

Abstract
Ongoing pain has been linked to ongoing activity (OA) in human C-fiber nociceptors, but rodent models of pain-related OA have concentrated on allodynia rather than ongoing pain, and on OA generated in non-nociceptive Aβ-fibers rather than C-fiber nociceptors. Little is known about how ongoing pain or nociceptor OA is generated. To define neurophysiological alterations underlying nociceptor OA, we have utilized isolated dorsal root ganglion neurons that continue to generate OA after removal from animals displaying ongoing pain. We subclassify OA as either spontaneous activity (SA) generated solely by alterations intrinsic to the active neuron or as extrinsically driven OA. Both types of OA were implicated previously in nociceptors in vivo and after isolation following spinal cord injury (SCI), which produces chronic ongoing pain. Using novel automated algorithms to analyze irregular changes in membrane potential, we have found, in a distinctive, non-accommodating type of probable nociceptor, induction by SCI of three alterations that promote OA: 1) prolonged depolarization of resting membrane potential, 2) a hyperpolarizing shift in the voltage threshold for action potential generation, and 3) an increase in the incidence of large depolarizing spontaneous fluctuations (DSFs). Can DSFs also be enhanced acutely to promote OA in neurons from uninjured animals? A low dose of serotonin failed to change resting membrane potential but lowered action potential threshold. When combined with artificial depolarization to model inflammation, serotonin also strongly potentiated DSFs and OA. These findings reveal nociceptor specializations for generating OA that may promote ongoing pain in chronic and acute conditions.
Keywords: spontaneous pain, spontaneous activity, resting membrane potential, action potential threshold, depolarizing spontaneous fluctuations, primary afferent neuron, spinal cord injury, serotonin
Summary
Neurophysiological specializations for generating ongoing electrical activity likely to promote ongoing pain have been defined in isolated primary nociceptors in neuropathic and inflammatory pain models.
1. Introduction
Ongoing (“spontaneous”) pain is often the worst complaint in neuropathic and inflammatory pain conditions, yet the mechanisms are unclear [12]. While central sensitization may contribute [50, 103, 116], ongoing activity (OA) in primary afferent neurons plays a prominent role [6, 36, 37, 72]. Primary afferent OA generated by low-threshold Aβ-fiber mechanosensory neurons appears to promote central sensitization and allodynia after nerve injury [24, 55, 71, 88, 95]. In C-nociceptors, OA occurs in inflammatory and neuropathic pain models [1, 11, 27, 35, 60, 61, 108, 115]. This OA is notable because nociceptor activity exhibits the clearest links to conscious pain in humans [15, 56, 57, 63, 81, 102] and to aversive pain-like states in animal models [9, 23, 27, 100, 117]. In contrast to the detailed descriptions of processes generating OA in large A−neurons [2–4, 29, 48, 49, 75, 111, 112], little is known about how small nociceptors generate OA.
Defining the neurophysiological basis of OA in nociceptors requires direct observation and precise manipulation of underlying changes in membrane potential. Uniquely detailed neurophysiological investigations are possible in isolated dorsal root ganglion (DRG) neuronal somata [e.g., 33, 104], which exhibit some of the distinctive properties of the somata and peripheral terminals observed in vivo [5, 32, 38]. A property retained after dissociation is generation of OA within the soma, which is seen in large DRG neurons (primarily low-threshold mechanosensory neurons) and small DRG neurons (primarily C-nociceptors) in peripheral neuropathic pain models [11, 54, 87, 120]. Importantly, somal generation of OA also occurs in vivo after nerve injury [16, 42, 55, 85, 94, 113] and is implicated in human amputation pain [93]. In a central neuropathic pain model, spinal cord injury (SCI), approximately 50% of small neurons (primarily nociceptors) dissociated from rat DRGs exhibit OA [10, 107, 117]. This may represent spontaneous activity (SA) because it is probably generated by mechanisms intrinsic to the neurons. Following SCI, OA also occurs in C-fiber nociceptors in a skin-nerve preparation [18], and is generated in C-neurons within DRGs in vivo [11]. SCI-induced OA, like C-fiber OA in peripheral pain models [27, 35, 43, 73, 105, 108, 115], consists of low-frequency irregular discharge without high-frequency bursts. In contrast, OA in Aβ sensory neurons can occur in bursts driven by high-frequency sinusoidal oscillations of membrane potential [3, 4].
OA generated in the absence of sensory generator potentials or synaptic potentials could, in principle, be driven by three functional aspects of membrane potential: 1) prolonged depolarization of resting membrane potential (RMP), 2) a hyperpolarizing shift in the voltage threshold for action potential (AP) generation, and/or 3) an increase in transient, depolarizing spontaneous fluctuations (DSFs). Using an SCI neuropathic pain model, we have found that each of these intrinsic factors changes to promote OA within a distinct nociceptor type. Furthermore, nociceptors taken from uninjured animals show acute potentiation of OA by an inflammatory agent, serotonin (5-HT), that enhances DSFs and lowers AP threshold. Thus, nociceptors exhibit a comprehensive set of neurophysiological specializations enabling the OA associated with ongoing pain.
2. Methods
2.1. Animals
All procedures followed the guidelines of the International Association for the Study of Pain and were approved by the McGovern Medical School at UTHealth Animal Care and Use Committee. A total of 49 male Sprague-Dawley rats (20 naive, 5 sham, 24 SCI) were included in this study. Rats (250–300 g, 2 per cage) were allowed to acclimate to a controlled environment (12-hour reverse light/dark cycle, 21 ± 1 °C) inside the McGovern Medical School animal research facility for at least 4 days before beginning experiments. Rats were provided with food and water ad libitum.
2.2. SCI procedures
Surgeries were conducted as previously described [8, 11, 107, 117]. Rats were anesthetized with an intraperitoneal (i.p.) injection of ketamine (60 mg/kg), xylazine (10 mg/kg), and acepromazine (1 mg/kg), or with isoflurane (induction 4–5%; maintenance 1–2%). A T10 vertebral laminectomy was followed by a dorsal contusive spinal impact (150 kdyne, 1 s dwell time) using an Infinite Horizon Spinal Cord Impactor (Precision Systems and Instrumentation, LLC, Fairfax Station, VA). Sham-operated rats received the same surgical treatment minus the contusion. Rats were randomly assigned to SCI and sham groups. The analgesic buprenorphine hydrochloride (0.02 mg/kg in 0.9% saline 2 ml/kg; Buprenex, Reckitt Benckiser Healthcare Ltd., Hull, England, UK) and the antibiotic enrofloxacin (0.3 ml in 0.9% saline; Enroflox, Norbrook, Inc., Overland Park, KS) were injected i.p. twice daily for 5 days (buprenorphine) or 10 days (enrofloxin). SCI rats included in this study exhibited a score of either 0 or 1 for both hindlimbs on the Basso, Beattie, and Bresnahan (BBB) Locomotor Rating Scale [7] the day after surgery. SCI rats had hindlimb BBB scores of ≤14 at the time of euthanasia 1–6 months post-surgery.
2.3. Dissociation and culture of DRG neurons
Rats were euthanized using pentobarbital/phenytoin (0.9 ml; Euthasol, Virbac AH, Inc., Fort Worth, TX) followed by transcardial perfusion of ice-cold phosphate buffered saline (Sigma-Aldrich, St. Louis, MO). DRGs from spinal T11 to L6 levels were excised and incubated at 34°C for 40 minutes with trypsin (0.3 mg/ml) and collagenase D (1.5 mg/ml) enzymes in Dulbecco’s Modified Eagle Medium (DMEM; Sigma-Aldrich). Following digestion and washing the DRG fragments were mechanically triturated in DMEM with a fire-polished Pasteur pipette and plated on 8 mm glass coverslips coated with poly-L-ornithine (Sigma-Aldrich). Dissociated neurons were incubated overnight (<5% CO2, 95% humidity, 37°C) in DMEM without serum, growth factors, or other supplements.
2.4. Whole-cell recordings from dissociated DRG neurons
Small DRG neurons (soma diameter ≤ 30 μm) were recorded on glass coverslips at room temperature, 18–30 hours after dissociation, on either a Zeiss Axiovert 200M or Olympus IX71 inverted microscope with 40X or 20X magnification, respectively. The bath was filled with extracellular solution containing (in mM): 140 NaCl, 3 KCl, 1.8 CaCl2, 2 MgCl2, 10 HEPES, and 10 glucose, which was adjusted to pH 7.4 with NaOH and 320 mOsM with sucrose. HEKA EPC10 amplifiers (HEKA Elektronik, Lambrecht/Pfalz, Germany) were used for whole cell patch clamp recordings. Data were sampled at 20 kHz with PatchMaster v2×90.1 (HEKA Elektronik) and filtered with a 10 kHz Bessel filter. Borosilicate glass capillaries with outer diameter of 1.5 mm and inner diameter of 0.86 mm (Sutter Instrument Co., Novato, CA) were pulled using a Sutter P-97 Flaming/Brown Micropipette Puller. Fire-polished patch pipettes had electrode resistances of 3–8 MΩ after filling with intracellular-like solution containing (in mM): 134 KCl, 1.6 MgCl2, 13.2 NaCl, 3 EGTA, 9 HEPES, 1 Mg-ATP, and 0.3 Na-GTP, which was adjusted to pH 7.2 with KOH and 300 mOsM with sucrose. Only neurons that were not in visible contact (at 20–60x magnification without any staining) with the somata or neurites of other neurons, or debris, were selected for whole-cell recording. Membrane resistance and capacitance were measured under voltage clamp using 5 ms, 5 mV depolarizing pulses from a holding potential of −60 mV. To permit direct comparison with our previous papers and many others, the liquid junction potential (calculated to be ~4.3 mV) was not corrected. This means that actual membrane potentials were probably ~4 mV more negative than all values stated in this paper. To measure SA, neurons were recorded under current clamp at RMP (0 current injected) for at least 1 minute beginning at least 1 minute after switching from voltage clamp. Next, membrane potential was set at −60 mV with a constant holding current under current clamp while a series of depolarizing current injections (2-second steps every 4 seconds, +5 pA increments) were used to measure rheobase, latency to the first AP at rheobase, the membrane time constant (τ), the AP voltage threshold, and any repetitive firing at rheobase or 2x rheobase. In some experiments neurons were held at −45 mV under current clamp for ≥30 second to facilitate OA. A subset of neurons was held at −60 mV and single APs were evoked by 2 ms depolarizing pulses (+20 pA increments) to measure AP and afterhyperpolarization (AHP) properties (modified from [119]). In some experiments DRG neurons were pretreated with 100 nM serotonin (5-hydroxytryptamine; 5-HT; Sigma-Aldrich, St. Louis, MO) dissolved in extracellular solution for 10–30 min. 5-HT remained in the recording chamber for the duration of each experiment. At the end of some experiments neurons were superfused with 1 µM capsaicin (dissolved in extracellular solution) under voltage or current clamp. Non-peptidergic DRG neurons were identified by binding of isolectin B4 (IB4) extracted from Griffonia simplicifolia [86]. Coverslips were pretreated with 3 µg/mL IB4 for 5 minutes and washed for 3 minutes before beginning patching. Neurons with a continuous green ring around the perimeter of the soma were considered IB4-positive.
2.5. Quantifying spontaneous fluctuations of membrane potential and AP threshold
Published methods for quantifying spontaneous fluctuations (SFs) of membrane potential in DRG neurons rely on power spectral density analyses, which require that the SFs be oscillations or appear at regular intervals if not oscillatory [3, 4, 58, 87, 110, 113, 121]. We developed an automated script that identifies waveforms independent of frequency or regularity, inspired by the Ramer-Douglas-Peucker algorithm [30, 74] to identify curves, in order to quantify the irregular DSFs observed in our recordings. Our program, termed SFA.py, was written and tested using Python v3.5.2 (Python Software Foundation, Beaverton, OR) and Anaconda v4.1.1 (Continuum Analytics, Austin, TX) with dependency on matplotlib and NumPy libraries (script is available upon request). Time and voltage coordinate data for 30–50 second periods exported from PatchMaster were imported into the script. SFA.py then performed the following functions: 1) generate a linear regression model as an initial estimate of membrane potential; 2) group the runs of unidirectional residuals into discrete membrane fluctuations, and employ user-defined criteria to classify some of these as AP/AHP complexes; 3) exclude AP/AHPs from analysis, then calculate the RMP at each point as a sliding median of the raw data within a 1-second window centered on that point -- this accounts for slow, non-linear changes in RMP which would otherwise increase or decrease the estimated amplitude of a given fluctuation; 4) run the groups of unidirectional residuals as discrete fluctuations, then apply user-defined criteria for minimum amplitude and duration (1.5 mV and 10 ms for this study) to identify depolarizing spontaneous fluctuations (DSFs) or hyperpolarizing spontaneous fluctuations (HSFs); 5) quantify and report the following values: coordinates, amplitudes, and durations of identified APs, AHPs, DSFs, and HSFs. DSFs and HSFs ≥ 1.5 mV were measured as differences from the sliding median of membrane potential. DSFs were subdivided into small (>1.5 to ≤ 3 mV), medium-sized (3–5 mV, almost always subthreshold) and large, (>5 mV, often suprathreshold) DSFs as described in Section 3.4. All HSFs were ≥ 1.5 mV, and were not subdivided for further analysis. Descriptive data for the recordings also include standard deviation of the membrane potential, number of APs, AP frequency, number of DSFs and HSFs, and their frequencies. Color-coded line graphs with labeled APs, AHPs, DSFs, and HSFs were generated using the matplotlib library. Inspection of SA under our conditions indicated that most APs in NA neurons were triggered by suprathreshold DSFs. As a conservative estimate of the amplitude of suprathreshold DSFs, these were assigned an amplitude equal to the AP voltage threshold. This threshold was estimated for each neuron by 3 independent measures that together provided a more accurate estimate of AP threshold in our irregular system than commonly utilized analytic methods [80] that we tested (data not shown). To estimate threshold, we 1) marked the inflection point for apparent acceleration of the change in membrane potential at the base of the ascending limb of the AP, 2) identified the maximum subthreshold DSF found anywhere in the 1–2 second step depolarizations used to determine rheobase in the same neuron, and 3) measured the largest subthreshold DSF during recorded SA at RMP. The most depolarized of these three independent measurements was defined as the AP threshold for that neuron, and in all cases at least two of these three values were in good agreement with each other (within ~2 mV).
2.6. Data analysis
Statistical analyses of raw electrophysiological data and SFA.py output data were performed using Prism v7.03 (GraphPad Software, Inc., La Jolla, CA, USA). Data are presented as mean ± SEM or incidence (% of neurons sampled). All data sets were tested for normality with the Shapiro-Wilk test. Normally distributed data were tested with parametric tests: t-test or 1-way ANOVA followed by Tukey’s test for each pair-wise comparison. Data that were not normally distributed were tested with non-parametric tests: Mann-Whitney U test or Kruskal-Wallis test followed by Dunn’s test for each pair-wise comparison. Comparisons of incidence were made with Fisher’s exact test with appropriate Bonferroni corrections for multiple comparisons. Statistical significance was set at P < 0.05 and all reported values are two-tailed.
3. Results
3.1. Probable nociceptors exhibit two predominant electrophysiological types in vitro: rapidly accommodating (RA) and non-accommodating (NA)
The major goal of this study was to define physiological properties that enable sustained OA in nociceptors. Sensory generator potentials that transiently activate mechanosensitive nociceptors are often large, rapid, relatively brief depolarizations [40, 91]. However, any continuous depolarization that persistently promotes OA (as may be produced by chemical stimuli during inflammation lasting days, weeks, or longer) must be limited in amplitude because excessive depolarization will cause sufficient steady-state inactivation of voltage-gated Na+ channels to prevent continuing generation of APs over long periods of time [20, 68]. Therefore, we first asked whether small DRG neurons (15–30 µm) taken from uninjured (“naïve”) rats exhibit non-accommodating firing during relatively weak and prolonged depolarization 1 day after dissociation, which would suggest potential specialization for persistent OA. We found two distinct types of neurons, “Non-Accommodating” (NA, Fig. 1A) and “Rapidly Accommodating” (RA, Fig. 1B) that exhibited opposite electrophysiological response patterns to ascending series of 2-second depolarizing steps delivered when a neuron was held under current clamp at an initial membrane potential of −60 mV. The NA type represented 69% of sampled neurons. Characteristic features of NA neurons were a relatively low rheobase and repetitive firing in response to injecting current equal to 2x rheobase (Fig. 1A, Table 1). An unusual feature was the random latency to the first AP at rheobase, which could occur at any time during the 2-second step depolarization. This is evident in the ranked distribution of first AP latencies, which appear evenly distributed and form a nearly straight line from shortest to longest latency (Fig. 1 C). Some NA neurons at rheobase (Fig. 1 C) and most neurons at 2x rheobase (Fig. 1D) fired multiple, irregularly spaced APs during the depolarizing steps (Table 1). All tested NA neurons fired multiple APs to one or more of the steps between 1x and 2x rheobase, although not to all suprathreshold steps. In each NA neuron, these irregularly occurring APs were equally likely to occur at any time after the first AP during repetitive firing, as shown in the raster plots (Fig. 1C, 1D). The lack of any tendency for the interspike interval to increase during repetitive firing confirmed the lack of AP accommodation (Fig. 1E). This irregular, non-accommodating activity continued for as long as the neurons were depolarized (>60 seconds, data not shown).
Figure 1. Two electrophysiologically distinct types of nociceptor exhibit opposite response patterns to prolonged depolarization. DRG neurons from naïve rats (n = 18) were sampled using whole-cell recordings 18–30 hours after dissociation. (A) Representative AP discharge at rheobase and 2x rheobase in an NA neuron. An ascending series of 2-second depolarizing current steps was injected in 5 pA increments at 4-second intervals. A constant holding current that initially set membrane potential to −60 mV was continuously injected throughout the sequence. (B) Typical discharge in an RA neuron during the same test protocol. (C) Distribution of first-AP latencies (ranked from shortest to longest) and time of occurrence of additional APs at rheobase across the 2-second depolarizing step in 95 NA neurons (initial APs are leftmost red dots) and 43 RA neurons (APs are blue dots). Additional activity is indicated along the same row at the time of each AP (red dots) for each repetitively firing neuron. (D) Timing of APs in the same tests at 2x rheobase from the subsets of neurons in which the depolarizing steps reached this level (30 NA neurons and 14 RA neurons). Each row represents a single neuron. (E) Interspike intervals at 2x rheobase in NA neurons that fired ≥2 APs. Bars show the mean ± SEM, numbers in bars show neuronal sample size.
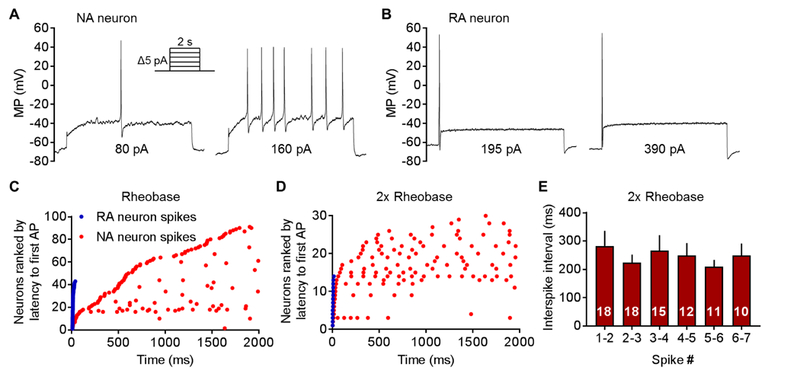
Table 1. Properties of NA and RA neurons.
| Property | NA Neurons | RA Neurons | Significance | Test |
|---|---|---|---|---|
| RMP (mV) | −62.1 ± 0.9 (96) | −65.2 ± 1.1 (43) | P < 0.05 | UPT |
| Rheobase (pA) | 88.3 ± 7.0 (96) | 169.2 ± 9.4 (43) | P < 0.0001 | MW |
| AP latency at rheobase (ms) | 812.6 ± 82.1 (96) | 14.6 ± 1.1 (43) | P < 0.0001 | MW |
| Number of APs at rheobase | 1.6 ± 0.1 (96) | 1.0 ± 0.0 (43) | P < 0.0001 | MW |
| Number of APs at 2x rheobase | 3.4 ± 0.6 (32) | 1.0 ± 0.0 (14) | P < 0.0001 | MW |
| Membrane time constant (t, ms) | 16.5 ± 1.8 (67) | 5.4 ± 0.5 (25) | P < 0.0001 | MW |
| Membrane resistance (MΩ) | 355.8 ± 24.8 (95) | 333.6 ± 29.6 (43) | P = 0.95 | MW |
| Membrane capacitance (pF) | 28.5 ± 0.9 (96) | 29.8 ± 1.0 (43) | P = 0.31 | MW |
| Soma diameter (µm) | 24.0 ± 0.5 (51) | 25.5 ± 0.5 (24) | P = 0.07 | MW |
| AP half-amplitude duration (ms) | 2.7 ± 0.3 (12) | 3.3 ± 0.2 (7) | P < 0.05 | MW |
| Capsaicin sensitivity (%) | 70% (43/61) | 67% (20/30) | P = 0.81 | F |
| IB4 Binding (%) | 49% (17/35) | 73% (8/11) | P = 0.19 | F |
Data were collected from DRG neurons taken from naïve rats (n = 18) 18–30 hours after dissociation. Sensitivity to 1 μM capsaicin was tested in neurons from 7 rats, and binding of IB4 was tested in neurons from 4 rats. Each value is the mean ± SEM followed in parentheses by number of neurons tested. Tests: UPT, unpaired t-test; MW, Mann-Whitney U; F, Fisher’s exact test.
In stark contrast, the RA type (31% of sampled neurons) never fired more than a single AP in these tests, which always occurred at the onset of the step depolarization (Fig. 1B, C, D). Interestingly, within the stimulation range of 1–3x rheobase, no RA neurons responded with multiple APs. Only at very high stimulus currents did some RA neurons fire a brief burst of 2 or 3 APs (not shown), and these were always confined to the onset of the stimulus. Compared to NA neurons, RA neurons showed significantly more hyperpolarized RMP, higher rheobase, much shorter latency to the first AP, and lower membrane time constant (Table 1). Individual APs and AHPs evoked by 2 ms depolarizing pulses were similar between NA and RA neurons. The only statistically significant difference found in these samples was for AP duration at half amplitude to be ~20% briefer in the NA neurons than in RA neurons (Table 1). No significant differences were found between NA and RA neurons in soma diameter or membrane capacitance (Table 1). Interestingly, far greater excitability was found in NA neurons than in RA neurons despite the NA neurons being more hyperpolarized following each of the larger depolarizing steps in the rheobase/repetitive firing test sequence. This is illustrated in Figures 1A and 1B. Although both neurons had the same −60 mV holding potential at the beginning of the series of depolarizing steps (not shown), membrane potential at the beginning of later steps in the series was more negative in NA neurons than in RA neurons; in the illustrated NA neuron this potential was ~ −70 mV versus ~ −65 mV in the RA neuron when rheobase and 2x rheobase were reached. This residual post-depolarization hyperpolarization resulted from there being insufficient time (2 seconds) between the larger 2-second depolarizing steps for recovery of membrane potential to −60 mV (a trade-off to allow numerous tests on each neuron).
Evidence that many of the NA and RA neurons are nociceptors was obtained by testing capsaicin sensitivity and binding of isolectin B4 (IB4). A majority of NA neurons and RA neurons tested with 1 µM capsaicin responded strongly under current clamp (Fig.2) or voltage clamp [not shown; see 107], indicating that large fractions of both types are TRPV1-expressing nociceptors (Table 1). In addition, about half of the sampled NA neurons and three quarters of the RA neurons bound IB4, suggesting that both types contain large fractions of non-peptidergic nociceptors (Table 1). While most small dissociated neurons and nearly all capsaicin-sensitive and IB4-binding neurons in rats are likely to be nociceptors [31, 32, 51, 69, 70, 83], a minority of small DRG neurons are not nociceptive [e.g., 31, 25], and we did not test most small DRG neurons for capsaicin sensitivity or IB4 binding. Thus, individual neurons selected for study in this paper were considered probable nociceptors, with the caveat that a minority of tested neurons would not have been nociceptive. Given the high incidence of capsaicin sensitivity and/or IB4 binding in NA and RA neurons, we contend that general properties established across sufficiently large samples of small DRG neurons under our culture conditions primarily represent the properties of small nociceptors, and that these include two physiologically defined classes, NA and RA.
Figure 2. NA and RA neurons are depolarized to a similar degree by superfusion of capsaicin, but only NA neurons exhibit repetitive discharge when depolarized by a high dose of capsaicin or by injected current that mimics depolarization produced by a low dose of capsaicin. (A) Representative example of depolarization and discharge evoked by a high dose of capsaicin (1 µM) in an NA neuron. (B) Example of depolarization evoked by the same dose of capsaicin in an RA neuron. Notice the lack of APs. (C) Examples showing part of the responses to prolonged depolarization (30 seconds) to −45 mV in RA and NA neurons similar to that produced by 10 nM capsaicin (see text). D) OA was promoted in NA neurons but not RA neurons by artificial depolarization to −45 mV. *P < 0.05, Fisher’s exact test. Neurons are from a subset (n = 12) of the naïve rats used for Figure 1.
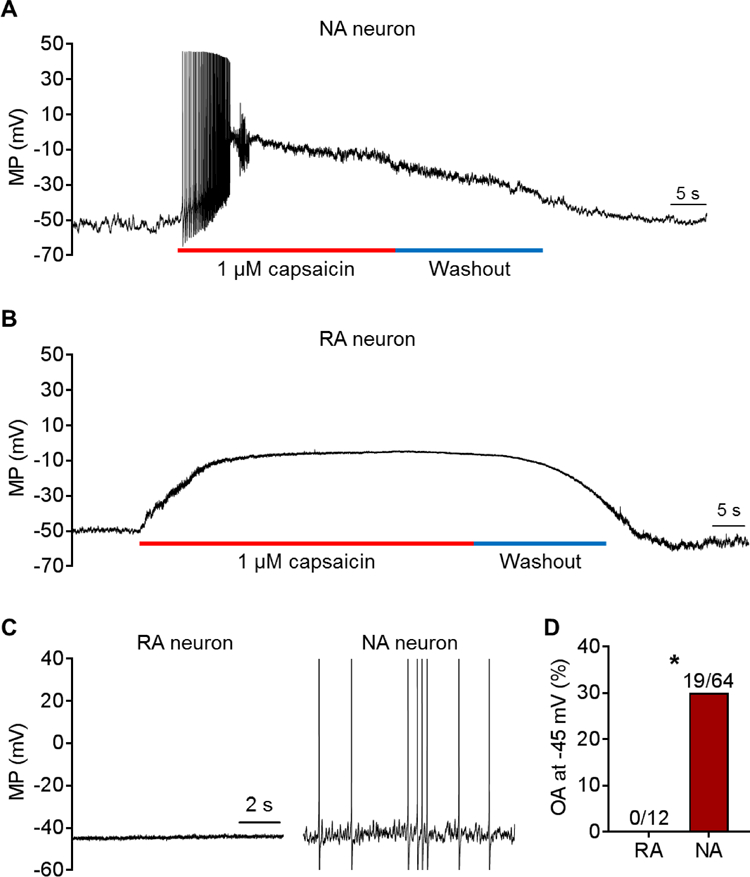
The responses of each electrophysiological type to capsaicin under current clamp provided additional evidence that NA neurons but not RA neurons are capable of OA. Perfusion of capsaicin (1 μM) evoked multiple APs in NA neurons under current clamp (Fig. 2A) [see also 107], whereas none of the tested RA neurons (n=5 from 3 rats) discharged any APs despite similar depolarization by capsaicin treatment (Fig. 2B). Our previous study showed that a low concentration of capsaicin (10 nM) could sometimes activate isolated small DRG neurons while depolarizing the neurons to between −50 and −45 mV [107]. To see if similar depolarization can produce OA like that produced by capsaicin in the earlier study, a 30-second step depolarization to −45 mV was produced by injecting current through the patch pipette. This evoked OA in 30% of the NA neurons but in none of the RA neurons (Fig. 2C).
Together, these findings indicate that many dissociated nociceptors have electrophysiological specializations to produce OA, and that this capability is restricted to the NA type. A major question raised by these observations concerns the neurophysiological basis of the stochastic characteristics of AP discharge evoked by prolonged depolarization of NA neurons.
3.2. Spinal cord injury (SCI) increases SA in NA but not RA neurons
Contusive SCI in rats induces persistent OA generated peripherally in primary nociceptors in a peripheral skin-nerve preparation [18] and within the DRG in probable C-fiber and Aδ nociceptors in vivo [11]. It also dramatically enhances the SA observed in small DRG neurons (primarily nociceptors) after dissociation [11]. We tested the prediction that SCI-induced SA in dissociated small DRG neurons occurs in neurons of the NA type but not the RA type. In addition, we asked whether sham surgery, which causes damage to deep tissues (including muscle and bone), causes any promotion of SA in NA neurons. SA was found in at least some of the NA neurons taken from naïve, sham, or SCI rats, but was not found in any RA neurons (Fig. 3A, B). As predicted by our earlier findings [8, 11, 106, 107, 117], the incidence of SA was significantly greater in neurons from SCI rats than in neurons from naïve or sham rats (Fig. 3B). In contrast to our earlier finding [11], we found that the incidence of SA in the sham group was modestly but significantly higher than in the naïve group (Fig. 3B). This finding and other evidence for persistent hyperexcitability in the sham group (see below and Table 2) differ from the earlier study. Unmasking of sham surgery effects may reflect improvements in our DRG extraction and dissociation procedures that reduced cellular stress, lowering the incidence of SA in the naïve group in the present study. These results indicate that SCI strongly enhances SA in NA nociceptors but not in RA nociceptors. In addition, tissue injury caused by sham surgery can produce a small increase in incidence of SA in NA neurons. While we cannot rule out the possibility that extrinsic factors in our neuronal cultures (either soluble factors or contact signals from small adjacent or underlying cells invisible with our microscopy methods) might contribute to the neuronal activity observed after SCI, we think it is highly likely that SA is produced by mechanisms intrinsic to NA neurons because we have observed no difference in the incidence of SCI-induced SA in cultures across a wide range of cell densities, whether the nearest neighboring cell is several hundreds of microns away or in clear contact with the sampled neuron, and because the incidence of SA is unchanged by rapid perfusion or no perfusion of the culture dish [8, 11, 106, 107] (and unpublished observations).
Figure 3. Injury-induced SA occurs in NA neurons but not RA neurons. Small DRG neurons from naïve (n = 18), sham (n = 5), and SCI rats (n = 13) were recorded 18–30 hours following dissociation under current clamp without injected current for ≥1 minute to measure SA. (A) Representative recordings of NA neurons from the indicated groups. (B) SA incidence in RA and NA neurons in each group. Fractions represent number of neurons with SA/total sample. Comparisons made using Fisher’s exact test (Bonferroni corrected), **P < 0.01, ****P < 0.0001.
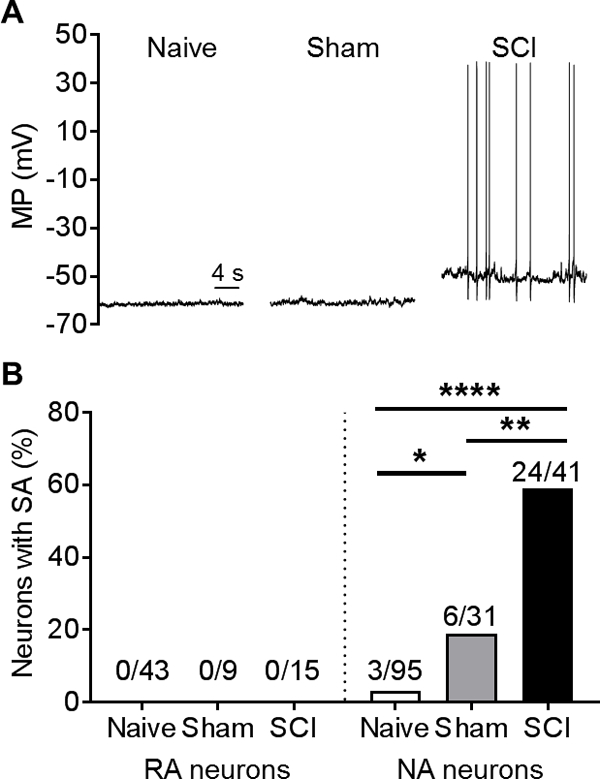
Table 2. Summary of SCI-induced alterations in excitability in NA neurons.
| Property | Naïve | Sham | SCI | Naïve vs Sham |
Naïve vs SCI |
Sham vs SCI |
Test |
|---|---|---|---|---|---|---|---|
| RMP (mV) | −61.9 ± 0.8 (95) | −60.1 ± 1.7 (31) | −54.2 ± 1.5 (41) | P = 0.99 | P < 0.001 | P < 0.05 | KW |
| AP voltage threshold (mV) | −34.0 ± 0.5 (94) | −33.6 ± 1.4 (31) | −37.1 ± 1.0 (39) | P = 0.94 | P < 0.05 | P < 0.05 | ANOVA |
| Rheobase (pA) | 89.1 ± 7.0 (95) | 56.6 ± 9.6 (31) | 44.9 ± 7.0 (41) | P < 0.05 | P < 0.001 | P = 0.99 | KW |
| AP latency at rheobase (ms) | 803.3 ± 82.5 (95) | 616.3 ± 94.4 (31) | 305.3 ± 48.3 (41) | P = 0.99 | P < 0.001 | P < 0.05 | KW |
| Number of APs at rheobase | 1.6 ± 0.1 (95) | 1.6 ± 0.3 (31) | 2.6 ± 0.4 (41) | P = 0.99 | P = 0.14 | P = 0.23 | KW |
| Number of APs at 2x rheobase | 4.0 ± 0.6 (32) | 4.1 ± 0.7 (20) | 6.5 ± 0.9 (22) | P = 0.99 | P < 0.05 | P = 0.17 | KW |
| Membrane resistance (MΩ) | 355.8 ± 24.8 (95) | 480.4 ± 52.6 (31) | 464.5 ± 30.9 (41) | P < 0.05 | P < 0.01 | P = 0.99 | KW |
| Summary of SCI-induced alterations in excitability in RA neurons | |||||||
| Property | Naïve | Sham | SCI |
Naïve vs Sham |
Naïve vs SCI |
Sham vs SCI |
Test |
| RMP (mV) | −65.2 ± 1.1 (43) | −64.9 ± 1.1 (9) | −61.8 ± 2.1 (15) | P = 0.99 | P = 0.25 | P = 0.55 | ANOVA |
| AP voltage threshold (mV) | −28.4 ± 0.9 (42) | −26.1 ± 2.0 (9) | −31.0 ± 1.6 (15) | P = 0.49 | P = 0.57 | P = 0.25 | ANOVA |
| Rheobase (pA) | 169.2 ± 9.4 (43) | 143.9 ± 17.3 (9) | 124.7 ± 22.0 (15) | P = 0.98 | P = 0.06 | P = 0.99 | KW |
| AP latency at rheobase (ms) | 14.6 ± 1.1 (43) | 14.7 ± 2.3 (9) | 24.4 ± 4.4 (15) | P = 0.99 | P = 0.10 | P = 0.93 | KW |
| Number of APs at rheobase | 1.0 ± 0.0 (43) | 1.0 ± 0.0 (9) | 1.0 ± 0.0 (15) | − | − | − | − |
| Number of APs at 2x rheobase | 1.0 ± 0.0 (14) | 0.8 ± 0.2 (5) | 1.0 ± 0.0 (10) | − | − | − | − |
| Membrane resistance (MΩ) | 333.6 ± 29.6 (43) | 785.8 ± 146.3 (7) | 295.9 ± 39.0 (14) | P < 0.01 | P = 0.99 | P < 0.01 | KW |
Data were collected 18–30 hours after dissociation of DRG neurons taken from naïve (n = 18), sham (n = 5), or SCI rats (n = 13). Comparisons were not made between groups for RA neurons in the cases of number of APs at rheobase or at 2x rheobase because repetitive firing did not occur in any RA neuron. Each value is the mean ± SEM followed in parentheses by number of neurons tested. Tests: KW, Kruskal-Wallis followed by Dunn’s tests; ANOVA, 1-way ANOVA followed by Tukey’s tests.
SCI-induced SA in dissociated small DRG neurons has been found up to 8 months post-SCI [11], but previous studies did not distinguish NA from RA neurons. To see if the occurrence of SA in NA neurons changed over the period of the present study, we compared the incidence of SCI-induced SA in neurons from rats tested 1–3 months and 3–6 months post-SCI. The mean incidence of NA neurons with SA at 1–3 months (63 ± 7%, n = 6 rats) was not significantly different from the incidence at 3–6 months (70 ± 3%, n = 5 rats) (P = 0.36, unpaired t-test). We also asked whether SCI might shift one type of probable nociceptor (RA or NA) into the other type. Very little difference was found in the ratio of NA to RA neurons in the naïve (see Section 3.1) or sham groups, so these were combined into a single control group. In this control group, 71% of 143 tested neurons were NA and 29% were RA. In the SCI group, 77% of 198 tested neurons were NA and 23% were RA. The small shift from RA to NA was not statistically significant (P = 0.098), but the possible trend suggests that further investigation is warranted into the question of whether in vivo injury or inflammation might promote a transition of one nociceptor type into the other. The NA/RA ratio was not affected by time post-SCI (82 ± 7% NA neurons at 1–3 months, n = 6 rats; 84 ± 5% NA neurons at 3–6 months, n = 5 rats; P = 0.86, unpaired t-test).
3.3. SCI persistently depolarizes RMP and lowers AP threshold in NA neurons
What are the neurophysiological mechanisms by which SCI promotes SA and OA in NA neurons? Two of the three intrinsic functional aspects of membrane potential that in principle can generate SA (and promote extrinsically driven OA) are prolonged depolarization of RMP and a hyperpolarizing shift in the voltage threshold for AP generation. Persistent SCI-induced depolarization of RMP was found previously in dissociated small DRG neurons [11], but AP voltage threshold was not measured, and whether either of these SA-promoting effects occurs in NA neurons after SCI has not been documented. Compared to NA neurons in the naïve and sham groups, NA neurons in the SCI group showed significant depolarization of RMP and significant reduction in voltage threshold for AP generation (Table 2). No significant differences in these properties were found between the naïve and sham groups. Three other measures also revealed significantly greater excitability in NA neurons in the SCI group versus the naïve group: rheobase dropped by 50%, repetitive firing in response to currents twice the rheobase value nearly doubled, and membrane resistance increased by 30% (Table 2). Interestingly, rheobase and membrane resistance in the sham group were significantly different from values in the naïve group, providing additional evidence for persistent hyperexcitability after sham surgery. No significant effects of SCI were found in RA neurons (Table 2). Fewer RA than NA neurons were examined, so it is possible that weak effects of SCI or sham treatment could be revealed by larger samples of RA neurons. These results show that two physiological alterations important for driving SA, persistent depolarization and reduction of AP voltage threshold, are induced in NA neurons by SCI. All of the measures of hyperexcitability were especially prominent in spontaneously active NA neurons taken from SCI rats (Table 3), consistent with a hyperexcitable state being induced by SCI that functions to promote SA [11]. In addition, sham surgery can also persistently increase excitability of NA neurons, expressed as lowered rheobase, but without substantial alteration of RMP or AP voltage threshold.
Table 3. Comparison of silent and spontaneously active NA neurons taken from SCI rats.
| Property | Silent | Exhibiting SA | Significance | Test |
|---|---|---|---|---|
| RMP (mV) | −61.6 ± 1.8 (17) | −49.0 ± 1.5 (24) | P < 0.0001 | MW |
| AP voltage threshold (mV) | −34.1 ± 1.0 (17) | −39.3 ± 1.5 (22) | P < 0.05 | UPT |
| Rheobase (pA) | 80.3 ± 11.1 (17) | 19.8 ± 4.4 (24) | P < 0.0001 | MW |
| AP latency at rheobase (ms) | 517.8 ± 84.4 (17) | 154.8 ± 32.2 (24) | P < 0.001 | MW |
| Number of APs at rheobase | 1.3 ± 0.2 (17) | 3.5 ± 0.6 (24) | P < 0.01 | MW |
| Number of APs at 2x rheobase | 3.4 ± 0.5 (5) | 7.5 ± 1.1 (17) | P = 0.07 | UPT |
| Membrane resistance (MΩ) | 411.3 ± 47.8 (17) | 502.3 ± 39.5 (24) | P = 0.10 | MW |
Data from a randomly selected subset of the SCI group in Table 2 (n = 8 rats). Each value is the mean ± SEM followed in parentheses by number of neurons tested. Tests: UPT, unpaired t-test; MW, Mann-Whitney U.
3.4. SCI persistently enhances depolarizing spontaneous fluctuations (DSFs) in NA neurons
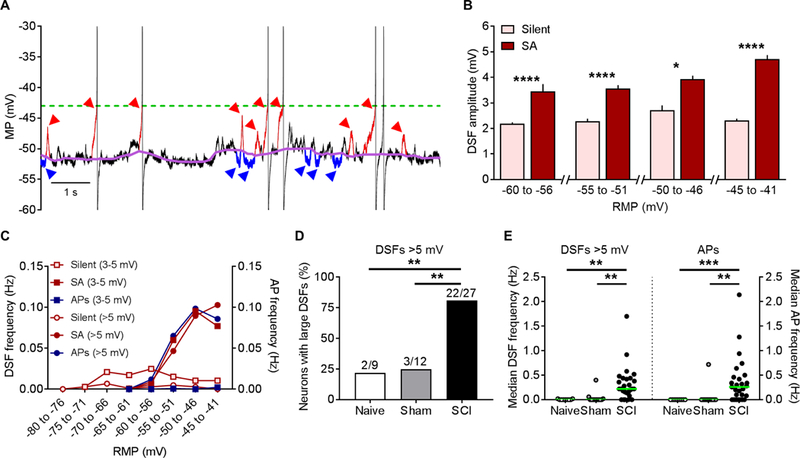
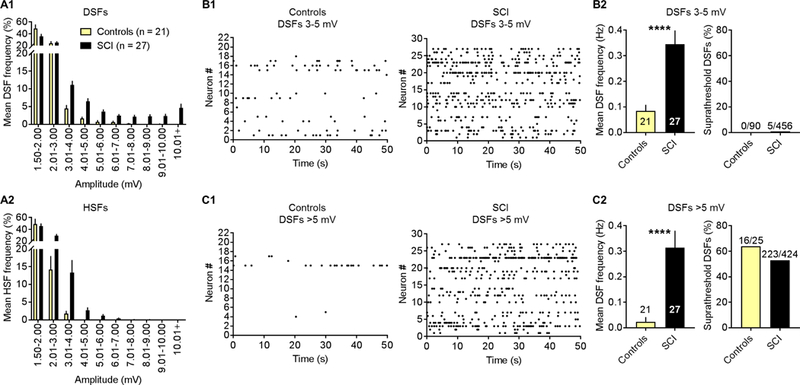
The third functional aspect of membrane potential that in principle can generate SA and promote extrinsically driven OA is an increase in the frequency of large DSFs that can reach AP threshold. Irregular spontaneous fluctuations of membrane potential have long been evident in published whole-cell patch recordings from dissociated small and medium-sized DRG neurons, but they have received remarkably little experimental attention. The most detailed study [87] found no obvious association between fluctuation amplitude and SA in a rat chronic constriction injury (CCI) model of neuropathic pain, but systematic quantitative measurements were not performed. We used two quantitative approaches to test whether SCI increases DSF amplitude and frequency in NA neurons. First, we asked whether total fluctuation amplitude (peak to peak) increased after SCI. Our preliminary results (not shown) indicated that, unlike the regular, sinusoidal oscillations in large and medium-sized DRG neurons that are enhanced by axotomy [2, 3, 54], the irregular fluctuations in small DRG neurons lack large sinusoidal components that contribute significantly to OA generated at RMP negative to −40 mV [see also 3], which is the RMP range where we have investigated SA and OA. Thus, as an alternative to fast Fourier transform analysis, we simply compared the standard deviation of all points (excluding APs and AHPs) in randomly selected 50-second samples in NA cells from each group. Standard deviation provides a symmetrical measure of dispersion of the fluctuations from the mean RMP. The standard deviations (SDs) of fluctuation amplitudes were significantly larger in the SCI group (mean of the fluctuation SDs for each neuron, 3.0 ± 0.3 mV, 27 neurons) than in the naïve group (1.2 ± 0.3 mV, 9 neurons) or sham group (1.1 ± 0.2 mV, 12 neurons) (Tukey’s multiple comparison P < 0.01 in each case). This result shows that SCI increases fluctuation amplitudes but does not distinguish between any differential effects of SCI on depolarizing and hyperpolarizing SFs (DSFs and HSFs).
Plotting all points in each trace relative to the median instead of the mean revealed a skew in the depolarizing direction, raising the possibility that SCI might selectively promote the generation of large DSFs in addition to (or instead of) enhancing oscillatory or hyperpolarizing fluctuations. This is important because HSFs as well as sinusoidal oscillations have been described in isolated DRG neurons [3, 58]. To rigorously test this prediction, we utilized a novel set of automated algorithms for measuring DSFs and HSFs, which were defined by reference to a sliding median of all points measured during 50-second samples (see Methods). An example of part of an analyzed trace is shown in Figure 4A. Note that DSFs are defined operationally and are unlikely to represent unitary events; indeed, there appears to be complex summation of smaller depolarizing (and possibly hyperpolarizing) events in many of the DSFs shown in this and the other illustrations in this paper. Analysis of DSFs in NA neurons exhibiting SA (from naïve, sham, and SCI groups) revealed that mean DSF amplitude was largest (~5 mV) when RMP was between −45 and −40 mV (Fig. 4B). Given that the voltage threshold for AP generation after SCI ranged from −28 to −50 mV, and RMP ranged between −70 and −40 mV (see also Table 2), we predicted that relatively large DSFs (>5 mV) could reach AP threshold often enough to contribute significantly to observed SA. Analysis of NA neurons exhibiting SA showed that the frequency within each trace of DSFs with amplitudes >5 mV (most of which initiated APs; see below) and medium-sized DSFs with amplitudes of 3–5 mV (which almost never evoked APs) showed striking parallels to the frequency of APs in the same neurons plotted as a function of RMP (Fig. 4C). This close parallel provides strong evidence that large DSFs play an important role in triggering APs in NA neurons. Importantly, significantly more NA neurons in the SCI group had large DSFs (>5 mV) than did neurons in the naïve or sham groups (Fig. 4D). Moreover, frequencies both of large DSFs and of APs within each recording were significantly greater in the SCI group (Fig. 4E).
Figure 4. SCI enhances the amplitudes and frequencies of DSFs in NA neurons. DSFs were quantified with an automated algorithm that estimates RMP via a sliding median function, and then identified SFs exceeding 1.5 mV above and below this continuously changing reference line (see Methods). (A) Sample recording of SA following SCI. Color labels: purple undulating line - sliding median, red arrowheads and red trace segments - subthreshold and suprathreshold DSFs ≥3 mV, blue arrowheads and blue trace segments – all HSFs ≥1.5 mV, green dashed line - AP threshold. (B) Neurons with SA (n = 27) showed enhanced DSF amplitudes compared to silent neurons at RMPs between −60 and −40 mV. DSFs were binned according to the RMP at DSF onset. DSF sample sizes left to right: 286, 68, 186, 386, 49, 568, 91, and 425. Data shown as mean ± SEM. Comparisons between silent and SA groups at each bin made using Mann-Whitney U tests. (C) The frequency of medium-amplitude DSFs (3–5 mV, squares) and large DSFs (>5 mV, circles) increased at more depolarized RMPs in neurons with SA (solid symbols) but not in silent neurons (open symbols), paralleling the increase in AP frequency (blue circles). Almost no APs were triggered by medium-sized DSFs (blue squares) in neurons with or without SA. DSFs and APs from neurons in naïve, sham, and SCI conditions were pooled together into silent and SA groups for analysis. Each point represents frequency (Hz) calculated by dividing the total number of DSFs or APs by the number of neurons per group (silent n = 21, SA n = 27) and the recording duration (50 s for each neuron). (D) Large DSF incidence was significantly greater following SCI. Fractions represent number of neurons exhibiting large DSFs/total sample. Comparisons made using Bonferroni-corrrected Fisher’s exact tests. (E) SCI increased the frequency of large DSFs and APs in each neuron. Green lines - medians. Overall significance assessed with Kruskal-Wallis test, multiple comparisons with Dunn’s tests. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Neurons are a randomly selected subset taken from the naïve (n = 2), sham (n = 3), and SCI rats (n = 8) used in Figure 3.
The effects of SCI on SFs are shown in greater detail in Figure 5. A frequency distribution for DSFs and for HSFs was obtained for each trace and averaged across all traces in each group. No apparent differences were found in the incidence or amplitude of either DSFs or HSFs in naïve compared to sham groups, so we pooled these two groups into a single control group for further analysis. Compared to the combined control group, SCI increased the frequency of occurrence of larger DSFs and HSFs, but most of the SCI effect on DSFs was on amplitudes from 3 to >10 mV (Fig. 5A1), whereas most of the effect on HSFs was on amplitudes between 2 and 4 mV (Fig. 5A2). Raster plots showed higher frequencies of medium-amplitude (3–5 mV) (Fig. 5B1, B2) and large (>5 mV) (Fig. 5C1, C2) DSFs in neurons from the SCI group compared to the control group. Almost none of the 3–5 mV DSFs triggered APs in neurons from the SCI or control groups (Fig. 5B2), whereas more than 50% of DSFs >5 mV triggered APs in neurons from SCI and control groups (Fig. 5C2).
Figure 5. SCI enhances the incidence of large and medium-amplitude DSFs and medium-amplitude HSFs in NA neurons. (A) SCI induced a rightward shift in the frequency distribution (% of total) of DSFs (A1) and HSFs (A2) of different amplitudes. Distributions obtained from each neuron for a 50-second period were averaged across neurons; bars represent the mean ± SEM for each amplitude bin. Naïve and sham groups were pooled together into a combined control group. (B1, C1) Medium-amplitude and large DSFs showed stochastic occurrence in control and SCI neurons. Each row represents one neuron and each dot a single DSF. (B2, C2) SCI increased the mean frequency of medium-amplitude and large DSFs in the 50-second samples, but not the fraction of large DSFs that evoked APs. Bars represent the mean ± SEM or fraction of the total sample. Significance tested with Mann-Whitney U or Fisher’s exact tests. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Neurons are the same as in Figure 4.
Large DSFs, like OA observed in vitro and in vivo in presumptive C-fiber nociceptors after SCI [11], occur randomly (Fig. 5C1) [see also 87]. Stochastic DSF occurrence was also seen during the 2-second depolarizations used to measure rheobase and repetitive firing (Fig. 1). A striking finding was the much longer latency to the first AP generated in the rheobase tests in NA compared to RA neurons (Table 1). This is consistent with the AP at rheobase in NA neurons being triggered by infrequent, randomly occurring, large DSFs. If so, the increase in frequency of large DSFs after SCI should increase the likelihood that large DSFs occur early during depolarizing test pulses, and this should decrease the latency to the first AP. Confirming this prediction, the mean latency to the first AP generated in NA neurons during rheobase measurement in the SCI group was much shorter than the latency in the naïve or sham groups (Table 2). Together, these findings show 1) that DSFs play a major role in generating the irregular SA found in NA neurons, and 2) that enhancement of DSF amplitude and large DSF frequency contributes to SCI-induced SA.
3.5. DSFs and OA can be potentiated acutely by an inflammatory mediator, serotonin
Is enhancement of DSFs and the consequent promotion of nociceptor activity solely a long-term phenomenon, perhaps unique to SCI, or can nociceptor DSFs also be enhanced acutely by extrinsic signals? In particular, could acute exposure to an inflammatory signal enhance DSFs and promote OA? To address this question, we used serotonin (5-HT), an inflammatory mediator that in the periphery can induce pain and hyperalgesia [e.g., 78, 84, 89]. 5-HT is interesting because it has complex effects on nociceptors [44, 53, 77, 79, 90, 118], one of which is to reduce AP voltage threshold [17]. In contrast to nearly all other studies of 5-HT’s actions on nociceptors, which utilized very high 5-HT concentrations (typically 10 μM), an early study showed that 10 nM 5-HT caused alterations in tetrodotoxin-resistant Na+ current that should lower AP threshold [34]. An implication of this observation is that a 5-HT concentration that modulates but does not activate NA nociceptors could potentiate depolarization-dependent OA in NA nociceptors. Potentiation of such OA would be much more likely if the same concentration of 5-HT also enhances DSFs. We tested this possibility in NA nociceptors dissociated from naïve rats.
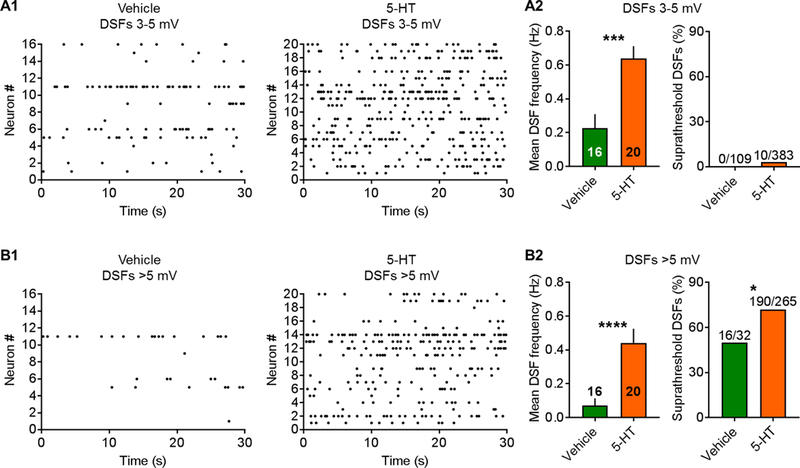
Treatment of each dish with 100 nM 5-HT for 10–30 minutes before and during recording produced no hint of sustained depolarization (Table 4). Furthermore, 5-HT did not induce OA at RMP (Fig. 6A, left panel). When a prolonged extrinsic depolarizing input (modeled by constant current injection through the patch pipette to hold the membrane potential at ~−45 mV for 30–60 seconds) was added to promote OA after vehicle treatment, no significant increase in the incidence of OA was found versus the incidence of SA at RMP (compare vehicle groups in left and right panels of Fig. 6A). In contrast, when 5-HT-treated nociceptors were depolarized to −45 mV, ~80% showed OA (Fig. 6A, right panel). When depolarized to −45 mV, AP firing rates during OA and the corresponding large DSF frequencies were significantly greater in 5-HT-treated neurons than vehicle-treated neurons (Fig. 6B). Amplitudes of DSFs ≥ 1.5 mV were also enhanced in 5-HT-treated neurons that were depolarized to −45 mV (Fig. 6C, left panel), and like the effects of SCI (Fig. 4B), the DSFs were largest in neurons with OA (Fig. 6C, right panel). Examples of DSFs and APs (OA) in NA neurons held at −45 mV with and without 5-HT treatment are shown in Figure 6D. DSFs occurred randomly after either vehicle treatment or 5-HT treatment (Fig. 6D, 7A1, 7B1). 5-HT increased the number of medium-amplitude (3–5 mV) and large (>5 mV) DSFs during each recording (Fig. 7A2, B2). The number of large DSFs paralleled the number of APs evoked during the same 30-second samples (Fig. 7B2). As predicted [17], 5-HT treatment also significantly (and substantially) lowered the voltage threshold for AP generation (Table 4). This likely contributed to the increased percentage of DSFs 3–5 mV and especially >5 mV that triggered APs (Fig. 7A2, B2). In addition, 5-HT treatment significantly decreased the rheobase (consistent with an increase in the frequency of large DSFs) (Table 4). In contrast to the effect of SCI on AP latency at rheobase, 5-HT did not decrease AP latency. However, because of the low frequency and stochastic occurrence of APs (and underlying DSFs), demonstrating possible effects on AP latency is likely to require a much larger sample size.
Table 4. Effects of 5-HT on NA neurons.
| Property | Vehicle | 5-HT | Significance | Test |
|---|---|---|---|---|
| RMP (mV) | −67.1 ± 1.6 (16) | −66.7 ± 1.3 (21) | P = 0.85 | UPT |
| AP voltage threshold (mV) | −34.0 ± 1.0 (16) | −40.0 ± 1.1 (21) | P < 0.001 | UPT |
| Rheobase (pA) | 92.5 ± 12.1 (16) | 50.2 ± 7.8 (21) | P < 0.01 | UPT |
| AP latency at rheobase (ms) | 651.5 ± 159.3 (16) | 750.8 ± 135.2 (21) | P = 0.46 | MW |
| Number of APs at rheobase | 1.1 ± 0.1 (16) | 1.3 ± 0.2 (21) | P = 0.25 | MW |
| Number of APs at 2x rheobase | 4.6 ± 1.3 (13) | 5.4 ± 0.8 (21) | P = 0.31 | MW |
| Membrane resistance (MΩ) | 350.5 ± 50.9 (16) | 298.5 ± 32.5 (21) | P = 0.66 | MW |
| Effects of 5-HT on RA neurons | ||||
| Property | Vehicle | 5-HT | Significance | Test |
| RMP (mV) | −68.7 ± 2.1 (10) | −63.0 ± 3.3 (9) | P = 0.15 | UPT |
| AP voltage threshold (mV) | −31.4 ± 1.6 (10) | −35.7 ± 1.8 (9) | P = 0.20 | MW |
| Rheobase (pA) | 154.0 ± 24.1 (10) | 123.9 ± 26.7 (9) | P = 0.59 | MW |
| AP latency at rheobase (ms) | 17.0 ± 3.1 (10) | 18.6 ± 3.7 (9) | P = 0.97 | MW |
| Number of APs at rheobase | 1.0 ± 0.0 (10) | 1.0 ± 0.0 (9) | − | − |
| Number of APs at 2x rheobase | 1.0 ± 0.0 (7) | 1.0 ± 0.0 (7) | − | − |
| Membrane resistance (MΩ) | 266.6 ± 37.5 (10) | 269.9 ± 35.1 (9) | P = 0.99 | MW |
Tests were conducted in the presence of 5-HT or vehicle applied 10–30 minutes earlier onto small DRG neurons taken from naïve rats (n = 4). Comparisons were not made between groups for RA neurons in the cases of number of APs at rheobase or at 2x rheobase because repetitive firing did not occur in any RA neuron.
Figure 6. Potentiation by 5-HT of OA in NA neurons. DRG neurons from naïve rats (n = 4) were treated with vehicle or 100 nM 5-HT for 10–30 minutes before and during each recording. Following measurement of any SA, extrinsically driven OA was modeled by depolarization to −45 mV under current clamp for 30–60 seconds. (A) Pretreatment with 5-HT did not induce OA at RMP but significantly increased OA at −45 mV (Fisher’s exact test). (B) In neurons tested at −45 mV, 5-HT significantly increased AP frequency during OA and large DSF frequency. Black lines - medians. Comparisons made using Mann-Whitney U tests. (C) 5-HT increased the amplitude of DSFs measured at −45 mV, and the neurons with OA showed larger DSFs than silent neurons. DSF sample sizes left to right: 1360, 2113, 1256, and 2217. Data shown as mean ± SEM. Comparisons between vehicle- and 5-HT-treatments or silent and OA groups made using Mann-Whitney U tests.P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (D) Representative recordings SFs and OA at −45 mV after treatment with vehicle or 5-HT. Insets: enlarged sections from each trace.
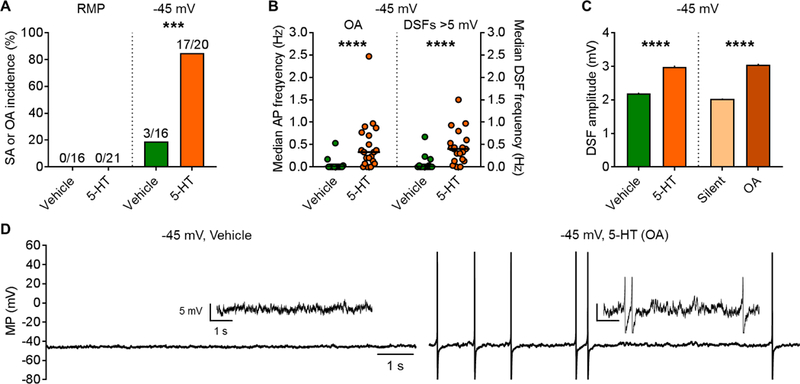
Figure 7. Treatment with 5-HT before and during recording at −45 mV increases the number of medium-amplitude and large DSFs in NA neurons from naïve rats. (A1, B1) Raster plots of medium-amplitude and large DSFs during depolarization to −45 mV. Each row represents one neuron and each point a single DSF. (A2, B2) At −45 mV, 5-HT increased the number of medium-amplitude and large DSFs, and the percentage of DSFs evoking APs. Bars represent the mean ± SEM or fraction in total sample, and significance was tested using Mann-Whitney U or Fisher’s exact tests. *P < 0.05, **P < 0.01, ****P < 0.0001. Neurons are from the naïve rats (n = 4) used in Figure 6.
In the RA neurons, pretreatment with 5-HT did not induce OA at RMP in any of the neurons tested, and comparisons between vehicle- and 5-HT-treated RA neurons did not reveal significant changes in excitability, although there was a trend for depolarization of RMP (Table 4). This suggests that RA neurons, unlike NA neurons, are not sensitive to low concentrations of 5-HT.
These findings show that acute enhancement of DSFs along with reduction of AP threshold by an inflammatory signal can strongly potentiate depolarization-dependent OA in NA nociceptors under conditions where the inflammatory signal does not by itself cause depolarization or produce OA.
4. Discussion
4.1. Definitions of OA and SA
Formal definitions of neuronal OA and SA are lacking in the pain research field. We think a useful distinction is implicit in everyday meanings of “ongoing” and “spontaneous.” Thus, we define OA generally as continuing discharge of APs driven by any intrinsic and/or extrinsic sources of ongoing excitation. We define SA as a subclass of OA that is generated solely by alterations intrinsic to the active neuron, which can only be demonstrated conclusively if the active neuron is isolated from extrinsic drivers of activity. OA observed in vivo may involve both extrinsically driven OA and intrinsic SA.
4.2. Neurons and physiological specializations mediating SA and OA
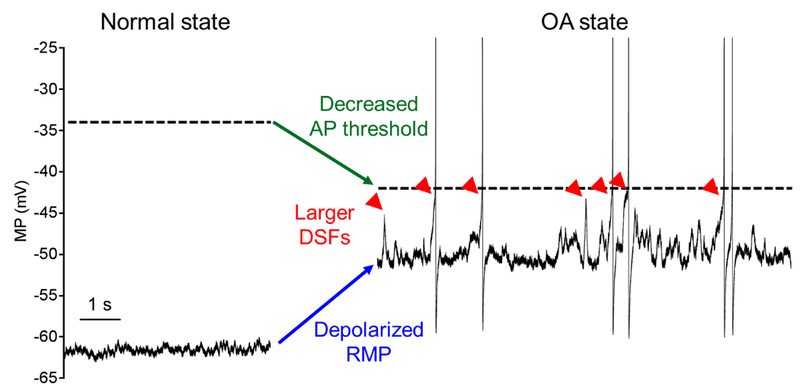
We found that SCI changed each of the three intrinsic functional aspects of membrane potential that in principle can drive SA and promote extrinsically driven OA. SCI promoted an OA state characterized by: 1) prolonged depolarization of RMP, 2) a hyperpolarizing shift in the voltage threshold for AP generation, and 3) an increase in the incidence of large transient DSFs (Fig. 8). Acute 5-HT treatment was found to produce the latter two changes. OA and all three physiological alterations promoting it was found only in NA neurons. Extensive examination of RMP, voltage threshold, and DSFs in RA neurons after SCI showed no hint of an effect on RMP or DSFs. However, a trend for reduction of AP threshold was found in RA neurons, which may have contributed to the significant reduction in rheobase of RA neurons after SCI. If reduction in RA and/or NA neuron AP threshold occurs near sensory terminals, this could contribute to the peripheral hypersensitivity observed after SCI that is likely to promote evoked pain [11, 18].
Figure 8. Summary of neurophysiological specializations that promote OA in NA nociceptors. OA can be entirely intrinsic and thus completely spontaneous (denoted as SA) or extrinsically driven. Nociceptor OA in vivo may be driven by acute or ongoing exposure to extrinsic drivers of activity, sometimes combined with long-lasting intrinsic alterations. Representative recordings from two NA neurons illustrate the normal inactive state (sample from a naïve rat) and the OA state (SA sample from an SCI rat). Compared to the normal state, the OA state is marked by depolarized RMP (blue arrow), decreased voltage threshold for APs (green arrow), and increased frequency of larger DSFs (red arrowheads indicate DSFs > 5 mV, which are highly likely to elicit APs). Both the inter-DSF intervals and interspike intervals between APs are irregular in the OA state and the discharge does not accommodate.
Under our in vitro conditions, NA neurons were twice as common as RA neurons. Despite dramatically different responses in firing evoked by depolarization and differences in RMP and membrane time constant, the two types shared many properties. No significant differences were found in soma size or membrane capacitance, although a trend for RA neurons to be slightly larger suggests this class may include some Aδ neurons. About 70% of sampled NA and RA neurons exhibited capsaicin sensitivity and/or IB4 binding, indicating that each type is largely nociceptive. Both types may also include non-nociceptive neurons and some nociceptors that could appear mechanically insensitive in vivo (as do many human C-fiber nociceptors displaying OA) [e.g., 64, 46, 82]. The similarities of NA and RA types raise the possibility that the two types could be different functional states rather than stable phenotypes. Indeed, there may have been a weak trend for SCI to increase the ratio of NA to RA neurons. It will be important to map these electrophysiologically defined types onto the large number of functionally and molecularly defined classes of nociceptors [e.g., 31, 92], and to test systematically the possibility that transitions can occur between NA and RA types.
A possibility that must be considered is that the NA type is enriched by the stressful procedures involved in DRG removal, neuronal dissociation, and whole-cell recording – either because NA neurons are more likely to survive these procedures or because injury-related stresses promote a transition to the NA state. Dissociation can increase repetitive firing and/or OA in invertebrate and mammalian nociceptors [52, 120]. However, the naïve group in the present study and our other SCI studies showed much lower SA incidence in vitro than did the SCI group. Moreover, a high incidence of OA after SCI was also found both in vitro and in vivo -- generated by C-fibers in or near the DRG [11]. This suggests that OA mechanisms defined in dissociated nociceptors are sufficiently similar to those operating in vivo to provide detailed insights that can guide in vivo investigations. Our unexpected finding that sham surgery (which injures deep tissues, including muscle and bone) moderately enhanced SA and hyperexcitability is consistent with the idea that diverse injury-related stresses can be detected by nociceptors to promote entry into a hyperactive state, with the probability of entry into this state increasing with the severity of injury [97].
A significant discovery was that enhancement of nociceptor DSFs can be a major contributor to OA. Large, randomly occurring fluctuations of RMP have long been noticed in small and medium-sized DRG neurons, and they were proposed to explain the irregular firing patterns characteristic of nociceptor OA more than two decades ago [87], which our observations confirm. However, that study attributed the generation of OA after CCI entirely to the observed reduction in AP threshold [87]. In contrast, our finding that SCI caused an increase in frequency of large DSFs that closely paralleled the frequency of APs during SA shows that enhancement of DSFs during a neuropathic pain condition plays a major role in OA. The biophysical and cell signaling mechanisms that generate and enhance DSFs have not been described and are under investigation. These mechanisms are likely to be complex, in part because they interact with complementary mechanisms that reduce AP threshold and depolarize RMP [8]. Identification of DSF mechanisms may help in the development of more targeted treatments for ongoing pain.
4.3. Functional implications of nociceptor specializations for OA
Our findings indicate that nociceptor specializations for OA are likely to contribute to forms of ongoing pain other than SCI pain. In particular, 5-HT in combination with extrinsic depolarization acutely potentiated OA by inducing two of the three physiological changes that can drive SA. This finding indicates that intrinsic mechanisms that generate SA chronically after SCI can also promote acute OA when engaged by extrinsic input. It is likely that combinations of all three physiological changes drive nociceptor OA in many ongoing pain conditions. Nociceptors are modulated by numerous inflammatory mediators, extrinsic damage-associated molecular patterns, and neuromodulators, some of which cause multiple sensitizing or excitatory effects [33, 41, 59, 98, 99]. Given the plethora of signals released for long periods during significant injury and/or inflammation, it seems likely that depolarization, lowering of AP threshold, and enhancement of DSFs are produced simultaneously by combined actions of extrinsic signals and intrinsic alterations to drive persistent OA in NA nociceptors and consequent ongoing pain in many conditions.
The selective expression within NA nociceptors of multiple physiological processes that intrinsically promote OA and synergize with extrinsic inflammatory inputs suggests that nociceptor OA in neuropathic conditions need not be a purely pathological side-effect of neuropathy, as is sometimes suggested [e.g., 21]. Nociceptor OA may represent a natural function mediated by complementary specializations within a distinct NA type (or NA state) that maintains OA under injury- and inflammation-related conditions. These specializations may also promote somally generated OA when the soma is disconnected traumatically from peripheral terminals. While purely pathological forms of nociceptor OA certainly exist [e.g., 67, 62, 47, 76], these are likely to engage OA mechanisms that evolved for adaptive functions. A plausible adaptive function for OA is to maintain continuous protective awareness of severely injured (and thus highly vulnerable) tissue [22, 96, 97]. Thus, persistent nociceptor OA, by continuously driving vigilance and guarding behavior, would complement the protection afforded by sensitization and allodynia.
4.4. Nociceptor OA is likely to promote ongoing pain
Most research on pain-related OA in rodent sensory neurons has measured allodynia rather than ongoing pain, and has focused on allodynic consequences of OA generated in Aβ-fibers that are usually non-nociceptive [24, 28, 55, 71, 88, 112]. In contrast, studies of OA in human sensory neurons have largely focused on nociceptor OA. Microneurographic recordings of intact peripheral nerves have shown strong associations between self-reports of ongoing pain and OA in C-fiber units in patients with peripheral neuropathy (including diabetic, chemotherapy-induced, systemic lupus erythematosus, and idiopathic) or fibromyalgia [46, 64, 67, 81, 82]. While the low signal-to-noise ratio of this technique has not allowed precise determination of the rates and patterns of firing, the human OA appears to be of low frequency and irregular pattern, and it resembles C-fiber OA measured with the same methods in rat neuropathy models [81]. These and other rodent neuropathy models, have revealed low-frequency, irregular C-fiber OA [1, 13, 14, 26, 27, 35, 45, 101, 105, 107, 109, 113]. Evidence that this OA drives ongoing pain in rodents comes from correlations between nociceptor OA and spontaneous foot lifting after inflammation or nerve injury [27], from conditioned preference for a place (CPP) paired with blockade of afferent activity generated at sites of injury or inflammation [19, 39, 65, 66], and from blocking CPP after SCI by knockdown of a sensory neuron-specific Na+ channel that is expressed primarily in nociceptors [117]. Some of the human and rodent nociceptor OA may arise within somata in DRGs, as we have found in our rat SCI model [11]. Suggestive evidence that injury-related ongoing pain in humans can be driven by nociceptor OA generated within DRGs has come from relief of amputation pain in patients produced by local delivery of lidocaine to the DRG [93]. Thus, mechanisms found to drive OA in dissociated rodent nociceptor somata may provide insight into mechanisms that promote ongoing pain in humans.
Supplementary Material
Acknowledgements
We thank Guo-Ying Xu, Kendra Wicks, and Tamara McGhee for technical assistance. This work was supported by National Institute of Neurological Diseases and Stroke Grant NS091759 to C.W.D. and E.T.W., US Army Medical Research Grant W81XWH-12–1-0504 to E.T.W., and a Zilkha Family Fellowship to M.A.O.
Footnotes
The authors declare no competing conflicts of interest.
References
- [1].Ali Z, Ringkamp M, Hartke TV, Chien HF, Flavahan NA, Campbell JN, Meyer RA. Uninjured C-fiber nociceptors develop spontaneous activity and alpha-adrenergic sensitivity following L6 spinal nerve ligation in monkey. J Neurophysiol 1999;81:455–466. [DOI] [PubMed] [Google Scholar]
- [2].Amir R, Liu CN, Kocsis JD, Devor M. Oscillatory mechanism in primary sensory neurones. Brain 2002;125:421–435. [DOI] [PubMed] [Google Scholar]
- [3].Amir R, Michaelis M, Devor M. Membrane potential oscillations in dorsal root ganglion neurons: role in normal electrogenesis and neuropathic pain. J Neurosci 1999;19:8589–8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Amir R, Michaelis M, Devor M. Burst discharge in primary sensory neurons: triggered by subthreshold oscillations, maintained by depolarizing afterpotentials. J Neurosci 2002;22:1187–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Baccaglini PI, Hogan PG. Some rat sensory neurons in culture express characteristics of differentiated pain sensory cells. Proc Natl Acad Sci U S A 1983;80:594–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Baron R, Hans G, Dickenson AH. Peripheral input and its importance for central sensitization. Ann Neurol 2013;74:630–636. [DOI] [PubMed] [Google Scholar]
- [7].Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma 1995;12:1–21. [DOI] [PubMed] [Google Scholar]
- [8].Bavencoffe A, Li Y, Wu Z, Yang Q, Herrera J, Kennedy EJ, Walters ET, Dessauer CW. Persistent Electrical Activity in Primary Nociceptors after Spinal Cord Injury Is Maintained by Scaffolded Adenylyl Cyclase and Protein Kinase A and Is Associated with Altered Adenylyl Cyclase Regulation. J Neurosci 2016;36:1660–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Beaudry H, Daou I, Ase AR, Ribeiro-da-Silva A, Séguéla P. Distinct behavioral responses evoked by selective optogenetic stimulation of the major TRPV1+ and MrgD+ subsets of C-fibers. Pain 2017;158:2329–2339. [DOI] [PubMed] [Google Scholar]
- [10].Bedi SS, Lago MT, Masha LI, Crook RJ, Grill RJ, Walters ET. Spinal cord injury triggers an intrinsic growth-promoting state in nociceptors. J Neurotrauma 2012;29:925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bedi SS, Yang Q, Crook RJ, Du J, Wu Z, Fishman HM, Grill RJ, Carlton SM, Walters ET. Chronic spontaneous activity generated in the somata of primary nociceptors is associated with pain-related behavior after spinal cord injury. J Neurosci 2010;30:14870–14882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bennett GJ. What is spontaneous pain and who has it. J Pain 2012;13:921–929. [DOI] [PubMed] [Google Scholar]
- [13].Bernal L, Lopez-Garcia JA, Roza C. Spontaneous activity in C-fibres after partial damage to the saphenous nerve in mice: Effects of retigabine. Eur J Pain 2016;20:1335–1345. [DOI] [PubMed] [Google Scholar]
- [14].Bove GM. Focal nerve inflammation induces neuronal signs consistent with symptoms of early complex regional pain syndromes. Exp Neurol 2009;219:223–227. [DOI] [PubMed] [Google Scholar]
- [15].Bromm B, Treede RD. Nerve fibre discharges, cerebral potentials and sensations induced by CO2 laser stimulation. Hum Neurobiol 1984;3:33–40. [PubMed] [Google Scholar]
- [16].Burchiel KJ. Effects of electrical and mechanical stimulation on two foci of spontaneous activity which develop in primary afferent neurons after peripheral axotomy. Pain 1984;18:249–265. [DOI] [PubMed] [Google Scholar]
- [17].Cardenas LM, Cardenas CG, Scroggs RS. 5HT increases excitability of nociceptor-like rat dorsal root ganglion neurons via cAMP-coupled TTX-resistant Na(+) channels. J Neurophysiol 2001;86:241–248. [DOI] [PubMed] [Google Scholar]
- [18].Carlton SM, Du J, Tan HY, Nesic O, Hargett GL, Bopp AC, Yamani A, Lin Q, Willis WD, Hulsebosch CE. Peripheral and central sensitization in remote spinal cord regions contribute to central neuropathic pain after spinal cord injury. Pain 2009;147:265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen J, Winston JH, Fu Y, Guptarak J, Jensen KL, Shi XZ, Green TA, Sarna SK. Genesis of anxiety, depression, and ongoing abdominal discomfort in ulcerative colitis-like colon inflammation. Am J Physiol Regul Integr Comp Physiol 2015;308:R18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Choi JS, Dib-Hajj SD, Waxman SG. Differential slow inactivation and use-dependent inhibition of Nav1.8 channels contribute to distinct firing properties in IB4+ and IB4- DRG neurons. J Neurophysiol 2007;97:1258–1265. [DOI] [PubMed] [Google Scholar]
- [21].Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci 2009;32:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Crook RJ, Hanlon RT, Walters ET. Squid have nociceptors that display widespread long-term sensitization and spontaneous activity after bodily injury. J Neurosci 2013;33:10021–10026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Daou I, Tuttle AH, Longo G, Wieskopf JS, Bonin RP, Ase AR, Wood JN, De Koninck Y, Ribeiro-da-Silva A, Mogil JS, Seguela P. Remote optogenetic activation and sensitization of pain pathways in freely moving mice. J Neurosci 2013;33:18631–18640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Devor M Ectopic discharge in Abeta afferents as a source of neuropathic pain. Exp Brain Res 2009;196:115–128. [DOI] [PubMed] [Google Scholar]
- [25].Djouhri L Electrophysiological evidence for the existence of a rare population of C-fiber low threshold mechanoreceptive (C-LTM) neurons in glabrous skin of the rat hindpaw. Neurosci Lett 2016;613:25–29. [DOI] [PubMed] [Google Scholar]
- [26].Djouhri L, Fang X, Koutsikou S, Lawson SN. Partial nerve injury induces electrophysiological changes in conducting (uninjured) nociceptive and nonnociceptive DRG neurons: Possible relationships to aspects of peripheral neuropathic pain and paresthesias. Pain 2012;153:1824–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Djouhri L, Koutsikou S, Fang X, McMullan S, Lawson SN. Spontaneous pain, both neuropathic and inflammatory, is related to frequency of spontaneous firing in intact C-fiber nociceptors. J Neurosci 2006;26:1281–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Djouhri L, Lawson SN. Abeta-fiber nociceptive primary afferent neurons: a review of incidence and properties in relation to other afferent A-fiber neurons in mammals. Brain Res Brain Res Rev 2004;46:131–145. [DOI] [PubMed] [Google Scholar]
- [29].Dong H, Fan YH, Wang YY, Wang WT, Hu SJ. Lidocaine suppresses subthreshold oscillations by inhibiting persistent Na(+) current in injured dorsal root ganglion neurons. Physiol Res 2008;57:639–645. [DOI] [PubMed] [Google Scholar]
- [30].Douglas DH, Peucker TK. Algorithms for the reduction of the number of points required to represent a digitized line or its caricature. Cartographica: The International Journal for Geographic Information and Geovisualization 1973;10:112–122. [Google Scholar]
- [31].Fang X, McMullan S, Lawson SN, Djouhri L. Electrophysiological differences between nociceptive and non-nociceptive dorsal root ganglion neurones in the rat in vivo. J Physiol 2005;565:927–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gold MS, Dastmalchi S, Levine JD. Co-expression of nociceptor properties in dorsal root ganglion neurons from the adult rat in vitro. Neuroscience 1996;71:265–275. [DOI] [PubMed] [Google Scholar]
- [33].Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nat Med 2010;16:1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci U S A 1996;93:1108–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gorodetskaya N, Constantin C, Janig W. Ectopic activity in cutaneous regenerating afferent nerve fibers following nerve lesion in the rat. Eur J Neurosci 2003;18:2487–2497. [DOI] [PubMed] [Google Scholar]
- [36].Gracely RH, Lynch SA, Bennett GJ. Painful neuropathy: altered central processing maintained dynamically by peripheral input. Pain 1992;51:175–194. [DOI] [PubMed] [Google Scholar]
- [37].Haroutounian S, Nikolajsen L, Bendtsen TF, Finnerup NB, Kristensen AD, Hasselstrom JB, Jensen TS. Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain 2014;155:1272–1279. [DOI] [PubMed] [Google Scholar]
- [38].Harper AA. Similarities between some properties of the soma and sensory receptors of primary afferent neurones. Exp Physiol 1991;76:369–377. [DOI] [PubMed] [Google Scholar]
- [39].Havelin J, Imbert I, Cormier J, Allen J, Porreca F, King T. Central Sensitization and Neuropathic Features of Ongoing Pain in a Rat Model of Advanced Osteoarthritis. J Pain 2016;17:374–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hu J, Lewin GR. Mechanosensitive currents in the neurites of cultured mouse sensory neurones. J Physiol 2006;577:815–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science 2016;354:572–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kajander KC, Wakisaka S, Bennett GJ. Spontaneous discharge originates in the dorsal root ganglion at the onset of a painful peripheral neuropathy in the rat. Neurosci Lett 1992;138:225–228. [DOI] [PubMed] [Google Scholar]
- [43].Kelly S, Dunham JP, Murray F, Read S, Donaldson LF, Lawson SN. Spontaneous firing in C-fibers and increased mechanical sensitivity in A-fibers of knee joint-associated mechanoreceptive primary afferent neurones during MIA-induced osteoarthritis in the rat. Osteoarthritis Cartilage 2012;20:305–313. [DOI] [PubMed] [Google Scholar]
- [44].Khomula EV, Ferrari LF, Araldi D, Levine JD. Sexual Dimorphism in a Reciprocal Interaction of Ryanodine and IP3 Receptors in the Induction of Hyperalgesic Priming. J Neurosci 2017;37:2032–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kirillova I, Rausch VH, Tode J, Baron R, Janig W. Mechano- and thermosensitivity of injured muscle afferents. J Neurophysiol 2011;105:2058–2073. [DOI] [PubMed] [Google Scholar]
- [46].Kleggetveit IP, Namer B, Schmidt R, Helås T, Rückel M, Ørstavik K, Schmelz M, Jørum E. High spontaneous activity of C-nociceptors in painful polyneuropathy. Pain 2012;153:2040–2047. [DOI] [PubMed] [Google Scholar]
- [47].Kleggetveit IP, Schmidt R, Namer B, Salter H, Helås T, Schmelz M, Jørum E. Pathological nociceptors in two patients with erythromelalgia-like symptoms and rare genetic Nav 1.9 variants. Brain Behav 2016;6:e00528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kovalsky Y, Amir R, Devor M. Subthreshold oscillations facilitate neuropathic spike discharge by overcoming membrane accommodation. Exp Neurol 2008;210:194–206. [DOI] [PubMed] [Google Scholar]
- [49].Kovalsky Y, Amir R, Devor M. Simulation in sensory neurons reveals a key role for delayed Na+ current in subthreshold oscillations and ectopic discharge: implications for neuropathic pain. J Neurophysiol 2009;102:1430–1442. [DOI] [PubMed] [Google Scholar]
- [50].Kuner R, Flor H. Structural plasticity and reorganisation in chronic pain. Nat Rev Neurosci 2016;18:20–30. [DOI] [PubMed] [Google Scholar]
- [51].Lawson SN, McCarthy PW, Prabhakar E. Electrophysiological properties of neurones with CGRP-like immunoreactivity in rat dorsal root ganglia. J Comp Neurol 1996;365:355–366. [DOI] [PubMed] [Google Scholar]
- [52].Liao X, Gunstream JD, Lewin MR, Ambron RT, Walters ET. Activation of protein kinase A contributes to the expression but not the induction of long-term hyperexcitability caused by axotomy of Aplysia sensory neurons. J Neurosci 1999;19:1247–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lin SY, Chang WJ, Lin CS, Huang CY, Wang HF, Sun WH. Serotonin receptor 5-HT2B mediates serotonin-induced mechanical hyperalgesia. J Neurosci 2011;31:1410–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Liu CN, Devor M, Waxman SG, Kocsis JD. Subthreshold oscillations induced by spinal nerve injury in dissociated muscle and cutaneous afferents of mouse DRG. J Neurophysiol 2002;87:2009–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Liu CN, Wall PD, Ben-Dor E, Michaelis M, Amir R, Devor M. Tactile allodynia in the absence of C-fiber activation: altered firing properties of DRG neurons following spinal nerve injury. Pain 2000;85:503–521. [DOI] [PubMed] [Google Scholar]
- [56].Lundberg LE, Jørum E, Holm E, Torebjörk HE. Intra-neural electrical stimulation of cutaneous nociceptive fibres in humans: effects of different pulse patterns on magnitude of pain. Acta Physiol Scand 1992;146:41–48. [DOI] [PubMed] [Google Scholar]
- [57].Marchettini P, Simone DA, Caputi G, Ochoa JL. Pain from excitation of identified muscle nociceptors in humans. Brain Res 1996;740:109–116. [DOI] [PubMed] [Google Scholar]
- [58].Mathers DA, Barker JL. Spontaneous voltage and current fluctuations in tissue cultured mouse dorsal root ganglion cells. Brain Res 1984;293:35–47. [DOI] [PubMed] [Google Scholar]
- [59].McMahon SB, La Russa F, Bennett DL. Crosstalk between the nociceptive and immune systems in host defence and disease. Nat Rev Neurosci 2015;16:389–402. [DOI] [PubMed] [Google Scholar]
- [60].Meyer RA, Raja SN, Campbell JN, Mackinnon SE, Dellon AL. Neural activity originating from a neuroma in the baboon. Brain Res 1985;325:255–260. [DOI] [PubMed] [Google Scholar]
- [61].Michaelis M, Blenk KH, Janig W, Vogel C. Development of spontaneous activity and mechanosensitivity in axotomized afferent nerve fibers during the first hours after nerve transection in rats. J Neurophysiol 1995;74:1020–1027. [DOI] [PubMed] [Google Scholar]
- [62].Namer B, Ørstavik K, Schmidt R, Kleggetveit IP, Weidner C, Mørk C, Kvernebo MS, Kvernebo K, Salter H, Carr TH, Segerdahl M, Quiding H, Waxman SG, Handwerker HO, Torebjörk HE, Jørum E, Schmelz M. Specific changes in conduction velocity recovery cycles of single nociceptors in a patient with erythromelalgia with the I848T gain-of-function mutation of Nav1.7. Pain 2015;156:1637–1646. [DOI] [PubMed] [Google Scholar]
- [63].Ochoa J, Torebjörk E. Sensations evoked by intraneural microstimulation of C nociceptor fibres in human skin nerves. J Physiol 1989;415:583–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ochoa JL, Campero M, Serra J, Bostock H. Hyperexcitable polymodal and insensitive nociceptors in painful human neuropathy. Muscle Nerve 2005;32:459–472. [DOI] [PubMed] [Google Scholar]
- [65].Okun A, DeFelice M, Eyde N, Ren J, Mercado R, King T, Porreca F. Transient inflammation-induced ongoing pain is driven by TRPV1 sensitive afferents. Mol Pain 2011;7:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Okun A, Liu P, Davis P, Ren J, Remeniuk B, Brion T, Ossipov MH, Xie J, Dussor GO, King T, Porreca F. Afferent drive elicits ongoing pain in a model of advanced osteoarthritis. Pain 2012;153:924–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Orstavik K, Namer B, Schmidt R, Schmelz M, Hilliges M, Weidner C, Carr RW, Handwerker H, Jorum E, Torebjork HE. Abnormal function of C-fibers in patients with diabetic neuropathy. J Neurosci 2006;26:11287–11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Patrick Harty T, Waxman SG. Inactivation properties of sodium channel Nav1.8 maintain action potential amplitude in small DRG neurons in the context of depolarization. Mol Pain 2007;3:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Petruska JC, Napaporn J, Johnson RD, Cooper BY. Chemical responsiveness and histochemical phenotype of electrophysiologically classified cells of the adult rat dorsal root ganglion. Neuroscience 2002;115:15–30. [DOI] [PubMed] [Google Scholar]
- [70].Petruska JC, Napaporn J, Johnson RD, Gu JG, Cooper BY. Subclassified acutely dissociated cells of rat DRG: histochemistry and patterns of capsaicin-, proton-, and ATP-activated currents. J Neurophysiol 2000;84:2365–2379. [DOI] [PubMed] [Google Scholar]
- [71].Pitcher GM, Henry JL. Cellular mechanisms of hyperalgesia and spontaneous pain in a spinalized rat model of peripheral neuropathy: changes in myelinated afferent inputs implicated. Eur J Neurosci 2000;12:2006–2020. [DOI] [PubMed] [Google Scholar]
- [72].Pitcher GM, Henry JL. Governing role of primary afferent drive in increased excitation of spinal nociceptive neurons in a model of sciatic neuropathy. Exp Neurol 2008;214:219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Pogatzki EM, Gebhart GF, Brennan TJ. Characterization of Adelta- and C-fibers innervating the plantar rat hindpaw one day after an incision. J Neurophysiol 2002;87:721–731. [DOI] [PubMed] [Google Scholar]
- [74].Ramer U An iterative procedure for the polygonal approximation of plane curves. Computer graphics and image processing 1972;1:244–256. [Google Scholar]
- [75].Ratté S, Zhu Y, Lee KY, Prescott SA. Criticality and degeneracy in injury-induced changes in primary afferent excitability and the implications for neuropathic pain. Elife 2014;3:e02370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sagafos D, Kleggetveit IP, Helas T, Schmidt R, Minde J, Namer B, Schmelz M, Jorum E. Single-Fiber Recordings of Nociceptive Fibers in Patients With HSAN Type V With Congenital Insensitivity to Pain. Clin J Pain 2016;32:636–642. [DOI] [PubMed] [Google Scholar]
- [77].Salzer I, Gantumur E, Yousuf A, Boehm S. Control of sensory neuron excitability by serotonin involves 5HT2C receptors and Ca(2+)-activated chloride channels. Neuropharmacology 2016;110:277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Schmelz M, Schmidt R, Weidner C, Hilliges M, Torebjork HE, Handwerker HO. Chemical response pattern of different classes of C-nociceptors to pruritogens and algogens. J Neurophysiol 2003;89:2441–2448. [DOI] [PubMed] [Google Scholar]
- [79].Scroggs RS. Up-regulation of low-threshold tetrodotoxin-resistant Na+ current via activation of a cyclic AMP/protein kinase A pathway in nociceptor-like rat dorsal root ganglion cells. Neuroscience 2011;186:13–20. [DOI] [PubMed] [Google Scholar]
- [80].Sekerli M, Del Negro CA, Lee RH, Butera RJ. Estimating action potential thresholds from neuronal time-series: new metrics and evaluation of methodologies. IEEE Trans Biomed Eng 2004;51:1665–1672. [DOI] [PubMed] [Google Scholar]
- [81].Serra J, Bostock H, Sola R, Aleu J, Garcia E, Cokic B, Navarro X, Quiles C. Microneurographic identification of spontaneous activity in C-nociceptors in neuropathic pain states in humans and rats. Pain 2012;153:42–55. [DOI] [PubMed] [Google Scholar]
- [82].Serra J, Collado A, Sola R, Antonelli F, Torres X, Salgueiro M, Quiles C, Bostock H. Hyperexcitable C nociceptors in fibromyalgia. Ann Neurol 2014;75:196–208. [DOI] [PubMed] [Google Scholar]
- [83].Shin DS, Kim EH, Song KY, Hong HJ, Kong MH, Hwang SJ. Neurochemical Characterization of the TRPV1-Positive Nociceptive Primary Afferents Innervating Skeletal Muscles in the Rats. J Korean Neurosurg Soc 2008;43:97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Sommer C Serotonin in pain and analgesia: actions in the periphery. Mol Neurobiol 2004;30:117–125. [DOI] [PubMed] [Google Scholar]
- [85].Song XJ, Zhang JM, Hu SJ, LaMotte RH. Somata of nerve-injured sensory neurons exhibit enhanced responses to inflammatory mediators. Pain 2003;104:701–709. [DOI] [PubMed] [Google Scholar]
- [86].Stucky CL, Lewin GR. Isolectin B(4)-positive and -negative nociceptors are functionally distinct. J Neurosci 1999;19:6497–6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Study RE, Kral MG. Spontaneous action potential activity in isolated dorsal root ganglion neurons from rats with a painful neuropathy. Pain 1996;65:235–242. [DOI] [PubMed] [Google Scholar]
- [88].Suter MR, Berta T, Gao YJ, Decosterd I, Ji RR. Large A-fiber activity is required for microglial proliferation and p38 MAPK activation in the spinal cord: different effects of resiniferatoxin and bupivacaine on spinal microglial changes after spared nerve injury. Mol Pain 2009;5:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Taiwo YO, Levine JD. Serotonin is a directly-acting hyperalgesic agent in the rat. Neuroscience 1992;48:485–490. [DOI] [PubMed] [Google Scholar]
- [90].Tappe-Theodor A, Constantin CE, Tegeder I, Lechner SG, Langeslag M, Lepcynzsky P, Wirotanseng RI, Kurejova M, Agarwal N, Nagy G, Todd A, Wettschureck N, Offermanns S, Kress M, Lewin GR, Kuner R. Gα(q/11) signaling tonically modulates nociceptor function and contributes to activity-dependent sensitization. Pain 2012;153:184–196. [DOI] [PubMed] [Google Scholar]
- [91].Treede RD. Transduction and transmission properties of primary nociceptive afferents. Ross Fiziol Zh Im I M Sechenova 1999;85:205–211. [PubMed] [Google Scholar]
- [92].Usoskin D, Furlan A, Islam S, Abdo H, Lönnerberg P, Lou D, Hjerling-Leffler J, Haeggström J, Kharchenko O, Kharchenko PV, Linnarsson S, Ernfors P. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 2015;18:145–153. [DOI] [PubMed] [Google Scholar]
- [93].Vaso A, Adahan HM, Gjika A, Zahaj S, Zhurda T, Vyshka G, Devor M. Peripheral nervous system origin of phantom limb pain. Pain 2014;155:1384–1391. [DOI] [PubMed] [Google Scholar]
- [94].Wall PD, Devor M. Sensory afferent impulses originate from dorsal root ganglia as well as from the periphery in normal and nerve injured rats. Pain 1983;17:321–339. [DOI] [PubMed] [Google Scholar]
- [95].Wall PD, Gutnick M. Properties of afferent nerve impulses originating from a neuroma. Nature 1974;248:740–743. [DOI] [PubMed] [Google Scholar]
- [96].Walters ET. Injury-related behavior and neuronal plasticity: an evolutionary perspective on sensitization, hyperalgesia, and analgesia. Int Rev Neurobiol 1994;36:325–427. [DOI] [PubMed] [Google Scholar]
- [97].Walters ET. Nociceptors as chronic drivers of pain and hyperreflexia after spinal cord injury: an adaptive-maladaptive hyperfunctional state hypothesis. Front Physiol 2012;3:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Walters ET. Neuroinflammatory contributions to pain after SCI: roles for central glial mechanisms and nociceptor-mediated host defense. Exp Neurol 2014;258:48–61. [DOI] [PubMed] [Google Scholar]
- [99].Walters ET. How is chronic pain related to sympathetic dysfunction and autonomic dysreflexia following spinal cord injury. Auton Neurosci 2018;209:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Wang S, Lim J, Joseph J, Wang S, Wei F, Ro JY, Chung MK. Spontaneous and Bite-Evoked Muscle Pain Are Mediated by a Common Nociceptive Pathway With Differential Contribution by TRPV1. J Pain 2017;18:1333–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Wang T, Hurwitz O, Shimada SG, Qu L, Fu K, Zhang P, Ma C, LaMotte RH. Chronic Compression of the Dorsal Root Ganglion Enhances Mechanically Evoked Pain Behavior and the Activity of Cutaneous Nociceptors in Mice. PLoS One 2015;10:e0137512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Wiesenfeld-Hallin Z, Hallin RG, Persson A. Do large diameter cutaneous afferents have a role in the transmission of nociceptive messages? Brain Res 1984;311:375–379. [DOI] [PubMed] [Google Scholar]
- [103].Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain 2011;152:S2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Woolf CJ, Ma Q. Nociceptors--noxious stimulus detectors. Neuron 2007;55:353–364. [DOI] [PubMed] [Google Scholar]
- [105].Wu G, Ringkamp M, Hartke TV, Murinson BB, Campbell JN, Griffin JW, Meyer RA. Early onset of spontaneous activity in uninjured C-fiber nociceptors after injury to neighboring nerve fibers. J Neurosci 2001;21:RC140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Wu Z, Li L, Xie F, Du J, Zuo Y, Frost JA, Carlton SM, Walters ET, Yang Q. Activation of KCNQ Channels Suppresses Spontaneous Activity in Dorsal Root Ganglion Neurons and Reduces Chronic Pain after Spinal Cord Injury. J Neurotrauma 2017;34:1260–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Wu Z, Yang Q, Crook RJ, O’Neil RG, Walters ET. TRPV1 channels make major contributions to behavioral hypersensitivity and spontaneous activity in nociceptors after spinal cord injury. Pain 2013;154:2130–2141. [DOI] [PubMed] [Google Scholar]
- [108].Xiao WH, Bennett GJ. Persistent low-frequency spontaneous discharge in A-fiber and C-fiber primary afferent neurons during an inflammatory pain condition. Anesthesiology 2007;107:813–821. [DOI] [PubMed] [Google Scholar]
- [109].Xiao WH, Bennett GJ. Chemotherapy-evoked neuropathic pain: Abnormal spontaneous discharge in A-fiber and C-fiber primary afferent neurons and its suppression by acetyl-L-carnitine. Pain 2008;135:262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Xie W, Strong JA, Kim D, Shahrestani S, Zhang JM. Bursting activity in myelinated sensory neurons plays a key role in pain behavior induced by localized inflammation of the rat sensory ganglion. Neuroscience 2012;206:212–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Xie W, Strong JA, Ye L, Mao JX, Zhang JM. Knockdown of sodium channel NaV1.6 blocks mechanical pain and abnormal bursting activity of afferent neurons in inflamed sensory ganglia. Pain 2013;154:1170–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Xie W, Strong JA, Zhang JM. Local knockdown of the NaV1.6 sodium channel reduces pain behaviors, sensory neuron excitability, and sympathetic sprouting in rat models of neuropathic pain. Neuroscience 2015;291:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Xie Y, Zhang J, Petersen M, LaMotte RH. Functional changes in dorsal root ganglion cells after chronic nerve constriction in the rat. J Neurophysiol 1995;73:1811–1820. [DOI] [PubMed] [Google Scholar]
- [114].Xing JL, Hu SJ, Jian Z, Duan JH. Subthreshold membrane potential oscillation mediates the excitatory effect of norepinephrine in chronically compressed dorsal root ganglion neurons in the rat. Pain 2003;105:177–183. [DOI] [PubMed] [Google Scholar]
- [115].Xu J, Brennan TJ. Guarding pain and spontaneous activity of nociceptors after skin versus skin plus deep tissue incision. Anesthesiology 2010;112:153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Xu Q, Yaksh TL. A brief comparison of the pathophysiology of inflammatory versus neuropathic pain. Curr Opin Anaesthesiol 2011;24:400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Yang Q, Wu Z, Hadden JK, Odem MA, Zuo Y, Crook RJ, Frost JA, Walters ET. Persistent pain after spinal cord injury is maintained by primary afferent activity. J Neurosci 2014;34:10765–10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Zeitz KP, Guy N, Malmberg AB, Dirajlal S, Martin WJ, Sun L, Bonhaus DW, Stucky CL, Julius D, Basbaum AI. The 5-HT3 subtype of serotonin receptor contributes to nociceptive processing via a novel subset of myelinated and unmyelinated nociceptors. J Neurosci 2002;22:1010–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Zhang H, Dougherty PM. Enhanced excitability of primary sensory neurons and altered gene expression of neuronal ion channels in dorsal root ganglion in paclitaxel-induced peripheral neuropathy. Anesthesiology 2014;120:1463–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Zheng JH, Walters ET, Song XJ. Dissociation of dorsal root ganglion neurons induces hyperexcitability that is maintained by increased responsiveness to cAMP and cGMP. J Neurophysiol 2007;97:15–25. [DOI] [PubMed] [Google Scholar]
- [121].Zhu YF, Henry JL. Excitability of Aβ sensory neurons is altered in an animal model of peripheral neuropathy. BMC Neurosci 2012;13:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.