

Abstract
Real-world studies include a broader patient population for a longer duration than randomised controlled trials (RCTs) and can provide relevant insights for clinical practice.
PASSPORT was a multicentre, prospective, post-authorisation study of patients who were newly prescribed pirfenidone and followed for 2 years after initiating treatment. Physicians collected data on adverse drug reactions (ADRs), serious ADRs (SADRs) and ADRs of special interest (ADRSI) at baseline and then every 3 months. Post hoc stepwise logistic regression models were used to identify baseline characteristics associated with discontinuing treatment due to an ADR.
Patients (n=1009, 99.7% with idiopathic pulmonary fibrosis) had a median pirfenidone exposure of 442.0 days. Overall, 741 (73.4%) patients experienced ADRs, most commonly nausea (20.6%) and fatigue (18.5%). ADRs led to treatment discontinuation in 290 (28.7%) patients after a median of 99.5 days. Overall, 55 (5.5%) patients experienced SADRs, with a fatal outcome in six patients. ADRSI were reported in 693 patients, most commonly gastrointestinal symptoms (38.3%) and photosensitivity reactions/skin rashes (29.0%). Older age and female sex were associated with early treatment discontinuation due to an ADR.
Findings were consistent with the known safety profile of pirfenidone, based on RCT data and other post-marketing experience, with no new safety signals observed.
Short abstract
Real-world safety results from 1009 patients in PASSPORT were consistent with the known pirfenidone safety profile http://ow.ly/oXjv30lrzAf
Introduction
Idiopathic pulmonary fibrosis (IPF) is a debilitating, progressive and fatal lung disease characterised by a chronic refractory cough, shortness of breath and exercise limitation [1, 2]. The prognosis for patients with IPF is poor, with a median survival of 2–3 years [1].
Pirfenidone is recommended for the treatment of the majority of patients with IPF in international treatment guidelines and is approved in many countries, including in the European Union (EU), the USA and Japan [3–6]. The safety and tolerability of pirfenidone have been well characterised [7–9], and the most common adverse events associated with pirfenidone are gastrointestinal and skin-related events, which are largely mild to moderate in severity and responsive to dose modification [8].
Real-world studies typically include a broader patient population treated in an uncontrolled environment for a longer duration than in a randomised controlled trial (RCT) setting, and can therefore provide some relevant insights for clinical practice [10] and a comprehensive understanding of a drug's benefit–risk profile [11]. Following the European marketing authorisation for pirfenidone, and as requested by the European Medicines Agency (EMA), a post-authorisation safety study (PASSPORT) [12] was conducted to collect and monitor adverse drug reactions (ADRs) in patients who received pirfenidone in a real-world setting. Here, we present the results from PASSPORT.
Methods
Study design
PASSPORT (ClinicalTrials.gov identifier NCT02699879) was a multicentre, prospective, observational, post-authorisation study that evaluated long-term safety in patients who were prescribed pirfenidone. The study was conducted in accordance with the pre-defined protocol and all applicable laws and regulations (including Good Pharmacoepidemiology Practices, ethical practices with an origin in the Declaration of Helsinki and applicable national guidelines). All patients provided informed consent according to local regulations for study eligibility.
PASSPORT included patients who had been prescribed pirfenidone (within 30 days of enrolment) as part of clinical practice in one of the following European countries: Austria, Denmark, Finland, France, Germany, Ireland, Italy, Norway, Sweden, Spain or the UK. Pirfenidone was prescribed to patients by the treating physicians (investigators) in accordance with the local access rules. Patients continued to receive pirfenidone at their physician's discretion and were followed for 2 years after treatment initiation (no minimum pirfenidone exposure was required to be considered a study completer) or until they discontinued the study.
The study population for the safety analysis included any patient who received one or more doses of pirfenidone, regardless of the duration of follow-up for that patient. Patients were excluded if they were receiving an investigational agent, had received pirfenidone treatment >30 days before the current treatment course or had absolute contraindications for treatment according to section 4.3 of the pirfenidone Summary of Product Characteristics [3]. If a physician, in collaboration with the patient, made a clinical decision to prescribe pirfenidone outside the terms of the Summary of Product Characteristics, patients who met the other criteria could be included in PASSPORT.
Pre-specified patient data were collected at baseline and every 3 months (±4 weeks) thereafter for the duration of participation in the study. Baseline data included patient demographics, medical and IPF history, lung function (at treatment initiation), concurrent diseases and medications, reason for pirfenidone treatment, initial prescribed dose of pirfenidone, and liver function tests (LFTs). Follow-up data were collected, including safety assessments, changes to pirfenidone treatment (including treatment discontinuation) and the reasons for change. Only deaths that were an outcome of an ADR or were the primary reason for study discontinuation were collected. Patients who had dose reductions and interruptions during the course of the study were also monitored.
Safety assessments
ADRs that occurred during PASSPORT were self-reported by patients at follow-up visits every 3 months. Training was provided to physicians on how to document the patient-reported events that qualified as ADRs. When events were reported, appropriate measures were taken to follow-up as needed; physicians could retrieve medical record information from their own clinic/practice. All serious ADRs (SADRs) and ADRs of special interest (ADRSI) were recorded. ADRSI comprised any important identified/potential risks listed in the EMA's Committee for Medicinal Products for Human Use (CHMP) assessment report for pirfenidone [12]. ADRSI included all related preferred terms from the Medical Dictionary for Regulatory Activities version 19.0 (www.meddra.org), e.g. the ADRSI of fatigue included the preferred terms of fatigue, asthenia, lethargy, somnolence and listlessness.
ADRs and ADRSI were analysed in the overall population and in the pre-specified patient subgroups, listed in the EMA's CHMP assessment report [12], which met the following criteria for baseline comorbidities or specific concurrent medication use: forced vital capacity (FVC) % pred <50%, use of concomitant immunosuppressives or other therapies for IPF, pirfenidone use for other conditions, predisposing conditions for liver dysfunction, pre-existing QT prolongation, secondary causes of pulmonary fibrosis, specific cardiac events and underlying severe cardiac, hepatic or pulmonary disease (excluding IPF). Other pre-specified subgroups in PASSPORT included patients who took warfarin at baseline and patients aged <18 years at enrolment.
Statistical analyses
A sample size of 1000 patients was planned to give sufficient statistical precision when expressed by a two-sided 95% confidence interval calculated around the expected incidence of ADRs. The expected incidence of ADRs was taken from observations made during 75 weeks of exposure among 789 patients treated with pirfenidone in five previous studies (incidence ranged from 2.5% to 22.0%). Precision was defined as half the width of the 95% confidence interval derived from normal approximation; therefore, the precision of estimates varied according to the rate of the ADR. For example, in a sample size of 1000, if the ADR had an expected rate of 2.5%, precision would be 0.97%; if the ADR had an expected rate of 5.0%, precision would be 1.35%.
Frequencies of ADRs were summarised and included calculation of drug exposure-adjusted reported event rates. Exposure summaries were based on prescribed dosing data rather than on actual pirfenidone intake, as the latter information was not collected. Exposure-adjusted event rates per 1000 person-years of exposure were calculated as: 1000×(number of reported events/total person-years of exposure). Person-years of exposure were calculated from the date of the first dose to the date of the last dose of pirfenidone in the study, summed over all patients.
All cases of and reasons for treatment discontinuation during the study period were recorded at 3-monthly follow-up visits. Post hoc stepwise logistic regression models were used to determine the odds of discontinuing treatment (after 6, 12 or 24 months) due to an ADR as a function of baseline patient characteristics. The model was built using forward/backward stepwise logistic regression models, with variables entered into the model at each step at the 0.15 significance level and removed at the 0.20 significance level.
Results
Patient disposition
Overall, 1009 patients (1006 with IPF (99.7%)) were enrolled from 99 pulmonary clinics across Europe: Germany (n=452), France (n=214), UK (n=184), Denmark (n=39), Italy (n=27), Norway (n=25), Finland (n=20), Ireland (n=19), Austria (n=17) and Sweden (n=12). In total, 655 (64.9%) patients discontinued PASSPORT early (i.e. prior to the end of 2-year follow-up); the most common reasons for permanent discontinuation were an ADR related to pirfenidone (n=282 (27.9%)) or death (n=160 (15.9%)) (figure 1). The UK had the highest proportion of patients discontinuing treatment early (n=146 (79.3%)) (supplementary table S1).
FIGURE 1.
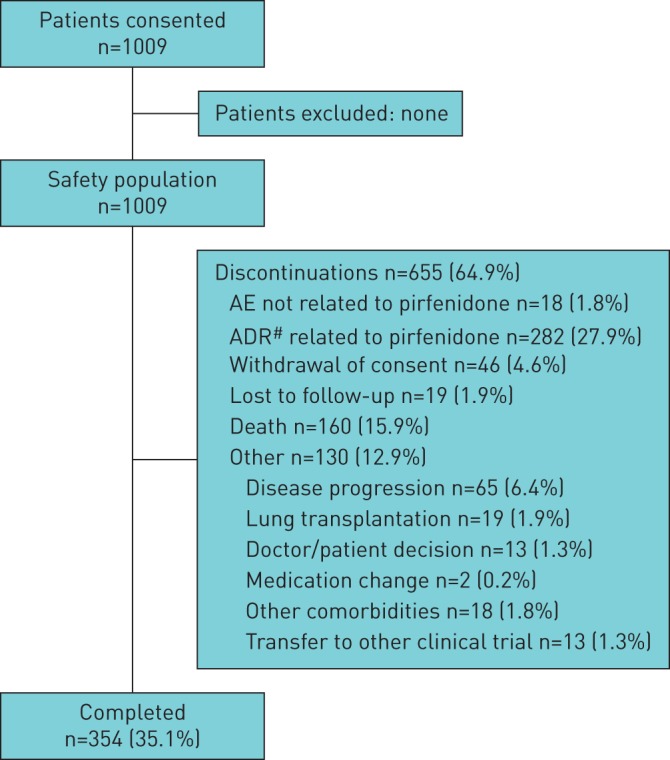
Patient disposition. AE: adverse event; ADR: adverse drug reaction. #: an ADR was defined as any safety event with a possible causal relationship to pirfenidone (the treating physician (investigator) made a clinical judgement to decide if the ADR was related to pirfenidone).
Most patients were male (n=807 (80.0%)), current/ex-smokers (n=627 (62.5%)), with a mean age of 69.6 years and a mean duration of IPF since diagnosis of 1.7 years. The mean FVC % pred at baseline was 66.0% and 144 (14.9%) patients had FVC % pred <50% at baseline (table 1). The majority of patients had at least one comorbidity, most commonly hypertension (n=423 (41.9%)) (table 1); chronic obstructive pulmonary disease and emphysema were found in 3.7% and 2.7% of patients, respectively. Among patients with a baseline LFT, most had values that were <1.5×upper limit of normal for alanine aminotransferase (98.5%), aspartate aminotransferase (98.8%) or total bilirubin (99.4%) (table 1).
TABLE 1.
Patient demographics and baseline characteristics
| Patients who provided data# | Result | |
| Age years | 1009 | 69.6±8.47 |
| Male | 1009 | 807 (80.0) |
| Ethnicity | 758 | |
| White | 753 (99.3) | |
| Other¶ | 5 (0.7) | |
| BMI kg·m−2 | 1006 | 27.8±4.57 |
| Smoking status | 1004 | |
| Current smoker | 25 (2.5) | |
| Ex-smoker | 602 (60.0) | |
| Never-smoker | 377 (37.5) | |
| Patients with IPF | 1009 | 1006 (99.7) |
| Time since IPF diagnosis years | 1006 | 1.7±3.0 |
| Patients with other reason for pirfenidone prescription+ | 1009 | 3 (0.3) |
| Supplementary oxygen use | 1006 | 277 (27.5) |
| FVC L | 970 | |
| Mean±sd | 2.6±0.78 | |
| Median (range) | 2.5 (0.7–5.0) | |
| FVC % pred | 968 | |
| Mean±sd | 66.0±16.1 | |
| Median (range) | 64.8 (21–121) | |
| FVC% pred <50% | 968 | 144 (14.9) |
| FEV1 L | 975 | |
| Mean±sd | 2.1±0.6 | |
| Median (range) | 2.1 (0.0–4.3) | |
| Liver function test | ||
| ALT | 961 | |
| <1.5×ULN | 947 (98.5) | |
| 1.5– <3.0×ULN | 14 (1.5) | |
| ≥3.0×ULN | 0 (0.0) | |
| AST | 745 | |
| <1.5×ULN | 736 (98.8) | |
| 1.5– <3.0×ULN | 9 (1.2) | |
| ≥3.0×ULN | 0 (0.0) | |
| Total bilirubin | 847 | |
| <1.5×ULN | 842 (99.4) | |
| 1.5– <3.0×ULN | 5 (0.6) | |
| ≥3.0×ULN | 0 (0.0) | |
| One or more ongoing medical conditions§ other than IPFƒ | 1009 | 921 (91.3) |
| Hypertension | 423 (41.9) | |
| Gastro-oesophageal reflux disease | 166 (16.5) | |
| Hypercholesterolaemia | 121 (12.0) | |
| Coronary artery disease | 106 (10.5) | |
| Diabetes mellitus | 97 (9.6) | |
| Type 2 diabetes mellitus | 87 (8.6) | |
| Sleep apnoea syndrome | 81 (8.0) | |
| Osteoarthritis | 63 (6.2) | |
| Cough | 56 (5.6) | |
| Benign prostatic hyperplasia | 55 (5.5) | |
| Hypothyroidism | 55 (5.5) | |
| Atrial fibrillation | 53 (5.3) | |
| Hyperlipidaemia | 52 (5.2) | |
| Obesity | 52 (5.2) | |
| Depression | 50 (5.0) | |
| Pre-baseline exposure to pirfenidone | ||
| Patients | 319 (31.6) | |
| Days | 15.0±9.3 |
Data are presented as n, mean±sd or n (%), unless otherwise stated. BMI: body mass index; IPF: idiopathic pulmonary fibrosis; FVC: forced vital capacity; FEV1: forced expiratory volume in 1 s; ALT: alanine aminotransferase; ULN: upper limit of normal; AST: aspartate aminotransferase. #: number of patients who had values for select parameters; ¶: includes Asian, Black or African-American, or any other ethnicities reported; +: includes two patients with extrinsic allergic alveolitis and one patient with nonspecific interstitial pneumonia; §: each medical condition was recorded by investigators using the Medical Dictionary for Regulatory Activities version 19.0 (www.meddra.org) system organ class and preferred term; ƒ: occurring in ≥5% of patients in PASSPORT.
Pirfenidone exposure
The median (interquartile range (IQR)) duration of pirfenidone exposure for all patients in PASSPORT was 442.0 (126.0–729.0) days. This included 319 (31.6%) patients who had received pirfenidone prior to the enrolment visit (table 1). Among the 354 patients who completed the 2-year study, the median (IQR) pirfenidone exposure was 741.5 (722.0–767.0) days. For the 655 patients who discontinued the study early, the median duration of pirfenidone exposure was 200.0 (73.0–426.0) days (due to an ADR: 99.5 (55.0–227.0) days; due to death: 303.0 (119.0–492.0) days; due to another reason: 304.0 (142.0–544.0) days).
ADRs and SADRs
As shown in table 2, 741 (73.4%) patients experienced 2167 ADRs, most commonly nausea (20.6%) and fatigue (18.5%). ADRs experienced by patients from different countries ranged from 69.5% to 82.2% (supplementary table S2). 373 (37.0%) patients had at least one dose adjustment (supplementary table S3).
TABLE 2.
Adverse drug reactions (ADRs)# in the overall population¶
| Total events | Patients with event | Exposure-adjusted event rate+ | Patients with event in first 30 days | Patients with events resulting in permanent discontinuation of pirfenidone | |
| Any ADR | 2167 | 741 (73.4) | 379 (37.6) | 290 (28.7) | |
| Most common ADRs (≥5% of patients) | |||||
| Nausea | 227 | 208 (20.6) | 181.2 | 113 (11.2) | 41 (4.1) |
| Fatigue | 194 | 187 (18.5) | 154.9 | 101 (10.0) | 15 (1.5) |
| Decreased appetite | 172 | 163 (16.2) | 137.3 | 69 (6.8) | 17 (1.7) |
| Decreased weight | 172 | 161 (16.0) | 137.3 | 59 (5.8) | 32 (3.2) |
| Rash | 135 | 123 (12.2) | 107.8 | 15 (1.5) | 32 (3.2) |
| Diarrhoea | 111 | 96 (9.5) | 88.6 | 44 (4.4) | 21 (2.1) |
| Dizziness | 69 | 65 (6.4) | 55.1 | 30 (3.0) | 5 (0.5) |
| Photosensitivity reaction | 61 | 59 (5.8) | 48.7 | 5 (0.5) | 15 (1.5) |
Data are presented as n or n (%). #: an ADR was defined as any safety event with a possible causal relationship to pirfenidone (the treating physician (investigator) made a clinical judgement to decide if the ADR was related to pirfenidone); ¶: n=1009 (person-years of observation=1252.8); +: exposure-adjusted event rate per 1000 person-years of exposure is calculated as: 1000×(number of reported events/total person-years of exposure).
Overall, 290 (28.7%) patients experienced an ADR that resulted in permanent discontinuation of pirfenidone, most commonly nausea (4.1%), decreased weight (3.2%) and rash (3.2%) (table 2). In total, 55 (5.5%) patients experienced 78 SADRs (table 3); six (0.6%) patients experienced an SADR that led to a fatal outcome (supplementary table S4).
TABLE 3.
Serious adverse drug reactions (SADRs)# in the overall population¶
| Total events | Patients with event | Exposure-adjusted event rate+ | |
| Any SADR | 78 | 55 (5.5) | |
| Most common SADRs (≥2 patients) | |||
| Diarrhoea | 6 | 6 (0.6) | 4.8 |
| Decreased weight | 5 | 5 (0.5) | 4.0 |
| Nausea | 4 | 4 (0.4) | 3.2 |
| IPF | 3 | 3 (0.3) | 2.4 |
| Erythema | 3 | 3 (0.3) | 2.4 |
| Decreased appetite | 2 | 2 (0.2) | 1.6 |
| Dyspnoea | 2 | 2 (0.2) | 1.6 |
| Pulmonary embolism | 2 | 2 (0.2) | 1.6 |
| Photosensitivity reaction | 2 | 2 (0.2) | 1.6 |
| Vomiting | 2 | 2 (0.2) | 1.6 |
Data are presented as n or n (%). IPF: idiopathic pulmonary fibrosis. #: an SADR was defined as an adverse drug reaction at any dose that resulted in death, disability or a congenital anomaly/birth defect, was life-threatening, required in-patient hospitalisation or prolonged an existing hospitalisation, but based upon appropriate medical judgement could have jeopardised the patient or may have required intervention to prevent one or more of the outcomes listed (the treating physician (investigator) made a clinical judgement to decide if the SADR was related to pirfenidone); ¶: n=1009 (person-years of observation=1252.8); +: exposure-adjusted event rate per 1000 person-years of exposure is calculated as: 1000×(number of reported events/total person-years of exposure).
Similar proportions of patients with an ADR had a dose adjustment versus no dose adjustment (48.3% versus 51.7%, respectively). More patients completed treatment following a dose adjustment (38.8%) than those who had no dose adjustment (26.1%) (figure 2).
FIGURE 2.

Dose adjustment and retention in patients who experienced an adverse drug reaction (ADR). #: an ADR was defined as any safety event with a possible causal relationship to pirfenidone (the treating physician (investigator) made a clinical judgement to decide if the ADR was related to pirfenidone).
In the overall population, 693 (68.7%) patients experienced ADRSI (table 4), with the most commonly reported special interest categories being gastrointestinal symptoms (38.3%) and photosensitivity reactions/skin rashes (29.0%). In the pre-specified subgroups, the ADRSI rates were generally consistent (table 5).
TABLE 4.
Adverse drug reactions of special interest (ADRSI)# in the overall population¶
| Total events | Patients with event | |
| Any ADRSI | 1577 | 693 (68.7) |
| Gastrointestinal symptoms | 591 | 386 (38.3) |
| Photosensitivity and skin rashes | 388 | 293 (29.0) |
| Fatigue | 257 | 244 (24.2) |
| Weight loss | 173 | 162 (16.1) |
| Dizziness | 79 | 72 (7.1) |
| Abnormal LFTs | 54 | 36 (3.6) |
| Angioedema | 10 | 9 (0.9) |
| Specific cardiac events | 9 | 7 (0.7) |
| Falls | 6 | 6 (0.6) |
| Blood dyscrasias | 4 | 4 (0.4) |
| Severe skin infections | 4 | 4 (0.4) |
| Drug interactions (including smoking) | 2 | 2 (0.2) |
| Increased platelet count | 0 | 0 (0.0) |
Data are presented as n or n (%). LFT: liver function test; EMA: European Medicines Agency; CHMP: Committee for Medicinal Products for Human Use. #: ADRSI included any important identified/potential risks identified in the EMA's CHMP assessment report for pirfenidone (important identified risks: photosensitivity reactions/skin rashes, abnormal LFTs, dizziness, weight loss, gastrointestinal symptoms, fatigue and angioedema; important potential risks: falls, drug interactions (particularly cytochrome P450 CYP1A2 inducers/inhibitors such as cigarette smoke and ciprofloxacin), increased platelet counts, specific cardiac events (including supraventricular tachyarrhythmia, atrioventricular block and sick sinus syndrome, ventricular arrhythmia, bundle branch block and aortic or pulmonic valvular incompetence)) [12]; in addition to these ADRSI included in the EMA's CHMP, this study also included angioedema, blood dyscrasias (specifically agranulocytosis, leukopenia and neutropenia), severe skin reactions and warfarin interactions as important/potential risks (the treating physician (investigator) made a clinical judgement to decide if the ADRSI was related to pirfenidone); ¶: n=1009.
TABLE 5.
Adverse drug reactions of special interest (ADRSI)# in the pre-specified subgroups
| Pre-specified subgroup | Patients | Total events | Patients with event |
| All patients | 1009 (100.0) | 1577 | 693 (68.7) |
| Baseline FVC % pred <50% | 144 (14.3) | 213 | 91 (63.2) |
| Concomitant immunosuppressive therapies | 272 (27.0) | 436 | 190 (69.9) |
| Concomitant other therapies for IPF | 586 (58.1) | 938 | 419 (71.5) |
| Pirfenidone use for conditions other than IPF | 3 (0.3) | 2 | 2 (66.7) |
| Predisposing conditions for liver disease | 190 (18.8) | 246 | 120 (63.2) |
| QT prolongation | 14 (1.4) | 18 | 11 (78.6) |
| Underlying specific cardiac events | 176 (17.4) | 326 | 126 (71.6) |
| Underlying hepatic disease | 43 (4.3) | 83 | 31 (72.1) |
| Underlying other forms of pulmonary disease | 271 (26.9) | 421 | 181 (66.8) |
| Warfarin use | 48 (4.8) | 76 | 34 (70.8) |
| Paediatric patients | 0 (0.0) | ||
| Secondary causes of pulmonary fibrosis | 0 (0.0) |
Data are presented as n (%) or n. FVC: forced vital capacity; IPF: idiopathic pulmonary fibrosis; EMA: European Medicines Agency; CHMP: Committee for Medicinal Products for Human Use. #: ADRSI included any important identified/potential risks identified in the EMA's CHMP assessment report for pirfenidone (important identified risks: photosensitivity reactions/skin rashes, abnormal liver function tests, dizziness, weight loss, gastrointestinal symptoms, fatigue and angioedema; important potential risks: falls, drug interactions (particularly cytochrome P450 CYP1A2 inducers/inhibitors such as cigarette smoke and ciprofloxacin), increased platelet counts, specific cardiac events (including supraventricular tachyarrhythmia, atrioventricular block and sick sinus syndrome, ventricular arrhythmia, bundle branch block and aortic or pulmonic valvular incompetence)) [12]; in addition to these ADRSI included in the EMA's CHMP, this study also included angioedema, blood dyscrasias (specifically agranulocytosis, leukopenia and neutropenia), severe skin reactions and warfarin interactions as important/potential risks (the treating physician (investigator) made a clinical judgement to decide if the ADRSI was related to pirfenidone).
In the overall population, factors associated with early discontinuation due to an ADR during the first 6 months of the study were older age, steroid use prior to the study and female sex (table 6). These factors remained associated with discontinuation during the first 12 months. When the entire study period (24 months) was included in this analysis, significant risk factors for discontinuation due to an ADR were older age, female sex, lower body mass index (BMI) and patients recruited from the UK (table 6).
TABLE 6.
Risk factors associated with discontinuation of pirfenidone due to an adverse drug reaction (ADR)# in the overall population
| Predictor of early discontinuation¶ | Comparison | OR (95% CI) | p-value |
| At 6 months | |||
| Age | Continuous | 1.06 (1.03–1.08) | <0.0001 |
| Female sex | Yes versus no | 1.59 (1.06–2.33) | 0.0228 |
| Steroid use prior to study | Yes versus no | 1.64 (1.10–2.43) | 0.0148 |
| At 12 months | |||
| Age | Continuous | 1.06 (1.04–1.08) | <0.0001 |
| Female sex | Yes versus no | 1.52 (1.04–2.17) | 0.0288 |
| Steroid use prior to study | Yes versus no | 1.48 (1.02–2.17) | 0.0406 |
| Underlying pulmonary disease+ | Yes versus no | 0.72 (0.50–1.03) | 0.0748 |
| During entire study | |||
| Age | Continuous | 1.03 (1.01–1.05) | 0.0009 |
| Female sex | Yes versus no | 1.54 (1.08–2.22) | 0.0193 |
| BMI | Continuous | 0.96 (0.92–0.99) | 0.0127 |
| UK patients | Yes versus no | 1.65 (1.14–2.39) | 0.0081 |
| Current alcohol use | Yes versus no | 0.73 (0.53–1.01) | 0.0555 |
| Years since IPF diagnosis | Continuous | 1.04 (0.99–1.09) | 0.1245 |
| Ex-/current smoker | Yes versus no | 1.30 (0.94–1.79) | 0.1111 |
BMI: body mass index; IPF: idiopathic pulmonary fibrosis; FVC: forced vital capacity. #: an ADR was defined as any safety event with a possible causal relationship to pirfenidone (the treating physician (investigator) made a clinical judgement to decide if the ADR was related to pirfenidone); ¶: following analysis of early discontinuation due to an ADR, the following baseline variables were included in the stepwise logistic regression model-building process: age, sex, BMI, smoking/alcohol status, steroid/azathioprine exposure, UK patient, alanine aminotransferase, FVC, years since IPF diagnosis, FVC % pred <50% and other pulmonary/hepatic/cardiovascular disease; +: other than IPF.
Cumulative probability of survival
The cumulative probability of survival is shown in supplementary figure S1. After 1 and 2 years, the percentages of patients surviving were 89.0% and 76.0%, respectively. The 10th percentile of time to death was 10.6 months and the 20th percentile was 19.6 months.
Discussion
This prospective, real-world, post-authorisation study aimed to assess the long-term safety and tolerability of pirfenidone. PASSPORT also monitored for any potentially unidentified safety or tolerability signals in a large population of patients. In this analysis of the final study data for PASSPORT, no new safety signals were identified and the safety profile was in line with that expected from clinical trials [7–9].
Entry criteria for PASSPORT were minimally restrictive, with patients enrolled from multiple countries, such that the results of the study could apply to a broad population. Generally, baseline characteristics of patients in PASSPORT were similar to those in the RCTs. However, the PASSPORT population included 144 (14.9%) patients with baseline FVC % pred <50%, whereas these patients were excluded from the RCTs at screening [7, 9]. In PASSPORT, the ADRSI rate was similar between patients with FVC % pred <50% (63.2%) and the overall population (68.7%). Additionally, the regression analysis did not identify FVC % pred <50% as a significant risk factor for discontinuation due to an ADR after 6, 12 or 24 months, although it is possible the inclusion of patients with a FVC % pred <50% led to a higher discontinuation due to death, as previously suggested in the interim analyses [13].
PASSPORT also differed from the RCTs in that self-reported ADRs were collected, with an ADR defined as any safety event with a possible causal relationship to pirfenidone, whereas all adverse events (regardless of their relationship to pirfenidone) were reported in the RCTs [7, 9]. The studies also differed in treatment duration and drug exposure: PASSPORT followed patients for 2 years (median exposure to pirfenidone 442.0 days), whereas the duration of the ASCEND trial was 52 weeks, and patients enrolled in CAPACITY received pirfenidone for between 72 and 120 weeks [7, 9]. Furthermore, PASSPORT included patients with a wider range of comorbidities than those who were included in the RCTs. These differences did not result in identification of any new safety issues.
Approximately half of patients with an ADR in PASSPORT experienced an ADR within the first 30 days (379 out of 741 (51.1%)). This corresponds with the RCT data, which demonstrated that adverse events tended to occur within the first 3 months of initiating treatment [14] and reinforces the need for close monitoring during the initial treatment phase. Approximately 29% of the patients in PASSPORT experienced ADRs leading to discontinuation after a median of 99.5 days. The ADRs that led to discontinuation of pirfenidone in this study were mostly gastrointestinal and skin-related, notably nausea (4.1%), decreased weight (3.2%) and rash (3.2%), as observed in the pirfenidone RCTs [7, 9]. Previous real-world studies reported similar pirfenidone discontinuation rates (21–29%); however, it should be noted that these numbers may not be directly comparable due to the differing lengths of follow-up [15, 16]. The real-world discontinuation rates tend to be higher than those observed in the pirfenidone RCTs [7, 9], although rates were still relatively low (<5%) for any single gastrointestinal-related ADR term or any single skin-related ADR. It is possible that the variation in discontinuation rates may have been due to inherent differences in study design. For example, compared with real-world studies, RCTs may be expected to have more stringent inclusion criteria that could favour a healthier population and more frequent follow-up visits to support continuation. Additionally, real-world studies tend to be of a longer duration versus RCTs.
Mitigation strategies to reduce the incidence and/or manage gastrointestinal and skin-related ADRs include taking pirfenidone during/after a meal, reducing/interrupting pirfenidone dose, avoiding sun exposure, wearing protective clothing and applying an ultraviolet A/B broad-spectrum sunscreen [3, 17]. An analysis of dose adjustment and discontinuation in patients with an ADR in PASSPORT found that fewer patients who had a dose adjustment discontinued treatment than those without a dose adjustment (61.2% versus 73.9%). This effect was primarily driven by a lower rate of discontinuation due to an ADR in patients with a dose modification.
In the regression analysis, factors found to be significantly associated (p<0.05) with early treatment discontinuation due to an ADR after 6 and 12 months were older age, use of steroids prior to the study and female sex. In the entire 2-year study, older age and female sex remained significantly associated with early treatment discontinuation. Furthermore, patients recruited in the UK and patients with a lower BMI were more likely to have discontinued treatment during the study. Patients aged ≥80 years have been shown to experience more adverse events when treated with pirfenidone versus patients aged <80 years [14, 18]. Older females tend to experience more adverse events of weight loss, nausea and vomiting, whereas older males tend to experience more adverse events of rash [18]. Similarly, older patients with BMI <25 kg·m−2 experienced greater frequency of fatigue, gastrointestinal and nutritional disorders than those with BMI ≥25 kg·m−2 [18]. It is therefore likely that older females, particularly those with a lower than average BMI, are at greater risk of pirfenidone ADRs and should be monitored closely. The reasoning behind UK patients being more likely to discontinue pirfenidone treatment due to ADRs is unclear and it is possible that this is an accidental finding. The cause for earlier discontinuation in patients with prior steroid use is also unclear, but may reflect heterogeneity of practice by centre or physician.
PASSPORT was conducted at the request of the EMA as a follow-up measure to the EU marketing authorisation received for pirfenidone in 2011 [3]. The study systematically monitored for potential new safety signals associated with pirfenidone, particularly among the pre-specified subgroups based on baseline comorbidities or concomitant medication use listed previously [12]. A large proportion of patients were included in the pre-specified subgroups based on baseline comorbidities and use of concomitant medications, such as immunosuppressive therapies and warfarin. Generally, the rate of ADRSI was similar in these pre-specified subgroups and the general population. Pirfenidone was used for the treatment of IPF in all except three patients and so PASSPORT was unable to provide evidence of safety in other conditions. A number of clinical trials are currently underway to assess the use of pirfenidone in other diseases, including unclassifiable interstitial lung disease (ILD) (ClinicalTrials.gov identifier NCT03099187), ILD associated with systemic sclerosis (ClinicalTrials.gov identifier NCT03221257) and ILD associated with rheumatoid arthritis (ClinicalTrials.gov identifier NCT02808871).
PASSPORT was an observational, open-label, single-arm study, which limits interpretation of the results. Moreover, ADRs were self-reported and only collected at 3-monthly visits, potentially leading to patient recall bias. Furthermore, 319 out of 1009 patients initiated pirfenidone up to 30 days prior to the baseline visit, meaning that some measurements collected at the baseline visit were not taken prior to the commencement of pirfenidone. It is also possible that ADRs resulted in discontinuation of therapy prior to patient enrolment in the study, thereby introducing a bias in the population recruited and ADRs being underreported. Other than warfarin use, medications for IPF and concomitant medications information was limited to medications taken at baseline or within 28 days prior to the onset of an ADR. Additionally, only deaths that were an outcome of an ADR or those that were the primary reason for discontinuation from the study were collected. Finally, data regarding efficacy (e.g. lung function or exercise capacity) were not collected in PASSPORT, as the study objective was to evaluate safety and tolerability.
In summary, no new safety signals were observed in PASSPORT when compared with the pirfenidone RCTs and other post-marketing experience. The safety findings in this study are consistent with the established safety profile of pirfenidone. Furthermore, dose adjustment had a favourable effect on treatment persistence. This study also provides new data on the safety findings of patients within pre-defined subgroups that support the established safety profile of pirfenidone. Identification of predictors of early treatment discontinuation due to an ADR, including older age, female sex and prior steroid use, allows better targeting of patient information to help manage ADRs in these patients.
Supplementary material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00084-2018_supp (216.8KB, pdf)
Footnotes
This article has supplementary material available from openres.ersjournals.com
This study is registered at ClinicalTrials.gov with identifier number NCT02699879. Qualified researchers may request access to individual patient-level data through the clinical study data request platform (www.clinicalstudydatarequest.com). Further details on Roche's criteria for eligible studies are available from https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Roche.aspx. For further details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm.
PASSPORT study group: Peter Cerkl (Hohenems, Austria), Holger Flick (Graz, Austria), Christian Geltner (Klagenfurt, Austria), Judith Löffler-Ragg (Innsbruck, Austria), Michael Studnicka (Salzburg, Austria), Elisabeth Bendstrup (Aarhus, Denmark), Helle Dall Madsen (Odense, Denmark), Saher B. Shaker (Hellerup, Denmark), Maritta Kilpelainen (Turku, Finland), Marjukka Myllaerniemi (Helsinki, Finland), Minna Purokivi (Kuopio, Finland), Seppo Saarelainen (Tampere, Finland), Emmanuel Bergot (Caen, France), Philippe Bonniaud (Dijon, France), Raphael Borie (Paris, France), Pascal Chanez (Marseille, France), Vincent Cottin (Bron, France), Francis Couturaud (Brest, France), Claire Dromer (Pessac, France), Frederic Gagnadoux (Angers, France), Anne-Sophie Gamez (Montpellier, France), Anne Gondouin (Besançon, France), Dominique Israel-Biet (Paris, France), Stephane Jouneau (Rennes, France), Romain Kessler (Strasbourg, France), Francois Lebargy (Reims, France), Sylvain Marchand-Adam (Tours, France), Borsi Melloni (Limoges, France), Jean-Marc Naccache (Paris, France), Christophe Pison (Grenoble, France), Gregoire Prevot (Toulouse, France), Martine Reynaud-Gaubert (Marseille, France), Abdellatif Tazi (Paris, France), Dominique Valeyre (Bobigny, France), Benoit Wallaert (Lille, France), Jost Achenbach (Lostau, Germany), Stefan Andreas (Immenhausen, Germany), Burkhard Bewig (Kiel, Germany), Reiner Bonnet (Bad Berka, Germany), Stephan Budweiser (Rosenheim, Germany), Ulrich Costabel (Essen, Germany), Martin Faehling (Esslingen, Germany), Joachim Ficker (Nuremberg, Germany), Juergen Fisher (Löwenstein, Germany), Sven Glaeser (Greifswald, Germany), Christian Grah (Berlin, Germany), Christian Grohe (Berlin, Germany), Andreas Günther (Giessen, Germany), Martin Hetzel (Stuttgart, Germany), Wolfgang Hohenforst-Schmidt (Coburg, Germany), David Jungck (Bochum, Germany), Peter Kardos (Frankfurt, Germany), Claus Keller (Frankfurt, Germany), Martin Kohlhaeufl (Gerlingen, Germany), Dirk Koschel (Coswig, Germany), Claus Kroegel (Jena, Germany), Rainer Kruegel (Treuenbrietzen, Germany), Berthold Michels (Frankfurt, Germany), Joachim Mueller-Quernheim (Freiburg, Germany), Claus Neurohr (Munich, Germany), Heinz-Theodor Pelzer (Würzburg, Germany), Michael Pfeifer (Donaustauf, Germany), Michael Prediger (Cottbus, Germany), Klaus F. Rabe (Großhansdorf, Germany), Winifried Randerath (Solingen, Germany), Nicolas Scheonfeld (Berlin-Zehlendorf, Germany), Robert Scheubel (Wangen, Germany), Jens Schreiber (Magdeburg, Germany), Hartwig Schutte (Berlin, Germany), Bernd Schoenhofer (Hannover, Germany), Barbara Wagener (Ballenstedt, Germany), Tobias Welte (Hannover, Germany), Michael Westhoff (Hemer, Germany), Heinrike Wilkens (Homburg an der Saar, Germany), Hubert Wirtz (Leipzig, Germany), Michael Henry (Cork, Ireland), Michael Keane (Dublin, Ireland), Anthony O'Regan (Galway, Ireland), Katherine O'Reilly (Dublin, Ireland), Carlo Albera (Orbassano, Italy), Fabrizio Luppi (Modena, Italy), Sandra Nutini (Firenze, Italy), Alberto Pesci (Monza, Italy), Venerino Poletti (Forlì, Italy), Elisabetta Rosi (Sassari, Italy), Paola Rottoli (Sienna, Italy), Thomas Eagan (Bergen, Norway), Durdica Kulosman (Trondheim, Norway), Tone Blorg Sjaheim (Oslo, Norway), Karl Axel Karlsson (Uppsala, Sweden), Magnus Sköld (Stockholm, Sweden), Sanjay Agrawal (Leicester, UK), Kesavan Suresh Babu (Cosham, UK), Stephen Bianchi (Sheffield, UK), Nazia Chaudhuri (Manchester, UK), Sophie Fletcher (Southampton, UK), Michael Gibbons (Exeter, UK), Simon Hart (Kingston upon Hull, UK), Gisli Jenkins (Nottingham, UK), Toby Maher (London, UK), Ann Millar (Bristol, UK), Joanna Porter (London, UK), Nicky Simler (Cambridge, UK), Lisa Spencer (Liverpool, UK), Monica Spiteri (Stoke-on-Trent, UK) and Melissa Wickremasinghe (London, UK).
Conflict of interest: V. Cottin has received consultancy and lectures fees, and support for travel to medical meetings from Actelion and Roche; has received personal fees for development of educational presentations, consultancy and lectures fees, and support for travel to medical meetings from Boehringer Ingelheim; has received consultancy fees from Bayer and Galapagos; has received personal fees from service on an adjudication committee from Gilead; consultancy fees from GlaxoSmithKline (in 2015) and MSD; has received consultancy and lecture fees from Novartis and Sanofi; is the chair of the data and safety monitoring board (DSMB) of Promedior; and serves on the DSMB of Celgene, all outside the submitted work.
Conflict of interest: D. Koschel has received consultancy or speaker fees from AstraZeneca, Boehringer Ingelheim, Grifols, Novartis, Roche/Intermune, Sanofi-Aventis and Teva.
Conflict of interest: A. Günther has received research funding from Sanofi, Inventiva and Roche; and consultancy or speaker fees from Boehringer Ingelheim, Roche and Teva.
Conflict of interest: C. Albera has received fees from Roche (formerly InterMune), Boehringer Ingelheim, Fibrogen, MSD, GlaxoSmithKline, Bayer and Sanofi for activity as an advisor, consultant or steering committee member; he has also received unrestricted grants from Boehringer Ingelheim.
Conflict of interest: A. Azuma has received personal fees from AFT Pharma, Boehringer Ingelheim, InterMune/Roche and Shionogi Co.
Conflict of interest: C.M. Sköld has received research funding from Boehringer Ingelheim, Roche and Sandoz; and consultancy or speaker fees from Almirall, AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, InterMune, Meda, Mundipharma, Novartis, Roche, Sandoz and Vicore Pharma.
Conflict of interest: S. Tomassetti has received speaker fees from Boehringer Ingelheim and Roche.
Conflict of interest: P. Hormel is an employee of Roche-Genentech and holds shares in that company.
Conflict of interest: J.L. Stauffer is an employee of Roche-Genentech and holds shares in that company.
Conflict of interest: I. Strombom is an employee of Roche-Genentech and holds shares in that company.
Conflict of interest: K-U. Kirchgaessler is an employee of Roche-Genentech and holds shares in that company.
Conflict of interest: T.M. Maher has received industry academic research funding from GlaxoSmithKline R&D and UCB Pharma; and consultancy or speaker fees from Apellis, AstraZeneca, aTyr Pharma, Bayer, Biogen Idec, Boehringer Ingelheim, Galapagos, GlaxoSmithKline R&D, ProMetic, Roche (and previously InterMune), Sanumed and UCB Pharma.
Support statement: This study was sponsored by F. Hoffmann-La Roche, Ltd. Medical writing support was provided by Gráinne Faherty on behalf of CMC AFFINITY (a division of Complete Medical Communications, Ltd, Glasgow, UK), funded by F. Hoffmann-La Roche, Ltd. T.M. Maher is supported by a National Institute for Health Research (NIHR) Clinician Scientist Fellowship (NIHR CS-2013-13-017) and a British Lung Foundation Chair in Respiratory Research (C17-3). Funding information for this article has been deposited with the Crossref Funder Registry.
Contributor Information
Collaborators: PASSPORT study group, Peter Cerkl, Holger Flick, Christian Geltner, Judith Löffler-Ragg, Michael Studnicka, Elisabeth Bendstrup, Helle Dall Madsen, Saher B. Shaker, Maritta Kilpelainen, Marjukka Myllaerniemi, Minna Purokivi, Seppo Saarelainen, Emmanuel Bergot, Philippe Bonniaud, Raphael Borie, Pascal Chanez, Vincent Cottin, Francis Couturaud, Claire Dromer, Frederic Gagnadoux, Anne-Sophie Gamez, Anne Gondouin, Dominique Israel-Biet, Stephane Jouneau, Romain Kessler, Francois Lebargy, Sylvain Marchand-Adam, Borsi Melloni, Jean-Marc Naccache, Christophe Pison, Gregoire Prevot, Martine Reynaud-Gaubert, Abdellatif Tazi, Dominique Valeyre, Benoit Wallaert, Jost Achenbach, Stefan Andreas, Burkhard Bewig, Reiner Bonnet, Stephan Budweiser, Ulrich Costabel, Martin Faehling, Joachim Ficker, Juergen Fisher, Sven Glaeser, Christian Grah, Christian Grohe, Andreas Günther, Martin Hetzel, Wolfgang Hohenforst-Schmidt, David Jungck, Peter Kardos, Claus Keller, Martin Kohlhaeufl, Dirk Koschel, Claus Kroegel, Rainer Kruegel, Berthold Michels, Joachim Mueller-Quernheim, Claus Neurohr, Heinz-Theodor Pelzer, Michael Pfeifer, Michael Prediger, Klaus F. Rabe, Winifried Randerath, Nicolas Scheonfeld, Robert Scheubel, Jens Schreiber, Hartwig Schutte, Bernd Schoenhofer, Barbara Wagener, Tobias Welte, Michael Westhoff, Heinrike Wilkens, Hubert Wirtz, Michael Henry, Michael Keane, Anthony O'Regan, Katherine O'Reilly, Carlo Albera, Fabrizio Luppi, Sandra Nutini, Alberto Pesci, Venerino Poletti, Elisabetta Rosi, Paola Rottoli, Thomas Eagan, Durdica Kulosman, Tone Blorg Sjaheim, Karl Axel Karlsson, Magnus Sköld, Sanjay Agrawal, Kesavan Suresh Babu, Stephen Bianchi, Nazia Chaudhuri, Sophie Fletcher, Michael Gibbons, Simon Hart, Gisli Jenkins, Toby Maher, Ann Millar, Joanna Porter, Nicky Simler, Lisa Spencer, Monica Spiteri, and Melissa Wickremasinghe
References
- 1.Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183: 431–440. [DOI] [PubMed] [Google Scholar]
- 2.Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis 2008; 3: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.European Medicines Agency. Esbriet (pirfenidone). Summary of Product Characteristics. 2017. www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002154/WC500103049.pdf Date last accessed: June 1, 2018.
- 4.US Food and Drug Administration. Esbriet. Highlights of Prescribing Information 2017. www.accessdata.fda.gov/drugsatfda_docs/label/2017/208780s000lbl.pdf Date last accessed: June 1, 2018.
- 5.Pharmaceuticals and Medical Devices Agency Japan. Pirfenidone Review Report 2008. www.pmda.go.jp/files/000153687.pdf Date last accessed: June 4, 2018.
- 6.Raghu G, Rochwerg B, Zhang Y, et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med 2015; 192: e3–e19. [DOI] [PubMed] [Google Scholar]
- 7.King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014; 370: 2083–2092. [DOI] [PubMed] [Google Scholar]
- 8.Lancaster L, Albera C, Bradford WZ, et al. Safety of pirfenidone in patients with idiopathic pulmonary fibrosis: integrated analysis of cumulative data from 5 clinical trials. BMJ Open Respir Res 2016; 3: e000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011; 377: 1760–1769. [DOI] [PubMed] [Google Scholar]
- 10.Saturni S, Bellini F, Braido F, et al. Randomized controlled trials and real life studies. Approaches and methodologies: a clinical point of view. Pulm Pharmacol Ther 2014; 27: 129–138. [DOI] [PubMed] [Google Scholar]
- 11.Nallamothu BK, Hayward RA, Bates ER. Beyond the randomized clinical trial: the role of effectiveness studies in evaluating cardiovascular therapies. Circulation 2008; 118: 1294–1303. [DOI] [PubMed] [Google Scholar]
- 12.European Medicines Agency. Committee for Medicinal Products for Human Use Assessment Report: Esbriet 2010. www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002154/WC500103073.pdf Date last accessed: June 5 2018.
- 13.Cottin V, Maher T, Azuma A, et al. Pirfenidone post-authorization safety registry (PASSPORT): update and concomitant use of NAC and/or corticosteroids. Chest 2015; 148: 364A. [Google Scholar]
- 14.Lancaster LH, de Andrade J, Zibrak JD, et al. Pirfenidone safety and adverse event management in idiopathic pulmonary fibrosis. Eur Respir Rev 2017; 26: 170057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galli JA, Pandya A, Vega-Olivo M, et al. Pirfenidone and nintedanib for pulmonary fibrosis in clinical practice: tolerability and adverse drug reactions. Respirology 2017; 22: 1171–1178. [DOI] [PubMed] [Google Scholar]
- 16.Hughes G, Toellner H, Morris H, et al. Real world experiences: pirfenidone and nintedanib are effective and well tolerated treatments for idiopathic pulmonary fibrosis. J Clin Med 2016; 5: E78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Costabel U, Bendstrup E, Cottin V, et al. Pirfenidone in idiopathic pulmonary fibrosis: expert panel discussion on the management of drug-related adverse events. Adv Ther 2014; 31: 375–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lancaster LH, Morrison LD, Auais A, et al. Pirfenidone in patients aged 80 years and older with idiopathic pulmonary fibrosis (IPF): safety findings from pooled trial databases. Am J Respir Crit Care Med 2017; 195: A5385. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary material 00084-2018_supp (216.8KB, pdf)