

Abstract
Verticillium wilt caused by the soil-borne fungus Verticillium dahliae is a common, devastating plant vascular disease notorious for causing economic losses. Despite considerable research on plant resistance genes, there has been little progress in modeling the effects of this fungus owing to its complicated pathogenesis. Here, we analyzed the transcriptional and metabolic responses of Arabidopsis thaliana to V. dahliae inoculation by Illumina-based RNA sequencing (RNA-seq) and nuclear magnetic resonance (NMR) spectroscopy. We identified 13,916 differentially expressed genes (DEGs) in infected compared with mock-treated plants. Gene ontology analysis yielded 11,055 annotated DEGs, including 2,308 for response to stress and 2,234 for response to abiotic or biotic stimulus. Pathway classification revealed involvement of the metabolic, biosynthesis of secondary metabolites, plant–pathogen interaction, and plant hormone signal transduction pathways. In addition, 401 transcription factors, mainly in the MYB, bHLH, AP2-EREBP, NAC, and WRKY families, were up- or downregulated. NMR analysis found decreased tyrosine, asparagine, glutamate, glutamine, and arginine and increased alanine and threonine levels following inoculation, along with a significant increase in the glucosinolate sinigrin and a decrease in the flavonoid quercetin glycoside. Our data reveal corresponding changes in the global transcriptomic and metabolic profiles that provide insights into the complex gene-regulatory networks mediating the plant’s response to V. dahliae infection.
Introduction
The soil-borne fungus Verticillium dahliae is responsible for widespread and devastating vascular disease in more than 200 species of dicotyledonous plants1. V. dahliae attacks susceptible plants through the roots, colonizes the plant vascular (xylem) system, and causes the death of aerial tissues2. The most typical symptom of Verticillium disease, generally referred to as Verticillium wilt, causes tremendous yield losses in many economically important crops3. Verticillium wilt is difficult to combat owing to the long-term survival of V. dahliae in the soil and the lack of fungicides with which to treat infected plants2. Currently, the preferred strategy to combat Verticillium wilt is the use of genetically improved Verticillium-resistant cultivars.
Plant resistance relies on the recognition of specific pathogen effector molecules by host plant resistance (R) proteins4. The first genetic locus found to be responsible for resistance against race 1 strains of V. dahliae, referred to as the Ve locus, was cloned in tomato (Solanum lycopersicum) and encodes cell surface receptor proteins5. The locus contains two closely linked and inversely oriented genes, Ve1 and Ve2, of which only Ve1 provides V. dahliae resistance in tomato6. The identification and functional characterization of Ve homologues was later extended to other plant species to include SlVe1 from S. lycopersicoides7, StVe from S. torvum Swartz8, mVe1 from Mentha longifolia9, GbVe from Gossypium barbadense10, VvVe from Vitis vinifera11, and NgVe1 from Nicotiana glutinosa12. Virus-induced gene silencing in tomato revealed that EDS1, NDR1, MEK2, and SERK3/BAK1 all act downstream of Ve1 and are required for resistance to V. dahliae6. The requirement for AtEDS1, AtNDR1, and AtSERK3/BAK1 for Verticillium resistance in Arabidopsis thaliana revealed that the critical signaling components used by Ve1 are conserved13.
Silencing of GhNDR1, GhMKK2, and GbEDS1 in cotton (Gossypium hirsutum) results in greater susceptibility to V. dahliae, suggesting that similar signaling cascades of Ve-mediated resistance exist in various species14,15. Gain- and loss-of-function mutations affecting a DNA-binding protein, AHL19, resulted in positive regulation of Verticillium wilt resistance in Arabidopsis16. A novel cotton subtilase, GbSBT1, recognizes a prohibitin-like protein secreted from V. dahliae and regulates Verticillium wilt resistance17. Further studies have been conducted on the role of transcription factors in Verticillium resistance. An ethylene-responsive GbERF1-like transcription factor contributes to resistance to V. dahliae in cotton by activating the expression of lignin biosynthesis genes18. GhATAF1, a NAC transcription factor, and GhMYB108 were both induced by V. dahliae infection and promote defense responses19,20.
Although various resistance genes have been functionally identified in the Verticillium resistance system, little is known about the complex molecular mechanisms underlying defense responses. Next-generation sequencing technologies offer fascinating opportunities to better understand the molecular networks of plant–pathogen interactions21. High-throughput RNA sequencing (RNA-seq), which does not require prior knowledge of genome sequences, has been used to obtain transcriptome changes in response to V. dahliae infection. RNA-seq analysis revealed 3,442 defense-responsive genes from the transcriptomic profiles of V. dahliae-infected cotton22. Further investigation of the expression of these genes revealed a critical role of lignin metabolism in the resistance of cotton to Verticillium wilt23. A comparison of RNA-seq results from infected sea-island and upland cotton to those from uninfected cotton revealed 44 differentially expressed genes (DEGs)24. A full-length cDNA library construction and expressed sequence tag (EST) sequencing in cotton challenged with V. dahliae identified 3,027 defense-related genes that are homologous to those in other plants, as well as 4,936 putative transcription factors25. Deep RNA sequencing of V. dahliae-infected N. benthamiana was performed to provide a catalog of transcripts produced by a Solanaceous model plant in response to pathogen attack26.
The use of a model plant-pathogen system could accelerate the discovery and understanding of the molecular mechanisms underlying Verticillium resistance. Arabidopsis possesses the first released genome sequence and the largest mutant collections. The conserved central components of the resistance signaling cascade have been reported, demonstrating that Arabidopsis is a suitable model to unravel the genetics of Verticillium resistance27–29. Therefore, the aim of this study was to use Arabidopsis as a model to identify transcriptome changes occurring during the process of V. dahliae infection. We examined Arabidopsis plants that had been infected with a highly toxic strain of V. dahliae, V991, at different time points after inoculation. We then performed transcriptomic analysis by RNA-seq and metabolomics analysis via NMR. We combined these data to analyze the expression of genes involved in signaling and metabolic pathways that are affected by V. dahliae inoculation.
Results
Establishment of experimental system
To minimize the impacts of any other fungus and bacteria, we sowed the Arabidopsis seeds on MS agar medium (Fig. 1a). We then inoculated four-to-six-true-leaf seedlings with Vd-GFP spore suspension and transferred the plants into MS medium as described in Materials and Methods (Fig. 1b). To avoid V. dahliae overgrowth, the MS medium was changed every 4 h. The environmental impact of the experimental system on the plants was minimal and mock-inoculated plants were included as a control.
Figure 1.

Colonization and infection of Vd-GFP on Arabidopsis roots. (a) The Arabidopsis seedlings were sown and grown on sterile MS agar medium. (b) Four-to-six-true-leaf seedlings were inoculated with 106 spores/mL and transplanted to MS medium. Confocal micrographs of the roots were taken at (c) 0 hpi, (d) 4 hpi, (e) 8 hpi, (f) 12 hpi, (g) 24 hpi, (h) 48 hpi, and (i) 56 hpi.
We observed the root surface of inoculated seedlings using a confocal microscope (Zeiss LSM 700, Jena, Germany) and found that the Vd-GFP spores were not attached to this surface (Fig. 1c). At 4 hour post inoculation (hpi), the conidia had colonized the root surface at random positions (Fig. 1d). At 8 hpi, a small quantity of spores had begun to germinate, with the germ tube forming from the merge of the conidium (Fig. 1e). At 12 hpi, many spores had germinated and the germ tube continued to elongate (Fig. 1f). At 24 hpi, germ tubes had developed into the longer mycelium, which extended over the surface of the root (Fig. 1g). At 48 and 56 hpi, the root was enclosed and entwined by a massive mycelium (Fig. 1h, i).
V. dahliae infection affects metabolism
To determine how V. dahliae affects metabolism, we performed NMR analysis using plant materials harvested at 0, 2, 8, 48, 96, and 144 hpi with V. dahliae to assess metabolic changes. We assayed 19 substrates, including 11 amino acids, sucrose, glucose, quercetin glycoside, fumaric acid, sinapoyl malate, feruloyl malate, sinigrin, and gallotanins. At the beginning, glutamic acid was the most abundant, while glutamine was second; aspartic acid, proline, arginine, alanine, threonine, valine, sucrose, and glucose were present at moderate levels and the rest at low levels (Fig. 2a).
Figure 2.

NMR analysis of inoculated plantlet from 0 hpi to 144 hpi. (a) Relative amounts of different compounds in the mock-treated sample at 0 hpi. (b) Dynamic changes in abundance of the compounds assayed over time. Color indicates the ratio of compound abundance in the inoculated samples to that in the mock-treated samples: red, 0; yellow, 1; and green, the maximum value (64.5, in sinigrin at 96 hpi). Single asterisk means p < 0.05; double asterisk means p < 0.01.
The NMR dynamics are demonstrated in Fig. 2b. After infection, the levels of tryptophan, aspartic acid, glutamine and glutamic acid, and arginine decreased over time. At 144 hpi, the tryptophan and aspartic acid levels in inoculated plants were around 10-fold lower than those in the mock-treated plants, arginine was 4-fold lower, and glutamine and glutamic acid were more than 20-fold lower. On the other hand, alanine and threonine levels increased as a result of V. dahliae infection, whereas proline, valine, leucine, and isoleucine did not show any marked variation.
The sucrose in the inoculated plantlets followed a more complex pattern, increasing from 0 to 96 hpi to a level around 5-fold higher than that in the mock-treated plants, but then decreasing until it dropped below that in mock-treated plants by 144 hpi. Furthermore, the glucose level in infected Arabidopsis did not show a substantial change in the period following inoculation with V. dahliae.
Along with the V. dahliae infection, the levels of quercetin glycoside, fumaric acid, and gallotanins in the plantlets decreased beginning at 96 hpi and were significantly lower than those in mock-treated plants at 144 hpi. Fumaric acid in the inoculated plantlet became undetectable at 144 hpi. Although the levels of sinapoyl malate and feruloyl malate also decreased, the decrease was not significant in comparison with the mock-treated plants. In addition, the level of sinigrin increased significantly, to almost 64-fold higher than that in the mock-treated plants (P < 0.01).
Data analysis by Illumina sequencing platform
To obtain a detailed transcriptomic profile in the early infection stage, we extracted RNA from the mock-treated and V. dahliae-inoculated seedlings at 0, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hpi. We obtained a total of 505,069,964 clean reads from ten samples using the Illumina sequencing platform after adaptors and any low-quality sequences were removed. Alignment with the Arabidopsis database (www.arabidopsis.org) showed that the mapping rates of these reads were up to 82% in genomic sequences and 90% in expressed genes. Through assembly into contigs and unigenes and alignment, 33,096 expressed genes were generated in total. The data are shown in Table 2.
Table 2.
Number of Arabidopsis reads used for the digital expression analysis.
| Sample name | Clean reads | Genome map rate | Gene map rate | Expressed genes |
|---|---|---|---|---|
| Mock | 50,449,400 | 84.26% | 93.81% | 28,125 |
| 0 hpi | 50,494,868 | 84.27% | 92.58% | 28,673 |
| 0.25 hpi | 50,496,898 | 83.36% | 93.22% | 28,143 |
| 0.5 hpi | 50,566,258 | 82.63% | 93.30% | 27,711 |
| 1 hpi | 50,524,584 | 82.42% | 90.15% | 28,277 |
| 2 hpi | 50,536,484 | 82.19% | 93.42% | 28,090 |
| 4 hpi | 50,516,548 | 83.33% | 93.37% | 27,989 |
| 8 hpi | 50,511,294 | 84.68% | 92.14% | 27,753 |
| 12 hpi | 50,565,214 | 84.01% | 93.30% | 27,942 |
| 24 hpi | 50,408,416 | 83.82% | 92.77% | 27,911 |
| Total number | 505,069,964 | 280,614 |
Differentially expressed gene (DEG) analysis in A. thaliana
To characterize Arabidopsis transcriptional responses to V. dahliae infection, we identified the unigenes whose expression levels changed significantly upon inoculation. The expression level was calculated in fragments per kilobase of transcript per million mapped reads (FPKM). To achieve this, we compared all the inoculated samples with the mock-treated samples. We found that transcription levels varied throughout the inoculation process. In response to the fungal stimulus and inoculation, a total of 13,916 genes showed differential expression (DEG) for at least one of the eight time points after inoculation. Some were identified as DEGs at more than one time points, and a subset demonstrated a different pattern at more than two time points. For example, the gene AT1G01100.1 was downregulated at 0.25 hpi, but upregulated at 1 hpi.
We separated the DEGs into downregulated and upregulated groups (Fig. 3). The highest level of DEG (>6,000) was achieved at 8hpi, of which the majority were downregulated. The smallest number of downregulated genes was found at 0 hpi, but that time point still had more than 3,000 DEGs.
Figure 3.
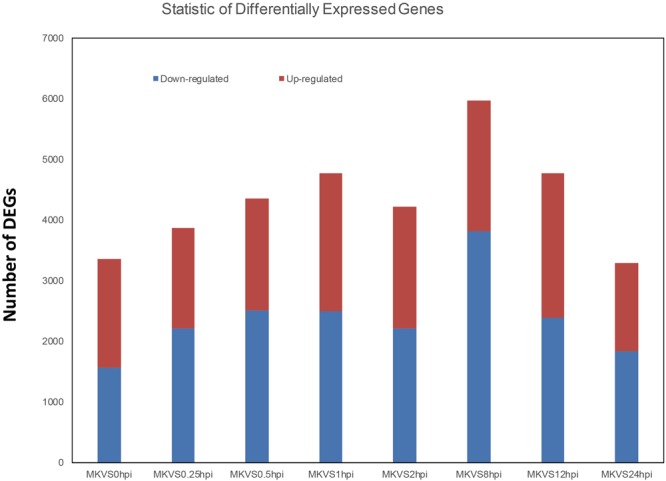
Numbers of DEGs down- and upregulated in the process of V. dahliae infection. MK, mock treatment; VS, versus; hpi, hour(s) post inoculation.
All of the DEG data are presented in Additional File 1.
Gene ontology (GO) enrichment analysis of DEGs
GO analysis was carried out to classify the functions of the differentially expressed genes. Based on Blast and SwissProt results, 11,055 DEGs were further annotated. All these annotated genes could be grouped into 46 categories according to their GOslim term. Among the top 10 categories, ‘response to stress’ containing 2,308 DEGs and ‘response to abiotic or biotic stimulus’ containing 2,234 DEGs ranked eighth and tenth, respectively (Fig. 4). Among the DEGs classified in ‘response to stress’, GO term identification showed that the most abundant was ‘response to salt stress’, with ‘respond to cold’ ranked second, ‘defense responds’ fourth, ‘defense response to fungus’ eighth, and ‘salicylic acid mediated signaling pathway’ tenth (Fig. 4b). Among the DEGs classified in ‘response to abiotic or biotic stimulus’, the ranking was quite similar, with ‘response to salt stress’ again ranked first, but ‘defense response to fungus’ ranked fifth and ‘salicylic acid mediated singling pathway’ sixth (Fig. 4c). All of the GO data are presented in Additional File 2.
Figure 4.

GO functional classification of the DEGs identified in this study. (a) Distribution of the DEGs among GOslim terms. (b) GO functional classification within the GOslim term ‘response to stress’. (c) GO functional classification within the GOslim term ‘response to abiotic or biotic stimulus’.
Pathway classification of DEGs
DEGs and all the genes assembled were analyzed with pathway tools in the KEGG database. From 0 hpi to 24 hpi, there were 2,008–3,636 DEGs annotated to be involved in 55 different biochemical pathways, as showed in Additional File 3. The top 10 enriched pathways at all nine sampling time points are shown in Fig. 5, while the top 20 were showed in Additional File 3. The biosynthesis of secondary metabolism was the category with the greatest enrichment, and metabolic pathway and plant–pathogen interaction were also very highly enriched. Plant hormone signal transduction, ribosome, starch and sucrose metabolism, tryptophan metabolism, circadian rhythm, glutathione metabolism, phenylalanine metabolism, carotenoid biosynthesis, and flavonoid biosynthesis also showed marked enrichment.
Figure 5.

Statistical analysis of pathway enrichment at all sampling time points. The top 10 most enriched pathways are shown. The percentage and the corresponding bar refer to the ratio of the number of detected genes to the total number of genes annotated in this pathway. MK, mock treatment; VS, versus; hpi, hour(s) post inoculation.
Among these pathways, those of amino acid metabolism were found. For example as mentioned above, the tryptophan metabolism pathway were highly enriched (included in the Top-20) in every time point checked, which coincided with the NMR data somehow. The enrichment in alanine, aspartate and glutamate metabolism pathway was also remarkable in some time points; on the other hand phenylalanine metabolism, and phenylalanine, tyrosine and trypophan biosynthesis were highly enrich as well.
Identification of putative transcription factors
Transcription factors mediate the expression levels of a series of genes and are tightly associated with pathways. Therefore, they can contribute both to the biological processes of normal development and to resistance against biotic and abiotic stimuli in plants30,31. Among the DEGs from 0 hpi to 24 hpi, we identified 477 independent genes as putative transcription factors by searching PlantTFDB2.0 from a comprehensive plant transcription factor database (PlantTFDB; http://planttfdb.cbi.pku.edu.cn). The most abundant family of transcription factors was MYB (15.72%), followed by MYB-related (12.79%), bHLH (10.48%), AP2-EREBP (10.06%), NAC (5.24%), WRKY (4.61%), C2H2 (3.56%), G2-like (2.31%), MADS (2.31%), and HSF (2.10%) (Fig. 6). All of the data for the transcription factors can be found in Additional File 4.
Figure 6.

DEGs predicted to encode transcription factors. Among the DEGs from 0 hpi to 24 hpi, 477 independent genes were identified as putative transcription factors (TFs). The top 10 putative TFs identified are shown in the chart. MK, mock treatment; VS, versus; hpi, hour(s) post inoculation.
V. dahliae-induced plant defense gene expression
Given that the salicylic acid (SA), jasmonic acid (JA), and ethylene (ET) signaling pathways mediate innate immune responses, we decided to investigate the expression of marker genes of these pathways in response to V. dahliae infection according to the RNA seq data as well as qRT-PCR. We picked the time point of 8 hpi, and PR-1 (AT2G14610.1), VSP2 (AT5G24770.1), and PDF1.2 (AT5G44420.1) as the marker genes for SA, JA and ET. All these three genes showed expression level elevation, validated by both methods, when the incretion assayed by qRT-PCR were generally higher than that of RNA-seq (Fig. 7). Later we chose four genes of up-regulated and another four of down regulated identified by RNA-seq, for further validation of qRT-PCR. The qRT-PCR results were in accordance with the RNA-seq result. The expression levels of these genes and their description were showed in Table 3.
Figure 7.
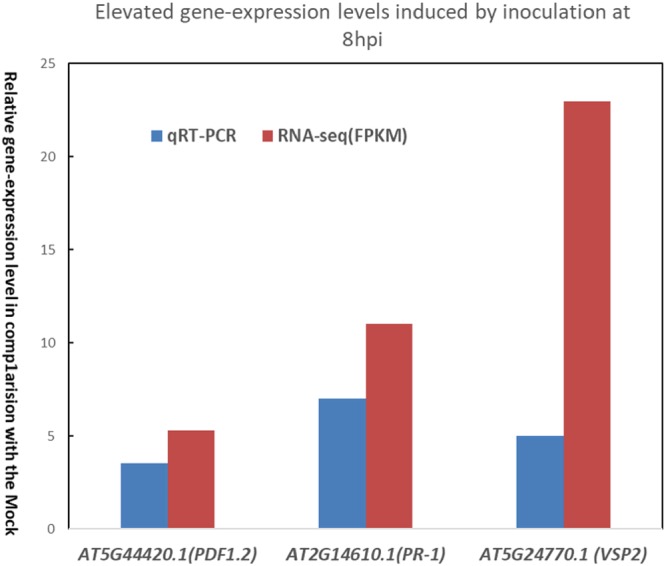
Expression levels of special genes in different treated groups. The salicylic acid (SA), jasmonic acid (JA), and ethylene (ET) signaling pathways mediate innate immune responses. The expression of the marker genes AT5G44420.1, T2G14610.1, and AT5G24770.1 in response to V. dahliae infection was monitored by qRT-PCR at 8 hpi time-point. AT5G44420.1 coding plant defensing (PDF1.2); AT2G14610.1 coding pathogenesis-related gene (PR-1); AT5G24770.1 coding vegetative storage protein 2 (VSP2).
Table 3.
The gene-expression level variation induced by inoculation.
| Gene ID | Time | Inoculation induced Variation | RNA-Seq | qRT-PCR |
|---|---|---|---|---|
| AT1G65960 | 12 hpi | Down | 0.77 | 0.59 |
| AT3G12120 | 1 hpi | Down | 0.97 | 0.43 |
| AT4G24460 | 8 hpi | Down | 0.55 | 0.92 |
| AT5G46490 | 2 hpi | Down | 0.26 | 0.37 |
| AT1G43640 | 8 hpi | Up | 20.00 | 6.29 |
| AT2G45220 | 12 hpi | Up | 16.40 | 9.21 |
| AT3G10720 | 1 hpi | Up | 1.30 | 8.43 |
| AT3G55770 | 12 hpi | Up | 1.46 | 2.15 |
Note: RNA-Seq means ratio between FPKM values of treatment and the mock, qRT-PCR means ratio between relative expression level of treatment and the mock assayed in qRT-PCR test; AT1G65960.2 coding glutamate decarboxylase 2; AT3G12120.1 coding fatty acid desaturase 2; AT4G24460.1 coding CRT (chloroquine-resistance transporter)-like transporter 2; AT5G46490.2 coding Disease resistance protein (TIR-NBS-LRR class) family; AT1G43640.1 coding tubby like protein 5; AT2G45220.1 coding pectin methylesterase 17; AT3G10720.1 coding plant invertase/pectin methylesterase inhibitor superfamily; AT3G55770.7 coding Arabidopsis LIM protein.
Discussion
Verticillium wilt causes significant economic losses worldwide owing to the lack of effective fungicides and the limited knowledge of Verticillium resistance mechanisms. The susceptibility of Arabidopsis to V. dahliae infection makes it possible to use this species as a model to provide increased insight into plant-pathogen recognition and the complex signaling networks involved in defense responses2,28. The high-throughput deep RNA sequencing (RNA-seq) of V. dahliae-infected Arabidopsis at different time points provided multiple insights into the genetic basis of Verticillium resistance. Interactions between the plant host and pathogen were observed through the whole infection. Even in the very early stage of infection, DEGs were detected. In 0 hpi, 0.25 hpi, 0.5 hpi and 1 hpi, the amount of DEGs kept increasing. It is remarkable because that fungal spores germinate and penetrate the epidermal cells of Arabidopsis roots within the first 12 hpi (Fig. 1d–f), which is visible and in accordance with previous data2,23. However, the DEGs data showed that the plant gave response in quite earlier stage. Even in 0 hpi when the plants were just inoculated in spore suspend after 2 min, there were more than 3000 DEGs taken into account. These DEGs might be caused by the staining (as descript in Materials and Methods), and it was also possibly the quick respond from Arabidopsis plantlet to the exogenous pathogen.
The initial step in the plant response to pathogen challenge is recognition of the non-self pathogen signals by the plant R protein. Up to date, only from tomato has a cell surface receptor, Ve1, been well characterized functionally, and this is thought to mediate Verticillium resistance5,6. However, the V. dahliae Ave1 effector induces various defense genes independently of Ve1 in tomato, suggesting that the signal might be transduced by another receptor32. In the present research, the pathway classification of DEGs identified a total of 2,308 DEGs involved in the plant’s response to stress. Of these, 453 DEGs are associated with defense response, 369 with regulation of the plant-type hypersensitive response, and 358 with the defense response to fungi. The largest group of DEGs was that comprising genes associated with response to salt stress, with other large groups being associated with cold stress response and the defense response to bacteria. GO annotation showed that many of the DEGs we identified encoded proteins with overlapping functions in multiple biotic or abiotic stresses. This implies that although some DEGs were identified as related to response to stresses other than fungi (such as salt, water deprivation, and wounding), they may also have unknown functions relevant to V. dahliae infection and may thus provide information that could help uncover other Ave1 receptors.
After the Ave1 effector is detected by the plant cell, signal transduction is carried on by phytohormone signaling pathways. SA is known to be effective against biotrophic pathogens while JA and ET are effective against necrotrophs33. The SA and JA signaling pathways are required for a form of Verticillium resistance induced by the nonpathogenic bacterium Paenibacillus alvei34. The pattern of increased expression of PR-1, VSP2, and PDF1.2 validated by RNA-seq as well as qRT-PCR confirmed the involvement of SA and JA signaling in defense responses to V. dahliae (Fig. 7). The RNA-seq data also showed ethylene biosynthetic gene AOC1 (AT4G35830) were triggered by the inoculation, its expression level kept up-regulated in almost all of the time-points. Among the DEGs from the entire infection process, we identified 413 genes involved in the SA hormone signaling pathway and 404 genes involved in JA signaling. Among the 413 DEGs associated with SA signaling, 162 are involved with SA-mediated systematic acquired resistance; among the DEGs associated with JA signaling, 11 are associated with JA-signal-mediated induced systematic resistance; and among the ethylene associated genes, 13 DEG are involved in jasmonic acid and ethylene-dependent systemic resistance. The expression of defense-related genes is mediated by transcription factors modulating the network of stress responses. A total of 477 putative transcription factor coding genes were identified among the DEGs, among which MYB and MYB-related transcription factors were the most abundant (Table 3). These transcription-factor-encoding DEGs could serve as a reference for the further analysis of V. dahliae and thereby contribute to the understanding of the complex signaling network of the plant’s response to pathogen invasion.
The host plant’s response to pathogens involves a systematic network. The pathogen-associated expression patterns of pivotal genes are closely related to primary and secondary metabolism. In the defense response to V. dahliae, for example, carbohydrate and nitrogen metabolism are reprogrammed and photosynthesis processes are depressed30. Xu et al. reported with PCR-select suppression subtractive hybridization, it is proved that V. dahliae inoculation on cotton resulted in DEGs mainly involved in metabolism, stress⁄defence response, cell structure and signal transduction22, when present research found quite similar phenomenon. Mo et al. found that Spermine biosynthesis is required for the SA-mediated signaling involved in adaptation to V. dahliae colonization by screening suppression subtractive hybridization and cDNA libraries of cotton (Gossypium) species tolerant to Verticillium wilt31. To date, there had been little information about the metabolomics of V. dahliae-infected plants. We therefore expected that an investigation of the relationships between metabolomics and resistance could promote understanding of Verticillium wilt resistance. In our present research, we used NMR analysis to monitor metabolic changes following inoculation with V. dahliae. Previous research had indicated that biotic and abiotic stress influences the homeostasis of free amino acids; in Arabidopsis, inoculation with P. syringae bacteria resulted in decreases in the abundance of branched-chain amino acids (valine, leucine, and isoleucine), aromatic amino acids (phenylalanine, tyrosine, and tryptophan), and aspartate35. In present research we did find the trend of decreasing in aromatic amino acid tryptophan after inoculation but not in the branched-chain amino acid. The metabolism variation assayed by NMR in present research showed that tryptophan content of the plantlet showed a dramatically dropdown due to the V. dahliae infection. Simultaneously, the transcriptomic data also showed that the tryptophan metabolic pathway genes were highly enriched in all of the time points. The gene (AT4G23100.2) coding the key enzyme (γ‐glutamylcysteine synthetase) of glutathione biosynthesis and tryptophan metabolism was the DEG of up regulated in 0 hpi, 0.25 hpi, 0.5 hpi, 2 hpi, 8 hpi and 12 hpi. As reported previously, glutathione is very important for plant resistant to diseases36,37. Simultaneously, aspartic acid, glutamine and glutamic acid were reduced especially in the late stage, but alanine increased. All these amino acid are involved in alanine, aspartate and glutamate metabolism pathway which is remarkably enriched in transcriptomic analysis (Additional File 3). The gene coding (ATG17290) alanine catalysis key enzyme alanine amino transferase, were highly expressed in 8 hpi. The variation in alanine, aspartate and glutamate metabolism could be induced by the fungal growth, but also possible due to the anaerobic stress38 in submerge treatment (as described in materials and methods part). Another metabolic pathway that has been found, in cotton, to be very important for Verticillium wilt resistance is lignin metabolism23. Lignin biosynthesis begins in the cytosol with the synthesis of glycosylated monolignols from phenylalanine. The RNA-seq data showed that phenylalanine metabolism highly enriched. Phenylalanine ammonia-lyse is an important plant enzyme to form trans-cinnamic acid, a precyrsor of lignin39. The genes (AT3G10340.1 and AT2G37040.1) encoding the enzyme showed elevated expression level in 8 hpi, 12 hpi and 24 hpi. We measured the levels of three precursors (fumaric acid, sinapoyl malate and Feruloyl malte) of lignin biosynthesis in V. dahliae-inoculated Arabidopsis. We found all of these three substances increased until 48 hpi and then kept decreasing difference. The transcriptome data showed the expression level of genes AT5G54160.1 and AT4G36220.1 were enhanced after inoculation (8 hpi, 12 hpi and 24 hpi as showed in Additional File 3). They encode caffeic acid/5-hydroxyferulic acid O-methyltransferase and ferulate 5-hydroxylase, which are both essential in lignin biosynthesis40. On the other hand, the level of the glucosinolate sinigrin increased and that of the flavonoid quercetin glycoside decreased, while flavonoid and glucosinolate biosynthesis were also markedly enriched in transcriptomic analysis.
Conclusion
Our present research provides comprehensive information on the metabolomic and transcriptomic changes occurring in Arabidopsis inoculated with V. dahliae. The metabolomic profile of the infected plantlets differed significantly from that of the mock-infected plantlets; in addition, a large number of DEGs were identified based on transcriptomic analysis. The dynamic events that follow fungal inoculation highlighted many functional genes and the pathways in which they are involved. The copious data we obtained, and the correspondence between the metabolome and transcriptome they imply, should serve as a useful reference in efforts to decipher the complexity of the interactions between plants and this devastating pathogen.
Materials and Methods
Fungal strain and inoculum preparation
The highly virulent V. dahliae strain V991 was kindly provided by Dr. Guiliang Jian of the Institute of Plant Protection, Chinese Academy of Agricultural Sciences (CAAS), Beijing, China. The pathogen was cultured in liquid complete medium (CM) with 50 mg/L ampicillin and 50 mg/L kanamycin at 25 °C. After five days, the culture was filtered through 0.4 µm mesh and centrifuged at 10,000 rpm for 5 min, and the supernatant was discarded. The number of spores was counted with a light microscope (OLYMPUS BX52, Tokyo, Japan) and the extract was diluted to 106 spores/mL to produce the spore suspension used for inoculation.
Plant materials and inoculation
Sterile seeds of Arabidopsis thaliana (ecotype Columbia 0) were sown on Murashige-Skoog (MS) agar medium and cultured at a photoperiod of 16 h light (24 °C) and 8 h darkness (21 °C). Four-to-six-true-leaf seedlings were transferred into liquid MS medium. After 48 h, the seedlings were submerged in the prepared spore suspension for 2 min. Subsequently, the seedlings were laid on sterile filter paper to eliminate excess solution and transferred into fresh liquid MS medium.
To obtain a detailed transcriptomic profile in the early infection stage, we extracted RNA from the mock-treated and V. dahliae-inoculated seedlings at 0, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hpi. Meanwhile, the plant materials were prepared for metabolomics analysis at 0, 2, 8, 48, 96, and 144 hpi. To minimize systematic bias, the data were collected three times.
cDNA preparation for RNA-seq
Twenty plants from each treatment were collected at 0, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 hours post inoculation (hpi). Uninfected wild-type seedlings were designated the mock-treatment group. The samples were washed with sterile water and then quickly frozen in liquid nitrogen and stored at −80 °C.
Total RNA was isolated from the frozen tissues using an RNA Extraction Kit (YPHBio, Tianjin, China) following the manufacturer’s instructions, including an on-column DNase I digestion. The mRNA was further purified using oligo(dT) magnetic beads and a magnet separator. The quality and concentration of mRNA were determined using an Agilent 2100 Bioanalyzer (Agilent, California, America). Next, the mRNA was broken into small fragments by heat treatment, and the fragments were reverse transcribed into first-strand cDNA with random hexamer primers. The second-strand cDNA was synthesized and sequenced using a HiSeqTM 2000 instrument (Illumina, America).
Analysis of RNA-seq data
The raw sequences were processed to remove low-quality reads (with ratio of unknown sequences ‘N’ > 10%) and adaptor sequences using the LUCY programs41. Meanwhile, possible Verticillium sequences were removed from the raw data (www.broadinstitute.org). After the quality-control mock treatment had been carried out, the clean data were assembled into contigs and aligned with Arabidopsis thaliana data (www.arabidopsis.org). After clustering and assembly, similarities between ESTs (expressed sequence tags) and sequences were identified using the NCBI BLAST program, version 2.2.626. Furthermore, the differentially expressed genes (DEGs) were subjected to gene ontology (GO) analysis42, pathway enrichment analysis24, transcription factor analysis, and cluster analysis43.
Preparation of plant material for nuclear magnetic resonance (NMR)
The plant material collected at 0, 2, 8, 48, 96, and 144 hpi was ground and subsequently freeze-dried. The mock-treated samples were prepared at the same time points but without infection. Then, 20 mg of freeze-dried ground tissues was added to 1 mL of deuterium solvents, CH3OH-d4-KH2O4 buffer in D2O, which includes trimethylsilyl propionic acid sodium salt-d4 (0.01%, w/w, TMSP), and then extracted by ultrasonication for 10 min and centrifuged at 13,000 rpm (10,968 g) for 1 min. From the supernatant, 250 µL of mixture was transferred into 3-mm NMR tubes and measured in an NMR instrument (600 MHz Bruker DMX-600 spectrometer) at 25 °C.
Analysis of NMR data
Deuterated methanol and water were purchased from Sigma-Aldrich (St. Louis, MO, USA). 1H NMR spectra were recorded at 25 °C on a 600 MHz Bruker DMX-600 spectrometer (Bruker, Karlsruhe, Germany) operating at a proton NMR frequency of 600.13 MHz for 13C. CD3OD was used as the internal control.
The 1H NMR spectra were automatically reduced to ASCII files. Spectral intensities were scaled to total or internal standards (TMSP or TMS signals at δ 0.0) and reduced to integrated regions of equal width (δ 0.04) corresponding to the region of δ 0.0–10.0. The regions of δ 4.7–4.9 and δ 3.28–3.34 were excluded from the analysis because of the residual signals of D2O and CD3OD, respectively. Bucketing was performed using AMIX software (ver. 3.0 Bruker) with scaling on total intensity. Projections to latent structures (PLS) and orthogonal PLS (OPLS) with scaling based on unit variance were performed with SIMCA-P + software (v. 13.3, Umetrics, Umeå, Sweden).
Expression level analysis of candidate genes by qRT-PCR
Total RNA was isolated from the Arabidopsis plants using RNA extraction kits (YPHBio, Tianjin, China) according to the manufacturer’s instructions. The cDNA was synthesized using reverse transcription kits (TransGen, Beijing, China). qRT-PCR was carried out with a 7500 Real Time PCR System (Applied Biosystems, Foster City, CA, USA), using TransStart Top Green qPCR SuperMix (TransGen, Beijing, China) as described by the manufacturer. Primers used to identify the transcripts of specific genes are listed in Table 1. The relative expression levels were calculated using the 2−∆∆Ct method25, with the Arabidopsis housekeeping gene (EF-1a, At5g60390) as the internal mock.
Table 1.
Primer sequences used in the qRT-PCR experiments.
| Gene | AGI | Sequences (5′-3′) |
|---|---|---|
| PDF1.2 | AT5G44420 | CTTGTTCTCTTTGCTGCTTTCG |
| CATGTTTGGCTCCTTCAAG | ||
| PR-1 | AT2G14610 | TTCTTCCCTCGAAAGCTCAA |
| AAGGCCCACCAGAGTGTATG | ||
| VSP2 | At5g24770 | GATACGGAACAGAGAAGACC |
| AGCTTCGAGATTGTCGAGAG | ||
| EF-1a | At5g60390 | TGAGCACGCTCTTCTTGCTTTC |
| GGTGGTGGCATCCATCTTGTTA | ||
| AT1G65960 | GGCGGTGGGAGTAGGGACAGTTGGT | |
| GGCTCCGGTGACAATGTTGGGTTTG | ||
| AT3G12120 | AGCGACTACCAATGGCTGGATGACA | |
| ATGCGTCCAAGAGGGTTGTTGAGGT | ||
| AT4G24460 | AGCTCACCACTTTCGGCTACGTCTT | |
| TCCATTTGGGTACATCCATCATTTC | ||
| AT5G46490 | ACTACCATTGCACGAGCTTTGTTTA | |
| ATCTATCTTTATGTTCGGCATCCTT | ||
| AT1G43640 | CATACGAGCTGAACTTGCTTGGAAC | |
| GTGAAATCTCCCGAGTGGTTGACTA | ||
| AT2G45220 | AAGCACGGCTCTAACCAATCTTGAC | |
| ACCAGGTTTGACCCACGAAGGGAAC | ||
| AT3G10720 | AAGATTCTTGGTGGTGGGAACTCAA | |
| ACGGACCGACGATCACTGCTTTACT | ||
| AT3G55770 | ACTTTAATTTGCATGTAACAGTCGA | |
| TAAGTGGTTGTAACTTCCCTTCTCC |
AGI, Arabidopsis Genome Initiative.
Electronic supplementary material
Acknowledgements
This research was supported by a grant from the National Natural Science Foundation of China (31772244), the special fund for agro-scientific research in the public interest (201503109) and the Agricultural Science and Technology Innovation Program of CAAS.
Author Contributions
H.G. and H.C. conceived and designed the experiments. X.S. and G.L. performed the experiments and wrote the manuscript. X.L. and K.Z. participated in data interpretation.
Data Availability
The NMR data and the RNA-seq information developed in present research are freely available to the research community. And the data generated or analysed during this study are included in Additional Files 1–4.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Xiaofeng Su and Guoqing Lu contributed equally.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-33743-x.
References
- 1.Agrios, G. N. Plant pathology. 4th ed. (1997).
- 2.Fradin EF, Thomma BPHJ. Physiology and molecular aspects of Verticillium wilt diseases caused by V. dahliae and V. albo-atrum. Molecular Plant Pathology. 2006;7:71. doi: 10.1111/j.1364-3703.2006.00323.x. [DOI] [PubMed] [Google Scholar]
- 3.Pegg, G. F., Brady, B. L., Pegg, G. F. & Brady, B. L. Verticillium wilts. (2002).
- 4.Martin GB, Bogdanove AJ, Sessa G. Understanding the functions of plant disease resistance proteins. Annual Review of Plant Biology. 2003;54:23. doi: 10.1146/annurev.arplant.54.031902.135035. [DOI] [PubMed] [Google Scholar]
- 5.Kawchuk LM, et al. Tomato Ve disease resistance genes encode cell surface-like receptors. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6511–6515. doi: 10.1073/pnas.091114198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fradin EF, et al. Genetic dissection of Verticillium wilt resistance mediated by tomato Ve1. Plant Physiology. 2009;150:320–332. doi: 10.1104/pp.109.136762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chai Y, et al. Molecular cloning of a potential Verticillium dahliae resistance gene SlVe1 with multi-site polyadenylation from Solanum licopersicoides. Mitochondrial DNA. 2003;14:375–384. doi: 10.1080/10425170310001605509. [DOI] [PubMed] [Google Scholar]
- 8.Fei J, et al. CDNA cloning and characterization of the Ve homologue gene StVe from Solanum torvum Swartz. Mitochondrial DNA. 2004;15:88–95. doi: 10.1080/1042517042000199942. [DOI] [PubMed] [Google Scholar]
- 9.Vining K, Davis T. Isolation of a Ve homolog, mVe1, and its relationship to Verticillium wilt resistance in Mentha longifolia (L.) Huds. Molecular Genetics and Genomics. 2009;282:173. doi: 10.1007/s00438-009-0454-6. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, et al. Cloning and characterization of a Verticillium wilt resistance gene from Gossypium barbadense and functional analysis in Arabidopsis thaliana. Plant Cell Reports. 2011;30:2085. doi: 10.1007/s00299-011-1115-x. [DOI] [PubMed] [Google Scholar]
- 11.Tang J, et al. Ectopic expression of a Ve homolog VvVe gene from Vitis vinifera enhances defense response to Verticillium dahliae infection in tobacco. Gene. 2016;576:492–498. doi: 10.1016/j.gene.2015.10.068. [DOI] [PubMed] [Google Scholar]
- 12.Song Y, et al. Broad taxonomic characterization of Verticillium wilt resistance genes reveals ancient origin of the tomato Ve1 immune receptor. Molecular Plant Pathology. 2016;18:195. doi: 10.1111/mpp.12390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fradin EF, et al. Interfamily transfer of tomato Ve1 mediates Verticillium resistance in Arabidopsis. Plant physiology. 2011;156:2255–2265. doi: 10.1104/pp.111.180067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao X, et al. Silencing GhNDR1 and GhMKK2 compromises cotton resistance to Verticillium wilt. The Plant Journal. 2011;66:293–305. doi: 10.1111/j.1365-313X.2011.04491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Su X, Qi X, Cheng H. Molecular cloning and characterization of enhanced disease susceptibility 1 (EDS1) from Gossypium barbadense. Molecular Biology Reports. 2014;41:3821–3828. doi: 10.1007/s11033-014-3248-9. [DOI] [PubMed] [Google Scholar]
- 16.Yadeta KA, et al. The Arabidopsis thaliana DNA-binding protein AHL19 mediates Verticillium wilt resistance. Molecular plant-microbe interactions: MPMI. 2011;24:1582. doi: 10.1094/MPMI-04-11-0090. [DOI] [PubMed] [Google Scholar]
- 17.Duan X, Zhang Z, Jin W, Zuo K. Characterization of a novel cotton subtilase gene GbSBT1 in response to extracellular stimulations and its role in Verticillium resistance. Plos One. 2016;11:e0153988. doi: 10.1371/journal.pone.0153988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo W, et al. An ethylene response-related factor, GbERF1-like, from Gossypium barbadense improves resistance to Verticillium dahliae via activating lignin synthesis. Plant Molecular Biology. 2016;91:305. doi: 10.1007/s11103-016-0467-6. [DOI] [PubMed] [Google Scholar]
- 19.He X, Zhu L, Xu L, Guo W, Zhang X. GhATAF1, a NAC transcription factor, confers abiotic and biotic stress responses by regulating phytohormonal signaling networks. Plant Cell Reports. 2016;35:2167–2179. doi: 10.1007/s00299-016-2027-6. [DOI] [PubMed] [Google Scholar]
- 20.Cheng HQ, et al. The cotton MYB108 forms a positive feedback regulation loop with CML11 and participates in the defense response against Verticillium dahliae infection. Journal of Experimental Botany. 2016;67:1935–1950. doi: 10.1093/jxb/erw016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ansorge WJ. Next-generation DNA sequencing techniques. New Biotechnology. 2009;25:195–203. doi: 10.1016/j.nbt.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 22.Xu L, et al. Differential gene expression in cotton defence response to Verticillium dahliae by SSH. Journal of Phytopathology. 2011;159:606–615. doi: 10.1111/j.1439-0434.2011.01813.x. [DOI] [Google Scholar]
- 23.Li X, et al. Lignin metabolism has a central role in the resistance of cotton to the wilt fungus Verticillium dahliae as revealed by RNA-Seq-dependent transcriptional analysis and histochemistry. Journal of Experimental Botany. 2011;62:5607. doi: 10.1093/jxb/err245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Q, et al. Analysis of sea-island cotton and upland cotton in response to Verticillium dahliae infection by RNA sequencing. BMC Genomics. 2013;14:852. doi: 10.1186/1471-2164-14-852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, et al. Transcriptome profiling of Gossypium barbadense inoculated with Verticillium dahliae provides a resource for cotton improvement. BMC genomics. 2013;14:637. doi: 10.1186/1471-2164-14-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faino L, de Jonge R, Thomma BP. The transcriptome of Verticillium dahliae-infected Nicotiana benthamiana determined by deep RNA sequencing. Plant signaling & behavior. 2012;7:1065–1069. doi: 10.4161/psb.21014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Polverari A, et al. Nitric oxide-mediated transcriptional changes in Arabidopsis thaliana. Molecular Plant-Microbe Interactions: MPMI. 2003;16:1094. doi: 10.1094/MPMI.2003.16.12.1094. [DOI] [PubMed] [Google Scholar]
- 28.Tjamos SE, Flemetakis E, Paplomatas EJ, Katinakis P. Induction of resistance to Verticillium dahliae in Arabidopsis thaliana by the biocontrol agent K-165 and pathogenesis-related proteins gene expression. Molecular Plant-Microbe Interactions: MPMI. 2005;18:555. doi: 10.1094/MPMI-18-0555. [DOI] [PubMed] [Google Scholar]
- 29.Veronese P, et al. Identification of a locus controlling Verticillium disease symptom response in Arabidopsis thaliana. Plant Journal for Cell & Molecular Biology. 2003;35:574. doi: 10.1046/j.1365-313X.2003.01830.x. [DOI] [PubMed] [Google Scholar]
- 30.Buhtz A, et al. Perturbations in the primary metabolism of tomato and Arabidopsis thaliana plants infected with the soil-borne fungus Verticillium dahliae. PLoS One. 2015;10:e0138242. doi: 10.1371/journal.pone.0138242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mo HJ, et al. Cotton S-adenosylmethionine decarboxylase-mediated spermine biosynthesis is required for salicylic acid- and leucine-correlated signaling in the defense response to Verticillium dahliae. Planta. 2016;243:1023–1039. doi: 10.1007/s00425-015-2463-5. [DOI] [PubMed] [Google Scholar]
- 32.Castroverde, C. D. M., Nazar, R. N. & Robb, J. Verticillium Ave1 effector induces tomato defense gene expression independent of Ve1 protein. 00–00 (2016). [DOI] [PMC free article] [PubMed]
- 33.Pieterse CM, Van DDD, Zamioudis C, Leon-Reyes A, Van Wees SC. Hormonal modulation of plant immunity. Annual Review of Cell & Developmental Biology. 2012;28:489. doi: 10.1146/annurev-cellbio-092910-154055. [DOI] [PubMed] [Google Scholar]
- 34.Gkizi D, et al. The innate immune signaling system as a regulator of disease resistance and ISR activity against Verticillium dahliae. Molecular Plant-Microbe Interactions: MPMI. 2016;29:313. doi: 10.1094/MPMI-11-15-0261-R. [DOI] [PubMed] [Google Scholar]
- 35.Zeier J. New insights into the regulation of plant immunity by amino acid metabolic pathways. Plant Cell & Environment. 2013;36:2085–2103. doi: 10.1111/pce.12122. [DOI] [PubMed] [Google Scholar]
- 36.Vincent P, et al. Identification of PAD2 as a γ-glutamylcysteine synthetase highlights the importance of glutathione in disease resistance of Arabidopsis. The Plant Journal. 2007;49:159–172. doi: 10.1111/j.1365-313X.2006.02938.x. [DOI] [PubMed] [Google Scholar]
- 37.Hiruma K, et al. Glutathione and tryptophan metabolism are required for Arabidopsis immunity during the hypersensitive response to hemibiotrophs. Proceedings of the National Academy of Sciences. 2013;110:9589–9594. doi: 10.1073/pnas.1305745110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Good AG, Muench DG. Purification and characterization of an anaerobically induced alanine aminotransferase from barley roots. Plant Physiology. 1992;99:1520–1525. doi: 10.1104/pp.99.4.1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jie K, et al. The effect of nitrogen application and planting density on the radiation use efficiency and the stem lignin metabolism in rapeseed (Brassica napus L.) Field Crops Research. 2016;199:89–98. doi: 10.1016/j.fcr.2016.09.025. [DOI] [Google Scholar]
- 40.Ruegger M, Chapple C. Mutations that reduce sinapoylmalate accumulation in Arabidopsis thaliana define loci with diverse roles in phenylpropanoid metabolism. Genetics. 2001;159:1741–1749. doi: 10.1093/genetics/159.4.1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chou HH, Holmes MH. DNA sequence quality trimming and vector removal. Bioinformatics. 2001;17:1093–1104. doi: 10.1093/bioinformatics/17.12.1093. [DOI] [PubMed] [Google Scholar]
- 42.Gotz S, et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Research. 2008;36:3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ye J, et al. WEGO: a web tool for plotting GO annotations. Nucleic Acids Research. 2006;34:W293–297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The NMR data and the RNA-seq information developed in present research are freely available to the research community. And the data generated or analysed during this study are included in Additional Files 1–4.