

CRISPR-Cas genome-editing nucleases hold substantial promise for human therapeutics1–6 but identifying unwanted off-target mutations is important for clinical translation7. A well-validated method that can reliably identify off-targets in vivo has not been described to date, leaving it unclear whether and how frequently these mutations occur. Here we describe Verification of In Vivo Off-targets (VIVO), a highly sensitive strategy that can robustly identify genome-wide CRISPR-Cas nuclease off-target effects in vivo. Using VIVO and a guide RNA (gRNA) deliberately designed to be promiscuous, we show that CRISPR-Cas nucleases can induce substantial off-target mutations in mice livers in vivo. More importantly, we used VIVO to show that appropriately designed gRNAs can direct efficient in vivo editing in mice livers with no detectable off-target mutations. VIVO provides a general strategy for defining and quantifying gene-editing nuclease off-target effects in whole organisms, thereby providing a blueprint to foster development of in vivo gene editing therapeutics.
VIVO consists of two steps (Fig. 1a). In an initial in vitro “discovery” step, a superset of potential off-target cleavage sites for a nuclease is identified using CIRCLE-seq8. This method is highly sensitive, avoids potential confounding effects associated with cell-based assays8, and can successfully identify supersets of sites that include bona fide off-targets in cultured human cells8. In a second in vivo “confirmation” step, sites identified by CIRCLE-seq are examined for indel mutations in target tissues that have been treated with the nuclease.
Figure 1. Overview and validation of VIVO.
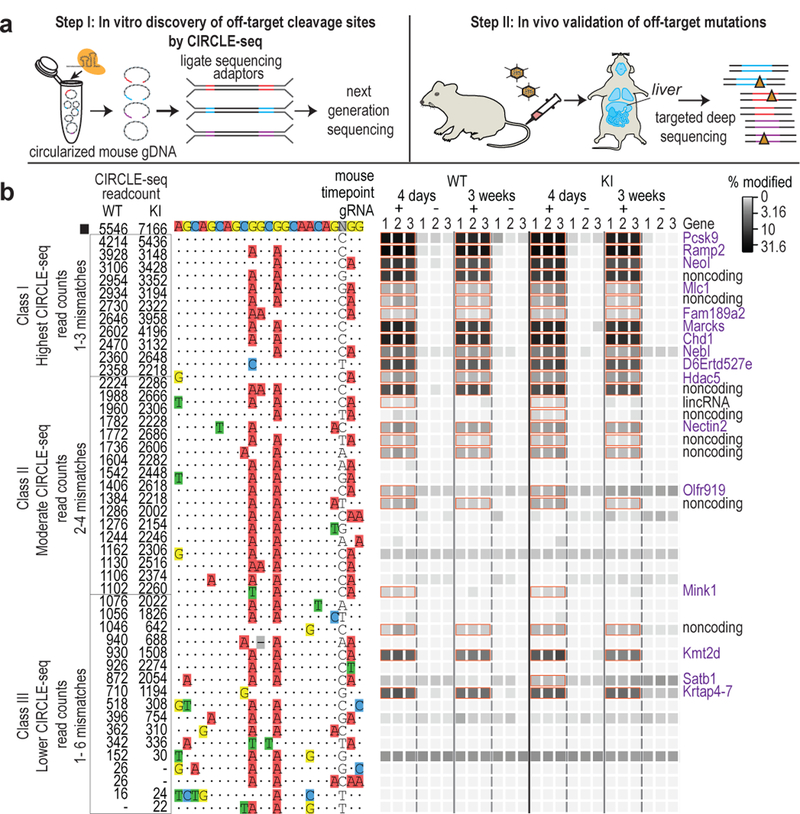
(a) Schematic illustrating the two-step VIVO method. In Step 1, CIRCLE-seq identifies off-target sites cleaved in vitro. In Step 2, the sites identified in Step 1 are assessed in vivo for indel mutations by targeted amplicon sequencing performed with genomic DNA isolated from the livers of nuclease-treated mice. (b) Assessment of in vivo off-target indels induced by gP/SpCas9. Indel frequencies as determined by targeted amplicon sequencing are presented as heat maps for the gP/SpCas9 on-target site (black square) and the Class I, Class II, and Class III off-target sites (identified from CIRCLE-seq experiments). Each locus was assayed in n=3 biologically independent WT and KI mice (1, 2, 3) using genomic DNA isolated from the liver of mice treated with experimental adenoviral vector encoding gP/SpCas9 (gRNA +) or control adenoviral vector GFP/SpCas9 (gRNA -). Mismatches relative to the on-target site are shown with colored boxes and spacer sequence is numbered from 1 (most PAM-proximal) to 20 (most PAM-distal). CIRCLE-seq read count numbers for each site are shown. Sites that showed a significant difference between the experimental gRNA + and gRNA - samples are outlined with orange boxes and labelled by genomic locus with coding regions shown in purple text. P values and significance were obtained by fitting a negative binomial generalized linear model (for source data and P values, see Supplementary Table 2).
To test VIVO, we designed a Streptococcus pyogenes Cas9 (SpCas9) gRNA targeted to the mouse Pcsk9 gene that we expected would have a high likelihood of inducing multiple off-target mutations in the mouse genome (Online Methods, Extended Data Fig. 1a and Extended Data Table 1). To deliver this promiscuous gRNA (gP) and SpCas9 to mouse livers in vivo, we infected cohorts of mice with an adenoviral vector encoding these components or a negative control vector encoding GFP and SpCas9. Adenoviral vectors have a known biodistribution, which includes efficient delivery to the liver (Extended Data Fig. 2). Two strains of mice were infected: a wild-type C57BL/6N strain (“WT” mice) and littermates that harbor a single copy of a human PCSK9 open reading frame knocked into the Rosa26 locus (“KI” mice; Carreras A & Pane LS et al., manuscript submitted; Extended Data Fig. 3; Online Methods). gP/SpCas9 induced stable and efficient mutation of the on-target Pcsk9 site and reductions in plasma Pcsk9 protein levels in both WT and KI mice (Extended Data Figs. 1b and 1c).
Having established efficacy of gP/SpCas9 for in vivo on-target Pcsk9 modification, we conducted the first screening step of VIVO by performing CIRCLE-seq with gP/SpCas9 on liver genomic DNA from WT and KI mice (Fig. 1a; Online Methods). We identified many off-target cleavage sites in vitro: 3107 and 2663 sites with WT and KI mice genomes, respectively (Extended Data Fig. 4 and Supplementary Table 1). These sites represent only a small percentage of all sites in the genome that have seven or fewer mismatches relative to the on-target site (Extended Data Fig. 5). 2368 sites were identified in both mouse genomes and showed strong concordance (r2 = 0.902) in their CIRCLE-seq read counts (Extended Data Fig. 4), which semi-quantitatively reflect cleavage efficiency8. Sites identified in only one or the other genome generally had among the lowest CIRCLE-seq read counts Extended Data Fig. 4), consistent with the possibility that these sites may or may not be detected just by chance because they lie at the assay limit of detection (as we previously observed with analogous experiments in human cells in culture8). SNPs might also play a role for a very small subset of these sites (Extended Data Table 2). The 20 sites with the highest CIRCLE-seq read counts all had three or fewer mismatches in the spacer sequence (Extended Data Fig. 4 and Supplementary Table 1). Many off-target sites contained protospacer adjacent motif (PAM) mismatches, with NAG as the most prevalent (Extended Data Fig. 4 and Supplementary Table 1), most likely because gP has an unusually high relative number of closely matched sites in the mouse genome with an NAG PAM (Extended Data Table 3), a known alternative PAM for SpCas98, 9.
To perform the second step of VIVO, we assessed whether gP off-target cleavage sites identified by CIRCLE-seq showed evidence of indels in vivo in the livers of WT and KI mice treated with gP/SpCas9 and control GFP/SpCas9 adenoviral vectors. Because of the very large number of sites identified by CIRCLE-seq, we performed targeted amplicon sequencing at four days and three weeks post-infection on only the following subsets of sites: the Pcsk9 on-target site, 11 “Class I” off-target sites with the highest CIRCLE-seq read counts (harboring one to three mismatches relative to the on-target site), 17 “Class II” sites with moderate CIRCLE-seq read counts (harboring two to four mismatches), and 17 “Class III” sites with lower CIRCLE-seq read counts (harboring one to six mismatches) (Extended Data Fig. 4 and Fig. 1b). The Pcsk9 on-target site was efficiently mutagenized and remained stably mutated in both WT and KI mice (mean indel frequencies ranging ~23 to 30%) ( Fig. 1b and Supplementary Table 2 ). 19 of the 45 sites we examined showed significant indels (mean frequencies ranging 41.9% to 0.13%) in WT and KI mice livers at four days and three weeks post-infection (Fig. 1b and Supplementary Table 2). Notably, higher CIRCLE-seq read counts generally correlated well with the likelihood of finding indels in vivo – 11 of the 11 Class I off-target sites, 5 of the 17 Class II sites, and 3 of the 17 Class III sites harbored indels (Fig. 1b); also, all 19 of these sites had three or fewer mismatches relative to the on-target site, with the majority located within gene coding sequences (Fig. 1b). Three additional sites, including two sites each containing three mismatches in the spacer and one mismatch in the PAM, showed evidence of significant indel mutations at only the four-day time point, but were no longer significant by three weeks; the mutation frequencies observed at these sites were around 0.13%, close to the limit of detection (0.1%) for next-generation sequencing10. Collectively, these data show that SpCas9 with a promiscuous gRNA can generate stable (and sometimes high-frequency) off-target mutations in vivo and that VIVO can identify such mutations even with frequencies as low as 0.13%.
Because therapeutic applications would not use a promiscuous gRNA with so many closely matched genomic sites, we sought to assess the in vivo off-target profiles of SpCas9 gRNAs designed to be more orthogonal to the mouse genome. We constructed two additional gRNAs (gM11 and gMH) targeted to mouse Pcsk9 (Extended Data Fig. 6a) but that have relatively few closely matched sites in the C57BL6/N mouse genome (Extended Data Table 1). gMH should also target a site (with one mismatch) in the human PCSK9 ORF present in the KI mouse genome (Extended Data Fig. 6a). Delivery of gM/SpCas9 and gMH/SpCas9 by adenoviral vectors induced expected genetic alterations in the mouse Pcsk9 gene and human PCSK9 transgene as well as corresponding decreases in plasma Pcsk9 and PCSK9 proteins (Extended Data Figs. 6b and 6c).
We conducted the first in vitro CIRCLE-seq step of VIVO with gM/SpCas9 and gMH/SpCas9 on WT and KI mouse genomic DNAs. The on-target mouse Pcsk9 was identified in all four experiments and the human PCSK9 transgene site only in the experiment with gMH on KI mouse DNA (Fig. 2). We identified fewer off-target sites with gM/SpCas9 and gMH/SpCas9 (Fig. 2, Supplementary Tables 3 and 4) than we had with gP/SpCas9. For gM, we found 182 off-target sites – 129 with WT mouse DNA and 145 with KI mouse DNA and with 92 sites common to both. For gMH, we found 529 off-target sites – 333 with WT mouse DNA and 394 with KI mouse DNA and with 198 sites common to both. All but two of the 711 off-target sites identified with these gRNAs had three or more mismatches, consistent with the higher orthogonality of these gRNAs relative to the mouse genome. For both gRNAs, there were good concordances in CIRCLE-seq read counts between the WT and KI mice when considering all off-target sites identified (r2 = 0.747 and 0.841 for gM and gMH, respectively) (Fig. 2). Sites found in only one mouse genome or the other again generally had low CIRCLE-seq read counts (Fig. 2) and analysis of gM CIRCLE-seq and targeted amplicon sequencing data suggests that these differences are not due to SNPs (Extended Data Table 2 and Supplementary Table 5).
Figure 2. Characterization of Pcsk9-targeted gRNAs designed to be orthogonal to the mouse genome by CIRCLE-seq.
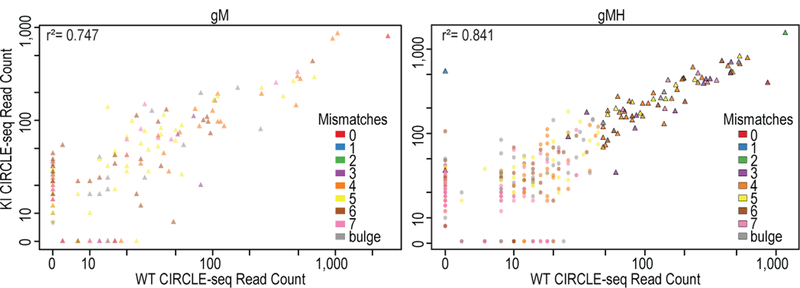
Scatterplots of CIRCLE-seq read counts for off-target cleavage sites identified in vitro with gM/SpCas9 and gMH/Cas9 on genomic DNA from WT (n=1) and KI (n=1) mice (for source data, see Supplementary Tables 3 and 4). Each site is color-coded for the number of mismatches it has relative to the on-target site. Sites represented as triangles were chosen for targeted amplicon sequencing. Correlation R2 values shown were obtained by linear regression performed using all values in each scatterplot.
We conducted the second in vivo step of VIVO for gM and gMH by performing targeted amplicon sequencing of sites found by CIRCLE-seq from liver genomic DNA of adenovirus-treated WT and KI mice. For gM, we comprehensively examined 181 of the 182 off-target CIRCLE-seq cleavage sites (one site could be amplified but not sequenced, see Online Methods) and the on-target site from liver DNA harvested at 4 days or 3 weeks following adenovirus infection. Strikingly, only the gM on-target site showed significant evidence of indels (ranging 12.6% to 18.5%) (Fig. 3 and Supplementary Table 6); no significant off-target indels were identified at any of the 181 off-target sites in both mice at either time point. For gMH, because CIRCLE-seq identified a large number of potential off-target sites (529 in total), we examined the on-target site and only a subset of 69 off-target sites that had the highest CIRCLE-seq read counts (and up to six mismatches). These 69 sites encompassed all but one of the CIRCLE-seq sites that had up to three mismatches (one site could be amplified but not sequenced, see Online Methods) and the human PCSK9 transgene site (with one mismatch) (Fig. 2). This choice was guided by our finding that none of the 19 stable in vivo off-target sites we found for gP/SpCas9 had four or more mismatches relative to the on-target site (Fig. 1b). Among the 69 sites, we found significant indel mutations at only two: the on-target mouse gMH site (27.4% to 43.6% indels) and the human PCSK9 transgene site bearing one mismatch (20.4% to 21.7% indels) (Extended Data Fig. 7; Supplementary Table 7).
Figure 3. Assessment of in vivo off-target indels induced by gM/SpCas9.

Indel frequencies determined by targeted amplicon sequencing for the gM/SpCas9 on-target site (black square) and 181 off-target sites identified by CIRCLE-seq. Each condition shown was assayed in n=3 biologically independent mice (1, 2, 3) using genomic DNA isolated from the liver of mice treated with experimental adenoviral vector encoding gM/SpCas9 (gRNA +) or control adenoviral vector GFP/SpCas9 (gRNA -). Data are presented as in Fig. 1b. The single site (the on-target site) that was significantly different between the experimental gRNA + and control gRNA – samples is highlighted with orange boxes. Additional closely matched sites in the mouse genome (not identified from the CIRCLE-seq experiments) examined for indel mutations are boxed in red. For source data and P values (negative binomial), see Supplementary Table 6.
To exclude the possibility that CIRCLE-seq might have missed any bona fide off-target sites, we also performed targeted amplicon sequencing of the most closely matched sites in the mouse C57BL/6N genome (harboring up to three mismatches in the spacer) not identified in our CIRCLE-seq experiments (4 sites for gM and 10 sites for gMH) (Extended Data Table 1). Three of the gM sites could not be individually selectively amplified and so these were assessed together as a pool (Online Methods). We did not observe significant indels at any of these sites in all treated mice at both time points (Fig. 3; Extended Data Fig. 7; Supplementary Tables 6 and 7).
Our report provides, to our knowledge, the first demonstration that CRISPR-Cas nucleases can robustly induce off-target mutations in vivo. Previous in vivo studies have reported no or very few off-target mutations but used the cell-based GUIDE-seq method12–14 or other in silico approaches that had not been validated to effectively identify these sites in vivo (see Supplementary Discussion). By contrast, VIVO enabled the robust and sensitive identification of off-target sites in vivo (with frequencies as low as ~0.13%). The high sensitivity of CIRCLE-seq is most likely what enabled identification of a superset of all potential off-target sites including those actually mutated in vivo (Supplementary Discussion). The detection limit of VIVO, like all existing methods, is bounded by the current error rate of next-generation sequencing for indels (~0.1%). In addition, we did not attempt to detect large-scale chromosomal rearrangements (translocations, inversions or large deletions) but we expect the frequencies of such alterations to be no higher than that of any off-target indels. Other methods15 might be used in future studies to test for these alterations.
VIVO defines a pathway for assessing the in vivo genome-wide specificities of CRISPR-Cas nucleases. Our work suggests that gRNAs should be designed to have the lowest possible number of closely matched genomic sites (with three or fewer mismatches), which can be done using existing in silico tools16. Such gRNAs can be assessed in VIVO Step 1 to identify those that have a reasonable number (<100–200) of in vitro off-target cleavage sites and then these sites can be comprehensively examined for indels in vivo using targeted amplicon sequencing in VIVO Step 2. We also recommend examination of any other closely matched genomic sites (<4 mismatches) not identified by CIRCLE-seq. Persistent off-target mutations might be further reduced using various strategies for improving the genome-wide specificities of CRISPR-Cas nucleases (Supplementary Discussion).
We believe VIVO sets an important standard for defining in vivo off-target effects of gene editing nucleases. The approach should be generalizable to non-CRISPR gene-editing nucleases, to non-mammalian organisms, and to other delivery methods (Supplementary Discussion). VIVO should also be useful for characterizing the in vivo specificities of engineered CRISPR-Cas9 variants and other CRISPR-Cas orthologues (Supplementary Discussion) and to assess the effectiveness of methods for reducing off-target effects of these nucleases. We expect our findings should accelerate the advancement of research and methods that will further spur the clinical translation of in vivo genome editing therapeutics.
Methods
Guide RNA design
The promiscuous gP gRNA was identified by searching for an on-target sequence within mouse Pcsk9 (ENSMUSG000044254) exons one to three that shows a high number of closely matched sites (two or fewer mismatches to the on-target site) in the mouse genome. gMH was designed by searching for a gRNA that can cleave both mouse Pcsk9 and human PCSK9 and that showed a perfect alignment to mouse Pcsk9 and up to two nucleotides mismatch to human PCSK9 (ENSG00000169174) at least eight nucleotides distal to the PAM. For both gRNA designs, AstraZeneca proprietary software was used as an in silico tool, which was developed based on Wellcome Trust Sanger Institute`s codebase (WGE: http://www.sanger.ac.uk/htgt/wge/)17 with the addition of the NAG PAM motif as well as NGG in the alignments for potential off-targets9. GRCm38/mm10 and GRCh38/hg38 genomes were used as reference for alignments. The gM gRNA targeted to mouse Pcsk9 has been previously described11.
Adenoviral constructs
Adenoviruses that express SpCas9 and gRNAs (Ad-Cas9-gM, Ad-Cas9-gMH, Ad-Cas9-gP) were generated by Vector Biolabs (Malvern, PA, USA). SpCas9 and gRNAs were expressed from chicken β-actin hybrid (CBh) and U6 promoters, respectively, in a replication-deficient adenoviral-serotype 5 (dE1/E3) backbone. A negative control adenovirus (Ad-Cas9-GFP) that expresses Cas9 and GFP from the CBh and CMV promoters, respectively, but no gRNA was also generated.
Animal studies
All animal experiments were approved by the AstraZeneca internal committee for animal studies as well as the Gothenburg Ethics Committee for Experimental Animals, (license number: 162–2015+) compliant with EU directives on the protection of animals used for scientific purposes.
C57BL/6N mice (Charles River, Sulzfeld, Germany) were individually housed in a temperature (21±2°C) and humidity (55±15%) controlled room with a 12:12 hours light:dark cycle. R3 diet (Lactamin AB, Stockholm, Sweden) and tap water were provided ad libitum. Cage bedding and enrichments include spen chips, shredded paper, gnaw sticks and a plastic house.
Humanized hypercholesterolemia mouse model was generated by liver-specific overexpression of human PCSK9 in C57BL/6N mice (Carreras A & Pane SL et al., manuscript submitted). Briefly, the KI mouse was generated by cloning human PCSK9 ORF downstream of the mouse albumin promoter from Albumin-Cre mice, in a vector designed to target the mouse Rosa26. Founder C57BL/6N hPCSK9 heterozygous males were crossed with C57BL/6N females to generate experimental animals, which are littermates with two genotypes: C57BL/6N hPCSK9KI+/− (referred as KI) and wild type C57BL/6N hPCSK9KI−/− (referred as WT) (Extended Data Figure 3).
For in vivo Pcsk9 gene editing, nine- to eleven-week-old male mice received a tail vein injection with a dose of 1 × 109 infection units (IFU) adenovirus (Ad-Cas9-gM, Ad-Cas9-gMH, Ad-Cas9-gP or Ad-Cas9-GFP) in 200 µl diluted with phosphate-buffered saline. Peripheral blood was sampled before virus administration (baseline), a week after virus administration, and at termination (four days or three weeks after virus administration). Animals were euthanized by cardiac puncture under isoflurane anesthesia at the experimental endpoint. The organs including liver, spleen, lungs, kidney, muscle, brain, and testes were dissected, snap-frozen in liquid nitrogen and stored at −80 until further analyses.
Ten mgs of frozen liver tissue was lysed to obtain 30–50 µg genomic DNA using the Gentra Puregene Tissue kit (Qiagen, Hilden, Germany). In vivo gene editing efficiency was evaluated using Surveyor mismatch cleavage assay (Integrated DNA Technologies, BVBA, Leuven, Belgium) (using primers: for gP GAGGCCGAAACCTGATCCTT and CTTAGAGACCACCAGACGGC, for gM GGAGGACACGTTTTCTGCAT and CTGCTGCTGTTGCTGCTAC, for gMH mouse locus GACTTTGTGAAGGCTGGGGA and TGCATGGAGCAATGCAGAGA, for gMH human transgene TAGCCTTGCGTTCCGAGGAG and CATTCTCGAAGTCGGTGACCA; with amplicon sizes 619, 415, 349, 495 base-pair, respectively) and targeted deep sequencing (primers listed in Supplementary Tables 2, 6, and 7).
Animals were randomized based on their weights measured prior to the experiments. The investigators were blinded to group allocation during data collection and analysis. No sample size calculation was performed. Sample size was determined as generation of triple independent samples for comparisons between groups that is sufficient to perform statistical tests.
Assessment of human PCSK9/mouse Pcsk9 protein levels in plasma
Peripheral blood was collected in EDTA-coated capillary tubes from vena saphena during the course of the study and by cardiac puncture at the time of termination. Samples were kept on ice for up to 2 hours prior to extraction of plasma by centrifugation at 10,000 rpm for 20 min at 4oC. Plasma was stored at −80°C until the samples were analyzed. Plasma human PCSK9 and mouse Pcsk9 levels were determined with a standard ELISA kit (DPC900 and MPC900; R&D Systems, Minneapolis, MN, USA) according to the manufacturer´s instructions. Prior to the assay, plasma samples were diluted 1:800 and 1:1000 for human PCSK9 and mouse Pcsk9, respectively.
Reference Genome for CIRCLE-seq, CRISPResso and Cas-OFFinder
Build 38 of the C57BL/6NJ genome, sequenced by the Sanger Mouse Genomes Project (http://csbio.unc.edu/CCstatus/pseudo2/C57BL6NJ_b38_f.fa.gz), was used as the reference genome for the experiments of this report. The human PCSK9 gene DNA sequence was inserted into the mouse genome as an extra chromosome in the reference and was named as “chrPCSK9KI”.
CIRCLE-seq
CIRCLE-seq was performed experimentally as previously described8. Data was processed using v1.1 of the CIRCLE-Seq analysis pipeline18 (https://github.com/tsailabSJ/circleseq) with parameters: “window_size: 3; mapq_threshold: 50; start_threshold: 1; gap_threshold: 3; mismatch_threshold: 7; merged_analysis: False, variant_analysis: True”. The gP off-target sites were adjusted for mapping artifacts in highly repetitive regions by consolidating contiguous sites whose mapping positions differed by 5 base pairs or less. The off-target sequences reported for these consolidated sites were obtained by performing the local alignment used in the CIRCLE-seq pipeline.
Targeted amplicon deep sequencing of off-targets
Genomic DNA from liver tissue of adenovirus injected mice was extracted at 4 days and 3 weeks post-treatment for indel analysis. As detailed in the text, to validate off-targets identified by CIRCLE-seq for gP we selected sites with read counts above 50% of the on-target and a variety of lower-ranked sites (containing up to 6 mismatches relative to the on-target) for targeted deep sequencing. In addition, to rule out that CIRCLE-seq was not missing potential off-target sites identified by in silico tools, we sequenced all sites containing up to 3 mismatches identified by Cas-OFFinder16 for gM and gMH. All sites analyzed were amplified from 150 ng of input genomic DNA (approximately 5 × 104 genomes) with Phusion Hot Start Flex DNA polymerase (New England Biolabs). PCR products were purified using magnetic beads made as previously described19, quantified using a QuantiFlor dsDNA System kit (Promega), normalized to 10 ng/µL per amplicon, and pooled. Pooled samples were end-repaired and A-tailed using an end prep enzyme mix and reaction buffer from NEBNext Ultra II DNA Library Prep Kit for Illumina, and ligated to Illumina TruSeq adapters using a ligation master mix and ligation enhancer from the same kit. Library-prepped samples were then purified with magnetic beads made as previously described19, size-selected using PEG/NaCl SPRI solution (KAPA Biosystems), quantified using droplet digital PCR (BioRad), and loaded onto an Illumina MiSeq for deep sequencing. To analyze amplicon sequencing of potential on- and off-targets, we used CRISPResso software20 v1.0.11 (https://github.com/lucapinello/crispresso) with the following parameters: ‘-q 30 --ignore_substitutions --hide_mutations_outside_window_NHEJ’.
For each of the 45 gP off-target sites we examined, we obtained 10,000 or more sequencing reads in at least two samples for treated and control samples at all time points (Supplementary Table 2). One potential gM off-target site (chr15:98037617–98037640) and one potential gMH off-target site (chr15:4878177–4878200) were amplified but could not be successfully sequenced. The problematic gM site was amplified with two different sets of primers and both amplicons failed to sequence. The gMH site is in a highly repetitive area with low complexity, and we were unable to differentiate this site from other sites in the genome so the site was removed from analysis. For all of the gM off-target sites we were able to sequence, we obtained 10,000 or more sequencing reads in at least two samples for treated and control samples at all time points (Supplementary Table 6). For all but one of the gMH off-target sites we were able to sequence, we obtained 10,000 or more sequencing reads in at least two samples for treated and control samples at all time points (Supplementary Table 7). One of the gMH off-target sites we sequenced (chr17:33501685–33501708) did not reach the 10,000 read threshold for any samples or time points but read counts ranged from 2509 to 9149. For the three sites that were identified in silico as being highly similar to the gM on-target site but that were not identified by CIRCLE-seq, we were unable to selectively amplify these sites individually due their sequence similarities: chr14:25878231–25878254, chr14:26018001–26018024 and chr14:26157615–26157638. Therefore, for these three sites, the read counts were pooled into one amplicon that encompasses all locations and that is labelled as “chr14:pooled” in Supplementary Table 6.
Targeted amplicon deep sequencing of WT and KI untreated mice
Genomic DNA from liver tissue of untreated mice (the same mice that CIRCLE-seq was performed on) was amplified at sites that contained no reads in either the KI or WT CIRCLE-seq for gM and deep sequenced to look for single nucleotide polymorphisms (Supplementary Table 5). Primers used are the same as in Supplementary Table 6. Sites were analyzed with CRISPRessoPooled with the following parameters: --cleavage_offset −7 –window_around_sgrna 13 in order to perform a focused variant analysis in the spacer.
Cas-OFFinder
Identification of potential off-targets by Cas-OFFinder16 (https://github.com/snugel/cas-offinder, version 2.4) was done using the off-line version allowing up to 7 mismatches and non-canonical PAMs. We then restricted the output to the sites with at most 6 mismatches in the spacer and at most 1 mismatch in the PAM.
Non-reference genetic variation
samtools (mpileup and bcftools, v1.3.121) was used to discover non-reference genetic variation at the off-target sites identified by CIRCLE-seq. Positions with a genotype quality score greater than 5 and depth of at least 3 were considered as potential variants if they did not fall adjacent to the cleavage site or at the edge of the reads and were not located in a highly repetitive region with poor mapping quality.
Statistical analysis of plasma protein levels
Data visualization and statistical analyses for plasma protein measurements were performed using GraphPad Prism 7.02. Protein levels after the adenoviral administration were normalized to baseline levels, and values for gRNA treatment groups were compared with the control treatment group. Comparisons between groups were performed using two-way ANOVA test followed by Sidak`s or Dunnett`s two-sided adjusted multiple comparisons test, depending on the number of comparison groups. P< 0.05 was considered to be statistically significant. The level of significance in all graphs is represented as follows: *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. Exact P values, confidence intervals and effect size are presented in the source data.
Statistical analysis of targeted amplicon deep sequencing data
P values were obtained by fitting a negative binomial generalized linear model (function MASS:glm.nb in R version 3.4.2 with parameter init.theta=1 and with the logarithm of the total number of reads as the offset) to the control and nuclease-treated samples for each evaluated site with at least one non-zero indel count among the nuclease-treated samples. In order to avoid convergence issues we added 1 to all the indel counts and confirmed that rerunning the models without the addition of the 1 did not result in any additional significant off-target sites. We adjusted for multiple comparisons using the Benjamini and Hochberg method (function p.adjust in R version 3.4.2). Multiple testing adjustment was performed within strata defined by guide, mouse background, and timepoint. We considered the indel percentage in the gRNA/SpCas9-treated replicates to be significantly greater than the indel percentage in the GFP/SpCas9-treated controls if the adjusted P value was less than 0.1, the nuclease-treatment coefficient was greater than zero, and the median indel frequency of the treated replicates was greater than 0.1%.
Extended Data
Extended Data Fig. 1. gP/SpCas9 efficiently mutates the mouse Pcsk9 gene and reduces Pcsk9 protein plasma levels in vivo.
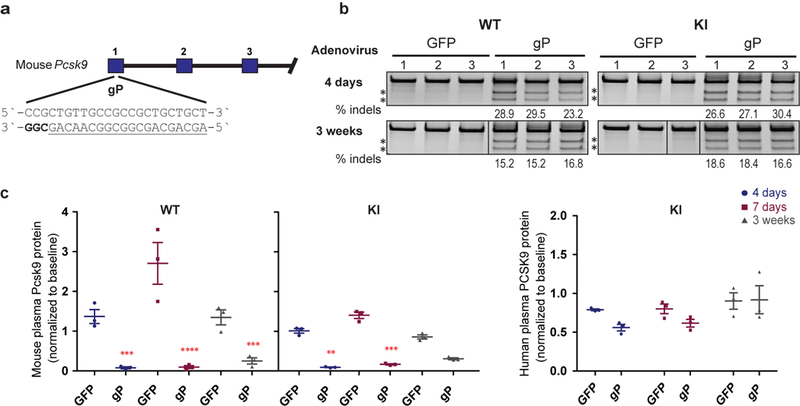
(a) The gP gRNA was designed to target a sequence within exon 1 of the mouse Pcsk9 gene that has many closely related genomic sites (i.e., those with one to three mismatches relative to the on-target site; Extended Data Table 1). Blue bars indicate exons for the mouse genomic region. (b) Surveyor assay and next-generation DNA sequencing data demonstrating efficient in vivo modification of the on-target mouse Pcsk9 gene site in mouse liver by gP/SpCas9. Assays were performed 4 days and 3 weeks after administration of adenoviral vectors encoding gP/SpCas9 (“gP”) or negative control GFP/SpCas9 (“GFP”). For each time point, the assays used genomic DNA isolated from livers of n=3 biologically independent wild-type C57BL/6N (WT) mice or C57BL/6N-derivative mice harbouring a single copy of the human PCSK9 ORF under Albumin promoter knocked into the Rosa26 locus (KI). Asterisks indicate cleaved PCR products expected following treatment with Surveyor nuclease. Percentages show the frequencies of indel mutations determined by targeted amplicon sequencing using next-generation sequencing (these are the same values shown for the on-target site in Fig. 1b). Lines divide lanes taken from different locations on the same gel. For source data for Surveyor assays and targeted amplicon sequencing, see Supplementary Figure 1 and Supplementary Table 2, respectively. (c) Plasma mouse Pcsk9 protein levels measured in n=3 biologically independent WT and KI mice and plasma human PCSK9 protein levels measured in n=3 biologically independent KI mice following nuclease treatment. Protein levels were assessed 4 days, 7 days, and 3 weeks following administration of gP or control GFP adenoviral vectors and normalized to baseline levels. Significant differences between experimental and control groups were determined using two-way ANOVA and Sidak’s two-sided adjusted multiple comparisons test, P: *<0.05, **<0.01, ***<0.001, ****<0.0001. See source data for Extended Data Figure 4b for exact adjusted P values. All values are presented as group means, error bars represent standard error of the mean (SEM). The enhanced reduction of plasma Pcsk9 relative to the frequency of Pcsk9 genetic alteration observed is consistent with previously published studies (Supplementary Discussion).
Extended Data Fig. 2. Bio-distribution studies of adenovirus-serotype 5 in mice.
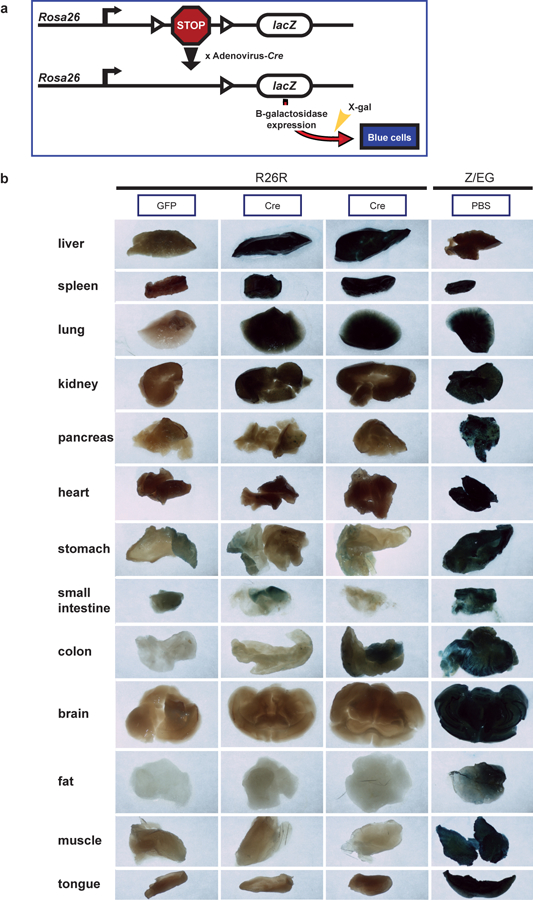
(a) Schematic of integrated reporter construct in R26R mice used to assess delivery of Cre recombinase using adenovirus-serotype 5 vector. Cre-mediated excision of a loxP-flanked transcriptional stop signal upstream of a lacZ gene results in expression of beta-galactosidase enzyme. Beta-galactosidase expression can be quantified by staining dissected tissues with X-gal, a compound that turns blue when cleaved by this enzyme. (b) Quantification of beta-galactosidase expression in sections of various dissected organs from n=2 biologically independent R26R mice intravenously injected with adenovirus-serotype 5 vector encoding Cre. Matched organs sections from a R26R mouse intravenously injected with an adenovirus-serotype 5 vector encoding GFP were used to determine background staining levels and serve as a negative control. Matched organ sections from Z/EG mice that constitutively express lacZ (beta-galactosidase) and intravenously injected with PBS (rather than adenovirus) were used to provide positive staining controls. All mice were evaluated one week after adenovirus or PBS injection. The experiment was performed one time.
Extended Data Fig. 3. Breeding strategy for generation of experimental mice containing human PCSK9 open reading frame knocked into the Rosa26 locus.
C57BL/6N-derivative mouse line harbouring a single copy of the human PCSK9 open reading frame knocked into the Rosa26 locus (C57BL/6N hPCSK9 KI +/−) are used for breeding with C57BL/6N mice. Offspring yielded experimental animals that are C57BL/6N hPCSK9 KI +/− (referred as “KI”) and C57BL/6N hPCSK9 KI −/− (referred as “WT”) males.
Extended Data Fig. 4. Scatterplot of CIRCLE-seq read counts for sites identified with gP/SpCas9 on genomic DNA from n=3 biologically independent WT and KI mice.
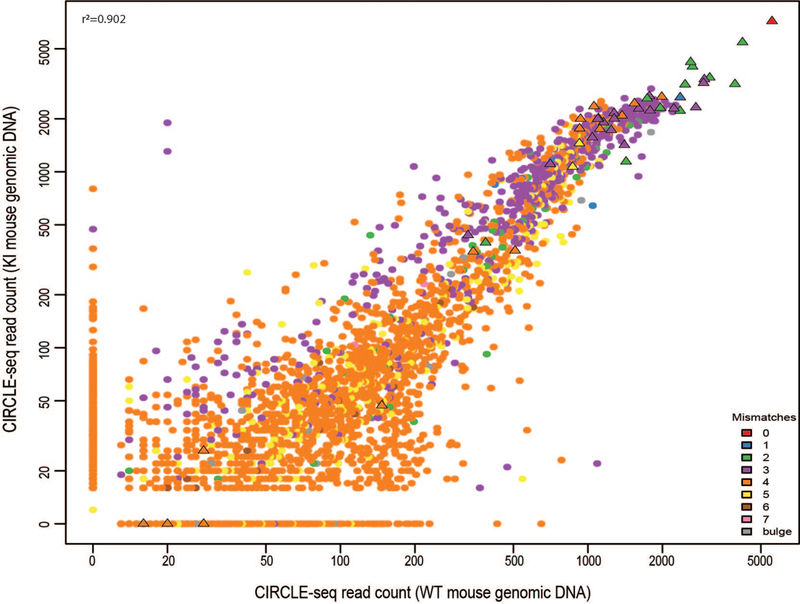
Read counts are shown on a log scale and colors indicate the number of mismatches in each off-target site relative to the on-target site. Sites shown as triangles were chosen for targeted amplicon sequencing. The correlation R2 value obtained using all values in the scatterplot is shown in the upper left-hand corner and was obtained using a linear regression.
Extended Data Fig. 5. Comparison of closely matched sites identified in silico and off-target cleavage sites identified by CIRCLE-seq.
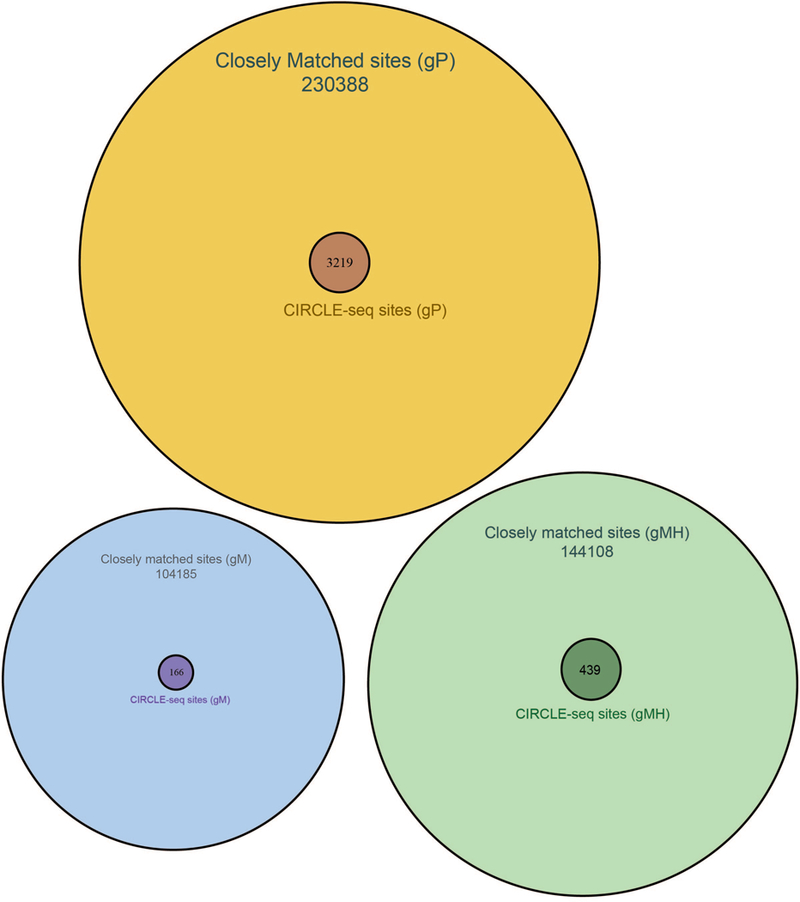
Venn diagrams comparing off-target cleavage sites in mouse genomic DNA identified by CIRCLE-seq experiments with closely matched sites (up to six mismatches relative to the on-target site) in the mouse genome identified in silico by Cas-OFFinder are shown for SpCas9 gRNAs gP, gM, and gMH.
Extended Data Fig. 6. Genetic and phenotypic alterations induced by delivery of gM/SpCas9 and gMH/SpCas9 in vivo.
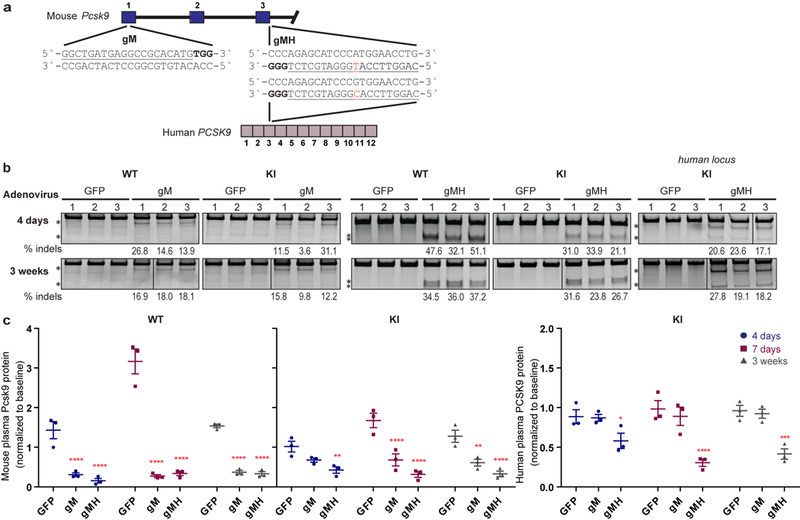
(a) Sequence and location of the SpCas9 gM (mouse) and gMH (mouse & human) gRNA target sites in the endogenous mouse Pcsk9 gene and human PCSK9 transgene inserted at the mouse Rosa26 locus. The single base position that differs between the gMH target sites in the mouse Pcsk9 gene and the human PCSK9 transgene is highlighted in red. Blue bars indicate exons for the mouse genomic region while purple bars represent exons for the human genomic locus; PAM sequence for the sites is in bold and the spacer sequence is underlined. (b) Surveyor assay and next-generation DNA sequencing data demonstrating efficient in vivo modification of the on-target endogenous mouse Pcsk9 site and human PCSK9 transgene in mouse liver. Assays were performed 4 days and 3 weeks following administration of adenoviral vectors encoding gM and SpCas9 (“gM”), gMH and SpCas9 (“gMH”) or GFP and SpCas9 (“GFP”) using genomic DNA isolated from livers of n=3 biologically independent WT and KI mice. Asterisks indicate cleaved PCR products expected following treatment with Surveyor nuclease. Percentages show the frequencies of indel mutations determined by targeted amplicon sequencing using next-generation sequencing (these are the same values shown for the on-target sites in Fig. 3 and Extended Data Fig. 7). Lines divide lanes taken from different locations on the same gel. For source data for Surveyor assays, see Supplementary Fig. 1. For source data for targeted amplicon sequencing, see Supplementary Tables 6 and 7 for gM and gMH, respectively. (b) Plasma mouse Pcsk9 protein levels in n=3 biologically independent WT and KI mice, and plasma human PCSK9 protein levels in n=3 biologically independent KI mice following CRISPR-Cas nuclease treatment. Plasma protein levels were assessed 4 days, 7 days, and 3 weeks following administration of gM, gMH, or control GFP adenoviral vectors and normalized to baseline levels at each timepoint. Significant differences between groups were determined using two-way ANOVA and Dunnett`s two-sided adjusted multiple comparisons test, p *<0.05, **<0.01, ***<0.001, ****<0.0001. See source data for Extended Data Figure 6 for exact adjusted P values. Values are presented as group means, error bars represent standard errors of the mean (SEM).
Extended Data Fig. 7. Assessment of in vivo off-target indel mutations induced by gMH/SpCas9.
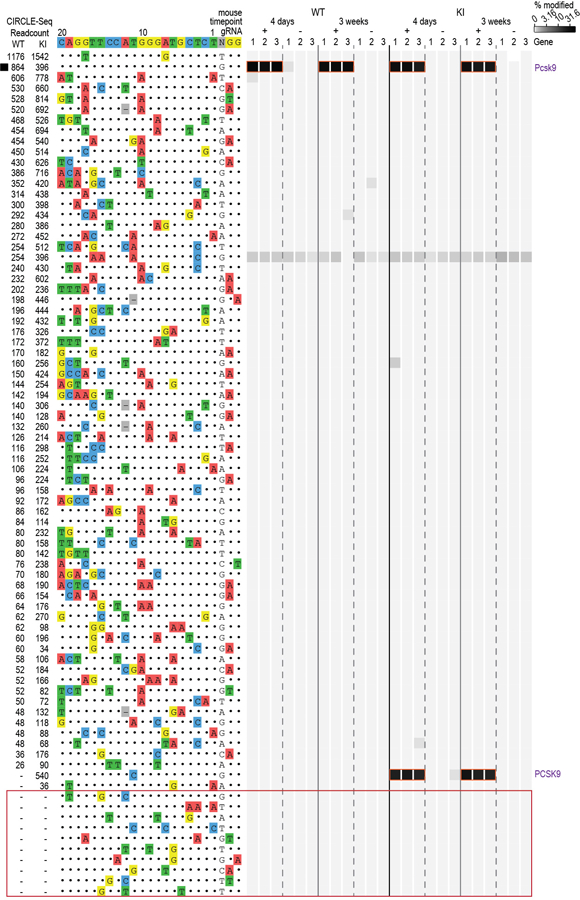
Indel mutation frequencies determined by targeted amplicon sequencing (using high-throughput sequencing) are presented as heat maps for the gMH/SpCas9 on-target site (black square) and 63 off-target sites identified from CIRCLE-seq experiments. Each locus was assayed in n=3 biologically independent mice (1, 2, 3) using genomic DNA isolated from the liver of WT and KI mice treated with experimental adenoviral vector encoding gMH/SpCas9 (gRNA +) or control adenoviral vector GFP/SpCas9 (gRNA -). For each site, mismatches relative to the on-target site are shown with colored boxes and bases in the spacer sequence are numbered from 1 (most PAM-proximal) to 20 (most PAM-distal). The number of read counts found for each site from the CIRCLE-seq experiments on WT and KI mouse genomic DNA are shown in the left columns (ranked from highest to lowest based on counts in the WT genomic DNA CIRCLE-seq experiment). Each box in the heatmap represents a single sequencing experiment. Sites that were significantly different between the experimental gRNA + and control gRNA – samples are highlighted with an orange outline around the boxes. Additional closely matched sites in the mouse genome (not identified from the CIRCLE-seq experiments) that were examined for indel mutations are boxed in red at the bottom of the figure. See Supplementary Table 7 for source data and P values (negative binomial).
Extended Data Table 1: Numbers of off-target sites for gP, gM, and gsMH gRNAs identified by Cas-OFFinder (in silico) and CIRCLE-seq (experimental).
For sites identified by CIRCLE-seq, the total number of sites found in the WT and KI mice are listed and just below that are the number of sites found in each of the two mice.
| Sites with canonical NGG PAM | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Number of spacer mismatches | |||||||||
| gRNA | Method | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
| gP | Cas-OFFinder | 1 | 5 | 41 | 355 | 1073 | 3347 | 21900 | 94051 |
| CIRCLE-seq (total WT&KI) | 1 | 5 | 38 | 231 | 121 | 44 | 15 | 4 | |
| CIRCLE-seq (WT mouse) | 1 | 5 | 38 | 226 | 117 | 43 | 13 | 2 | |
| CIRCLE-seq (Kl mouse) | 1 | 5 | 38 | 218 | 93 | 25 | 11 | 4 | |
| gM | Cas-OFFinder | 1 | 0 | 0 | 8 | 77 | 780 | 8315 | 55093 |
| CIRCLE-seq (total WT&KI) | 1 | 0 | 0 | 4 | 18 | 36 | 32 | 25 | |
| CIRCLE-seq (WT mouse) | 1 | 0 | 0 | 4 | 17 | 26 | 23 | 17 | |
| CIRCLE-seq (Kl mouse) | 1 | 0 | 0 | 3 | 17 | 30 | 23 | 14 | |
| gMH | Cas-OFFinder | 1 | 1 | 1 | 15 | 178 | 1609 | 10992 | 55363 |
| CIRCLE-seq (total WT&KI) | 1 | 1 | 1 | 9 | 52 | 65 | 82 | 159 | |
| CIRCLE-seq (WT mouse) | 1 | 0 | 1 | 8 | 46 | 46 | 45 | 81 | |
| CIRCLE-seq (Kl mouse) | 1 | 1 | 1 | 9 | 43 | 53 | 64 | 102 | |
| Sites with PAM harboring single mismatch | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Number of spacer mismatches | |||||||||
| gRNA | Method | 0 | 1 | 2 | 3 | 4 | 5 | 6 | |
| gP | Cas-OFFinder | 0 | 16 | 370 | 12028 | 9828 | 20097 | 70495 | |
| CIRCLE-seq (total WT&KI) | 0 | 14 | 279 | 2160 | 271 | 31 | 6 | ||
| CIRCLE-seq (WT mouse) | 0 | 14 | 272 | 1648 | 164 | 13 | 2 | ||
| CIRCLE-seq (Kl mouse) | 0 | 14 | 271 | 1904 | 262 | 31 | 4 | ||
| gM | Cas-OFFinder | 0 | 0 | 1 | 20 | 423 | 4911 | 34722 | |
| CIRCLE-seq (total WT&KI) | 0 | 0 | 1 | 3 | 18 | 18 | 10 | ||
| CIRCLE-seq (WT mouse) | 0 | 0 | 0 | 3 | 15 | 16 | 8 | ||
| CIRCLE-seq (Kl mouse) | 0 | 0 | 1 | 3 | 14 | 8 | 4 | ||
| gMH | Cas-OFFinder | 0 | 0 | 7 | 80 | 907 | 8285 | 67109 | |
| CIRCLE-seq (total WT&KI) | 0 | 0 | 3 | 19 | 21 | 9 | 17 | ||
| CIRCLE-seq (WT mouse) | 0 | 0 | 3 | 18 | 18 | 7 | 9 | ||
| CIRCLE-seq (Kl mouse) | 0 | 0 | 3 | 16 | 9 | 5 | 13 | ||
Extended Data Table 2. Off-target sites identified by CIRCLE-seq for gP, gM, and gMH that exhibit single nucleotide polymorphisms (SNPs) based on CIRCLE-seq data.
Mismatches relative to the on-target site are shown as lower-case letters. SNPs that differ from the C57BL/6N mouse strain are shown as red-colored lowercase letters. Targeted amplicon sequencing of sites for gM (first row) was performed with the same genomic DNA from WT and KI mice used for CIRCLE-seq experiments (data in Supplementary Table 7). N.D. = not done.
| Location of site | gRNA | Mouse | gRNA on-target site | Off-target sequence | Number of mismatches |
SNPs confirmed by targeted amplicon- sequencing |
|---|---|---|---|---|---|---|
| chr9:78832014-78832037 | gM | KI | GGCT GAT GAGGCCGCACATGNGG | atCAGAT aAaaCCaCACATGGag | 8 | No |
| chr4:129226173-129226196 | gMH | KI | CAGGTTCCATGGGATGCTCTNGG | agGGcT CacctGGATGCTCTGtG | 8 | N.D. |
| chr9:68916733-68916756 | gMH | KI | CAGGTTCCATGGGATGCTCTNGG | tAGGgagagaGGATGCTCTGaG | 8 | N.D. |
| chr3:19461541-19461564 | gMH | KI | CAGGTTCCATGGGATGCTCTNGG | CAtGTaCCAaGGGATGtTCTAcG | 5 | N.D. |
| chr13:37109353-37109376 | gP | KI | AGCAGCAGCGGCGGCAACAGNGG | a-cAGCAGCAGCAGCAACAACGA | 6 | N.D. |
| chr6:112201818-112201841 | gp | WT | AGCAGCAGCGGCGGCAACAGNGG | AGCAAcagcagcagcagcagtAG | 5 | N.D. |
Extended Data Table 3. Numbers of closely matched sites in the mouse genome with canonical NGG, alternate NAG, and other alternate non-NGG/non-NAG PAMs for gP, gM, and gMH.
Top panel shows the total numbers of these sites identified by Cas-OFFinder and CIRCLE-seq. In the bottom panel, the sites with alternate NAG PAMs identified by Cas-OFFinder are shown by the number of mismatches present in the spacer region for each gRNA.
| Guide RNA | Method for finding off-target sites | Canonical NGG PAM | NAG PAM | Non-canonical Non-NAG | Total sites |
|---|---|---|---|---|---|
| gp | Found in mouse genome by Cas-OFFinder | 120773 | 64538 | 78925 | 264236 |
| gp | Found by CIRCLE-seg | 472 | 2668 | 263 | 3403 |
| gM | Found in mouse genome by Cas-OFFinder | 64274 | 10285 | 37421 | 111980 |
| gM | Found by CIRCLE-seg | 130 | 25 | 28 | 183 |
| gMH | Found in mouse genome by Cas-OFFinder | 68160 | 16404 | 79049 | 163613 |
| aMH | Found by CIRCLE-seg | 448 | 52 | 30 | 530 |
| Sites with NAG PAM | |||
|---|---|---|---|
| Number of mismatches | gP sites | gMH sites | gM sites |
| 1 | 0 | 0 | 0 |
| 2 | 14 | 0 | 0 |
| 3 | 331 | 4 | 0 |
| 4 | 11349 | 26 | 8 |
| 5 | 7817 | 220 | 122 |
| 6 | 12416 | 1858 | 1676 |
| 7 | 32611 | 14296 | 8479 |
Supplementary Material
Acknowledgements
J.K.J. is supported by the Desmond and Ann Heathwood MGH Research Scholar Award. J.K.J., M.L.B., and J.G. were supported by a sponsored research agreement with AstraZeneca. L.P. is supported by a National Human Genome Research Institute (NHGRI) Career Development Award (R00HG008399). J.K.J., M.J.A. and J.M.L. are supported by a National Institutes of Health Maximizing Investigators’ Research Award (MIRA) (R35 GM118158). J.K.J., L.P. and K.C. are supported by the Defense Advanced Research Projects Agency (HR0011-17-2-0042). We thank Mike Snowden, Stefan Platz and Steve Rees for resource allocation from AstraZeneca Research Funds. We thank Jonathan Y. Hsu for discussions and input.
Footnotes
Data availability statement
Sequence data that support the findings of this study have been deposited with SRA accession number SRP151131 (https://trace.ncbi.nlm.nih.gov/Traces/sra_sub/sub.cgi?acc=SRP151131&focus=SRP151131&from=list&action=show:STUDY).
Supplementary Information is linked to the online version of the paper.
References
- 1.Musunuru K The Hope and Hype of CRISPR-Cas9 Genome Editing: A Review. JAMA cardiology 2, 914–919 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Fellmann C, Gowen BG, Lin PC, Doudna JA & Corn JE Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat Rev Drug Discov 16, 89–100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Komor AC, Badran AH & Liu DR CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 168, 20–36 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koo T & Kim JS Therapeutic applications of CRISPR RNA-guided genome editing. Brief Funct Genomics 16, 38–45 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Cornu TI, Mussolino C & Cathomen T Refining strategies to translate genome editing to the clinic. Nat Med 23, 415–423 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Dunbar CE et al. Gene therapy comes of age. Science 359 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Tsai SQ & Joung JK Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat Rev Genet 17, 300–312 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai SQ et al. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods 14, 607–614 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu PD et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31, 827–832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai SQ et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding Q et al. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res 115, 488–492 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yin H et al. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat Biotechnol 35, 1179–1187 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yin H et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol (2016). [DOI] [PMC free article] [PubMed]
- 14.Gao X et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 553, 217–221 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giannoukos G et al. UDiTaS, a genome editing detection method for indels and genome rearrangements. BMC Genomics 19, 212 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bae S, Park J & Kim JS Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodgkins A et al. WGE: a CRISPR database for genome engineering. Bioinformatics (Oxford, England) 31, 3078–3080 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsai SQ, Topkar VV, Joung JK & Aryee MJ Open-source guideseq software for analysis of GUIDE-seq data. Nat Biotechnol 34, 483 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Rohland N & Reich D Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res 22, 939–946 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pinello L et al. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat Biotechnol 34, 695–697 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.