

Abstract
Hydrogen peroxide (H2O2) is a reactive oxygen species that serves as an important signaling molecule in normal brain function. At the same time, excessive H2O2 concentrations contribute to myriad pathological consequences resulting from oxidative stress. Studies to elucidate the diverse roles that H2O2 plays in complex biological environments have been hindered by the lack of robust methods for probing dynamic H2O2 fluctuations in living systems with molecular specificity. Background-subtracted fast-scan cyclic voltammetry (FSCV) at carbon-fiber microelectrodes provides a method of detecting rapid H2O2 fluctuations with high temporal and spatial resolution in brain tissue. However, H2O2 fluctuations can be masked by local changes in pH (ΔpH), because the voltammograms for these species can have significant peak overlap, hindering quantification. We present a method for removing ΔpH-related contributions from complex voltammetric data. By employing two distinct potential waveforms per scan, one in which H2O2 is electrochemically silent and a second in which both ΔpH and H2O2 are redox active, a clear distinction between H2O2 and ΔpH signals is established. A partial least squares regression (PLSR) model is used to predict the ΔpH signal and subtract it from the voltammetric data. The model has been validated both in vitro and in vivo using k-fold cross-validation. The data demonstrate that the double waveform PLSR model is a powerful tool that can be used to disambiguate and evaluate naturally occurring H2O2 fluctuations in vivo.
Keywords: principal component, predictive modeling, training set, carbon-fiber microelectrode, reactive oxygen species, fast-scan cyclic voltammetry
Graphical Abstract
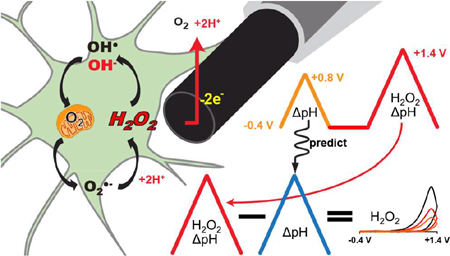
Introduction
Hydrogen peroxide (H2O2) is an important, membrane permeable, reactive oxygen species (ROS) present in the brain that is commonly regarded as a potential toxin, as it can react to form aggressive hydroxyl radicals that can irreversibly alter DNA, lipids, and protein structures.1 H2O2 also serves as an indicator of the upstream production of more detrimental ROS by mitochondria, where elevated concentrations are indicative of cellular malfunction. Pathological oxidative stress leads to neuronal degeneration that is symptomatic of several neurodegenerative disease states and aging.2–6 Importantly, H2O2 also serves as a subcellular signaling molecule in redox signaling pathways important to normal cell function, as well as a modulator of dopamine transmission in the dorsolateral striatum.7–9 Finally, the electrochemical detection of H2O2 is also of great interest because H2O2 can be exploited to serve as a reporter molecule in the indirect detection of non-electroactive species such as glucose, glutamate, and acetylcholine using oxidase based biosensors.10–14
H2O2 oxidation is electrocatalyzed at platinum, which is widely used to construct electrodes for these applications.15,16 However, recent work from our group has demonstrated the utility of fast-scan cyclic voltammetry (FSCV) coupled with carbon-fiber microelectrodes for the detection of H2O2 at a biosensor surface,17 as well as for the detection of endogenous H2O2 fluctuations in brain slice and in vivo.14,18 This approach offers both sensitivity and some selectivity, making it ideal for monitoring molecular fluctuations in vivo. By employing a waveform extended to +1.4 V (vs. Ag/AgCl), the carbon surface is oxidized, rendering it more sensitive to many analytes, including H2O217,19–21 However, endogenous H2O2 events are often accompanied by other chemical events that are detected by the electrode, including small shifts in pH, that render quantification of in vivo data difficult. Much work has gone into developing paradigms to improve selectivity in voltammetric experiments. These range from the incorporation of modified data collection strategies to the incorporation of statistical paradigms to deconvolute complex signals. For instance, electrodes can be coated with ion-selective polymers,22–24 a double electrochemical waveform can be employed,25 or the voltammetric sweep can be combined with a small potential step to help remove unwanted signal.26 Alternatively, voltammograms for individual chemical contributors to complex FSCV signals can be resolved using multivariate data analysis paradigms, such as principal component regression (PCR), elastic net regression,27 or multivariate curve resolution.28 One example that has been generally accepted by the field is PCR, a combination of principal component analysis with inverse least squares regression.29–37 PCR utilizes information collected across the entire potential window for a training set comprised of cyclic voltammograms (CVs) of known analytes (and concentrations) to determine principal components (PCs), or basis eigenvectors, that describe the variance in the training data.34,37 Then, concentration dynamics for these species can be predicted for unknown data by projecting the data onto the PCs to extrapolate the contribution of each analyte contained in the training set. This has been shown to work well for discriminating DA from other signals, including ΔpH, in data collected in vivo.27,29,36 Yet, PCR struggles when two analytical signals have similar sources of variance; i.e. have similar voltammetric features.34,35 Such is the case for H2O2 and ΔpH signals. The voltammetric signature for H2O2 consists of a single, well defined peak at ~1.3 V. By contrast, that for ΔpH exhibits current across the entire potential window, including a peak at ~1.3 V. To further confound the situation, the ΔpH signal varies widely across electrodes due to a dependence on the chemical composition of the recording environment and the surface state of the carbon, which contains many oxygen-containing functional groups.38,39 Thus, PCR is not always adequate to reliably distinguish current contributions from H2O2 and ΔpH, as these are not reliably defined in factor space.
Herein, we employ a double voltammetric waveform to more accurately quantify H2O2 fluctuations that occur at the electrode surface simultaneously with small shifts in pH. To accomplish this, a smaller waveform in which H2O2 is electrochemically silent is scanned immediately prior to the standard, larger waveform that collects current contributions from both H2O2 and ΔpH. PLSR is a logical and easy to understand variant of PCR that is commonly used in analytical chemistry due to readily available software and advantages in predictive power in some applications.40–45 It is used herein to reduce the dimensionality of calibration sets of CVs collected using both waveforms. Calibration sets collected with the small waveform (sWF) contain predictor variables to describe response variables that are evident in calibration sets collected with the large waveform (lWF). Thus, PLSR was used to exploit information on ΔpH collected with the small waveform and to predict and subtract the current response due to ΔpH from voltammograms collected with the larger waveform, without altering information related to H2O2 dynamics. The validity of the DW-PLSR was assessed using 5-fold cross validation and cumulative variance. Finally, the model was used to subtract ΔpH (and dopamine, DA) contributions from complex signals recorded in the dorsal striatum, clearly revealing endogenous and stimulated H2O2 and fluctuations. The model reliably discriminates between H2O2 and ΔpH signals both in vitro and in vivo. Thus, DW-PLSR promises to serve as a reliable tool to increase confidence in the quantification of endogenous H2O2 dynamics.
Experimental Section
Chemicals
All chemicals (≥95%, HPLC assay) were purchased from Sigma-Aldrich Co. (St. Louis, MO), unless otherwise stated, and used without additional processing. In vitro electrochemical experiments were performed in TRIS buffered saline (15 mM 2-amino 2-hydroxymethylpropane-1,3-diol, 3.25 mM KCl, 1.20 mM CaCl2, 1.2 mM MgCl2, 2 mM Na2SO4, 1.25 mM NaH2PO4, and 145 mM NaCl) adjusted to pH 7.4 with 1 M NaOH and 1 M HCl. All aqueous solutions were made using ultrapure 18.2 MΩ water.
Microelectrode Fabrication
Cylindrical carbon-fiber microelectrodes were fabricated using T-650 carbon fibers (Cytec Industries, Inc., Woodland Park, NJ) as previously described.46 In short, a single carbon fiber was aspirated into a glass capillary, a glass seal was created using a micropipette puller (Narishige, Tokyo, Japan), and the fiber extending past the seal was cut to 100 μm in length. An electrical connection to the carbon fiber was established with conductive silver epoxy and a wire lead. For experiments in freely moving-animals, electrodes were insulated in polyimide-coated, fused-silica tubing (Polymicro Technologies, Phoenix, AZ; 164.7 μm outer diameter/98.6 μm inner diameter), as previously described.47 Briefly, the tubing was cut to 4 cm in length and placed in an aqueous 70% isopropyl alcohol solution. A T-650 carbon fiber was inserted and allowed to dry. An epoxy seal was created at one end and an electrical connection to the fiber was completed via conductive silver epoxy. Electrodes were then cured at 100°C for 20 min. Finally, the exposed carbon fiber was cut to length.
Flow-Injection System
Working electrodes were positioned in a custom electrochemical flow cell using a micromanipulator (World Precision Instruments, Sarasota, FL). A syringe pump (New Era Pump Systems, Wantagh, NY) supplied a continuous buffer stream (1 mL min−1) across the working and reference electrodes (Ag/AgCl pellet, World Precision Instruments, Inc., Sarasota, FL). Two-second bolus injections of analyte were accomplished with a six-port HPLC valve mounted on a two-position actuator controlled by a digital pneumatic solenoid (Valco Instruments, Houston, TX). The entire apparatus was enclosed in a custom-built, grounded Faraday cage.
Data Acquisition
The double triangular waveform consisted of a small cyclic waveform ranging from −0.4 V to +0.8 V, and a larger cyclic waveform ranging from −0.4 to +1.4 V, applied using a scan rate of 400 V s−1 and separated by a 12 msec hold at −0.4 V (Figure 3A). The waveform was applied with an application frequency of 10 Hz and a sampling frequency of ~37 kHz, using a custom-built instrument for potential application and current transduction (Universal Electrochemistry Instrument, University of North Carolina at Chapel Hill, Department of Chemistry, Electronics Facility). This resulted in small and large CVs consisting of 223 and 334 data points, respectively. High Definition Cyclic Voltammetry software (HDCV, University of North Carolina at Chapel Hill) was used for waveform output, signal processing, and data analysis, in conjunction with data acquisition cards (National Instruments, Austin TX) used for measuring current and synchronizing the electrochemical cell with the flow-injection system. Analog filtering was accomplished with a 2-pole Sallen-Key, low-pass filter at 2 kHz. Data were then digitally filtered with a first-order low-pass Bessel filter; cutoff frequencies of 2 and 100 kHz in HDCV.
Figure 3:

A double waveform-partial least squares regression (DW-PLSR) model. A) Double waveform containing a small triangular waveform that scans from −0.4 to +0.8 V and back at 400 V s−1, and a larger one that extends to +1.4 V. B) Representative CVs collected with the small (left) and large (right) waveforms for H2O2 (top), basic ΔpH (middle), and known mixtures of these two analytes. Note that H2O2 (top) does not generate significant current using the sWF. C) Cross validation and cumulative percent variance. The number of components retained is plotted on the ordinate. Left: Cumulative variance for the sWF-predictor (black) and lWF-response (red) CVs. A dashed line indicates the position of 99.5% variance. Right: average mean-squared error (nA2/CV) for the sWF-predictor (gray) and lWF response (pink) CVs. 5-fold cross-validation was done by training the model without data from each electrode. D) ΔpH-subtracted CVs for a single electrode, using a five component PLSR model for H2O2 (top), ΔpH (middle), and known mixtures (bottom). Inset are coefficients of determination (n=15 CVs) for the H2O2 (B, top-right) and the ΔpH-subtracted CVs collected using the lWF.
Animal Experiments
Animal care and use was in accordance with North Carolina State University Institutional Animal Care and Use Committee (IACUC) guidelines. Male Sprague−Dawley rats (250−300 g) were purchased with or without a unilateral 6-hydroxydopamine (6-OHDA) lesion (Charles River Laboratories, Raleigh, NC), individually housed on a 12:12 hr light/dark cycle with free access to food and water, and allowed to acclimate to the facility for several days. On surgery day, animals designated for freely-moving experiments were anesthetized with 4% isoflurane during an induction period, then isoflurane was adjusted to 2.5% for electrode placement. A silica-insulated working electrode was placed in the dorsal striatum of the lesioned hemisphere (+1.2 mm AP, +2.0 mm ML, +5.0 mm DV relative to bregma) using a flat-skull stereotaxic apparatus (Kopf Instrumentation; Tujunga, CA), as previously described.46 A Ag/AgCl reference electrode was placed in the contralateral forebrain. Electrodes were permanently affixed with dental cement, and animals were allowed to recover for 4 weeks. On experiment day, the electrodes were connected to a head-mounted amplifier and electrical connections passed through a commutator that allowed free movement in an open field chamber.
Animals designated for acute experiments were deeply anesthetized with urethane (1.5 g kg−1), a heating pad (Harvard Apparatus, Holliston, MA) was used to maintain body temperature at 37°C, and electrodes were implanted as described above with the following exceptions: the carbon-fiber working electrode positioned in the dorsal striatum was insulated with glass, and a bipolar stimulation electrode was implanted in the medial forebrain bundle (+2.7 mm AP, +1.7 mm ML,−7.5 mm DV, relative to bregma). Electrical stimulations consisted of trains of 100 biphasic, 300 μA pulses at a frequency of 60 Hz with a pulse width of 2 ms. All working electrodes were conditioned at 30 Hz for 15 min with the double waveform prior to data collection with the same waveform.
Data Processing and Analysis
The first 10 CVs (1 sec) of each 30 sec data file were averaged to obtain the background voltammogram used to produce background-subtracted data for that file. For in vitro experiments, the maximum oxidation current was evaluated for each analyte (H2O2 and pH). Voltammograms (and corresponding small waveform voltammograms) containing the maximal current, as well as two CVs immediately before and after, were averaged to produce a set of CVs for calibration, and also training data for PLSR. In vivo data were examined manually in HDCV using the tool described in Figure 4 to extract CVs in the same manner as the training set.
Figure 4:

Training set selection tool. A) Correlation plots describe current collected at any two potentials in the voltammetric detection of ΔpH (top) and H2O2 (bottom). Large waveform potentials are plotted on the ordinate and abscissa. Red coloring indicates that currents collected at two potentials are correlated, i.e., they simultaneously increase in intensity with increasing analyte concentration. Blue indicates little to no correlation. For clarity, example current traces (linear voltammograms) for each analyte are shown along the ordinate and abscissa. Stars indicate positions of significantly contrasting correlation discussed in the text. B) Normalized current at +0.6 and +1.3 V extracted from the ΔpH calibration data for one electrode. The magnitude of the pH shift is labeled next to each data cluster. Inset is a CV with arrows that indicate the potentials investigated. C) Displays the normalized currents collected at +0.6 V and +1.3 data for standards of ΔpH (red), H2O2 (blue), and two sets of mixtures of these two analytes (purple).
PLSR was performed using the ‘plsregress’ function in MATLAB R2014a (Mathworks, Natick, MA), along with the SIMPLS algorithm to center the data. This approach was computationally efficient, and served to maximize covariance between the predictor and response variables.48 Mean-squared error was determined using a 5-fold cross validation. All statistical analyses and graphical depiction of the data were carried out using GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA) and MATLAB.
Results and Discussion:
FSCV Detection of H2O2 and ΔpH
The standard approach to the voltammetric detection of H2O2 using FSCV at a carbon-fiber microelectrode employs a cyclic triangular waveform that scans from a −0.4 V holding potential to a +1.4 V switching potential and back to −0.4 V at a constant scan rate of 400 V s−1, applied at a frequency of 10 Hz.17,18,49 This generates a large but stable background signal composed predominately of non-faradaic capacitive (charging) current,50 contributions arising from oxidation and reduction of groups inherent to the electrode surface,39,51 and faradaic contributions from electroactive species that are statically present in the extracellular space.46 The background current can be subtracted to reveal faradaic current generated in response to an electroactive species transiently present in the immediate vicinity of the electrode. This current is plotted versus the applied potential to create a CV, where peak position(s) serve as a chemical identifier and the intensity is used for quantification. Due to the volume of data collected, CVs are concatenated with respect to time to create a ‘color plot’, which displays three dimensions of data in a simplified manner. Figure 1A contains representative color plots and inset CVs for bolus injections of H2O2 (left), basic ΔpH (middle), and a mixture of H2O2 and basic ΔpH (right). The overall shape of the CVs for H2O2 and ΔpH differ, as evidenced by the patterns visible in the color plots. Importantly, shifts in pH generate current across the entire potential window.38 However, the peak current is generated at ~1.3 V (reverse scan) for both H2O2 and ΔpH, convolving identification of the species contributing to the mixed voltammogram and hindering reliable calibration.
Figure 1:

A) Representative color plots and CVs (inset) for of 80 μM H2O2 (left), +0.25 basic ΔpH (middle) and a mixture of both (right). Time and potential are plotted as the ordinate and abscissa, respectively. Current is depicted in false color, as indicated by the scale bar. CVs are plotted with positive potentials to the right. Arrows indicate the direction of the scan. Representative CVs (left) and a calibration plot (right) for B) H2O2, and C) ΔpH (calibration carried out using the peak at ~0.6 V). Data are presented as mean ± standard deviation (n=5 electrodes). All data presented were collected using the larger scan of the double waveform.
In voltammetry, peak current is directly proportional to analyte concentration, and the slope of a regression line through the calibration data can be used as a calibration factor to describe sensitivity to a given analyte. Figure 1B–C displays representative CVs (left) collected in vitro for physiologically relevant concentrations of H2O2 and ΔpH,17,18,30,38 as well as calibration plots (right) where the slopes of the regression models are 0.131 ± 0.001 nA/μM for H2O2, and 53 ± 3 nA/ΔpH unit. However, univariate calibration is not adequate for quantification when multiple unresolved signals overlap, which is often the case when making measurements in complex in vivo environments. The current produced by individual analytes is additive at each potential with contributions proportional to the concentration of each species, as long as there is no chemical reaction or competing surface interaction for the constituents. If the ΔpH were known it would be straightforward to determine and subtract its contribution at a specific potential. However, if the contributions of both ΔpH and H2O2 are unknown, it becomes impossible to accurately assign values for each component using univariate calibration.
Principal Component Regression: Complications in Distinguishing H2O2 and ΔpH
To use PCR with FSCV, principal component analysis (PCA) is performed by compiling a training set composed of CVs for each analyte expected to contribute to the signal, with intensities encompassing the relevant magnitudes.33,34 PCA calculates a ranked set of principal component vectors describing the entirety of the training set, with the first component capturing the maximum variance inherent to the training set; and remaining components each describing the remaining variance, always orthogonal to the previous component.37,52 Then scores, or scaled projections of the training CVs onto the individual components, can be regressed against concentration. This allows signal contributions from different analytes, whose voltammograms differ in factor (component) space, to be individually quantified. A useful tool to evaluate the efficacy of PCR models is the Cook’s score (distance) plot.30,34,53 A good PCR model distinguishes each analyte well in factor space, and will generally result when the training set consists of analytes with voltammograms that are sufficiently unique. Such is the case for a training set composed of H2O2 and DA CVs. Example Cook’s plots for two PCR training sets are displayed in Figures 2A (H2O2 and DA) and 2B (H2O2 and ΔpH). The data shown in Figure 2A were recorded using a conventional catecholamine waveform spanning −0.4 to +1.3 V with a scan rate of 400 V s−1. For two analytes, one expects to observe two score vectors, or linear groups of data. An ideal model would produce components that are analyte-pure, with sample scores resulting in orthogonal (uncorrelated) linear groups lying on the coordinates, rather than within the quadrants. This would indicate that each component describes the variance of one individual analyte.52–54 This PCR model for H2O2 and DA results in nearly orthogonal (dashed line) score vector components (∠83°), as shown in Figure 2A. DA’s score vector lies close to the abscissa, indicating that most of the variance is captured by PC1, and H2O2’s score vector spreads predominately along the ordinate. In both cases, the adjacent PC describes some of the variance, signifying that the voltammograms for these species share at least some common variance. Figure 2C plots the two PC loadings versus the applied potential. PC1 (black) is similar to dopamine’s CV, and PC2 (red) resembles the CV for H2O2. This indicates that the model sufficiently separated the analytes in factor space.
Figure 2:

Principal component regression is insufficient to resolve individual contributions to the voltammetric signal from ΔpH and H2O2. A) Score plot for a training set comprised of voltammograms for H2O2 (red) and DA (blue) collected using the catecholamine waveform (−0.4 to +1.3 V, 400 V s−1). The sample scores on the first and second components are plotted on the abscissa and ordinate, respectively. The angle between the two score vectors (red and blue lines) is inset. B) Score plot for a training set comprised of voltammograms for H2O2 (red) and ΔpH (blue) collected using the H2O2 waveform (−0.4 to +1.4 V, 400 V s−1). C,D) Principal component loadings (ordinate) plotted vs. potential (abscissa) for the first (black) and second (red) components in (A) and (B), respectively.
On the other hand, if voltammograms for two analytes are not sufficiently dissimilar (i.e. there are similar sources of variance), it is likely that the two analytes cannot be accurately resolved using PCR. Significant deviations from orthogonal sample score vectors indicate multi-collinearity, and thus highly correlated factors.54 In the case of voltammograms for ΔpH and H2O2 recorded using the standard H2O2 waveform spanning −0.4 to +1.4 V, at least one component contains significant contributions from both analytes. Figure 2B shows that the sample scores for both analytes lie well within a quadrant, indicating that the first two PCs contain substantial contributions from both species. Additionally, the angle between the two score vectors (∠60°) deviates significantly from orthogonality, indicating highly correlated factors. Indeed, the first PC (Figure 2D, black) resembles the voltammogram for ΔpH; it is not visually discernable that it contains contributions from H2O2. By contrast, the second PC (red) contains characteristics of both analytes. Ultimately this confounds reliable separation of contributions from ΔpH from those of H2O2 using PCR. In an effort to more accurately distinguish and quantify voltammetric signals comprised of both ΔpH and H2O2, alternate strategies were investigated.
A Double Waveform-Partial Least Squares Regression Model
A model to reliably identify, predict, and subtract current contributions due to local shifts in pH requires gathering information specific to the ΔpH signal. To achieve this, we employed a novel voltammetric waveform that incorporates two triangular sweeps of differing magnitude, as depicted in Figure 3A. Figure 3B displays representative CVs collected using the small (left) and large (right) waveforms for H2O2 (top), ΔpH (middle), and a mixture of the two analytes (bottom). The sWF scans from −0.4 V to +0.8 V and back at 400 V s−1, as H2O2 is electrochemically silent in this potential window and ΔpH generates substantial current. Thus, the sWF captures information specific to ΔpH that can serve as a predictor of the ΔpH contribution to voltammograms collected 12 msec later with the lWF, which scans to +1.4 V at 400 V s−1, generating CVs that contain contributions from both analytes.
Data collected using the double waveform were used to produce a multivariate calibration model to determine the ΔpH contribution to voltammograms subsequently collected using the lWF. PCR and PLSR are similar in that they both utilize predictor components to describe the observed data, but they differ in the manner in which these components are constructed. PCR creates PCs to describe variance in the training set voltammograms (predictor) without considering the response (analyte concentration), and is therefore considered to be an unsupervised dimensionality reduction technique.52,55 In the context of the double waveform described herein, predictor-sWF and response-lWF voltammograms differ in length (and number of data points). Therefore PCR is not suitable, as the size of the sWF predictor differs from that of the lWF-response. By contrast, PLSR is a supervised dimensionality reduction technique that projects both the sWF-predictor and lWF-response variables onto a new vector space to find components that maximize the covariance of the projected structures.55,56 This generally allows PLSR models to describe the training data more efficiently with fewer components (than PCR), resulting in an output prediction model that is often (but not always) more reliable when adjusted.40,42,56
When using PLSR, the total number of components produced by the model will be one less than the size of the training set. However, only a fraction of the components will describe the ‘real’ signal (PCs), with the remaining components describing noise.37,52 Supervised predictive modeling techniques, such as PLSR, generally employ cross-validation approaches to determine the proper number of PCs to retain.41 As such, k-fold cross validation was used to determine the number of PCs retained in the PLSR analysis. This was done by training the model following removal of a fraction of the training set. The removed data were then used to compute the mean-square error (MSE) of the predicted lWF-response. Finally, cumulative variance was used as a figure of merit to assess the level of noise in the data.44,57 A training set was constructed of ΔpH (including ΔpH=0.0) voltammograms collected with both the sWF and the corresponding lWF using five electrodes in vitro. Representative training set data are depicted in Figure 3B (middle). Figure 3C displays the 5-fold cross validation and cumulative percent variance for both the sWF predictor (black and gray) and lWF response (red and pink). The MSE associated with both the sWF-predictor and lWF-response variables show no significant improvement in predictive power by retaining greater than five components. Therefore, five components were retained for this in vitro model, which capture an excess of 99.5% of the training set variance. To further validate the model, sWF voltammograms from every electrode for ΔpH, H2O2, and mixtures (+0.25 ΔpH with five H2O2 concentrations) were input into the model. The PLSR-predicted ΔpH signal was subtracted from voltammograms collected using the lWF. ΔpH-subtracted CVs for a single electrode are displayed in Figure 3D for H2O2 (top), ΔpH (middle), and known mixtures (bottom). Importantly, the model effectively removed ΔpH contributions to signals that contained a known contribution from ΔpH. The coefficient of determination (R2) between the training CVs for H2O2 collected using the lWF and the ΔpH-subtracted H2O2 and mixture CVs are 0.988 ± 0.008 and 0.96 ± 0.02, respectively (Figure 3D, inset text). In fact, calibration data for H2O2 (Figure 1B), and for ΔpH-subtracted H2O2 and mixture samples were not significantly different (data not shown, slope: F(2,39)=0.025, p=0.98, intercept: F(2,41)=0.86, p=0.43). Furthermore, the model effectively removed ΔpH signals that were ~20x larger than the magnitude of the smallest H2O2 contributions. This is important because H2O2 signals recorded in vivo are often smaller in magnitude than voltammetric signals for local shifts in pH. Thus, the DW-PLSR model should serve as a useful tool in vivo.
Construction of an In Vivo Training Set
Whenever possible, a training set or calibration should be generated in a recording environment that matches the experimental environment so as to eliminate matrix effects, or changes in the analytical signal caused by anything in the sample other than the analyte.58 Constructing a training set for in vitro studies is straightforward, as the experimenter knows what constitutes each sample. However, constructing an appropriate training set of data collected in vivo presents a bigger challenge, as the chemical complexity of living tissue is immense, dynamic, and has not been accurately replicated. In traditional in vivo studies, DA-specific voltammograms have been obtained by electrical stimulation of dopaminergic afferents to evoke DA release in the vicinity of the electrode, generating a well-characterized CV that is easily recognized.59 Electrical stimulation also generates a hemodynamic response that includes a large contribution from a local pH change,60 but also likely contains contributions from other electroactive substances including H2O2, O2, and adenosine. Thus, a method to aid in selection of CVs collected in vivo that best represent pure shifts in pH was devised by examining relationships inherent to the ΔpH data collected in vitro. Figure 4A displays correlation plots (n=5 electrodes) for current collected at any two potentials of the lWF for ΔpH (top) and H2O2 (bottom). Red indicates that currents collected at two distinct potentials simultaneously increase in intensity (disregarding sign) with increasing analyte concentration, and thus are correlated; blue indicates regions in the voltammogram in which there is little to no correlation. ΔpH generates a strong signal at potentials less than +0.8 V, and there is a high correlation (R2 ~0.99) between currents collected at +0.6 V (forward-scan) and +1.3 V (reverse scan). Little to no current is generated at +0.6 V in the voltammetric detection of H2O2, and thus the correlation between currents collected at these specific potentials is much weaker for H2O2 (R2~0.5). As such, the relationship between currents collected at +0.6 V (forward-scan) and +1.3 V (reverse scan) can be used to differentiate voltammograms resulting from a local shift in pH from those due to local fluctuations in H2O2.
To demonstrate this, in vitro data were collected for standards of ΔpH, H2O2. and mixtures of these two species, and the currents recorded at +0.6 V and +1.3 V were normalized to the maximum currents recorded at these potentials (across all samples). Figure 4B displays the results for the ΔpH voltammograms. The current at both potentials increases as ΔpH increases, resulting in a ΔpH-only vector distributed about the ordinate and abscissa. Figure 4C displays the results for all samples: ΔpH (red), H2O2 (blue), H2O2/+0.1 ΔpH (dark purple), and H2O2/+0.2 ΔpH (light purple). In short, the data group nicely. Increases along the ordinate indicate pure shifts in pH. By contrast, H2O2 is electrochemically silent below +0.6 V, and thus has little spread along the ordinate. Therefore, these relationships can be used to facilitate selection of ΔpH CVs collected in vivo, and to exclude voltammograms that most likely result from mixtures of ΔpH and H2O2.
Applying the DW-PLSR Model to In Vivo Data
The DW-PLSR model was used to evaluate H2O2 fluctuations in the dorsal striatum of an awake, freely-moving, 6-OHDA lesioned rat. This is a common model for Parkinson’s disease, and data were recorded on the lesioned side which contains very few dopaminergic terminals.61,62 Local shifts in pH can generate significant current and, as a result, H2O2 fluctuations can be easily masked. Thus, it is necessary to reliably subtract ΔpH contributions in order to more accurately assess underlying H2O2 dynamics. The in vivo data were investigated (as described above) to select appropriate ΔpH voltammograms to serve as ‘standards’ in the training set. Figure 5A shows voltammograms for acidic ΔpH (left; 12 CVs) and blanks (right; 3 CVs) collected using both the sWF (top) and lWF (bottom). These CVs and their inversions were used for PLSR, as inverting the voltammogram for an acidic pH shift results in the voltammogram for a basic shift in pH.
Figure 5:

In vivo training set for PLSR model. A) sWF (top) and corresponding lWF (bottom) CVs were extracted from in vivo data for acidic ΔpH (left; 12 CVs), and blank (right; 3 CVs) signals. The inversion (not shown) of each training CV was also included to produce a training set of 30 CVs. B) Cross validation and cumulative percent variance. The number of components retained is plotted on the ordinate. Left: Cumulative variance for the sWF-predictor (black) and lWF-response (red) CVs. A dashed line indicates the position of 99.5% variance. Right: average mean-squared error (nA2/CV) for the sWF-predictor (gray) and lWF-response (pink) CVs. 5 fold cross validation was performed by randomly dividing up the training data.
Figure 5B displays the cross validation results used to determine the number of PCs to retain. The MSE for both the sWF-predictor (gray) and lWF-response (pink) variables were not improved when greater than five components were retained. Consequently, five components were kept for this in vivo model. This five-component model also contained in excess of 99.5% of the variance inheret to the original in vivo dataset. As further evidence of proper variable selection, the sWF-predictor and lWF-response loadings for the first five components are plotted versus the corresponding waveform potentials in Figure S-1A. Loadings for the first PC (top) resemble the ΔpH training data. Loadings for PCs 2–5 (bottom) for the sWF predictor (left) appear to be predominately noise, whereas those for the lWF-response (right) contain prominent features near +1.3 V. Additionally, the residual signal for each training CV, not captured by the first five components, appears to be noise and does not contain features reminiscent of either analyte (Figure S-1B).
Figure 6 summarizes the results obtained for ΔpH subtraction from data collected in vivo. Figures 6A and 6B (top) show color plots for 30 sec recordings – for simplicity, only the data collected using the lWF are presented. The data presented in Figure 6A contain information on H2O2 fluctuations, as indicated by the blue/green/purple colors at ~+1.3 V. The data presented in Figure 6B are similar, but also contain significant contributions from ΔpH – note the different current scale. The PLSR-predicted, lWF response (describing the ΔpH contribution) is presented in the middle row, and the ΔpH-subtracted color plots are displayed at the bottom. In Figure 6A, where no interfering signal was evident, the raw data and ΔpH-subtracted color plots are visually similar. They describe the H2O2 activity, which is minimal in this recording. In Figure 6B, where an interfering ΔpH signal was evident, the raw data and ΔpH-subtracted color plots are substantially different, highlighting the ability to model and subtract contributions from ΔpH and retain information selectively describing H2O2 dynamics. Voltammograms extracted from the raw data (black), the PLSR-predicted ΔpH response (blue), and the-ΔpH subtracted data (red) are compared in Figures 6C and 6D. The raw and ΔpH-subtracted voltammograms in Figure 6C are similar, as the model determined that there is not significant interference from ΔpH. By contrast, the PLSR-predicted response in Figure 6B is similar to the raw voltammogram, which contains a significant ΔpH contribution. The ΔpH-subtracted voltammogram for the time point indicated by the dashed vertical line reveals a well-defined, H2O2 signal. Similarly, the corrected color plot provides for easy visualization and quantification of the H2O2 dynamics across this 30 sec period.
Figure 6:

Application of DW-PLSR model to in vivo data collected in the striatum of a freely-moving, 6 OHDA lesioned rat. A) and B) contain color plots for two different 30 sec recordings. For simplicity, only the data collected using the large waveform are shown. Raw data (top), the PLSR ΔpH-predicted response (middle), and the ΔpH-subtracted data (bottom) are shown. Colormap scaling is shown above each set. C) and D) display voltammograms extracted from color plots in A) and B), respectively. Dashed white lines indicate the position of the extracted CVs.
When working in the striatum, DA is commonly monitored. Thus, it is important to demonstrate that the DW-PLSR model is effective for elucidating H2O2 dynamics, even in the presence of DA. Striatal recordings from an intact, anesthetized rat were used to construct a ΔpH/DA training set for PLSR, as done in Figure 6. The training set CVs (8 ΔpH, 8 DA, and 3 blank CVs), PC loadings, cumulative variance, and cross-validation data are displayed in Figure S 2. The cross-validation shows that the MSE begins to level off when greater than four components are retained. Thus, four PCs were retained for analysis of the data shown in Figure 7. It is evident from the PC loadings that PC1 is predominately indicative of a ΔpH signal, PC2 contains features indicative of both ΔpH and DA, and PC3 5 resemble noise. Figure 7A displays the raw data (top), the PLSR-predicted ΔpH/DA response (middle), and ΔpH/DA-subtracted color plots (bottom) for a 60 sec recording. The ΔpH/DA-subtracted data retain features clearly indicative of H2O2, and the PLSR predicted, lWF response contains features inherent to ΔpH and DA. Figure 7B displays a comparison of CVs extracted immediately following an electrical stimulation (top), as well as from a later time point (bottom). DA and contributions from ΔpH are evident following the stimulation (at the time indicated by the white line in panel A), and the model clarifies the H2O2 component at this time point (top, panel B). A more substantial ΔpH signal is observed ~30 sec later (black line, panel A). The DW-PLSR can also predict and subtract this component from the data, clarifying the H2O2 information (bottom, panel B). Thus, the DW-PLSR model is applicable for the analysis of complex data containing multiple chemical contributions.
Figure 7:

Application of the DW-PLSR model to data collected in the striatum of an intact rat. A) Color plot for a 60 sec recording, the time of electrical stimulation is indicated by the arrow. Raw data (top), the PLSR-predicted ΔpH/DA response (middle), and the ΔpH/DA-subtracted data (bottom) are shown. B) CVs extracted from color plots in A), at the positions indicated by the vertical dashed lines.
Conclusions
In all electrochemical studies carried out in vivo, chemical selectivity is a primary concern. Herein, we demonstrate that PLSR can be combined with a double voltammetric waveform to provide an effective means of quantitatively resolving the electrochemical contribution of an analyte in a complex mixture. This can be done even in the presence of an interferent that also exhibits substantial redox activity at the same potential. By using a sWF that captures information specific to the primary interferents (in this case ΔpH and DA) as a predictor variable, the contribution of these species in the analytical region of interest can be subtracted non-subjectively from the lWF signal, revealing the underlying signal for the analyte of interest. A major advantage of this technique is that it simplifies time consuming experimental protocols for building in vivo training sets-researchers are no longer required to search in vivo recordings for voltammograms representative of all analytes present; only interferent CVs are necessary. As such, the inclusion of a stimulating electrode to elicit an analyte response may no longer be necessary, simplifying surgical procedures and limiting tissue damage. Importantly, the model does not subtract information in the absence of the interfering species. Finally, the potential limits of the double waveform can be easily adjusted for broad applicability. This data analysis tool should prove useful in distinguishing the contribution of interferents from that of any other analyte with redox activity that appears outside the potential window encompassed by the smaller waveform.
Supplementary Material
Acknowledgement
The authors would like to thank Leslie Wilson, Karen Butler, and Christie Lee for their assistance with data collection. This work was supported by the U.S. National Institutes of Health (R01-NS076772 to L.A.S). C.J.M. is supported by an NSF Graduate Research Fellowship (DGE-1252376).
Footnotes
Associated Content
Supplemental Figures S-1 and S-2. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests
References
- (1).Halliwell BJ Neurochem. 2006, 97, 1634–1658. [DOI] [PubMed] [Google Scholar]
- (2).Adam-Vizi V Antioxid. Redox Signal. 2005, 7, 1140–1149. [DOI] [PubMed] [Google Scholar]
- (3).Fridovich IJ Biol. Chem 1997, 272, 18515–18517. [DOI] [PubMed] [Google Scholar]
- (4).Mattson MP; Liu D Neuromolecular Med. 2002, 2, 215–231. [DOI] [PubMed] [Google Scholar]
- (5).Zhang Y; Dawson VL; Dawson TM Neurobiol. Dis 2000, 7, 240–250. [DOI] [PubMed] [Google Scholar]
- (6).Kishida KT; Klann E Antioxid. Redox Signal. 2007, 9, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bao L; Avshalumov MV; Patel JC; Lee CR; Miller EW; Chang CJ; Rice ME J. Neurosci 2009, 29, 9002–9010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Stone JR; Yang S Antioxid. Redox Signal. 2006, 8, 243–270. [DOI] [PubMed] [Google Scholar]
- (9).Rhee SG Science 2006, 312, 1882–1883. [DOI] [PubMed] [Google Scholar]
- (10).Burmeister JJ; Gerhardt GA Anal. Chem 2001, 73, 1037–1042. [DOI] [PubMed] [Google Scholar]
- (11).Parikh V; Kozak R; Martinez V; Sarter M Neuron 2007, 56, 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wassum KM; Tolosa VM; Wang J; Walker E; Monbouquette HG; Maidment NT Sensors 2008, 8, 5023–5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wilson GS; Johnson MA Chem. Rev 2008, 108, 2462–2481. [DOI] [PubMed] [Google Scholar]
- (14).Smith SK; Lee CA; Dausch ME; Horman BM; Patisaul HB; McCarty GS; Sombers LA ACS Chem. Neurosci 2017, 8, 272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zhang Y; Wilson GS J. Electroanal. Chem 1993, 345, 253–271. [Google Scholar]
- (16).Hall SB; Khudaish EA; Hart AL Electrochim. Acta 1999, 44, 2455–2462. [Google Scholar]
- (17).Sanford AL; Morton SW; Whitehouse KL; Oara HM; Lugo Morales LZ; Roberts JG; Sombers LA Anal. Chem 2010, 82, 5205–5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Spanos M; Gras-Najjar J; Letchworth JM; Sanford AL; Toups JV; Sombers LA ACS Chem. Neurosci 2013, 4, 782–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Aoki K; Ishida M; Tokuda K; Hasebe KJ Electroanal. Chem. Interfacial Electrochem 1988, 251, 63–71. [Google Scholar]
- (20).Nowall WB; Kuhr WG Electroanalysis 1997, 9, 102–109. [Google Scholar]
- (21).Roberts JG; Toups JV; Eyualem E; McCarty GS; Sombers LA Anal. Chem 2013, 85, 11568–11575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Vreeland RF; Atcherley CW; Russell WS; Xie JY; Lu D; Laude ND; Porreca F; Heien ML Anal. Chem 2015, 87, 2600–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Szentirmay MN; Martin CR Anal. Chem 1984, 56, 1898–1902. [Google Scholar]
- (24).Gerhardt GA; Oke AF; Nagy G; Moghaddam B; Adams RN Brain Res 1984, 290, 390–395. [DOI] [PubMed] [Google Scholar]
- (25).Jang DP; Kim I; Chang SY; Min HK; Arora K; Marsh MP; Hwang SC; Kimble CJ; Bennet KE; Lee KH Analyst 2012, 137, 1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Johnson JA; Hobbs CN; Wightman RM Anal. Chem 2017, 89, 6166–6174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kishida KT; Saez I; Lohrenz T; Witcher MR; Laxton AW; Tatter SB; White JP; Ellis TL; Phillips PEM; Montague PR Proc. Natl. Acad. Sci 2016, 113, 200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Johnson JA; Gray JH; Rodeberg NT; Wightman RM Anal. Chem 2017, 89, 10547–10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Keithley RB; Carelli RM; Wightman RM Anal. Chem 2010, 82, 5541–5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Keithley RB; Wightman RM ACS Chem. Neurosci 2011, 2, 514–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Keithley RB; Mark Wightman R; Heien ML TrAC, Trends Anal. Chem 2009, 28, 1127–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Keithley RB; Wightman RM; Heien ML TrAC, Trends Anal. Chem 2010, p 110. [Google Scholar]
- (33).Rodeberg NT; Johnson JA; Cameron CM; Saddoris MP; Carelli RM; Wightman RM Anal. Chem 2015, 87, 11484–11491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Johnson JA; Rodeberg NT; Wightman RM ACS Chem. Neurosci 2016, 7, 349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Heien MLA V; Johnson, M. A.; Wightman, R. M. Anal. Chem. 2004, 76, 5697–5704. [DOI] [PubMed] [Google Scholar]
- (36).Hermans A; Keithley RB; Kita JM; Sombers LA; Wightman RM Anal. Chem 2008, 80, 4040–4048. [DOI] [PubMed] [Google Scholar]
- (37).Kramer R Chemometric techniques for quantitative analysis; CRC Press: New York, NY, 1998. [Google Scholar]
- (38).Takmakov P; Zachek MK; Keithley RB; Bucher ES; McCarty GS; Wightman RM Anal. Chem 2010, 82, 9892–9900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).McCreery RL Chem. Rev 2008, 108, 2646–2687. [DOI] [PubMed] [Google Scholar]
- (40).Wentzell PD; Montoto LV Chemom. Intell. Lab. Syst 2003, 65, 257–279. [Google Scholar]
- (41).Geladi P; Kowalski BR Anal. Chim. Acta 1986, 185, 1–17. [Google Scholar]
- (42).Godoy JL; Vega JR; Marchetti JL Chemom. Intell. Lab. Syst 2014, 130, 182–191. [Google Scholar]
- (43).Saurina J; Hernández Cassou S; Saurina J; Fàbregas E; Alegret S Analyst 1999, 124, 733–737. [Google Scholar]
- (44).Lavine B; Workman J Anal. Chem 2008, 80, 4519–4531. [DOI] [PubMed] [Google Scholar]
- (45).Sjöström M; Wold S; Lindberg W; Persson JÅ; Martens H Anal. Chim. Acta 1983, 150, 61–70. [Google Scholar]
- (46).Roberts JG; Lugo-Morales LZ; Loziuk PL; Sombers LA In Leading Methods in Dopamine Research; Kabbani N, Ed.; Humana Press, New York, NY, 2013. [Google Scholar]
- (47).Clark JJ; Sandberg SG; Wanat MJ; Gan JO; Horne EA; Hart AS; Akers CA; Parker JG; Willuhn I; Martinez V; Evans SB; Stella N; Phillips PE M. Nat. Methods 2010, 7, 126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).de Jong S Chemom. Intell. Lab. Syst 1993, 18, 251–263. [Google Scholar]
- (49).Roberts JG; Hamilton KL; Sombers LA Analyst 2011, 136, 3550–3556. [DOI] [PubMed] [Google Scholar]
- (50).Bard AJ; Faulker LR Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley & Sons: New York, NY, 2001. [Google Scholar]
- (51).Takmakov P; Zachek MK; Keithley RB; Walsh PL; Donley C; McCarty GS; Wightman RM Anal. Chem 2010, 82, 2020–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Härdle W; Simar L Applied Multivariate Statistical Analysis Applied Multivariate Statistical Analysis, 6th ed.; Pearson: New York, 2003. [Google Scholar]
- (53).Cook RD Technometrics 1977, 19, 15–18. [Google Scholar]
- (54).Loehlin JC Latent Variable Models: An Introduction to Factor, Path, and Structural Equation Analysis, 4th ed.; Taylor & Francis Group: London, 2003. [Google Scholar]
- (55).Maitra S; Yan J Casualty Actuar. Soc 2008, Applying Multivariate Statistical Models, 79–90. [Google Scholar]
- (56).Mehmood T; Liland KH; Snipen L; Sæbø S Chemom. Intell. Lab. Syst 2012, 118, 62–69. [Google Scholar]
- (57).Palermo G; Piraino P; Zucht HD Adv. Appl. Bioinforma. Chem 2009, 2, 57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Harris DC Quantitative Chemical Analysis, 9th ed.; MacMillan: London, 2015. [Google Scholar]
- (59).Rodeberg NT; Sandberg SG; Johnson JA; Phillips PEM; Wightman RM ACS Chem. Neurosci 2017, 8, 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Venton BJ; Michael DJ; Wightman RM J. Neurochem 2003, 84, 373–381. [DOI] [PubMed] [Google Scholar]
- (61).Deumens R; Blokland A; Prickaerts J Exp. Neurol 2002, 175, 303–317. [DOI] [PubMed] [Google Scholar]
- (62).Hoffman AF; van Horne CG; Eken S; Hoffer BJ; Gerhardt GA Exp. Neurol 1997, 147, 130–141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.