to-event outcome. Bernhardt and others (2014) discussed a multiple imputation method for handling left-truncated longitudinal variables used as covariates in AFT survival models. Król and others (2016) considered joint models of a left-truncated longitudinal variable, recurrent events, and a terminal event. The truncated values of the longitudinal variable were assumed to follow the same normal distributions as the untruncated values. Other recent work includes Fu and Gilbert (2017), Barrett and others (2015), Elashoff and others (2015), Chen and others (2014), Taylor and others (2013), Rizopoulos (2012b), and Zhu and others (2012). Analysis of HIV vaccine trial data offers the following new challenges: (i) some longitudinal data may be left truncated by a lower limit of quantification (LLOQ) of the biomarker assay, and the common approach of assuming that truncated values follow parametric distributions is unverifiable and may be unreasonable for vaccine trial data; (ii) the longitudinal multivariate biomarker response data are intercorrelated and may be of mixed types such as binary and continuous; (iii) the longitudinal data may exhibit periodic patterns over time, due to repeated administrations of the HIV vaccine; (iv) some longitudinal biomarkers may have measurement errors and missing data; and (v) the computation associated with likelihood inference can be very intensive and challenging. A comprehensive statistical analysis of HIV vaccine trial data requires us to address all the foregoing issues simultaneously. Therefore, despite extensive literature on joint models, new statistical models and methods are in demand.
In this article, we propose innovative models and methods to address the above issues. The contributions of the paper are: (i) when longitudinal data are left truncated, we propose a new method that does not assume any parametric distributions for the truncated values, which is different from existing approaches in the literature that unrealistically assume truncated values to follow the same distributions as those for the observed values; (ii) for longitudinal data with left truncation, the observed values are assumed to follow a truncated normal distribution; (iii) we incorporate the associations among several longitudinal responses of mixed types by linking the longitudinal models with shared and correlated random effects (Rizopoulos, 2012b); and (iv) we address the computational challenges of likelihood inference by proposing a computationally very efficient approximate method based on the h-likelihood method (Lee and others, 2006). It is known that, when the baseline hazard in the Cox survival model is completely unspecified, the standard errors of the parameter estimates in the joint models may be underestimated (Rizopoulos, 2012a; Hsieh and others, 2006). To address this issue, we also propose a new approach to estimate the standard errors of parameter estimates.
The article is organized as follows. In Section 2, we introduce the HIV vaccine trial data that motivates the research. In Section 3, we describe the proposed models and methods, which address all of the issues discussed above simultaneously. Section 4 presents analysis of the HIV vaccine trial data. Section 5 shows simulation studies to evaluate the proposed models and methods. We conclude the article with some discussion in Section 6.
2. The HIV vaccine data
Our research is motivated by the VAX004 trial, which is a 36-month efficacy study of a candidate vaccine to prevent HIV-1 infection, which contained two recombinant gp120 Envelope proteins: the MN and GNE8 HIV HIV-1 strains (Flynn and others, 2005). One of the main objectives in the trial was to assess immune response biomarkers measured in vaccine recipients for their association with the incidence of HIV infection. It was addressed using plasma samples collected at the immunization visits, 2 weeks after the immunization visits, and the final visit (i.e. months 0, 0.5, 1, 1.5, 6, 6.5, …, 30, 30.5, 36) to measure several immune response variables in vaccine recipients. Eight immune response variables were measured in total, most of which are highly correlated with each other and some have up to 16% missing data. We focus on a subset of these variables that may be representative and have low rates of missing data. In particular, we use the NAb and the MNGNE8 variables, where NAb is the titer of neutralizing antibodies to the MN strain of the HIV-1 gp120 Env protein and MNGNE8 is the average level of binding antibodies (measured by ELISA) to the MN and GNE8 HIV-1 gp120 Env proteins (Gilbert and others, 2005). Due to space limitation, the details of the clinical research questions, participant selection procedure, and other immune response variables are described in Section 1 in the Supplementary material available at Biostatistics online.
The data set we consider has 194 participants in total, among whom 21 participants acquired HIV infection during the trial with time of infection diagnosis ranging from day 43 to day 954 and event rate of 10.8%. The average number of repeated measurements over time is 12.6 per participant. Moreover, NAb has a LLOQ of 1.477, and about 27% of NAb measurements are below the LLOQ (left-truncated). To minimize the potential for bias due to missing data on the immune response biomarkers, the models adjust for the dominant baseline prognostic factor for HIV-1 infection—baseline behavioral risk score, which is grouped into three categories: 0, 1, or 2 for risk score 0, 1–3, 4–7, respectively.
Figure 1 shows the longitudinal trajectories of the immune responses of a few randomly selected participants, where the left truncated values in NAb are substituted by the LLOQ. The value of an immune response typically increases sharply right after each vaccination, and then starts to decrease about 2 weeks after the vaccination. Such patterns are shown as the reverse sawtooth waves in Figure 1. We see that participants HIV-infected later on seem to have lower values of MNGNE8 and NAb than those uninfected by the end of the study. In particular, for MNGNE8, there seem to be clear differences between HIV-infected and uninfected participants, separated by the median value. The figures show that the longitudinal patterns of some immune responses seem to be associated with HIV infection, motivating inference via joint models of the longitudinal and survival data. In addition, some immune responses are highly correlated over time and are of mixed types, so we should also incorporate the associations among different types of longitudinal variables. Moreover, due to substantial variations across subjects, mixed effects models may be useful. The random effects in mixed effects models can serve several purposes: (i) they represent individual variations or individual-specific characteristics of the participants; (ii) they incorporate the correlation among longitudinal measurements for each participant; and (iii) they may be viewed as summaries of the individual profiles. Therefore, mixed effects models seem to be a reasonable choice for modeling the HIV vaccine trial data.
Fig. 1.

Longitudinal trajectories of two immune response variables for a few randomly selected VAX004 vaccine recipients, where the solid lines represent pre-infection trajectories of participants who acquired HIV infected and the dashed lines represent trajectories for participants who never acquired HIV infection. The left truncated values in NAb are substituted by the LLOQ of 1.477.
More results of the exploratory data analysis are given in Sections 2 and 3 in the Supplementary material available at Biostatistics online, including the summary statistics of the immune responses, Kaplan–Meier plot of the time to HIV infection, and longitudinal trajectories of NAb and MNGNE8 with the time variable shifted and aligned at the event times.
3. Joint models and inference
3.1. The longitudinal, truncation, and survival models
3.1.1. Models for longitudinal data of mixed types.
In the following, we denote by 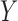 a random variable,
a random variable, 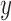 its observed value,
its observed value, 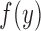 a generic density function, with similar notation for other variables. For simplicity of presentation, we consider two correlated longitudinal variables,
a generic density function, with similar notation for other variables. For simplicity of presentation, we consider two correlated longitudinal variables, 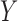 and
and 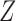 , where
, where 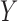 is continuous and subject to left truncation due to LLOQ, and
is continuous and subject to left truncation due to LLOQ, and 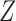 is binary or count (e.g. dichotomized variable or number of CD4 T cells). The models can be easily extended to more than two longitudinal processes. Let
is binary or count (e.g. dichotomized variable or number of CD4 T cells). The models can be easily extended to more than two longitudinal processes. Let 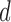 be the LLOQ of
be the LLOQ of 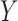 and
and 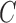 be the truncation indicator of
be the truncation indicator of 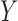 such that
such that 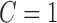 if
if 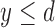 and
and 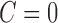 otherwise.
otherwise.
For the continuous longitudinal variable 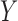 , after possibly some transformations such as a log-transformation, we may assume that the untruncated data of
, after possibly some transformations such as a log-transformation, we may assume that the untruncated data of 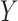 follow a truncated normal distribution. We consider a linear or nonlinear mixed effects (LME or NLME) model for the observed values of
follow a truncated normal distribution. We consider a linear or nonlinear mixed effects (LME or NLME) model for the observed values of 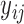 given that
given that 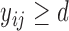 , that is,
, that is,
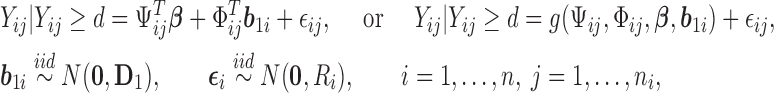 |
(3.1) |
where 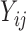 is the longitudinal variable of participant
is the longitudinal variable of participant 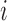 at time
at time 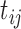 ,
, 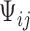 and
and 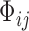 are vectors of covariates,
are vectors of covariates, 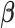 contains fixed parameters,
contains fixed parameters, 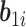 contains random effects,
contains random effects, 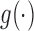 is a known nonlinear function,
is a known nonlinear function, 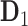 and
and 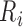 are covariance matrices, and
are covariance matrices, and 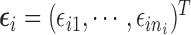 are random errors independent of
are random errors independent of 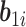 . A LME model is usually an empirical model while an NLME model is a mechanistic model widely used in HIV viral dynamics (Wu, 2009). We assume that
. A LME model is usually an empirical model while an NLME model is a mechanistic model widely used in HIV viral dynamics (Wu, 2009). We assume that 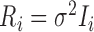 , i.e. the within-individual repeated measurements are independent conditional on the random effects.
, i.e. the within-individual repeated measurements are independent conditional on the random effects.
In model (3.1), the observed 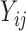 ’s given the random effects and the condition “
’s given the random effects and the condition “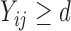 ” (or
” (or 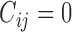 ) are assumed to be normally distributed, so it is reasonable to assume the
) are assumed to be normally distributed, so it is reasonable to assume the 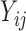 follow a truncated normal distribution (Mehrotra and others, 2000). For the truncated
follow a truncated normal distribution (Mehrotra and others, 2000). For the truncated 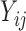 values (i.e.
values (i.e. 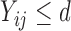 ), any parametric distributional assumptions are unverifiable, although most existing literature makes such assumptions for convenience of likelihood inference. Moreover, the truncated values are unlikely to follow normal distributions in most cases, since the
), any parametric distributional assumptions are unverifiable, although most existing literature makes such assumptions for convenience of likelihood inference. Moreover, the truncated values are unlikely to follow normal distributions in most cases, since the 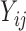 values at least must be positive while a normal random variable can take any real values. Thus, it is more reasonable to assume the truncated normal distribution for the observed
values at least must be positive while a normal random variable can take any real values. Thus, it is more reasonable to assume the truncated normal distribution for the observed 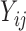 values and leave the distribution of the truncated
values and leave the distribution of the truncated 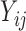 values completely unspecified. The density function of “
values completely unspecified. The density function of “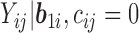 ” is given as Mehrotra and others (2000)
” is given as Mehrotra and others (2000)
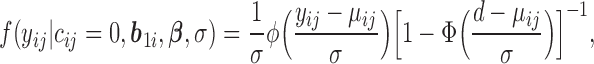 |
(3.2) |
where 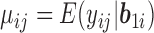 ,
, 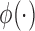 is the probability density function of the standard normal distribution
is the probability density function of the standard normal distribution 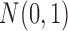 and
and 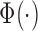 is the corresponding cumulative distribution function.
is the corresponding cumulative distribution function.
For the discrete longitudinal variable 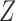 , we consider the following generalized linear mixed effects model (GLMM)
, we consider the following generalized linear mixed effects model (GLMM)
 |
(3.3) |
where 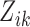 is the longitudinal variable of participant
is the longitudinal variable of participant 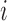 at time
at time 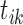 ,
, 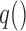 is a known link function,
is a known link function, 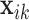 and
and 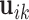 are vectors of covariates,
are vectors of covariates, 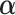 are fixed parameters,
are fixed parameters, 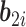 is a vector of random effects with
is a vector of random effects with 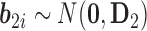 , and
, and 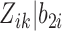 is assumed to follow a distribution in the exponential family.
is assumed to follow a distribution in the exponential family.
The longitudinal data may contain intermittent missing data and dropouts. We assume that the intermittent missing data and dropouts are missing at random. The fact that the missing data are biomarkers measuring immune responses to the vaccine (and not variables such as toxicity that could obviously be related to missed visits or dropout), and that the vaccine has a large safety data base showing it is not toxic, makes this assumption plausible.
3.1.2. A new approach for truncated longitudinal data.
When the 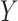 values are truncated, a common approach in the literature is to assume that the truncated values continue to follow the normal distribution assumed for the observed values (Hughes, 1999; Wu, 2002). However, such an assumption is unverifiable and may be unreasonable in some cases, as noted earlier. In particular, when the truncation rate is high, the normality assumption is even less reasonable as the truncation rate can be much larger than the left-tail probability of the normal distribution for the observed data. For example, Figure 2 displays histograms of NAb for two participants, where the left truncated data are substituted by the LLOQ of 1.477. The truncation rates, 27% for participant 1 and 33% for participant 2, seem much lager than the left-tail probabilities of the assumed distributions for the observed data.
values are truncated, a common approach in the literature is to assume that the truncated values continue to follow the normal distribution assumed for the observed values (Hughes, 1999; Wu, 2002). However, such an assumption is unverifiable and may be unreasonable in some cases, as noted earlier. In particular, when the truncation rate is high, the normality assumption is even less reasonable as the truncation rate can be much larger than the left-tail probability of the normal distribution for the observed data. For example, Figure 2 displays histograms of NAb for two participants, where the left truncated data are substituted by the LLOQ of 1.477. The truncation rates, 27% for participant 1 and 33% for participant 2, seem much lager than the left-tail probabilities of the assumed distributions for the observed data.
Fig. 2.

Histograms of NAb of two VAX004 vaccine recipients, where the left-truncated data are substituted by the LLOQ of 1.477.
Here we propose a different approach: we do not assume any parametric distributions for the truncated values, but instead we conceptually view the truncated values as a point mass or cluster of unobserved values below the LLOQ without any distributional assumption. Note that, although the truncation status 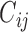 can be determined by the
can be determined by the 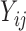 values, in HIV vaccine studies, many biomarkers are measured infrequently over time, due to both budget and practical considerations, while some other variables can be measured more frequently. For this reason, when the
values, in HIV vaccine studies, many biomarkers are measured infrequently over time, due to both budget and practical considerations, while some other variables can be measured more frequently. For this reason, when the 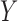 values are not measured, we can roughly predict the truncation status of the
values are not measured, we can roughly predict the truncation status of the 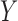 value based on other measured variables that are associated with
value based on other measured variables that are associated with 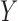 , including time. It is important to predict the truncation status of
, including time. It is important to predict the truncation status of 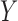 values, since left-truncated
values, since left-truncated 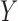 values have important implications (e.g. a positive immune response may be needed for protection by vaccination). A model for the truncation indicator
values have important implications (e.g. a positive immune response may be needed for protection by vaccination). A model for the truncation indicator 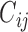 can help make reasonable predictions of
can help make reasonable predictions of 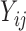 when such predictions are needed. Therefore, we assume the following model for the truncation indicator
when such predictions are needed. Therefore, we assume the following model for the truncation indicator 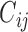 :
:
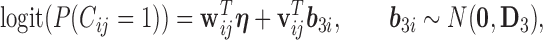 |
(3.4) |
where 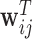 and
and 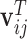 contain covariates,
contain covariates, 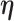 contains fixed parameters, and
contains fixed parameters, and 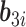 contains random effects. The contribution of the longitudinal data of
contains random effects. The contribution of the longitudinal data of 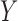 for individual
for individual 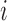 to the likelihood given the random effects is
to the likelihood given the random effects is 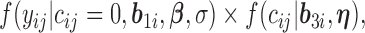 where
where 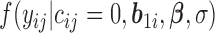 is given by (3.2) and
is given by (3.2) and 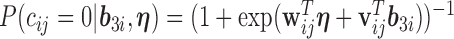 .
.
Another use of model (3.4) is modeling non-ignorable or informative missing data in the longitudinal  data. When longitudinal data have both left-truncated data and non-ignorable missing data, we should consider two separate models similar to (3.4). Here we do not consider the issue of non-ignorable missing data, but the models and methods can be easily extended to handle missing data. In fact, left truncated data may be viewed as non-ignorable missing data.
data. When longitudinal data have both left-truncated data and non-ignorable missing data, we should consider two separate models similar to (3.4). Here we do not consider the issue of non-ignorable missing data, but the models and methods can be easily extended to handle missing data. In fact, left truncated data may be viewed as non-ignorable missing data.
3.1.3. Association between mixed types of longitudinal variables.
Different immune response variables are typically highly correlated and may be of different types, such as one being continuous and another one being binary. The exact structures of the associations among different longitudinal variables may be complicated. However, we can reasonably assume that the variables are associated through shared or correlated random effects from different models. This is a reasonable assumption, since the random effects represent individual deviations from population averages and can be interpreted as unobserved or latent individual characteristics, such as individual genetic information or health status, which govern different longitudinal processes. This can be seen from Figure 1 where different immune response variables within the same individual exhibit similar patterns over time, including the truncation process. Therefore, we assume that 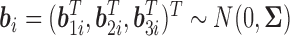 , where
, where 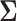 is an arbitrary covariance matrix. Note that we allow the random effects in the longitudinal models to be different since the longitudinal trajectories of different variables may exhibit different between-individual variations (as measured by random effects), especially for different types of longitudinal variables such as binary and continuous variables.
is an arbitrary covariance matrix. Note that we allow the random effects in the longitudinal models to be different since the longitudinal trajectories of different variables may exhibit different between-individual variations (as measured by random effects), especially for different types of longitudinal variables such as binary and continuous variables.
3.1.4. A Cox model for time-to-event data.
The times to HIV infection may be related to the longitudinal patterns of the immune responses and left-truncated statuses. The specific nature of this dependence may be complicated. There are several possibilities: (i) the infection time may depend on the current immune response values at infection times; (ii) the infection time may depend on past immune response values; and (iii) the infection time may depend on summaries or key characteristics of the longitudinal or truncation trajectories. Here we consider case (iii) for the following reasons: (a) the random effects may be viewed as summaries of individual-specific longitudinal trajectories; (b) the immune response values may be truncated due to lower detection limits; and (c) this approach is also widely used in the joint model literature. Since the random effects in the longitudinal models may be interpreted as “summaries” or individual-specific characteristics of the longitudinal processes, we may use random effects from the longitudinal models as “covariates” in the survival model. Such an approach is commonly used in the literature and is often called “shared parameter models” (Wulfsohn and Tsiatis, 1997; Rizopoulos, 2012b).
Let 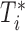 be the time to HIV infection,
be the time to HIV infection, 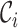 be the right-censoring time,
be the right-censoring time, 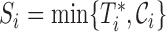 be the observed time, and
be the observed time, and 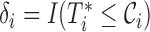 be the event indicator. We assume the censoring is non-informative and consider a Cox model for the observed survival data
be the event indicator. We assume the censoring is non-informative and consider a Cox model for the observed survival data 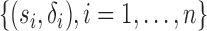 ,
,
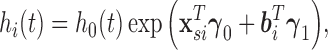 |
(3.5) |
where 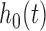 is an unspecified baseline hazard function,
is an unspecified baseline hazard function, 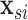 contains baseline covariates of individual
contains baseline covariates of individual 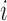 , and
, and 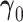 and
and 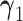 are vectors of fixed parameters. In model (3.5), the parameters
are vectors of fixed parameters. In model (3.5), the parameters 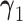 link the risk of HIV infection at time
link the risk of HIV infection at time 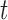 to the random effects in the longitudinal or truncation models, which allow us to check if individual-specific characteristics of the longitudinal immune responses are associated with the risk of HIV infection. We assume that the survival data and the longitudinal data are conditionally independent given the random effects.
to the random effects in the longitudinal or truncation models, which allow us to check if individual-specific characteristics of the longitudinal immune responses are associated with the risk of HIV infection. We assume that the survival data and the longitudinal data are conditionally independent given the random effects.
3.2. An approximate method for likelihood inference
We consider the likelihood method for parameter estimation and inference for the above models. Let 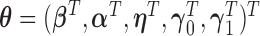 be the collection of all mean parameters and
be the collection of all mean parameters and 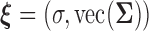 be the collection of variance–covariance (dispersion) parameters. The (joint) likelihood for all the observed longitudinal data and time-to-infection data is given by
be the collection of variance–covariance (dispersion) parameters. The (joint) likelihood for all the observed longitudinal data and time-to-infection data is given by
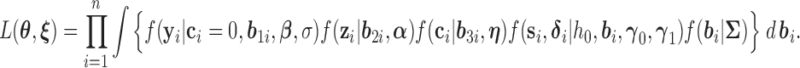 |
Since the dimension of the random effects 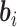 is often high and some density functions can be highly complicated, evaluation of the above integral can be a major challenge. The common approach based on the Monte Carlo EM algorithm can offer potential difficulties such as very slow or even non-convergence (Hughes, 1999). Numerical integration methods such as the Gaussian Hermite (GH) quadrature method can also be very tedious. Therefore, in the following we consider an approximate method based on the h-likelihood, which can be computationally much more efficient while maintaining reasonable accuracy (Lee and others, 2006; Ha and others, 2003; Molas and others, 2013). Its performance in the current context will be evaluated by simulations later.
is often high and some density functions can be highly complicated, evaluation of the above integral can be a major challenge. The common approach based on the Monte Carlo EM algorithm can offer potential difficulties such as very slow or even non-convergence (Hughes, 1999). Numerical integration methods such as the Gaussian Hermite (GH) quadrature method can also be very tedious. Therefore, in the following we consider an approximate method based on the h-likelihood, which can be computationally much more efficient while maintaining reasonable accuracy (Lee and others, 2006; Ha and others, 2003; Molas and others, 2013). Its performance in the current context will be evaluated by simulations later.
Essentially, the h-likelihood method uses Laplace approximations to the intractable integral in the likelihood. A first-order Laplace approximation can be viewed as the GH quadrature method with one node. So a Laplace approximation can be less accurate than the GH quadrature method with more than one node. However, when the dimension of the integral is high, a Laplace approximation can be computationally much less intensive than the GH quadrature method whose computational intensity grows exponentially with the dimension of the integral. Moreover, it produces approximate MLEs for the mean parameters and approximate restricted maximum likelihood estimates (REMLs) for the variance–covariance (dispersion) parameters. For the models (3.1), (3.3)–(3.5) in the previous section, the log h-likelihood function is given by
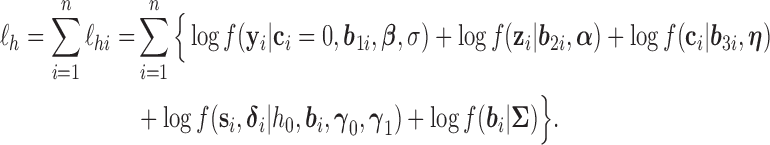 |
(3.6) |
Based on Ha and others (2003) and Molas and others (2013), we propose the following estimation procedure via the h-likelihood. Beginning with some starting values 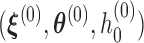 , we iterate the steps below:
, we iterate the steps below:
Step 1: At iteration
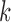 , given
, given 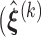 ,
, 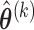 ,
, 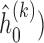 , obtain updated estimates of the random effects
, obtain updated estimates of the random effects 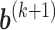 by maximizing
by maximizing 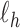 in (3.6) with respect to
in (3.6) with respect to 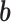 ;
;- Step 2: Given
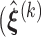 ,
, 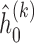 ,
, 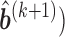 , obtain updated estimates of the mean parameters
, obtain updated estimates of the mean parameters 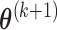 by maximizing the following adjusted profileh-likelihood as in Lee and Nelder (1996) with respect to
by maximizing the following adjusted profileh-likelihood as in Lee and Nelder (1996) with respect to 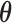 :
:
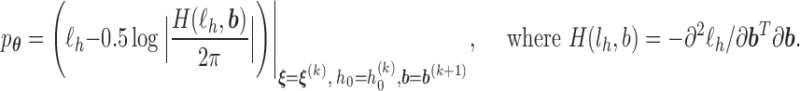
- Step 3: Given (
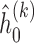 ,
, 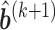 ,
, 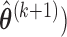 , obtain updated estimates of the variance-covariance
, obtain updated estimates of the variance-covariance 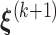 by maximizing the following adjusted profile h-likelihood,
by maximizing the following adjusted profile h-likelihood,
where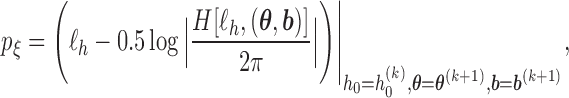
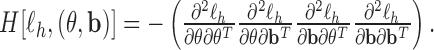
- Step 4: Given
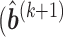 ,
, 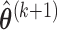 ,
, 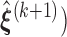 , obtain an updated nonparametric estimate of the baseline hazard
, obtain an updated nonparametric estimate of the baseline hazard 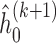 as follows
as follows
where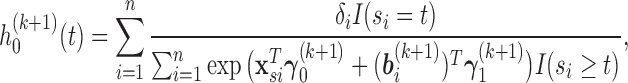
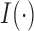 is an indicator function.
is an indicator function.
By iterating the above four steps until convergence, we can obtain approximate MLEs for the mean parameters, approximate REMLs for the variance–covariance parameters, empirical Bayes estimates of the random effects, and a nonparametric estimate of the baseline hazard function. To set starting values, we may first fit the models separately and then choose the resulting parameter estimates as the starting values for  . More details are described in Section 4.3. The standard errors of the parameter estimates can be obtained based on
. More details are described in Section 4.3. The standard errors of the parameter estimates can be obtained based on
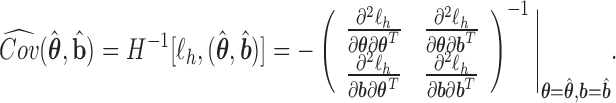 |
That is, the estimated variances of 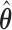 can be chosen to be the diagonal elements of the top left corner of the matrix
can be chosen to be the diagonal elements of the top left corner of the matrix 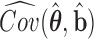 (Lee and Nelder, 1996; Ha and others, 2003).
(Lee and Nelder, 1996; Ha and others, 2003).
As mentioned in Section 1, the standard errors of parameter estimates may be under-estimated when the baseline hazard 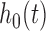 is unspecified (Rizopoulos, 2012a; Hsieh and others, 2006). A bootstrap method for obtaining standard errors is a good choice, but it is computationally intensive. Thus, here we propose a new approach to estimate the standard errors of parameter estimates based on the adaptive Gauss–Hermite (aGH) method (Rizopoulos, 2012a; Hartzel and others, 2001; Pinheiro and Bates, 1995). The basic idea is as follows. After convergence of the above steps, we can approximate the score function of
is unspecified (Rizopoulos, 2012a; Hsieh and others, 2006). A bootstrap method for obtaining standard errors is a good choice, but it is computationally intensive. Thus, here we propose a new approach to estimate the standard errors of parameter estimates based on the adaptive Gauss–Hermite (aGH) method (Rizopoulos, 2012a; Hartzel and others, 2001; Pinheiro and Bates, 1995). The basic idea is as follows. After convergence of the above steps, we can approximate the score function of 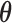 for subject
for subject 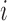 by the following:
by the following:
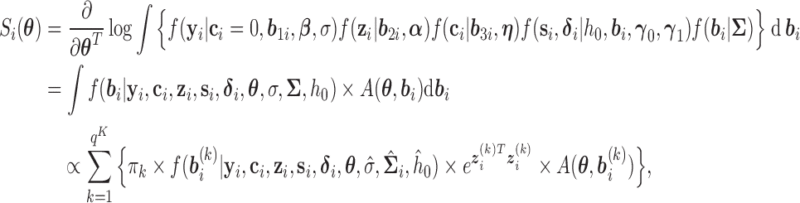 |
where 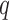 is the dimension of the random effects,
is the dimension of the random effects, 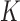 is the number of quadrature points for each random effect,
is the number of quadrature points for each random effect, 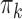 are weights for the original GH nodes
are weights for the original GH nodes 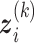 ,
, 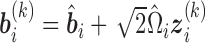 with
with 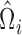 being the upper triangular factor of the Cholesky decomposition of
being the upper triangular factor of the Cholesky decomposition of 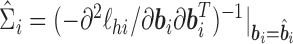 , and
, and 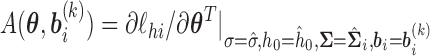 . Then, the standard errors of the parameter estimates can be estimated based on
. Then, the standard errors of the parameter estimates can be estimated based on 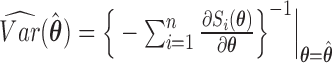 . In practice, we can calculate
. In practice, we can calculate 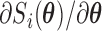 numerically using the central difference approximation (Rizopoulos, 2012a).
numerically using the central difference approximation (Rizopoulos, 2012a).
4. Data analysis
4.1. HIV vaccine data and new time variables
In this section, we analyze the VAX004 data set described in Section 2, based on the proposed models and methods. Our objective is to check if individual-specific longitudinal characteristics of immune responses are associated with the risk of HIV infection. A comprehensive analysis may be infeasible due to space limitation, but we will focus on the essential features of the data. Since the immune response variables are mostly highly correlated, we choose two variables, “MNGNE8” and “NAb”, which may represent the key features of the longitudinal immune response data. Note that some variables are often conveniently converted to binary data for simpler clinical interpretations. Here, we let 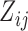 be the dichotomized MNGNE8 data such that
be the dichotomized MNGNE8 data such that 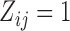 if the MNGNE8 value of individual
if the MNGNE8 value of individual 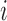 at time
at time 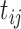 is larger than the sample median 0.57 and
is larger than the sample median 0.57 and 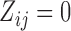 otherwise. Let
otherwise. Let 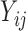 be the original NAb value of individual
be the original NAb value of individual 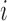 at time
at time 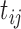 . Recall that 27% of the original NAb values are below this variable’s LLOQ (i.e. left truncated).
. Recall that 27% of the original NAb values are below this variable’s LLOQ (i.e. left truncated).
A unique feature of vaccine trial data is that the longitudinal immune response data typically exhibit periodic patterns, due to repeated administration of the vaccine. This can be clearly seen in Figure 1. Statistical modeling must incorporate these features. Here we use a simple periodic function 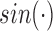 to empirically capture the periodic patterns and further define the following time variables (in months): (i) the time from the beginning of the study to the current scheduled measurement time, denoted by
to empirically capture the periodic patterns and further define the following time variables (in months): (i) the time from the beginning of the study to the current scheduled measurement time, denoted by 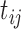 ; (ii) the time from the most recent immunization to the current scheduled measurement time, denoted by
; (ii) the time from the most recent immunization to the current scheduled measurement time, denoted by 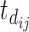 (so
(so 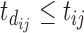 ); and (iii) the time between two consecutive vaccine administrations, denoted by
); and (iii) the time between two consecutive vaccine administrations, denoted by 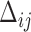 , so there will be at least one
, so there will be at least one 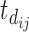 and one
and one 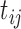 between
between 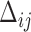 and
and 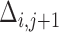 . For measurement time
. For measurement time 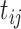 scheduled after the final vaccination, we define
scheduled after the final vaccination, we define 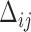 as the time between the final vaccination and the final measurement time. These different time variables are needed in modeling the longitudinal trajectories. Figure 3 gives an example of how different time variables are defined for a randomly chosen participant
as the time between the final vaccination and the final measurement time. These different time variables are needed in modeling the longitudinal trajectories. Figure 3 gives an example of how different time variables are defined for a randomly chosen participant 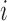 . Recall that vaccinations are scheduled at months 0, 1, 6, 12, 18, 24, 30, and the study ends at month 36. For this participant
. Recall that vaccinations are scheduled at months 0, 1, 6, 12, 18, 24, 30, and the study ends at month 36. For this participant 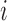 , s/he receives the first four vaccinations, but then drops out from the study before receiving the fifth vaccination. There are eight measurements over time in total, denoted by the cross symbols, where the measurement times may be different from the vaccination times. Suppose that the sixth measurement is taken at month 9, i.e.
, s/he receives the first four vaccinations, but then drops out from the study before receiving the fifth vaccination. There are eight measurements over time in total, denoted by the cross symbols, where the measurement times may be different from the vaccination times. Suppose that the sixth measurement is taken at month 9, i.e. 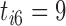 , then we have
, then we have 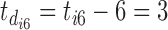 , the difference between the sixth measurement time and the latest vaccination time (Vac 3 at month 6) for this participant, and
, the difference between the sixth measurement time and the latest vaccination time (Vac 3 at month 6) for this participant, and 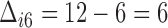 , since the sixth measurement happens between the third vaccination (Vac 3 at month 6) and the fourth vaccination (Vac 4 at month 12). To avoid very large or small parameter estimates, we also re-scale the times as follows:
, since the sixth measurement happens between the third vaccination (Vac 3 at month 6) and the fourth vaccination (Vac 4 at month 12). To avoid very large or small parameter estimates, we also re-scale the times as follows: 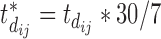 (in weeks) and
(in weeks) and 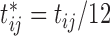 (in years).
(in years).
Fig. 3.

Illustration of three time variables in VAX004. The cross symbols indicate the measurement times of subject  . The dashed vertical lines show the scheduled times of vaccinations and the end of study (i.e. month 0, 1, 6, 12, 18, 24, 30, 36), where the black dashed lines represent the times when subject
. The dashed vertical lines show the scheduled times of vaccinations and the end of study (i.e. month 0, 1, 6, 12, 18, 24, 30, 36), where the black dashed lines represent the times when subject  received vaccines and the gray dashed lines represent the times when subject
received vaccines and the gray dashed lines represent the times when subject 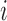 missed the scheduled vaccinations. The arrow lines represent the time periods
missed the scheduled vaccinations. The arrow lines represent the time periods 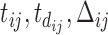 of the sixth and eighth measurements with
of the sixth and eighth measurements with 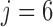 and
and 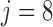 , respectively.
, respectively.
4.2. Models
Based on rationales discussed in Sections 3 and 4.1, we consider empirical models for the continuous and binary longitudinal data and survival model. The longitudinal models are selected based on AIC values (see details in Section 3 in the Supplementary material available at Biostatistics online). For the NAb data with 27% truncation, we model the untruncated data by using the LME model
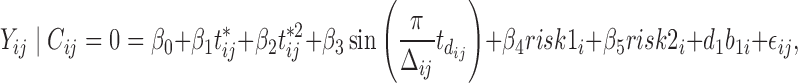 |
(4.1) |
where 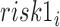 and
and 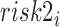 are categories 1 and 2 of baseline behavioral risk score, the random effects
are categories 1 and 2 of baseline behavioral risk score, the random effects 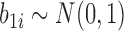 ,
, 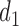 is the variance parameter,
is the variance parameter, 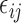 follows a truncated normal distribution with mean 0 and variance
follows a truncated normal distribution with mean 0 and variance 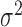 , and
, and 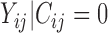 follows a truncated normal distribution. To ensure identifiability of the models, we assume that
follows a truncated normal distribution. To ensure identifiability of the models, we assume that 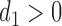 . We only consider a random intercept in the model because adding more random effects does not substantially reduce AIC values while making the models more complicated.
. We only consider a random intercept in the model because adding more random effects does not substantially reduce AIC values while making the models more complicated.
We also model the truncation indicator, 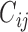 , of NAb to find possible associations of truncation with the time variables and other covariates and to predict the truncation status of NAb at times when NAb values are unavailable. The selected model is given as below,
, of NAb to find possible associations of truncation with the time variables and other covariates and to predict the truncation status of NAb at times when NAb values are unavailable. The selected model is given as below,
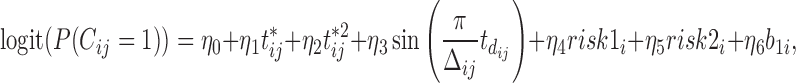 |
(4.2) |
which shares the same random effect as the NAb model (4.1), since these two processes seem to be highly correlated with each other. In many studies, the 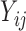 and
and 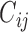 values are measured sparsely and we can use model (4.2) to predict the truncation status of
values are measured sparsely and we can use model (4.2) to predict the truncation status of 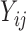 at times when Y-measurements are unavailable.
at times when Y-measurements are unavailable.
For the binary MNGNE8 data, variable selections by AIC values lead to the model
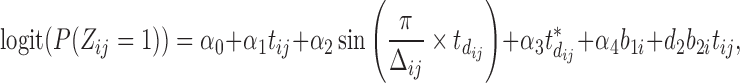 |
(4.3) |
where 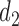 is the variance parameter with
is the variance parameter with 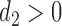 and the individual characteristics are incorporated via random slope
and the individual characteristics are incorporated via random slope 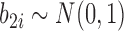 and random intercept
and random intercept 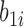 shared by models (4.1)–(4.2).
shared by models (4.1)–(4.2).
The association among the longitudinal models is incorporated through shared and correlated random effects from different models: 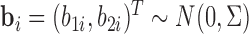 , with
, with 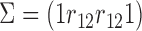 and
and 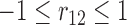 . Note that the random effect
. Note that the random effect 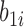 is shared by all the longitudinal models, since all the immune response longitudinal data exhibit similar individual-specific patterns and the random effect
is shared by all the longitudinal models, since all the immune response longitudinal data exhibit similar individual-specific patterns and the random effect 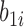 for the continuous NAb data best summarizes these patterns. For example, when a participant has a high baseline measurement of NAb, s/he likely also has a high baseline value of MNGNE8 and a low baseline probability that NAb is left truncated.
for the continuous NAb data best summarizes these patterns. For example, when a participant has a high baseline measurement of NAb, s/he likely also has a high baseline value of MNGNE8 and a low baseline probability that NAb is left truncated.
The survival model for the time to HIV infection is given by the “shared-parameter” model
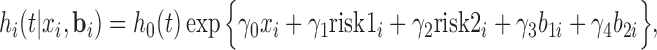 |
(4.4) |
where 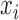 is the measurement of GNE8_CD4 (i.e. blocking of the binding of the GNE8 HIV-1 gp120 Env protein to soluble CD4) for individual
is the measurement of GNE8_CD4 (i.e. blocking of the binding of the GNE8 HIV-1 gp120 Env protein to soluble CD4) for individual 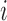 on the first day of the study after the first immunization, rescaled to have a mean of 0 and a standard deviation of 1. We call
on the first day of the study after the first immunization, rescaled to have a mean of 0 and a standard deviation of 1. We call 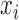 the standardized baseline GNE8_CD4. Since the analysis in this section is exploratory in nature, for simplicity we ignore other covariates.
the standardized baseline GNE8_CD4. Since the analysis in this section is exploratory in nature, for simplicity we ignore other covariates.
4.3. Parameter estimates, model diagnostics, and new findings
We estimate model parameters using the proposed h-likelihood method. As a comparison, we also use the two-step method, which fits each longitudinal model separately and obtains random effect estimates in the first step and then in the second step the random effects in the Cox model are simply substituted by their estimates from the first step. The results of the two-step method are obtained using the R packages lme4 and survival. The drawbacks of the two-step method are: (i) it may under-estimate the standard errors of the parameter estimates in the survival model, since it fails to incorporate the estimation uncertainty in the first step; (ii) it fails to incorporate the associations among the longitudinal variables; and (iii) it may lead to biased estimates of longitudinal model parameters when longitudinal data are terminated by event times and/or truncated longitudinal data are simply replaced by the LLOQ or half this limit (Wu, 2009). Table 1 summarizes estimation results based on the above two methods. Algorithms based on the h-likelihood method were terminated when the relative change became less than 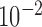 in the estimates or
in the estimates or 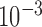 in the approximated log-likelihood. Since our main objective is to investigate if individual-specific characteristics of the longitudinal immune responses are associated with the risk of HIV infection, we mainly focus on
in the approximated log-likelihood. Since our main objective is to investigate if individual-specific characteristics of the longitudinal immune responses are associated with the risk of HIV infection, we mainly focus on 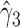 and
and 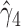 in the survival model (4.4) as these parameters link the random effects to the hazard of HIV infection.
in the survival model (4.4) as these parameters link the random effects to the hazard of HIV infection.
Table 1.
Estimates of all model parameters in VAX004
| Model | Par | Two-step method | H-likelihood method | ||||
|---|---|---|---|---|---|---|---|
| Est | SE | p-value | Est | SE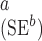
|
p-value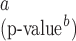
|
||
| Estimates of mean parameters | |||||||
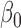
|
1.57 | 0.05 |
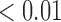
|
2.35 |
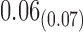
|
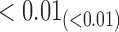
|
|
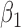
|
1.89 | 0.05 |
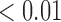
|
0.95 |
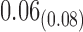
|
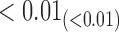
|
|
| LME model (4.1) |
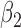
|
 0.54 0.54 |
0.02 |
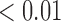
|
 0.31 0.31 |
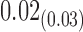
|
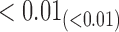
|
| for NAb |
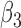
|
0.55 | 0.05 |
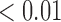
|
1.46 |
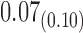
|
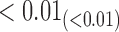
|
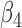
|
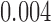
|
0.04 | 0.93 |
 0.04 0.04 |
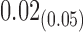
|
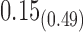
|
|
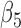
|
 0.27 0.27 |
0.12 | 0.03 |
 0.10 0.10 |
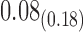
|
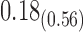
|
|
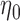
|
2.09 | 0.17 |
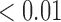
|
1.94 |
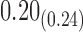
|
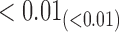
|
|
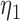
|
 6.52 6.52 |
0.31 |
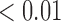
|
 6.27 6.27 |
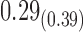
|
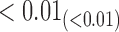
|
|
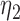
|
1.71 | 0.11 |
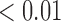
|
1.65 |
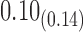
|
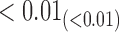
|
|
| Truncation model (4.2) |
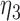
|
 0.64 0.64 |
0.22 |
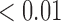
|
 0.61 0.61 |
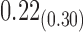
|
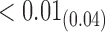
|
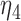
|
 0.09 0.09 |
0.14 | 0.50 |
 0.03 0.03 |
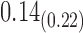
|
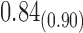
|
|
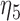
|
1.15 | 0.38 |
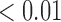
|
0.96 |
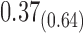
|
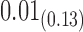
|
|
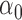
|
 1.60 1.60 |
0.10 |
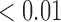
|
 1.68 1.68 |
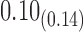
|
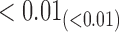
|
|
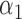
|
0.11 | 0.01 |
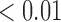
|
0.14 |
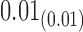
|
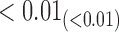
|
|
| GLMM (4.3) |
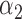
|
1.76 | 0.19 |
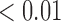
|
1.82 |
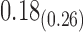
|
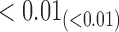
|
| for MNGNE8 |
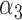
|
 0.05 0.05 |
0.01 |
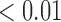
|
 0.05 0.05 |
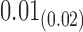
|
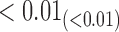
|
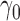
|
 0.72 0.72 |
0.23 |
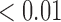
|
 0.79 0.79 |
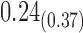
|
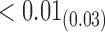
|
|
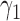
|
0.70 | 0.56 | 0.21 |
 0.28 0.28 |
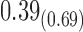
|
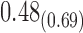
|
|
| Survival model (4.4) |
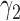
|
1.46 | 1.14 | 0.20 | 1.86 |
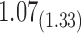
|
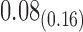
|
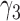
|
 0.01 0.01 |
0.24 | 0.96 |
 1.69 1.69 |
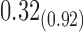
|
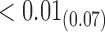
|
|
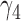
|
0.13 | 0.23 | 0.58 | 2.37 |
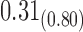
|
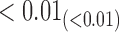
|
|
| Estimates of variance-covariance parameters | |||||||
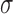
|
0.66 | 0.48 | |||||
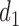
|
0.22 | 0.54 | |||||
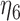
|
 0.89 0.89 |
 1.60 1.60 |
|||||
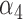
|
0.43 | 0.004 | |||||
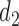
|
0.05 | 0.19 | |||||
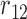
|
0.37 | 0.76 | |||||
SE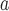 and p-value
and p-value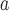 : Standard error and p-value based on the h-likelihood method.
: Standard error and p-value based on the h-likelihood method.
SE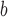 and p-value
and p-value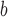 : Standard error and p-value based on the newly proposed method with 4 quadrature points.
: Standard error and p-value based on the newly proposed method with 4 quadrature points.
From Table 1, we see that the two methods lead to quite different results, especially the estimates of 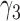 and
and 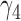 in the survival model that are our main focus in this analysis. For the two-step method, the estimates of
in the survival model that are our main focus in this analysis. For the two-step method, the estimates of 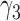 and
and 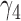 are near zero with confidence intervals including zero, not supporting that individual-specific immune response longitudinal trajectories are associated with the risk of HIV infection. However, these parameter estimates based on the proposed joint model with the h-likelihood method lead to different conclusions. Both
are near zero with confidence intervals including zero, not supporting that individual-specific immune response longitudinal trajectories are associated with the risk of HIV infection. However, these parameter estimates based on the proposed joint model with the h-likelihood method lead to different conclusions. Both 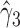 and
and 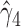 are highly significant based on the standard errors estimated by the joint model with the h-likelihood method (denoted as SE
are highly significant based on the standard errors estimated by the joint model with the h-likelihood method (denoted as SE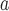 ), suggesting that individual-specific immune response longitudinal trajectories are highly associated with the risk of HIV infection. Since the standard errors based on the h-likelihood method may be under-estimated (Hsieh and others, 2006; Rizopoulos, 2012b), as discussed earlier, we also calculate the standard errors using the proposed method based on the aGH method, and the results with four quadrature points are given as SE
), suggesting that individual-specific immune response longitudinal trajectories are highly associated with the risk of HIV infection. Since the standard errors based on the h-likelihood method may be under-estimated (Hsieh and others, 2006; Rizopoulos, 2012b), as discussed earlier, we also calculate the standard errors using the proposed method based on the aGH method, and the results with four quadrature points are given as SE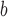 in the table. We see that, based on the new standard errors, the p-value for testing
in the table. We see that, based on the new standard errors, the p-value for testing 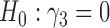 is slightly larger than
is slightly larger than 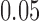 while that for testing
while that for testing 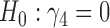 is still highly significant. Therefore, we may conclude that individual-specific immune response longitudinal trajectories are associated with the risk of HIV infection. This conclusion is unavailable based on the two-step method.
is still highly significant. Therefore, we may conclude that individual-specific immune response longitudinal trajectories are associated with the risk of HIV infection. This conclusion is unavailable based on the two-step method.
The negative estimate of 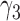 suggests that higher NAb values are associated with a lower risk of HIV infection, and the positive estimate of
suggests that higher NAb values are associated with a lower risk of HIV infection, and the positive estimate of 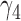 suggests that large increases in MNGNE8 over time are associated with a higher risk of HIV infection. Specifically, there is an estimated 81.6% decrease (i.e.
suggests that large increases in MNGNE8 over time are associated with a higher risk of HIV infection. Specifically, there is an estimated 81.6% decrease (i.e. 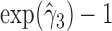 ) in the hazard/risk with a one unit increase in the individual effect
) in the hazard/risk with a one unit increase in the individual effect 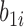 and an estimated 10.6 times increase (i.e.
and an estimated 10.6 times increase (i.e. 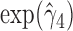 ) in the hazard/risk with a one unit increase in the individual-specific slope
) in the hazard/risk with a one unit increase in the individual-specific slope 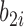 , holding other covariates constant. These findings are original, since they are unavailable based on the two-step method, and show the important contribution of the proposed joint model and the h-likelihood method.
, holding other covariates constant. These findings are original, since they are unavailable based on the two-step method, and show the important contribution of the proposed joint model and the h-likelihood method.
The joint model method and the two-step method have consistent significances of the parameters in the longitudinal models (4.1)–(4.3), except for 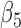 and
and 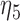 . By the two-step method, the tests for
. By the two-step method, the tests for 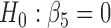 and for
and for 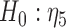 yield significant p-values, suggesting that participants with baseline behavioral risk score in category 2 (i.e. risk2 = 1) have significantly lower NAb values than other participants. By the joint model method, on the other hand, such a negative association is not statistically significant. For model (4.1), the mean square error (MSE) based on the joint model is 0.296, while the MSE based on the two-step method is 0.403.
yield significant p-values, suggesting that participants with baseline behavioral risk score in category 2 (i.e. risk2 = 1) have significantly lower NAb values than other participants. By the joint model method, on the other hand, such a negative association is not statistically significant. For model (4.1), the mean square error (MSE) based on the joint model is 0.296, while the MSE based on the two-step method is 0.403.
The model diagnostics are conducted to check the assumptions and goodness-of-fit of the models. The results are listed in Section 4 in the Supplementary material available at Biostatistics online. Overall, the assumptions hold and the models fit the data well. The data used in this example may be requested through a concept proposal to the owner of the data—Global Solutions in Infectious Diseases.
5. Simulation studies
In this section, we conduct three simulation studies to evaluate the proposed joint model with the h-likelihood method. The models and their true parameter values in the simulation studies are chosen to be similar to the estimated values in the models for real data in the previous section.
5.1. Simulation study 
Conditional on the random effects, the binary data 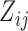 are generated from a Bernoulli distribution with probabilities
are generated from a Bernoulli distribution with probabilities 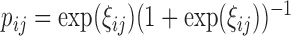 , where
, where 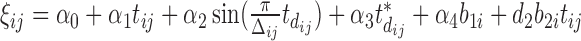 . For the continuous data
. For the continuous data 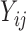 , we first randomly generate
, we first randomly generate 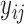 from a normal distribution
from a normal distribution 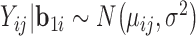 with
with 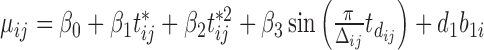 . Then we create truncations so that
. Then we create truncations so that 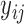 is observed if
is observed if 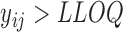 and truncated otherwise and we choose LLOQ = 2. The random effects
and truncated otherwise and we choose LLOQ = 2. The random effects 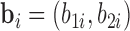 are generated from a multivariate normal distribution
are generated from a multivariate normal distribution 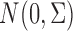 . The true values of the parameters are set to be:
. The true values of the parameters are set to be: 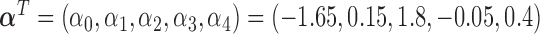 ,
, 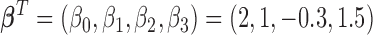 ,
, 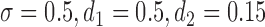 ,
, 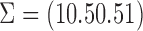 . The survival times
. The survival times 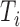 are generated from a Weibull distribution with shape parameter of
are generated from a Weibull distribution with shape parameter of 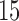 and scale parameter of
and scale parameter of 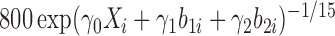 , where
, where 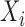 is a baseline covariate generated from the standard normal distribution. The non-informative censoring times
is a baseline covariate generated from the standard normal distribution. The non-informative censoring times 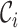 are generated from a Weibull distribution with shape parameter of 5 and scale parameter of
are generated from a Weibull distribution with shape parameter of 5 and scale parameter of 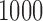 . The true parameter values are given as
. The true parameter values are given as 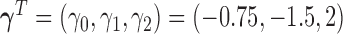 .
.
5.2. Simulation studies 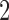 and
and 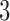
To better evaluate the performance of the h-likelihood method, we conducted two additional simulation studies: (i) a joint model with higher dimensions of random effects (Study 2, four random effects); and (ii) a joint model with a parametric survival model (Study 3, a Weibull survival model). For the parametric joint model in Study 3, we also estimate the model parameters using the aGH method as comparison. Due to space limitation, we put the details of these two simulation studies in Sections 5 and 6 in the Supplementary material available at Biostatistics online.
5.3. Simulation results and discussions
We compare the performance of the methods based on the relative bias and MSE of the parameter estimates, which are defined as follows (say, for parameter 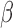 ):
):
relative bias (%) of
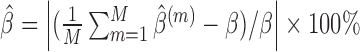 ,
,relative MSE (%) of
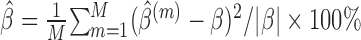 ,
,
where 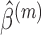 is the estimate of
is the estimate of 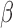 in simulation iteration
in simulation iteration 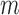 ,
, 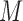 is the total number of repetitions, and
is the total number of repetitions, and 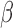 is the true parameter value.
is the true parameter value.
Table 2 summarizes the results of Simulation Study 1 (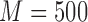 ) when the longitudinal measurements are collected bi-weekly. The proposed h-likelihood method outperforms the two-step method as it returns much less biased estimates for most of the parameters. As for the bias in
) when the longitudinal measurements are collected bi-weekly. The proposed h-likelihood method outperforms the two-step method as it returns much less biased estimates for most of the parameters. As for the bias in 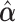 , it is known that the h-likelihood method may perform less satisfactorily for logistic mixed effects models (Kuk and Cheng, 1999; Waddington and Thompson, 2004). The standard errors of
, it is known that the h-likelihood method may perform less satisfactorily for logistic mixed effects models (Kuk and Cheng, 1999; Waddington and Thompson, 2004). The standard errors of 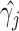 ’s seem to be underestimated by the h-likelihood method. This problem has been reported elsewhere (Hsieh and others, 2006; Rizopoulos, 2012b). However, our newly proposed method based on the aGH method with 4 quadrature points returns coverage probabilities much closer to the nominal coverage probabilities. The results of Simulation Studies 2 and 3 are given in Tables S5 and S6 in the Supplementary material available at Biostatistics online. The main conclusions are consistent with those from Table 2. In Simulation Study 3, the
’s seem to be underestimated by the h-likelihood method. This problem has been reported elsewhere (Hsieh and others, 2006; Rizopoulos, 2012b). However, our newly proposed method based on the aGH method with 4 quadrature points returns coverage probabilities much closer to the nominal coverage probabilities. The results of Simulation Studies 2 and 3 are given in Tables S5 and S6 in the Supplementary material available at Biostatistics online. The main conclusions are consistent with those from Table 2. In Simulation Study 3, the 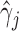 ’s based on the h-likelihood method are much less biased, though with slightly larger MSEs, than those based on the aGH method, while for
’s based on the h-likelihood method are much less biased, though with slightly larger MSEs, than those based on the aGH method, while for 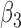 ,
, 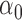 , and
, and 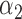 , the aGH method has less biased estimates than the h-likelihood method (see Table S6 in the Supplementary material available at Biostatistics online). Synthesizing the simulation results, we conclude that the proposed h-likelihood method, with the new approach of estimating the standard errors, performs reasonably well. Its performance remains consistent with higher dimensions of random effects and parametric survival models. Although it is sometimes less accurate than the aGH method, it is computationally much more efficient.
, the aGH method has less biased estimates than the h-likelihood method (see Table S6 in the Supplementary material available at Biostatistics online). Synthesizing the simulation results, we conclude that the proposed h-likelihood method, with the new approach of estimating the standard errors, performs reasonably well. Its performance remains consistent with higher dimensions of random effects and parametric survival models. Although it is sometimes less accurate than the aGH method, it is computationally much more efficient.
Table 2.
Simulation results with bi-weekly longitudinal measurements based on the two-step (TS) method and the h-likelihood (HL) method
| Model | Par | True | Estimate | SSE | rBias (%) | rMSE (%) | Coverage probability (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| value | TS | HL | TS | HL | TS | HL | TS | HL | TS | HL
|
HL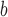
|
||
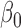
|
2.00 | 2.14 | 2.01 | 0.04 | 0.06 | 7.05 | 0.55 | 1.07 | 0.16 | 15 | 94 | 98 | |
| (4.1) |
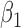
|
1.00 | 0.88 | 1.00 | 0.04 | 0.04 | 11.55 | 0.20 | 1.47 | 0.20 | 13 | 94 | 99 |
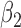
|
 0.30 0.30 |
 0.26 0.26 |
 0.30 0.30 |
0.02 | 0.02 | 13.07 | 0.00 | 0.60 | 0.12 | 32 | 94 | 99 | |
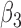
|
1.50 | 1.41 | 1.49 | 0.02 | 0.02 | 5.70 | 0.37 | 0.52 | 0.04 | 1 | 94 | 98 | |
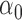
|
 1.65 1.65 |
 1.64 1.64 |
 1.64 1.64 |
0.10 | 0.10 | 0.57 | 0.59 | 0.62 | 0.63 | 94 | 95 | 99 | |
| (4.3) |
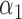
|
0.15 | 0.15 | 0.15 | 0.02 | 0.02 | 2.02 | 2.45 | 0.18 | 0.17 | 94 | 89 | 96 |
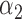
|
1.80 | 1.80 | 1.78 | 0.11 | 0.10 | 0.26 | 0.84 | 0.61 | 0.61 | 96 | 95 | 99 | |
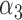
|
 0.05 0.05 |
 0.05 0.05 |
 0.05 0.05 |
0.01 | 0.01 | 1.80 | 2.68 | 0.06 | 0.06 | 93 | 93 | 97 | |
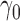
|
 0.75 0.75 |
 0.68 0.68 |
 0.74 0.74 |
0.16 | 0.17 | 9.44 | 0.74 | 4.00 | 3.76 | 88 | 85 | 97 | |
| (4.4) |
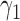
|
 1.50 1.50 |
 1.10 1.10 |
 1.44 1.44 |
0.22 | 0.29 | 26.91 | 3.99 | 13.96 | 5.92 | 38 | 65 | 84 |
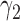
|
2.00 | 1.53 | 2.02 | 0.23 | 0.31 | 23.40 | 1.10 | 13.70 | 4.88 | 34 | 69 | 87 | |
HL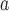 : Coverage probability based on the h-likelihood method.
: Coverage probability based on the h-likelihood method.
HL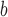 : Coverage probability based on the newly proposed method for standard errors with 4 quadrature points.
: Coverage probability based on the newly proposed method for standard errors with 4 quadrature points.
6. Discussion
In this article, we have considered a joint model for mixed types of longitudinal data with left truncation and a survival model and proposed a new method to handle the left-truncation in longitudinal data. A main advantage of this method, compared with existing methods in the literature (e.g. Hughes, 1999), is that it does not make any untestable distributional assumption for the truncated data that are below a measurement instruments LLOQ. Different types of longitudinal data are assumed to be associated via shared and correlated random effects. We have also proposed an h-likelihood method for approximate joint likelihood inference, which is computationally much more efficient than the aGH method. Moreover, we have proposed a new method to better estimate the standard errors of parameter estimates from the h-likelihood method. Based on a MacBook Pro Version 10.11.4, the average computing times of the h-likelihood method were 2.7 min for the semiparametric joint model with 2 random effects and 21.9 min for the semiparametric joint model with 4 random effects, respectively. For the parametric joint model with 2 random effects, the average running time of the h-likelihood method was 9.1 min, much faster than the aGH method that takes 28.4 min.
Analysis of the real HIV vaccine data based on the proposed method shows that the individual-specific characteristics of longitudinal immune response, summarized by random effects in the models, are highly associated with the risk of HIV infection. This finding is quite interesting and helpful to designing future HIV vaccine studies. We have also proposed a model for the left-truncation indicator of the longitudinal immune response data and showed that the left-truncation status follows certain patterns as functions of time. Such a model can be used to predict the left-truncation status (below LLOQ status) of some longitudinal immune response values when measurement schedules are infrequent or sparse.
The joint model in this article may be extended in several directions. For example, the Cox model may be replaced by an accelerated failure time model or survival model for interval censored data or competing risks data. The association among different types of longitudinal processes may also be modeled in other ways such as shared latent processes. In addition, the dropouts in the real data may be associated with longitudinal patterns, so we may consider incorporating missing data mechanisms into the joint models in future research. Research for these extensions will be reported separately.
7. Software
Software in the form of R code and a sample input data set are available at https://github.com/oliviayu/HHJMs.
Supplementary material
Supplementary material is available at http://biostatistics.oxfordjournals.org.
Supplementary Material
Acknowledgments
The authors thank the reviewers for the thoughtful comments to help improve the article greatly. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or BMGF. The authors thank the participants, investigators, and sponsors of the VAX004 trial, including Global Solutions for Infectious Diseases. Conflict of Interest: None declared.
Funding
National Institute Of Allergy And Infectious Diseases of the National Institutes of Health (NIH) (Award Numbers R37AI054165 and UM1AI068635); and Bill and Melinda Gates Foundation (BMGF) (Award Number OPP1110049).
References
- Barrett J., Diggle P., Henderson R. and Taylor-Robinson D. (2015). Joint modelling of repeated measurements and time-to-event outcomes: flexible model specification and exact likelihood inference. Journal of the Royal Statistical Society: Series B 77, 131–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt P. W., Wang H. J., and Zhang D. (2014). Flexible modeling of survival data with covariates subject to detection limits via multiple imputation. Computational Statistics and Data Analysis, 69, 81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., May R. C., Ibrahim J. G., Chu H., and Cole S. R. (2014). Joint modeling of longitudinal and survival data with missing and left-censored time-varying covariates. Statistics in Medicine, 33, 4560–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elashoff R. M., Li G., and Li N. (2015). Joint Modeling of Longitudinal and Time-to-Event Data. Boca Raton, FL: Chapman & Hall/CRC. [Google Scholar]
-
Flynn N., Forthal D., Harro C., Judson F., Mayer K., Para M., and Gilbert P..
The rgp120
 Vaccine Study Group (2005). Placebo-controlled phase 3 trial of recombinant glycoprotein 120 vaccine to prevent HIV-1 infection.Journal of Infectious Diseases, 191, 654–65. [DOI] [PubMed] [Google Scholar]
Vaccine Study Group (2005). Placebo-controlled phase 3 trial of recombinant glycoprotein 120 vaccine to prevent HIV-1 infection.Journal of Infectious Diseases, 191, 654–65. [DOI] [PubMed] [Google Scholar] - Fu R. and Gilbert P. B. (2017). Joint modeling of longitudinal and survival data with the cox model and two-phase sampling. Lifetime Data Analysis, 23, 136–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert P. B., Peterson M. L., Follmann D., Hudgens M. G., Francis D. P., Gurwith M., Heyward W. L., Jobes D. V., Popovic V., Self S. G.,. et al. (2005). Correlation between immunologic responses to a recombinant glycoprotein 120 vaccine and incidence of hiv-1 infection in a phase 3 hiv-1 preventive vaccine trial. Journal of Infectious Diseases, 191, 666–677. [DOI] [PubMed] [Google Scholar]
- Ha I. D., Park T., and Lee Y. (2003). Joint modelling of repeated measures and survival time data. Biometrical Journal, 45, 647–658. [Google Scholar]
- Hartzel J., Agresti A., and Caffo B. (2001). Multinomial logit random effects models. Statistical Modelling, 1, 81–102. [Google Scholar]
- Hsieh F., Tseng Y.-K., and Wang J.-L. (2006). Joint modeling of survival and longitudinal data: likelihood approach revisited. Biometrics, 62, 1037–1043. [DOI] [PubMed] [Google Scholar]
- Hughes J. P. (1999). Mixed effects models with censored data with application to hiv rna levels. Biometrics, 55, 625–629. [DOI] [PubMed] [Google Scholar]
- Król A., Ferrer L., Pignon J.-P., Proust-Lima C., Ducreux M., Bouché O., Michiels S., and Rondeau V. (2016). Joint model for left-censored longitudinal data, recurrent events and terminal event: predictive abilities of tumor burden for cancer evolution with application to the ffcd 2000–05 trial. Biometrics, 72, 907–916. [DOI] [PubMed] [Google Scholar]
- Kuk A. Y. and Cheng Y. W. (1999). Pointwise and functional approximations in monte carlo maximum likelihood estimation. Statistics and Computing, 9, 91–99. [Google Scholar]
- Lawrence Gould A., Boye M. E., Crowther M. J., Ibrahim J. G., Quartey G., Micallef S., and Bois F. Y. (2015). Joint modeling of survival and longitudinal non-survival data: current methods and issues. report of the dia bayesian joint modeling working group. Statistics in Medicine, 34, 2181–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. and Nelder J. A. (1996). Hierarchical generalized linear models. Journal of the Royal Statistical Society: Series B, 58, 619–678. [Google Scholar]
- Lee Y., Nelder J. A., and Pawitan Y. (2006). Generalized Linear Models with Random Effects: Unified Analysis via H-likelihood. Boca Raton, FL: Chapman & Hall/CRC. [Google Scholar]
- Mehrotra K. G., Kulkarni P. M., Tripathi R. C., and Michalek J. E. (2000). Maximum likelihood estimation for longitudinal data with truncated observations. Statistics in Medicine, 19, 2975–2988. [DOI] [PubMed] [Google Scholar]
- Molas M., Noh M., Lee Y., and Lesaffre E. (2013). Joint hierarchical generalized linear models with multivariate gaussian random effects. Computational Statistics and Data Analysis, 68, 239–250. [Google Scholar]
- Pinheiro J. C. and Bates D. M. (1995). Approximations to the log-likelihood function in the nonlinear mixed-effects model. Journal of Computational and Graphical Statistics, 4, 12–35. [Google Scholar]
- Rizopoulos D. (2012a). Fast fitting of joint models for longitudinal and event time data using a pseudo-adaptive gaussian quadrature rule. Computational Statistics and Data Analysis, 56, 491–501. [Google Scholar]
- Rizopoulos D. (2012b). Joint Models for Longitudinal and Time-to-Event Data: With Applications in R. Boca Raton, FL: Chapman & Hall/CRC. [Google Scholar]
- Rizopoulos D., Verbeke G., and Lesaffre E. (2009). Fully exponential laplace approximations for the joint modelling of survival and longitudinal data. Journal of the Royal Statistical Society: Series B, 71, 637–654. [Google Scholar]
- Taylor J. M., Park Y., Ankerst D. P., Proust-Lima C., Williams S., Kestin L., Bae K., Pickles T., and Sandler H. (2013). Real-time individual predictions of prostate cancer recurrence using joint models. Biometrics, 69, 206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington D. and Thompson R. (2004). Using a correlated probit model approximation to estimate the variance for binary matched pairs. Statistics and Computing, 14, 83–90. [Google Scholar]
- Wu L. (2002). A joint model for nonlinear mixed-effects models with censoring and covariates measured with error, with application to aids studies. Journal of the American Statistical Association, 97, 955–964. [Google Scholar]
- Wu L. (2009). Mixed Effects Models for Complex Data. Boca Raton, FL: Chapman & Hall/CRC. [Google Scholar]
- Wulfsohn M. S. and Tsiatis A. A. (1997). A joint model for survival and longitudinal data measured with error. Biometrics, 53, 330–339. [PubMed] [Google Scholar]
- Zhu H., Ibrahim J. G., Chi Y.-Y., and Tang N. (2012). Bayesian influence measures for joint models for longitudinal and survival data. Biometrics, 68, 954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.