

Abstract
Protein, DNA, and RNA methyltransferases have an ever‐expanding list of novel substrates and catalytic activities. Even within families and between homologs, it is becoming clear the intricacies of methyltransferase specificity and regulation are far more diverse than originally thought. In addition to specific substrates and distinct methylation levels, methyltransferase activity can be altered by complex formation with close homologs. We work with the N‐terminal methyltransferase homologs NRMT1 and NRMT2. NRMT1 is a ubiquitously expressed distributive trimethylase. NRMT2 is a monomethylase expressed at low levels in a tissue‐specific manner. They are both nuclear methyltransferases with overlapping consensus sequences but have distinct enzymatic activities and tissue expression patterns. Co‐expression with NRMT2 increases the trimethylation rate of NRMT1, and here we aim to understand how this occurs. We use analytical ultracentrifugation to show that while NRMT1 primarily exists as a dimer and NRMT2 as a monomer, when co‐expressed they form a heterotrimer. We use co‐immunoprecipitation and molecular modeling to demonstrate in vivo binding and map areas of interaction. While overexpression of NRMT2 increases the half‐life of NRMT1, the converse is not true, indicating that NRMT2 may be increasing NRMT1 activity by stabilizing the enzyme. Accordingly, the catalytic activity of NRMT2 is not needed to increase NRMT1 activity or increase its affinity for less preferred substrates. Monomethylation can also not rescue phenotypes seen with loss of trimethylation. Taken together, these data support a model where NRMT2 expression activates NRMT1 activity, not through priming, but by increasing its stability and substrate affinity.
Keywords: NRMT1, NRMT2, N‐terminal methylation, methyltransferase, homologs
Introduction
The initial defining feature of methyltransferase enzymes was onto which type of substrate they catalyzed the transfer of methyl groups from S‐adenosyl‐methionine (SAM). DNA methyltransferases have been further characterized into those that place 5′methylcytosine de novo or maintain it at hemimethylated CpG sites.1 Protein methyltransferases have been further characterized by the amino acid they methylate and the level (mono, di, or tri) of methylation they catalyze.2 Likewise, the RNA methyltransferases have been divided into four superfamilies based on their structure and the nucleotide they methylate.3 It is becoming clear, however, that in addition to these intrinsic methyltransferase qualities, there is an additional layer of functional regulation in their expression patterns and complex formation.
A common theme emerging is complex formation between methyltransferase homologs. In Trypanosoma brucei, the major type I arginine methyltransferase, PRMT1, has a very narrow substrate specificity and weak activity in vitro.4 However, co‐expression with its close paralog PRMT3 increases PRMT1 stability, and subsequently, its activity.4 PRMT3 is missing key catalytic residues and does not exhibit in vitro enzymatic activity and has been characterized as a prozyme.4 Activation of a catalytically weak enzyme by an inactive prozyme paralog has also been seen in T. brucei polyamine biosynthesis and described as the prozyme paradigm.5 On average, the enzyme/prozyme complex typically exceeds the activity of the active enzyme alone by 2000‐fold.5, 6
Similarly, the recently discovered N6‐methyladenosine (m6A) RNA methyltransferase METTL3 forms a heterodimer with its close homolog METTL14.7 Unlike T. brucei PRMT1 and PRMT3, METTL3 and METTL14 each show weak methyltransferase activity in vitro.7, 8 However, m6A methylase activity is significantly increased upon heterodimer formation, and this binding also promotes reciprocal stability of the homologs.7, 9 Although these data suggest METTL14 is not strictly an inactive prozyme, it appears its catalytic activity is not required for in vivo activity of the complex.10 The crystal structure of the heterodimer catalytic domains also indicates the METTL14 catalytic site is relatively occluded and does not bind SAM.10 The current model is that the primary role of METTL14 is to provide structural support to METTL3 and interact with RNA substrate.8, 10
The mammalian methyltransferase EZH2 is part of the PRC2 complex that represses transcription by di‐ and trimethylating histone H3 on lysine 27 (H3K27me2/3).11 EZH2 has one close homolog, EZH1, which also interacts with members of the PRC2 complex.12 PRC2‐EZH1 can also repress transcription, but EZH1 has weak H3K27me2/3 activity.12 In contrast to PRC2‐EZH2, PRC2‐EZH1 represses transcription by binding and compacting nucleosomes.12 The PRC2‐EZH2 and PRC2‐EZH1 complexes are thought to be primarily mutually exclusive, but data indicate EZH1 and EZH2 can interact with each other both in vitro and in vivo.12, 13 It is still functionally unclear why a PRC2 complex that contains EZH2 would also contain EZH1,13 but as they accomplish the same goal by different mechanisms, there may be certain circumstances when synergistic activity would be beneficial. Unlike T. brucei PRMT1 and PRMT3 and mammalian METTL14 and METTL3, Ezh1 and Ezh2 have very different expression patterns,12 and the need for complexed EZH1 and EZH2 may be specific to the tissues and developmental times where their expression patterns overlap.
We work on the N‐terminal methyltransferase homologs NRMT1 (N‐terminal RCC1 methyltransferase 1) and NRMT2 (N‐terminal RCC1 methyltransferase 2), which following cleavage of the initiating methionine, methylate the α‐amine of the first N‐terminal residue of their substrates. NRMT1 and NRMT2 are 50% identical and 75% similar and share an N‐terminal X‐P‐K consensus sequence.14, 15 Although structurally similar, they differ in their catalytic activities. In vitro, NRMT1 exhibits distributive trimethylase activity, and in vivo, complete knockout of NRMT1 via homologous recombination or CRISPR/Cas9 abolishes N‐terminal trimethylation.14, 16, 17 NRMT2 exhibits monomethylase activity in vitro,14 but its knockout phenotypes remain to be characterized due to lack of an antibody that successfully detects it endogenously.
There are over 300 predicted NRMT targets15 and over a dozen verified targets, including RCC1, the tumor suppressor Retinoblastoma protein (Rb), the histone chaperone SET, the centromere proteins CENP‐A and CENP‐B, Poly(ADP‐Ribose) Polymerase 3 (PARP3), Damaged DNA‐binding protein 2 (DDB2), and a variety of myosin light chains and ribosomal proteins.18, 19, 20, 21, 22 For the verified targets, N‐terminal methylation seems to predominantly regulate protein–DNA interactions. Loss of N‐terminal methylation of RCC1 disrupts its binding to DNA, its localization to chromatin, and its ability to properly establish mitotic spindles.23 N‐terminal trimethylation of CENP‐B enhances its binding to α‐satellite DNA,19 and N‐terminal trimethylation of CENP‐A is also required for its recruitment to the centromere.24 DDB2 that cannot get N‐terminally methylated cannot be as efficiently recruited to UV‐induced cyclobutane dimers and these dimers cannot be as effectively repaired.21
NRMT1 and NRMT2 have distinct mRNA expression patterns. NRMT1 is expressed ubiquitously in all tissues, while NRMT2 has much lower, tissue‐specific expression, with its highest expression found in the skeletal muscle and the liver.14 This indicates NRMT1 can function independently without NRMT2. In addition, NRMT1 knockout does not significantly affect monomethylation levels,16 indicating that even at low levels, NRMT2 can function without NRMT1 present. Taken together these data indicate both NRMT1 and NRMT2 can function independently without the other present. However, it is unknown if N‐terminal mono‐ and trimethylation are functionally redundant, and in vitro co‐expression of NRMT2 with NRMT1 is able to increase the trimethylation activity of NRMT1,14 indicating the two may also work synergistically under certain conditions.
Here we aimed to better understand the interdependence of NRMT1 and NRMT2 and determine if they function in a complex. First, we used analytical ultracentrifugation to show that purified recombinant NRMT1 and NRMT2 interact and determined the stoichiometry of interaction as 2:1 NRMT1 to NRMT2. Next, we used co‐immunoprecipitation (co‐IP) to show NRMT1 and NRMT2 can be found complexed together in vivo and molecular modeling to map their areas of interaction. We also used cycloheximide experiments to determine that while the half‐life of NRMT1 increases in the presence of overexpressed NRMT2, the converse is not true. We used kinetic assays to quantitatively determine the effect of NRMT2 expression on NRMT1 activity and site directed mutagenesis to determine that NRMT2 enzymatic activity is not needed for an increase in NRMT1 trimethylase activity or substrate affinity. Finally, to determine if N‐terminal mono‐ and trimethylation are functionally redundant, we tagged the NRMT substrate SETα with Dendra2 photoswitchable protein and showed its half‐life is significantly decreased in NRMT1 CRISPR/Cas9 knockout cells. This decrease in half‐life can be rescued by NRMT1 overexpression but not by overexpression of NRMT2. From these data, we conclude that NRMT1 and NRMT2 are most similar to their family members METTL3 and METTL14, which both exhibit methyltransferase activity in vitro but the activity of METTL3 predominates in vivo. Stability and substrate affinity are increased by complex formation and monomethylase priming activity is not required. These data do not rule out an additional unique function of N‐terminal monomethylation by NRMT2, but show one of its functions when expressed is to promote N‐terminal trimethylation.
Results
NRMT1 and NRMT2 form heterotrimers
Like METTL3 and METTL14, NRMT1 (METTL11A), and NRMT2 (METTL11B) are part of the Methyltransferase like (METTL) family of class I methyltransferases containing seven‐beta‐strand methyltransferase motifs and Rossman folds for binding SAM.25, 26 Although the enzymes in the METTL family have a wide range of substrates, including protein, RNA, and small molecules, they are structurally similar.27 Given that METTL3 and METTL14 must form a heterodimer for optimal m6A methylation activity,7 and NRMT1 has been shown to have increased N‐terminal trimethylation activity when co‐expressed with NRMT2,14 we wanted to determine if NRMT1 and NRMT2 also heterodimerize.
His‐tagged recombinant human NRMT1 and NRMT2 were expressed and purified from E. coli and analyzed individually and combined by analytical ultracentrifugation. Ultrascan 3 analysis identified a pool of NRMT1, that when analyzed alone, sediments as a monomer of approximately 33 kD [Fig. 1(a)]. This is slightly larger than the predicted molecular mass of 25 kD for a NRMT1 monomer. The majority of NRMT1, however, sedimented as a 52 kD dimer [Fig. 1(a)]. NRMT2 showed an opposite pattern when run alone. There was a pool of NRMT2 that sedimented as a 52.6 kD dimer, but the majority sedimented as a 35.7 kD monomer, which is close to its predicted molecular mass of 32 kD [Fig. 1(b)]. When run in combination at equimolar concentrations, monomers of each NRMT1 and NRMT2 were still present, but a new species of 83.8 kD was now visible, representing a heterotrimer formed from a NRMT1 dimer and a NRMT2 monomer [Fig. 1(c)]. The pool of NRMT1 dimer was completely depleted, suggesting that if NRMT2 monomer is present the NRMT1 dimer will bind it. In addition, as there was excess monomeric NRMT1 and NRMT2 present and no indication of a NRMT1 and NRMT2 heterodimer, these data also suggest NRMT1 cannot bind NRMT2 unless it is already homodimerized and a pool of NRMT2 monomer can exist independent of the heterotrimer.
Figure 1.
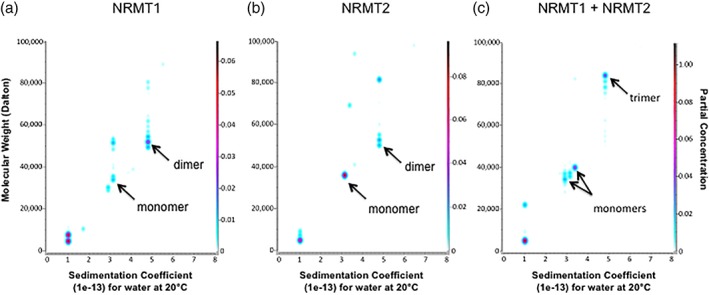
NRMT1 and NRMT2 form heterotrimers. (a) Ultrascan 3 analysis shows a minor pool of His‐tagged NRMT1 sediments as a monomer of approximately 33 kD and a major pool sediments as a homodimer of approximately 52 kD. (b) The majority of His‐tagged NRMT2 sediments as a monomer of approximately 35.7 kD, with a minor pool sedimenting as a homodimer of approximately 52.7 kD. (c) An equimolar mixture of NRMT1 and NRMT2 shows the formation of an 83.8 kD heterotrimeric species, representing a NRMT1 dimer and NRMT2 monomer. NRMT1 dimers are no longer present, but monomers of both NRMT1 and NRMT2 can still be seen.
NRMT1 and NRMT2 interact in vivo
Now knowing that NRMT1 and NRMT2 can interact in vitro, we wanted to next determine if the homologs also interact in vivo. Full length C‐terminally‐tagged human NRMT1‐FLAG and NRMT2‐GFP were co‐expressed in human embryonic kidney cells (HEK293) by transient transfection. Twenty‐four hours post‐transfection, NRMT1‐FLAG was immunoprecipitated (IP) out of the cell lysate and western blots were used to determine if NRMT2‐GFP co‐immunoprecipitated. As expected, full length NRMT1‐FLAG was able to co‐IP NRMT2‐GFP [Fig. 2(a)]. To test if the reciprocal was true, full length human NRMT2‐FLAG and human NRMT1‐GFP were also co‐expressed in HEK293 cells by transient transfection. While NRMT1‐GFP did show some non‐specific binding to beads alone, it was enriched in the co‐IP lane, indicating full length NRMT2‐FLAG is able to co‐IP NRMT1‐GFP [Fig. 2(b)].
Figure 2.
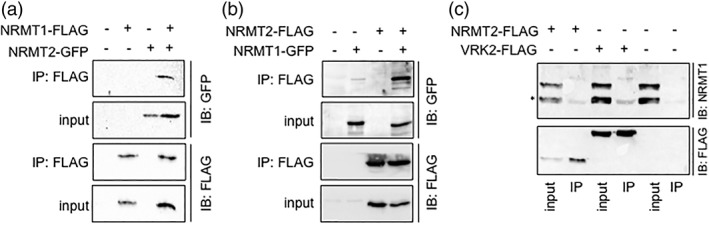
NRMT1 and NRMT 2 interact in vivo. (a) Western blots showing NRMT2‐GFP co‐immunoprecipitates with NRMT1‐FLAG after co‐transfection into HEK293 cells. (b) Reciprocally, NRMT1‐GFP co‐immunoprecipitates with NRMT2‐FLAG. (c) Endogenous NRMT1 co‐immunoprecipitates with both NRMT2‐FLAG and VRK2‐FLAG (a known binding partner of NRMT1, used as a positive control).15 * denotes NRMT1 band. Upper band is a non‐specific band detected by NRMT1 antibody.22
As both these co‐IPs were done with overexpressed tagged proteins, we next wanted to determine if endogenous NRMT1 and NRMT2 interact. We have previously developed a rabbit antibody against human NRMT1 that successfully works for western blot but not for IP.22 Although NRMT1 and NRMT2 are 50% identical, the 14 amino acid peptide antigen used to produce the NRMT1 antibody only had three amino acids in common and does not appear to detect NRMT2.22 As we have yet to find a NRMT2 antibody that consistently works for either western blot or IP from cell lysate, we expressed full length NRMT2‐FLAG in HEK cells, immunoprecipitated it with anti‐FLAG agarose and assayed for the presence of endogenous NRMT1 by western blot. As a control, we also expressed VRK2‐FLAG, which has been shown to interact with endogenous NRMT1.15 We found that endogenous NRMT1 did co‐IP with both VRK2‐FLAG and NRMT2‐FLAG [Fig. 2(c)]. Taken together, these data indicate that NRMT1 and NRMT2 can interact both in vitro and in vivo.
Modeling of NRMT1 and NRMT2 interactions
We next wanted to determine which domains on NRMT1 and NRMT2 are responsible for heterotrimer formation. The crystal structures of NRMT1 and NRMT2 (PDB codes 2EX4 and 5UBB) have been determined to 1.75 and 2 Å, respectively,28, 29 and as predicted by homology modeling,14 are almost identical barring the additional N‐terminal domain on NRMT2, which was not included in the structure.29 The core of both enzymes is a seven‐stranded β sheet (β1‐7) flanked by five α‐helices (α1‐5).30 In addition, there are several other structural elements including three N‐terminal helices and a pair of β hairpins (βAB) inserted between β5 and α6.30
To help predict which domains of NRMT1 and NRMT2 interact, we first used PyMOL molecular visualization software to model the NRMT1 dimer. The space‐filling model [Fig. 3(a)] and ribbon diagram [Fig. 3(b)] indicate NRMT1 homodimerization is facilitated by α5 of one monomer and the loop between α1/β1 on the second monomer [Fig. 3(b)]. Neither of these regions are involved in either the substrate or SAM‐binding motifs30 but are exposed on the outside of the protein and would be available for interaction [Fig. 3(a)]. The model specifically predicts interactions between Ser157 and Gly156 of the α5 helix with Ser64 and Arg68 of the α1/β1 loop, which are 4.2 and 3.9 Å apart, respectively [Fig. 3(c)].
Figure 3.
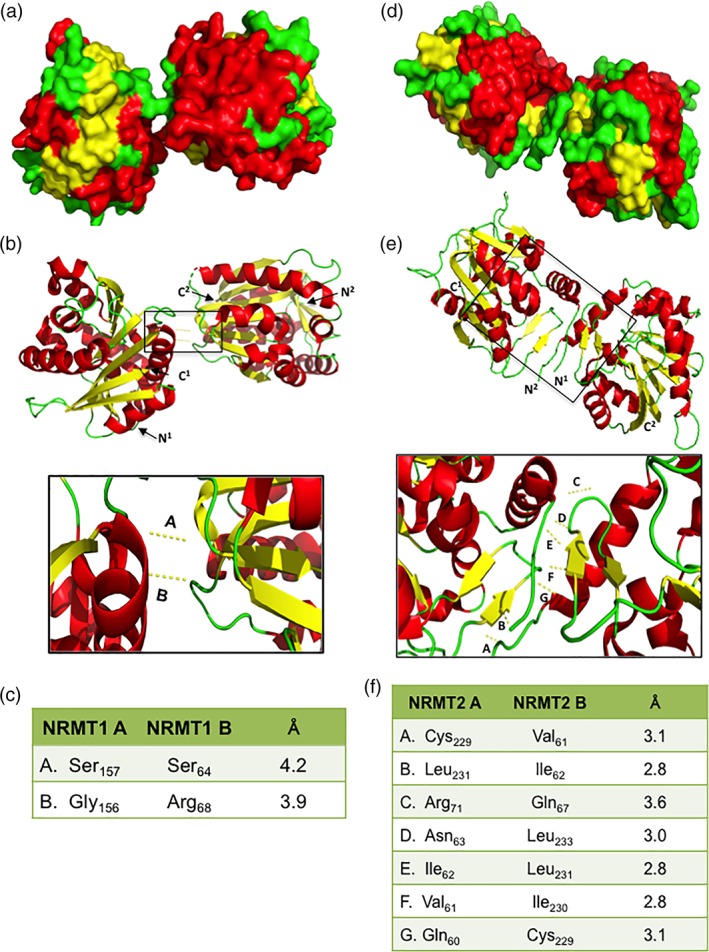
Modeling of NRMT1 and NRMT2 dimers. Space‐filling model of (a) NRMT1 and (d) NRMT2 homodimers showing regions of dimerization (green overlap, middle). Ribbon diagrams of (b) NRMT1 and (e) NRMT2 homodimers and detail of regions of interaction between monomers. N‐ and C‐termini of each monomer are labeled. All measurements between residues fall within accepted Van Der Waals distances for interaction. Boxed regions are enlarged below each model. Measured distances between interacting residues from monomers of (c) NRMT1 and (f) NRMT2. Each interaction is denoted in the enlargement above by the corresponding letter. All interactions were modeled with PyMOL using PDB accessions codes 2EX4 and 5UBB.
Though our analytical ultracentrifugation results suggest NRMT2 is predominantly found as a monomer, it can also be found as a homodimer [Fig. 1(b)], and these dimerization interactions could provide insights into how it heterotrimerizes with NRMT1. The NRMT2 crystal structure (PDB code 5UBB) and our PyMOL models of NRMT2 dimerization [Fig. 3(d,e)], indicate its homodimerization is facilitated by the unstructured N‐termini (minus the 59 amino acid region unique to NRMT2, which is not included in the crystal structure) and the βAB hairpins of each monomer. Our model specifically predicts Gln60, Val61, and Ile62 of the N‐terminus interact with Cys229, Ile230, and Leu231 of the βAB hairpin, respectively [Fig. 3(f)]. The extreme N‐terminus is not currently predicted to contribute to substrate or SAM binding (Wu, 2015), but it is hard to predict how the additional 59 amino acid NRMT2 N‐terminal tail could affect the overall structure. The βAB hairpins are predicted to contribute to the substrate‐binding pocket,30 which indicates binding of NRMT2 to NRMT1 could alter the ability of NRMT2 to bind substrate.
We next used PyMOL to model the possible interactions between a NRMT1 dimer and a NRMT2 monomer [Fig. 4(a)]. We found that if the previously modeled interactions between NRMT1 monomers are maintained, the NRMT2 monomer primarily interacts within the first 50 amino acids of NRMT1, with a secondary set of interactions between Arg79, Pro83, andTyr104 of NRMT1 and Leu231, Asp232, and Gln60 of NRMT2 [Fig. 4(a–c)]. The NRMT2 residues involved are almost identical to those involved in homodimerization [Fig. 4(c)]. As predicted, the model suggests that NRMT2 interacts with the NRMT1 dimer as it would with another NRMT2 monomer. As the N‐terminus of NRMT1 is not needed for its dimerization, both NRMT1 monomers would theoretically be available for binding to NRMT2. However, NRMT1 dimerization places both N‐termini in the same plane, so steric hindrance my preclude binding of more than one NRMT2 monomer to the NRMT1 dimer [Fig. 4(d)].
Figure 4.
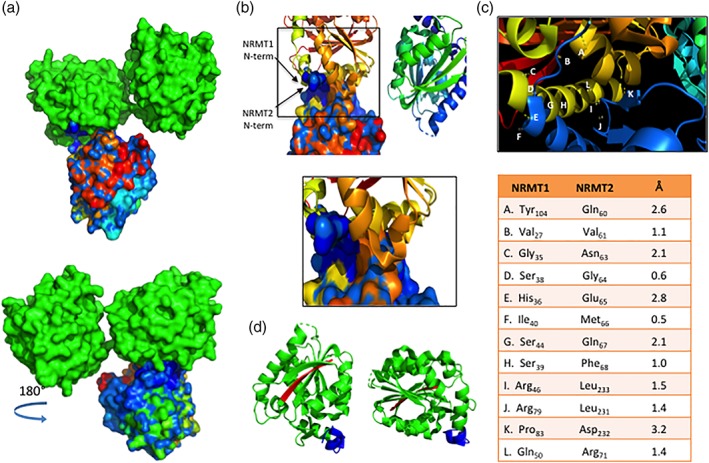
Modeling of NRMT1 and NRMT2 heterotrimer. (a) Top—Space‐filling model showing area of interaction between the NRMT1 dimer (green) and NRMT2 monomer (multicolored). Bottom—180° rotation of the heterotrimer model. The NRMT2 monomer is shaded with a blue‐to‐red gradient, representing the N‐terminal to C‐terminal orientation of the amino acid sequence. The majority of interactions between the NRMT1 dimer and NRMT2 monomer are predicted to involve the blue (N‐terminus) shaded region of NRMT2. (b) Ribbon diagram of the NRMT1 dimer interacting with the space‐filling model of the NRMT2 monomer. The N‐termini of both NRMT1 and NRMT2 are labeled. Boxed region is enlarged below and shows the majority of residues of interaction are found in the N‐terminus of both enzymes. (c) Diagram of and measured distances between interacting residues of NRMT1 and NRMT2. Each interaction is denoted in the enlargement above by the corresponding letter. (d) Ribbon diagram of NRMT1 homodimer, indicating N‐termini of each monomer (blue) are oriented in the same plane. C‐termini are colored red. All interactions were modeled with PyMOL using PDB accessions codes 2EX4 and 5UBB.
To test these models, we performed co‐IPs with N‐ and C‐terminal fragments of NRMT1 and NRMT2 [Fig. 5(a)]. To determine if it is primarily the unstructured N‐terminal tail of NRMT2 that interacts with full‐length NRMT1‐FLAG, we made constructs consisting of the first 112 amino acids of NRMT2 (containing important predicted residues) or amino acids 77–223 of NRMT2 (directly succeeding the important predicted residues) fused to GFP. An additional C‐terminal fragment of NRMT2 (amino acids 207–283) was attempted but did not express. NRMT1‐FLAG and NRMT21–112‐GFP or NRMT277–223‐GFP were co‐expressed in HEK293 cells, and NRMT1‐FLAG was immunoprecipitated out using anti‐FLAG antibody conjugated to agarose beads. Contrary to the model, the first 112 amino acids of NRMT2 did not strongly interact with NRMT1, though amino acids 77–223 did [Fig. 5(b,c)]. The main difference between the co‐IPs and the models was that the first 112 amino acids in the co‐IPs contained the extra 59 amino acid N‐terminal tail that was not included in the crystal structure or subsequent model. These data indicate that the presence of the additional N‐terminal amino acids of NRMT2 affect its binding to NRMT1.
Figure 5.
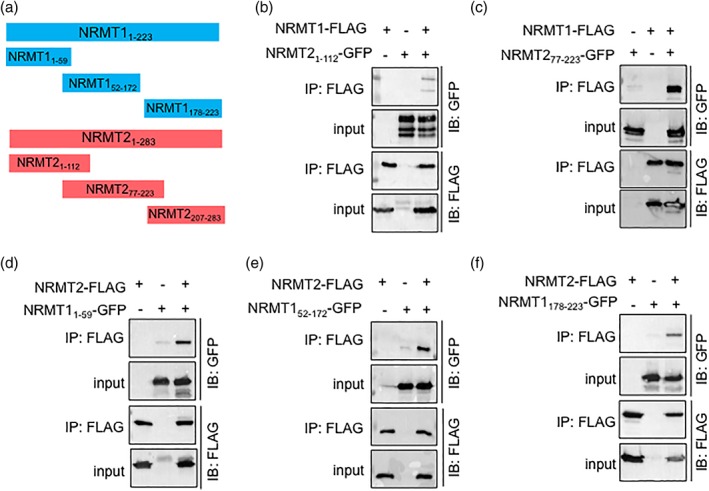
NRMT1 and NRMT2 truncation mutants. (a) Diagram showing full‐length and truncation constructs of NRMT1 and NRMT2. Subscript indicates the amino acid (aa) sequence from N‐ to C‐terminus of each construct. (b) Full‐length NRMT1‐FLAG only weakly interacts with the first 112 aa of NRMT2, (c) though strongly interacts with a NRMT2‐GFP fragment (amino acids 77–223) that is missing the 59 aa tail that is also lacking from the crystal structure and subsequent model. (d) Full‐length NRMT2‐FLAG strongly interacts with the first 59 aa of NRMT1, as well as, (e) amino acids 52–172. (f) There is a decreased interaction of NRMT2‐FLAG with the C‐terminal fragment of NRMT1 (amino acids 178–223).
To determine if it is primarily the first 50 amino acids of NRMT1 that interact with full‐length NRMT2‐FLAG, we made constructs consisting of the first 59 amino acids, amino acids 52–172, or amino acids 178–223 of NRMT1 fused to GFP [Fig. 5(a)]. NRMT2‐FLAG and NRMT11–59‐GFP, NRMT152–172‐GFP, or NRMT1178–223‐GFP were co‐expressed in HEK293 cells, and NRMT2‐FLAG was immunoprecipitated out as previously. Supporting the model, the first 59 amino acids of NRMT1 did strongly interact with NRMT2 [Fig. 5(d)]. However, amino acids 52–172 also interacted with NRMT2 [Fig. 5(e)], as did amino acids 178–223, though to a lesser degree than the first two constructs [Fig. 5(f)]. These data indicate that the first 50 amino acids of NRMT1 are sufficient to promote interaction with NRMT2, though the predicted interactions of Arg79, Pro83, and Tyr104 may also be playing a role.
NRMT2 expression stabilizes NRMT1
As both PRMT1 and METTL3 stability is enhanced by binding to its homolog partner,4, 7, 9 we wanted to next determine if the half‐life of NRMT1 is enhanced by the expression of NRMT2 or vice versa. To first measure the half‐life of endogenous NRMT1 under basal conditions [i.e., low levels of NRMT2 as determined by qRT‐PCR (unpublished data)], we treated HCT116 human colorectal carcinoma cells with 100 μg/mL cycloheximide and harvested cell lysates at six time points, up to 12 h after treatment. Western blots of the lysates were probed with our anti‐NRMT1 antibody22 and anti‐GAPDH as a loading control. NRMT1 levels were normalized to GAPDH and calculated using Image Lab software. The experimentally calculated half‐life for endogenous NRMT1 was approximately 8.8 h [Fig. 6(a)].
Figure 6.
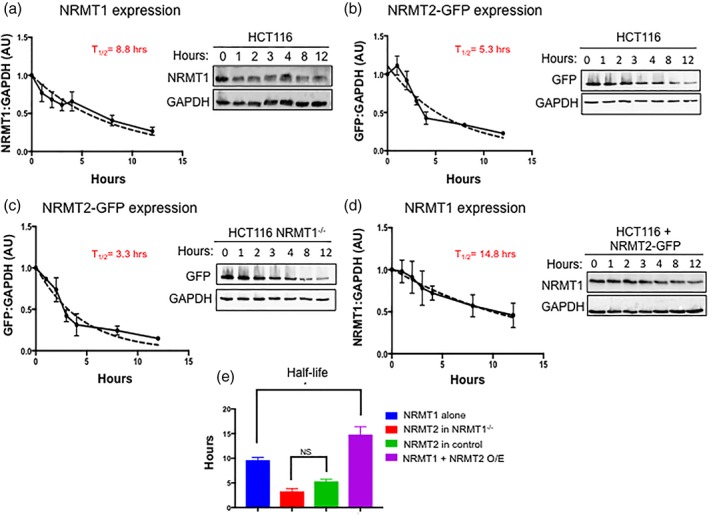
NRMT2 expression stabilizes NRMT1. HCT116 cells were treated for 0–12 h with 100 μg/mL cycloheximide to inhibit protein synthesis. (a) Half‐life of endogenous NRMT1 protein was calculated to be 8.8 h. (b) Half‐life of NRMT2‐GFP protein was calculated to be 5.3 h. (c) HCT116 cells with CRISPR/Cas9‐mediated knockout of NRMT1 (HCT116 NRMT1−/−) were also treated for 0–12 h with 100 μg/mL cycloheximide. The half‐life of NRMT2‐GFP in the absence of NRMT1 was slightly lower at 3.3 h. (d) Finally, HCT116 cells overexpressing NRMT2‐GFP were treated for 0–12 h with 100 μg/mL cycloheximide. The half‐life of endogenous NRMT1 in the presence of overexpressed NRMT2 was found to be a significantly longer 14.8 h. (e) Comparison of the half‐lives of each group. Blots are representative images of three independent experiments. Pixel densitometry was quantified using Bio‐Rad Image Lab software, with GAPDH serving as the loading control for each lane. NRMT1 and/or GFP bands were internally normalized to T = 0 lane and set to 1. Half‐life was calculated using a one‐phase decay non‐linear regression and constraining Y 0 to 1 and the plateau to 0 (dashed lines). * denotes P < 0.05 as determined by ordinary one‐way ANOVA to compare groups using GraphPad Prism.
Due to the lack of an antibody that specifically detects endogenous NRMT2 in cell lysate, we used NRMT2‐GFP to determine the half‐life of NRMT2 under basal conditions. HCT116 cells were transduced with lentivirus expressing NRMT2‐GFP at an MOI of 1. About 48 h post‐transduction, successfully transduced cells were selected for by puromycin treatment. This was to ensure all cells in the experiment were expressing a homogenously low level of NRMT2‐GFP. Again, cells were treated with 100 μg/mL cycloheximide and harvested at six time points, up to 12 h after treatment. Western blots of the lysates were probed with anti‐GFP and anti‐GAPDH and analyzed as above. The experimentally calculated half‐life for NRMT2 under basal conditions (i.e., with NRMT1 present) was approximately 5.3 h [Fig. 6(b)].
To determine if NRMT2 expression is altered in the absence of NRMT1, we first transduced CRISPR/Cas9‐mediated NRMT1 knockout HCT116 cells16 with lentivirus expressing NRMT2‐GFP at an MOI of 1. After puromycin selection, cells were treated with 100 μg/mL cycloheximide and harvested as previously. The experimentally calculated half‐life for NRMT2 in cells lacking NRMT1 expression was approximately 3.3 h [Fig. 6(c)]. This was not statistically significant from the half‐life of NRMT2‐GFP in control cells [Fig. 6(e)], indicating the stability of NRMT2 is not dependent on NRMT1 expression. This also highlights the short half‐life of NRMT2, and indicates its expression may be tightly regulated and confined to times of specific need.
Next, we aimed to measure if NRMT2 overexpression affects the stability of endogenous NRMT1. As previously, HCT116 cells were transduced with lentivirus expressing NRMT2‐GFP at an MOI of 1. They were then treated with 100 μg/mL cycloheximide and harvested as previously. Interestingly, in the presence of overexpressed NRMT2, the half‐life of NRMT1 increases to approximately 14.8 h [Fig. 6(d)]. This is a significant increase in NRMT1 half‐life as compared with its half‐life of 8.8 h under basal NRMT2 conditions [Fig. 6(e)]. These data fit with a model of NRMT1 providing basal housekeeping N‐terminal methyltransferase activity, and NRMT2 being induced to enhance NRMT1 activity when needed during times of stress or increased substrate burden.
Catalytic activity of NRMT2 is not necessary for increased trimethylation by NRMT1
To determine if NRMT2 enhances NRMT1 activity through a priming activity as previously hypothesized,14 or if it is simply providing enhanced stability, we performed in vitro methylation assays with different combinations of wild type (WT) NRMT1, WT NRMT2, and NRMT2 V224L, a mutant initially identified in human breast cancer that is almost completely devoid of methyltransferase activity.16 When using as substrate a peptide consisting of the first 15 amino acids of RCC1 (SPKRIAKRRSPPADA), NRMT1 recombinant enzyme alone exhibited a K m of 1.1 μM [Fig. 7(a)], WT NRMT2 recombinant enzyme alone exhibited a K m of 55.6 μM [Fig. 7(b)], and NRMT2 V224L had no activity [Fig. 7(c)]. In combination with WT NRMT2, the K m of NRMT1 decreased to 0.9 μM [Fig. 7(d)], and in combination with NRMT2 V224L, the K m of NRMT1 decreased to 0.1 μM [Fig. 7(e)], indicating the increased activity of NRMT1 is not dependent on the catalytic activity of NRMT2.
Figure 7.
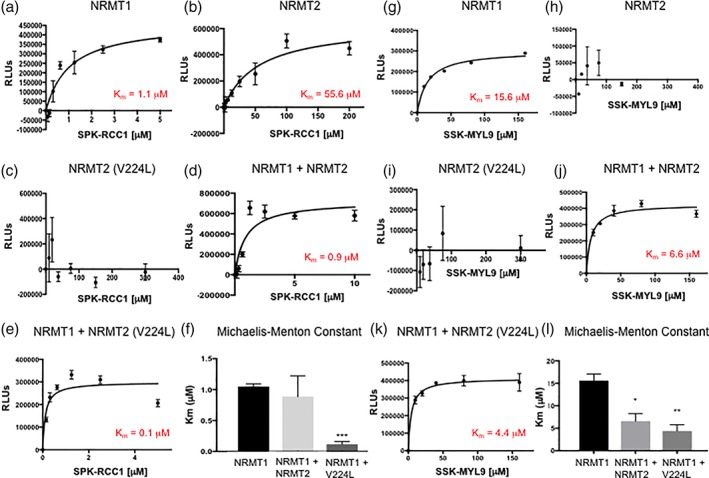
Catalytic activity of NRMT2 is not necessary for increased trimethylation by NRMT1. (a) Wildtype NRMT1 shows a K m of 1.1 μM when trimethylating the SPK‐RCC1 peptide substrate, (b) while the K m of NRMT2 is 55.6 μM. (c) The catalytically inactive NRMT2 V224L mutant is unable to methylate the same substrate. (d) Combining NRMT1 and NRMT2 lowers the K m of NRMT1 to 0.9 μM, while (e) the combination of NRMT1 and catalytically inactive NRMT2 V224L further lowers the K m of NRMT1 to 0.1 μM. (f) Comparison of Michaelis–Menton constants for NRMT1 alone, NRMT2 alone, or NRMT1 in combination with NRMT2 or NRMT2 V224L with SPK‐RCC1 peptide as substrate. Only the combination of NRMT1 with NRMT2 V224L was significantly different from NRMT1 alone. Calculated constants are the mean of three independent experiments ±SEM. *** denotes P < 0.001 as determined by unpaired Student's t‐test. Using a less preferred, but still enzymatically favorable substrate SSK‐MYL9, (g) increases the K m of NRMT1 to 15.6 μM but shows no enzymatic activity with (h) NRMT2 or (i) NRMT2 V224L. (j) Combining NRMT1 and NRMT2 decreases the K m of NRMT1 to 6.6 μM, and (k) the combination of NRMT1 and NRMT2 V224L further lowers the K m of NRMT1 to 4.4 μM. (l) Comparison of Michaelis–Menton constants for NRMT1 alone, NRMT2 alone, or NRMT1 in combination with NRMT2 or NRMT2 V224L with SSK‐MYL9 peptide as substrate. The constants for both the combinations of NRMT1 with NRMT2 and NRMT1 with NRMT2 V224L were significantly different from NRMT1 alone. Calculated constants are the mean of three independent experiments ±SEM. * and ** denote P < 0.05 and 0.01, respectively, as determined by unpaired Student's t‐test.
However, only the decrease in K m with NRMT2 V224L was statistically significant from the K m of NRMT1 alone [Fig. 7(f)]. As the Ser–Pro–Lys consensus sequence of the RCC1 peptide is a preferred substrate for NRMT1,15 we wanted to test if we could see a greater decrease in K m with NRMT2 addition when using a less preferred substrate. We have previously seen that the K m of NRMT1 is considerably higher when using the Ser–Ser–Lys consensus sequence of myosin regulatory light chain 9 (MYL9) (unpublished data), so we repeated the experiments above using a peptide of the first 14 amino acids of MYL9 (SSKRAKAKTTKKRP) as substrate. NRMT1 recombinant enzyme alone exhibited a K m of 15.6 μM [Fig. 7(g)]. Neither WT NRMT2 nor NRMT2 V224L had any activity on the MYL9 peptide [Fig. 7(h,i)]. In combination with WT NRMT2, the K m of NRMT1 decreased to 6.6 μM [Fig. 7(j)], and in combination with NRMT2 V224L, the K m of NRMT1 decreased to 4.4 μM [Fig. 7(k)]. Using this Ser–Ser–Lys substrate, both WT NRMT2 and NRMT2 V224L were able to significantly lower the K m of NRMT1 [Fig. 7(l)], further verifying the catalytic activity of NRMT2 is not needed and indicating NRMT2 is not increasing NRMT1 activity by serving as a primer but by providing stability and altering substrate affinity.
To understand why NRMT2 V224L was able to significantly decrease the NRMT1 K m with the RCC1 peptide, and not WT NRMT2, we performed molecular modeling of the NRMT2 V224L mutant [Supporting Information Fig. S1(a)]. It is easy to see how V224L might disrupt NRMT2 catalytic activity, as Val224 is in the substrate binding pocket and is directly adjacent to Asn223, which forms critical hydrogen bonds and electrostatic interactions with substrate.14, 16 When mutated to Leu, this residue extends much further into the pocket, potentially disrupting nearby interactions with substrate [Supporting Information Fig. S1(b)]. Interestingly, another residue critical for NRMT2 catalytic activity, Asp232,14 is also nearby in this pocket, and is one of the predicted secondary sites of interaction with NRMT1 [Fig. 4(c) and Supporting Information Fig. S1(c)]. Though they are not directly adjacent to each other they are connected by a short, unstructured loop, and it is possible to envision how a change in the position of residue 224 could indirectly affect the position of Asp232 [Supporting Information Fig. S1(c)]. Mutation of Val224 to Leu could impact NRMT2 function via two different mechanisms, first by disrupting substrate binding and subsequent catalytic activity, and second by promoting Asp232 accessibility and subsequent complex formation with NRMT1.
NRMT2 overexpression cannot rescue NRMT1 knockout phenotypes
The above data lead to a model where the NRMT1 homodimer normally functions alone as the primary N‐terminal methyltransferase in cells, but can act synergistically with NRMT2 to increase N‐terminal trimethylation levels at times of high need. This model implies that NRMT2 is not simply a functionally redundant homolog of NRMT1 but an important activator. To further confirm NRMT1 and NRMT2 are not functionally redundant, we wanted to test if NRMT2 expression could rescue NRMT1 knockout phenotypes. It has been shown that N‐terminal methylation protects the N‐terminus of cytochrome c557 from digestion in Crithidia oncopelti.31 We wanted to see if N‐terminal mono‐ or trimethylation differentially affected this protection and affect the rate of decay, using a mammalian substrate with a known consensus sequence for both NRMT1 and NRMT2, the α isoform of SET (SETα).15, 22
Human SETα was C‐terminally tagged with Dendra2 photoswitchable protein and lentivirally expressed in HCT116 human colorectal carcinoma cells at an MOI of 1 [Fig. 8(b)]. Before excitation, Dendra2 fluoresces green, but when activated by UV–violet or blue light, fluoresces red [Fig. 8(a)].32 The red form is highly stable independently, but when fused to a protein of interest, its signal can be followed to determine the rate of decay of the protein.32 We first measured the rate of decay of SETα‐Dendra2 in control HCT116 cells [Fig. 8(c), blue]. We then showed the rate of decay of SETα‐Dendra2 in HCT116 NRMT1−/− cells was significantly quicker [Fig. 8(c), red].16 Overexpression of NRMT1 in the HCT116 NRMT1−/− cells restored the original rate of decay of SETα‐Dendra2 [Fig. 8(c), green]. However, overexpression of NRMT2 in the HCT116 NRMT1−/− cells was not able to rescue the SETα‐Dendra2 rate of decay, and it remained significantly different from the HC116 control [Fig. 8(c), purple]. These data indicate N‐terminal mono‐ and trimethylation are not functionally redundant and suggest that, unlike EZH1 and EZH2, NRMT1 and NRMT2 are not serving the same function through different mechanisms. Our data point to the stabilization of NRMT1 and an increase in substrate binding as important functions of NRMT2 but do not rule out other specific roles for N‐terminal monomethylation that are distinct from those regulated by N‐terminal trimethylation.
Figure 8.
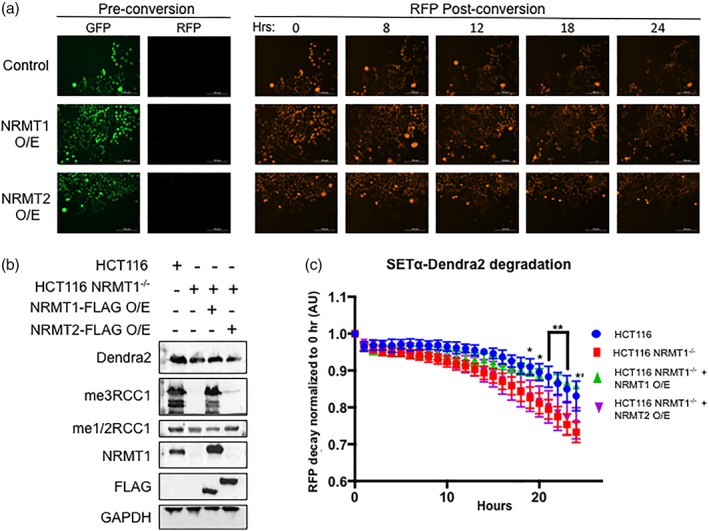
NRMT2 overexpression cannot rescue NRMT1 knockout phenotypes. (a) HCT116 NRMT1−/− cells transduced with empty pCDH virus (control), virus overexpressing NRMT1 (NRMT1 O/E), or virus overexpressing NRMT2 (NRMT2 O/E) were also transduced with virus expressing SETα‐Dendra2. Pre conversion SETα‐Dendra2 expresses in the GFP channel, post‐conversion it expresses in the RFP channel. The RFP signal then decreases over time and a rate of decay can be calculated. (b) Western blots showing HCT116 cells express endogenous NRMT1 and were fully mono/dimethylated (me1/2RCC1) and trimethylated (me3RCC1) (lane 1). In contrast, HCT116 NRMT1−/− cells do not express NRMT1 or exhibit N‐terminal trimethylation, but retain mono/dimethylation (lane 2). In HCT116 NRMT1−/− cells overexpressing NRMT1‐FLAG, both NRMT1 protein and trimethylation were restored (lane 3). In HCT116 NRMT1−/− cells overexpressing NRMT2‐FLAG, a slight increase in mono/dimethylation can be seen, with no restoration of NRMT1 or trimethylation (lane 4). FLAG blot shows both NRMT1‐FLAG and NRMT2‐FLAG are expressing. Dendra2 blot shows SETα‐Dendra2 expression levels are similar between lines. GAPDH was used as a loading control. (c) First, the rate of decay of SETα‐Dendra2 was measured in control HCT116 cells (blue). This rate of decay significantly increased in HCT116 NRMT1−/− cells (red). Overexpressing NRMT1‐FLAG in HCT116 NRMT1−/− cells restores the original rate of decay (green), and there was no statistically significant difference between control and NRMT1 overexpression cell lines. Overexpressing NRMT2‐FLAG in HCT116 NRMT1−/− cells cannot rescue the decay (purple), which remains significantly faster than control. Curves were constructed by comparing loss of RFP signal over time as compared with time 0 and are the mean of three independent experiments ±SEM. *, **, *** denotes P < 0.05, 0.01, and 0.001, respectively, as determined by 2‐way ANOVA.
Discussion
Our work is the first to demonstrate a physical interaction between the N‐terminal methyltransferase homologs, NRMT1 and NRMT2. Here we show that NRMT1 homodimers interact with NRMT2 monomers to form heterotrimers. Heterotrimer formation increases the stability of NRMT1 but has no significant effect on the stability of NRMT2. Though we have previously seen that co‐expression with NRMT2 increases NRMT1 activity,14 we now show that NRMT2 catalytic activity is not necessary for this increase in NRMT1 activity and propose that NRMT2 is not priming for NRMT1, but instead increasing its stability. We also show that N‐terminal mono‐ and trimethylation are not functionally redundant, at least in regards to promoting protein stability, indicating that NRMT1 and NRMT2 are also not functionally redundant. The sole role of NRMT2 may be to stabilize and activate NRMT1, or it may have other distinct functions.
In addition to stabilizing METTL3, METTL14 has been proposed to enhance interaction with RNA substrates.8, 10 We have seen that NRMT1 has varying affinities for the different NRMT N‐terminal consensus sequences,15 and it may be that binding of NRMT2 to the NRMT1 homodimer, strengthens its affinity for certain less favored consensus sequences. Our in vitro methyltransferase data support this model, as NRMT2 binding had little effect on the K m of NRMT1 when methylating a Ser–Pro–Lys peptide, but significantly decreased the K m of NRMT1 when methylating the less preferred Ser–Ser–Lys peptide. We had originally proposed that NRMT2 was a primer for NRMT1 that was expressed at times of high substrate burden,14 but it may be that NRMT2 is expressed at times when less preferred substrates require methylation. In this way, it is both providing stability to NRMT1 and altering its substrate preference.
These data indicate that, while overall unique, the interaction between NRMT1 and NRMT2 has the most in common with the interactions of PRMT1/3 and METTL3/14. Like PRMT1/3 and METTL3/14, binding of the homologs increases the stability of the active enzyme.4, 7, 9 Similar to METTL14, but unlike PRMT3, NRMT2 has weak catalytic activity in vitro.4, 7, 9 However, SAM was visualized in the active site of the NRMT2 crystal, and we have seen that CRISPR‐mediated knockout of NRMT1 results in a complete loss of N‐terminal trimethylation, but N‐terminal monomethylation remains the same [Fig. 8(b)].16 This indicates that, unlike METTL14, NRMT2 also has some activity in vivo. What the function of this in vivo N‐terminal monomethylation is remains to be determined.
If charge or bulk of the N‐terminus is the main regulatory aspects of N‐terminal PTMs, it seems intuitive that N‐terminal monomethylation could not functionally replace trimethylation, as it is much smaller and does not produce a pH‐independent positive charge.33 However, similar to lysine monomethylation, N‐terminal monomethylation could still have its own distinct function. RNA polymerase II (Pol II) pausing is regulated by the switch between mono‐ and trimethylation of histone H4 lysine 20 (H4K20me1 and H4K20me3, respectively).34 While H4K20me3 promotes pausing by inhibiting the recruitment of the male‐specific lethal (MSL) complex, H4K20me1 recruits MSL and promotes the release of Pol II into elongation.34 One way subtle changes in the degree of methylation can affect protein function so dramatically, is that each can be recognized by different methyl‐reading chromodomains. H4K20me1 can efficiently recruit the MSL complex to chromatin due to the chromo‐barrel domain of MSL3,35, 36 whereas H4K20me3 is preferentially bound by the double tudor domain of the histone demethylase jumonji domain containing 2A (JMJD2A).37 Therefore, it is possible that N‐terminal mono‐ and trimethylation could similarly recruit distinct proteins with different binding domains, and in doing so, differentially affect many cellular processes. It will be interesting to determine if families of such N‐terminal methylation “readers” exist.
Though the interactions between NRMT1 and NRMT2 seem the most distinct from the interactions between EZH1 and EZH2, there are also some similarities. Most striking is the tissue distribution of the homologs. NRMT1, like EZH1, is ubiquitously expressed in most adult tissue types.12, 14 While NRMT2 has very low basal expression in adult mice, we do see enrichment in both skeletal muscle and the liver.14 EZH2 also has very low basal expression in adult tissue, with some enrichment in the spleen, but seems to be highly expressed in the proliferative tissue of the kidney during development.12 It has been hypothesized that these disparate expression patterns represent an evolutionary sub‐functionalization of the homologs.12 EZH1 is the general methyltransferase directly involved in transcriptional repression, while EZH2 is dedicated to placing repressive marks during replication.12 Though EZH1 and EZH2 are catalytically distinct, they are functionally redundant in regards to transcriptional repression, and their differential expression patterns could allow for a tighter regulation of this repression.12 The function of PRC2 complexes expressing both EZH1 and EZH2 remains to be determined,12, 13 but like PRMT1/3, METTL3/14, and NRMT1/2, co‐expression might result in a more active complex, useful in times of high demand.
Unlike EZH1/2, NRMT1 and NRMT2 do not seem to be functionally redundant. Their evolution may have led to neo‐functionalization instead of sub‐functionalization.38 We propose a model where each has a distinct function, but at times when they are co‐expressed, can form heterotrimers with increased N‐terminal trimethylation activity. In this way, regulated NRMT2 expression would serve to promote its developmental or cell type‐specific role and increase N‐terminal methylation when needed. It will be interesting to see if NRMT2 expression is regulated during development or in proliferating tissues and continue to explore potential unique roles for N‐terminal monomethylation. Also of note is the V224L NRMT2 mutation found in human breast cancer.16 While NRMT1 has been shown to have cancer type‐specific roles as a tumor suppressor or oncogene,16, 39 there is as yet no data demonstrating NRMT2 misregulation in any cancer type. Therefore, extensive characterization of this mutation has not occurred. However, these data demonstrating it not only abolishes NRMT2 activity but also promotes NRMT1 activity make it an interesting candidate for a driver mutation in cancers that overexpress NRMT1 or a protective mutation in cancers that downregulate NRMT1. Future studies will determine how expression of this mutant affects oncogenic phenotypes in such cancer cell lines.
Materials and Methods
Constructs and antibodies
For recombinant protein production, full‐length human NRMT1, NRMT2, and NRMT2 V224L were cloned into pet15b (Merck Millipore, Burlington, MA) and purified as described previously.14, 16, 40 The full‐length human NRMT1‐FLAG, NRMT1‐GFP, NRMT2‐FLAG, and NRMT2‐GFP used in the co‐IPs were cloned as previously described.14 All truncated constructs were sub‐cloned from these full‐length ORFs using primer sets that combined the primers used for full‐length cloning and the following unique primers:
NRMT11–59 Reverse: 5′‐GGGAATTCTTGTTCGGGCCTTCCCTCA‐3′,
NRMT152–172 Forward: 5′‐GGTCTAGAATGTTTTTGAGGGAAGGCCCGAAC‐3′.
NRMT152–172 Reverse: 5′‐GGGAATTCTCCTGGGCCATGTTGTCTTTGATGAC‐3′.
NRMT1178–223 Forward: 5′‐GGTCTAGAATGGACGTGGACAGCAGCGTGTGC‐3′.
NRMT21–112 Reverse: 5′‐GGGGATCCCCAGGCCCCCCAACAAATTT‐3′.
NRMT277–223 Forward: 5′‐GGTCTAGAATGCAAGAAGTACCAGCCACAGAA‐3′.
NRMT277–223 Reverse: 5′‐GGGGATCCGTCCTTCAATATGATGATGCC‐3′.
To make SETα‐Dendra2‐pCDH, human SETα was amplified from our previously published SETα‐FLAG construct22 to introduce 5′ Xho1 and 3′ HindIII restriction sites and sub‐cloned into the pDendra2‐N vector (Clontech, Mountain View, CA). The SETα‐Dendra2 construct was then amplified to introduce 5′ Xba1 and 3′ Swa1 restriction sites and sub‐cloned into the lentiviral vector pCDH‐CMV‐EF1‐Puro (pCDH, Addgene, Cambridge, MA). Lentivirally expressed NRMT2‐GFP, NRMT1‐FLAG, and NRMT2‐FLAG were first amplified from the corresponding mammalian expression vectors to introduce 5′ Xba1 and 3′ Swa1 restriction sites and sub‐cloned into pCDH. Primary antibodies for western blots were used at the following dilutions: rabbit anti‐FLAG‐HRP (1:1000, Sigma‐Aldrich, St. Louis, MO), rabbit anti‐GFP (1:1000, Cell Signaling Technologies, Danvers, MA), rabbit anti‐NRMT1 (1:1000),22 rabbit anti‐me3‐RCC1 (1:10,000),22 rabbit anti‐me1/2‐RCC1 (1:5000),22 rabbit anti‐GAPDH (1:2000, Trevigen, Gaithersburg, MD), and mouse anti‐Dendra2 (1:500, Origene, Rockville, MD).
Cell culture and lentivirus production
Human embryonic kidney cells (HEK293, ATCC, Manassas, VA) cells were maintained in Dulbecco's Modified Eagle Medium (DMEM, Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals) and 1% penicillin–streptomycin (P/S; Life Technologies). HCT116 human colorectal carcinoma cell lines (generous gift from Dr. Ian Macara) were maintained in McCoy's 5a Modified Medium (Life Technologies) supplemented with 10% FBS and 1% P/S. Lentivirus expressing empty pCDH vector, NRMT2‐GFP‐pCDH, SETα‐Dendra2‐pCDH, NRMT1‐FLAG‐pCDH, or NRMT2‐FLAG pCDH was made by co‐transfecting HEK293 cells with 50 μg pCDH containing the appropriate cDNA, 37.5 μg psPAX2 packaging vector, and 15 μg pMD2.G viral envelope plasmid using calcium phosphate transfection.41 Forty‐eight hours post‐transfection, supernatant was collected and virus concentrated using 100 kD ultrafilters (EMD Millipore). Virus was titered in HEK293 cells and HCT116 cells were transduced by addition of a volume of virus equal to a multiplicity of infection (MOI) of 1. Three days post‐transduction, transduced cells were selected for by addition of 2 μg/mL puromycin. For Dendra2 experiments, cells were first transduced with overexpression virus. Transduced cells were selected for with puromycin after 3 days, and then retransduced with SETα‐Dendra2 expressing virus.
Co‐immunoprecipitations (co‐IPs)
For co‐IP experiments, 1 × 106 HEK293 cells were seeded in 60 mm tissue culture dishes 24 h prior to calcium phosphate transfection with 2 μg each of appropriate constructs. About 24 h post‐transfection, cells were scraped directly into 100 μL lysis buffer (50 mM Tris pH 7.5, 300 mM NaCl, 5 mM MgCl2, 1% NP‐40, 7 mM BME) plus protease inhibitors for protein isolation. 20 μL of cell lysate was retained for input control, and the remainder mixed with 20 μL M2 agarose beads (Sigma‐Aldrich). The bead/protein mixture was rotated 1–2 h at 4°C and washed 3× with PBS + 0.1% NP‐40. The immunoprecipitated proteins were eluted from the beads in 5× Laemmli buffer and boiled for 5 min at 95°C. The bead‐free supernatant from IP and input samples were run on 12% SDS‐PAGE gels. Western blotting was performed using the above antibodies.
Analytical ultracentrifugation
Sedimentation velocity experiments were carried out in a Beckman Coulter ProteomeLab XL‐A analytical ultracentrifuge (Beckman Coulter Inc., Brea, CA) at 20°C and 40,000 rpm in standard 2 sector cells. Buffer density and viscosity (50 mM Tris, 50 mM KCl at pH 8.0) were calculated using Sednterp (free software: Biomolecular Interaction Technologies Center at the University of New Hampshire, http://jphilo.com/sdtr0601.exe). Data were analyzed with the program Ultrascan 3.0 (free software: http://www.ultrascan.uthscsa.edu/) using the “combine pseudo‐3D distributions” operation. The partial specific volumes of NRMT1 and NRMT2 were calculated from the amino acid compositions (0.734 mL/g for NRMT1 and 0.737 mL/g for NRMT2) using the Protparam tool in ExPASy (free software: web.http://expasy.org).
Cycloheximide experiments
1 × 106 HCT116 cells, HCT116 cells transduced with lentivirus expressing NRMT2‐GFP, HCT116 cells with CRISPR/Cas9‐mediated knockout of NRMT1 (HCT116 NRMT1−/−),16 or HCT116 NRMT1−/− cells transduced with lentivirus expressing NRMT2‐GFP were plated 24 h prior to the start of the cycloheximide chase experiment. At the start of each time point, the McCoy's 5a Modified Medium was replaced with fresh media containing 100 μg/mL cycloheximide (Sigma‐Aldrich). Cells were harvested in 100 μL cell lysis buffer (see above) and protein was quantified using the Pierce 660 nM Protein Assay (Thermo‐Fisher Scientific, Waltham, MA). 20 μg total protein per time point was separated on 12% SDS‐PAGE gels and subjected to Western blotting using antibodies against NRMT1, GFP, and GAPDH as a loading control. Pixel density and normalization for each band was quantified using Image Lab software (Bio‐Rad, Hercules, CA).
Dendra2 experiments
As described above, HCT116 or HCT116 NRMT1−/− cells were transduced with control (empty pCDH vector), NRMT1‐FLAG, or NRMT2‐FLAG expressing lentivirus at an MOI of 1. Three days post‐transduction cells were selected with puromycin treatment. Three days post‐puromycin treatment, cells were retransduced with lentivirus expressing SETα‐Dendra2 at an MOI of 1. 72 h post‐transduction, 2000 cells per well of each experimental condition were plated at an N = 6 on a 96‐well plate and allowed to adhere and grow for 48–72 h. Kinetic fluorescent analysis using the Cytation5 Imaging System (BioTek, Winooski, VT) was started by taking pre‐conversion images in the GFP and RFP channel for each well. Each well was then exposed to the DAPI channel (UV) at maximum intensity for 16 seconds for photoconversion of the Dendra2. Post‐conversion RFP kinetic imaging was performed once per hour for 24 h, with cells maintained at 5% CO2 and 37°C. Imaging settings remained constant throughout. The raw fluorescence value of the RFP channel for each well was then normalized to the 0‐h time point and fluorescent decay between cell types was measured.
In vitro methylation assays
Methyltransferase assays were conducted using the MTase‐Glo Methyltransferase Assay (Promega, Madison, WI) and following the given protocol. Briefly, all assays used 0.4 μM recombinant enzyme, 20 μM S‐adenosyl methionine (SAM), and varied concentrations of SPK‐RCC1 (SPKRIAKRRSPPADA) or SSK‐MYL9 (SSKRAKAKTTKKRPK) peptides as substrate. Substrates were serially diluted in a 96‐well plate and reactions were started with the addition of the appropriate recombinant enzyme. All reactions were carried out at room temperature and stopped after 20 min with the addition of 0.5% trifluoroacetic acid. The MTase‐Glo detection reagents were then added and luminescence was quantified using the Cytation5 Imaging System (BioTek).
Protein modeling and statistical analysis
Molecular modeling and the associated images were produced using the PyMOL Molecular Graphics System, Version 2.0.6 (Schrödinger, LLC). PDB accession numbers are 2EX4 (NRMT1) and 5UBB (NRMT2). Modeling was performed by molecular alignment of monomeric species of NRMT2 (5UBB) and dimeric NRMT1 (2EX4). Each species was rotated to minimal root‐mean‐square deviation (RMSD) scores using a method adapted from Coutsias et al.42 and potential interacting residues were measured using the “measurement wizard,” natively incorporated into the PyMOL user interface. All measured residues of interaction had a distance >4 Å, satisfying the maximum Van der Waals radius between atoms involved. All statistical analyses were performed using GraphPad Prism Software for Mac Version 7.0d (La Jolla, California). The specific statistical test is noted in the respective figure captions and results are presented as mean ± standard error, n = 3 unless specifically stated.
Conflict of Interest
The authors declare that they have no conflicts of interest related to this article.
Supporting information
Appendix S1: Supplementary Fig. 1
Acknowledgments
This work was supported by a grant to CST from the National Institutes of Health (grant number R01GM112721).
Impact Statement: This work describes the biochemical interactions between the two N‐terminal methyltransferase homologs, NRMT1 and NRMT2. It posits complex formation with close homologs as a widespread mechanism for methyltransferase regulation and illustrates how mutations in NRMT2 could alter the oncogenic properties of NRMT1.
References
- 1. Holliday R, Pugh JE (1975) DNA modification mechanisms and gene activity during development. Science 187:226–232. [PubMed] [Google Scholar]
- 2. Boriack‐Sjodin PA, Swinger KK (2016) Protein methyltransferases: A distinct, diverse, and dynamic family of enzymes. Biochemistry 55:1557–1569. [DOI] [PubMed] [Google Scholar]
- 3. Motorin Y, Helm M (2011) RNA nucleotide methylation. Wiley Interdiscip Rev RNA 2:611–631. [DOI] [PubMed] [Google Scholar]
- 4. Kafkova L, Debler EW, Fisk JC, Jain K, Clarke SG, Read LK (2017) The major protein arginine methyltransferase in Trypanosoma brucei functions as an enzyme‐prozyme complex. J Biol Chem 292:2089–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Willert EK, Fitzpatrick R, Phillips MA (2007) Allosteric regulation of an essential trypanosome polyamine biosynthetic enzyme by a catalytically dead homolog. Proc Natl Acad Sci U S A 104:8275–8280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nguyen S, Jones DC, Wyllie S, Fairlamb AH, Phillips MA (2013) Allosteric activation of trypanosomatid deoxyhypusine synthase by a catalytically dead paralog. J Biol Chem 288:15256–15267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, Dai Q, Chen W, He C (2014) A METTL3‐METTL14 complex mediates mammalian nuclear RNA N6‐adenosine methylation. Nat Chem Biol 10:93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, Gong Z, Wang Q, Huang J, Tang C, Zou T, Yin P (2016) Structural basis of N(6)‐adenosine methylation by the METTL3‐METTL14 complex. Nature 534:575–578. [DOI] [PubMed] [Google Scholar]
- 9. Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC (2014) N6‐methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol 16:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang P, Doxtader KA, Nam Y (2016) Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell 63:306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G (2007) Genome regulation by polycomb and trithorax proteins. Cell 128:735–745. [DOI] [PubMed] [Google Scholar]
- 12. Margueron R, Li G, Sarma K, Blais A, Zavadil J, Woodcock CL, Dynlacht BD, Reinberg D (2008) Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 32:503–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Oliviero G, Brien GL, Waston A, Streubel G, Jerman E, Andrews D, Doyle B, Munawar N, Wynne K, Crean J, Bracken AP, Cagney G (2016) Dynamic protein interactions of the polycomb repressive complex 2 during differentiation of pluripotent cells. Mol Cell Proteom 15:3450–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Petkowski JJ, Bonsignore LA, Tooley JG, Wilkey DW, Merchant ML, Macara IG, Schaner Tooley CE (2013) NRMT2 is an N‐terminal monomethylase that primes for its homologue NRMT1. Biochem J 456:453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Petkowski JJ, Schaner Tooley CE, Anderson LC, Shumilin IA, Balsbaugh JL, Shabanowitz J, Hunt DF, Minor W, Macara IG (2012) Substrate specificity of mammalian N‐terminal alpha‐amino methyltransferase NRMT. Biochemistry 51:5942–5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shields KM, Tooley JG, Petkowski JJ, Wilkey DW, Garbett NC, Merchant ML, Cheng A, Schaner Tooley CE (2017) Select human cancer mutants of NRMT1 alter its catalytic activity and decrease N‐terminal trimethylation. Protein Sci 26:1639–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bonsignore LA, Tooley JG, Van Hoose PM, Wang E, Cheng A, Cole MP, Schaner Tooley CE (2015) NRMT1 knockout mice exhibit phenotypes associated with impaired DNA repair and premature aging. Mechan Age Dev 146‐148:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bailey AO, Panchenko T, Sathyan KM, Petkowski JJ, Pai PJ, Bai DL, Russell DH, Macara IG, Shabanowitz J, Hunt DF, Black BE, Foltz DR (2013) Posttranslational modification of CENP‐A influences the conformation of centromeric chromatin. Proc Natl Acad Sci U S A 110:11827–11832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dai X, Otake K, You C, Cai Q, Wang Z, Masumoto H, Wang Y (2013) Identification of novel alpha‐n‐methylation of CENP‐B that regulates its binding to the centromeric DNA. J Proteome Res 12:4167–4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dai X, Rulten SL, You C, Caldecott KW, Wang Y (2015) Identification and functional characterizations of N‐terminal alpha‐N‐methylation and phosphorylation of serine 461 in human poly(ADP‐ribose) polymerase 3. J Proteome Res 14:2575–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cai Q, Fu L, Wang Z, Gan N, Dai X, Wang Y (2014) alpha‐N‐methylation of damaged DNA‐binding protein 2 (DDB2) and its function in nucleotide excision repair. J Biol Chem 289:16046–16056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tooley CE, Petkowski JJ, Muratore‐Schroeder TL, Balsbaugh JL, Shabanowitz J, Sabat M, Minor W, Hunt DF, Macara IG (2010) NRMT is an alpha‐N‐methyltransferase that methylates RCC1 and retinoblastoma protein. Nature 466:1125–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen T, Muratore TL, Schaner‐Tooley CE, Shabanowitz J, Hunt DF, Macara IG (2007) N‐terminal alpha‐methylation of RCC1 is necessary for stable chromatin association and normal mitosis. Nat Cell Biol 9:596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sathyan KM, Fachinetti D, Foltz DR (2017) alpha‐amino trimethylation of CENP‐A by NRMT is required for full recruitment of the centromere. Nat Commun 8:14678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Iyer LM, Zhang D, Aravind L (2016) Adenine methylation in eukaryotes: Apprehending the complex evolutionary history and functional potential of an epigenetic modification. Bioessays 38:27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schapira M, Ferreira de Freitas R (2014) Structural biology and chemistry of protein arginine methyltransferases. Medchemcomm 5:1779–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Richon VM, Johnston D, Sneeringer CJ, Jin L, Majer CR, Elliston K, Jerva LF, Scott MP, Copeland RA (2011) Chemogenetic analysis of human protein methyltransferases. Chem Biol Drug Des 78:199–210. [DOI] [PubMed] [Google Scholar]
- 28. Min J, Wu H, Loppnau P, Sundstrom M, Arrowsmith CH, Edwards AM, Bochkarev A, Plotnikov AN (2005) The crystal structure of human AD‐003 protein in complex with S‐adenosyl‐L‐homocysteine. http://www.rcsb.org/structure/2EX4.
- 29. Dong C, Zhu L, Tempel W, Dong A, Bountra C, Arrowsmith CH, Edwards AM, Min J (2017) Crystal structure of human alpha N‐terminal protein methyltransferase. Motif E.C. 2.1.1.299: http://www.rcsb.org/structure/5UBB
- 30. Wu R, Yue Y, Zheng X, Li H (2015) Molecular basis for histone N‐terminal methylation by NRMT1. Genes Dev 29:2337–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smith GM, Pettigrew GW (1980) Identification of N,N‐dimethylproline as the N‐terminal blocking group of Crithidia oncopelti cytochrome c557. Eur J Biochem 110:123–130. [DOI] [PubMed] [Google Scholar]
- 32. Zhang L, Gurskaya NG, Merzlyak EM, Staroverov DB, Mudrik NN, Samarkina ON, Vinokurov LM, Lukyanov S, Lukyanov KA (2007) Method for real‐time monitoring of protein degradation at the single cell level. Biotechniques 42:446, 448–450. [DOI] [PubMed] [Google Scholar]
- 33. Stock A, Clarke S, Clarke C, Stock J (1987) N‐terminal methylation of proteins: structure, function and specificity. FEBS Lett 220:8–14. [DOI] [PubMed] [Google Scholar]
- 34. Kapoor‐Vazirani P, Vertino PM (2014) A dual role for the histone methyltransferase PR‐SET7/SETD8 and histone H4 lysine 20 monomethylation in the local regulation of RNA polymerase II pausing. J Biol Chem 289:7425–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moore SA, Ferhatoglu Y, Jia Y, Al‐Jiab RA, Scott MJ (2010) Structural and biochemical studies on the chromo‐barrel domain of male specific lethal 3 (MSL3) reveal a binding preference for mono‐ or dimethyllysine 20 on histone H4. J Biol Chem 285:40879–40890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim D, Blus BJ, Chandra V, Huang P, Rastinejad F, Khorasanizadeh S (2010) Corecognition of DNA and a methylated histone tail by the MSL3 chromodomain. Nat Struct Mol Biol 17:1027–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Garske AL, Craciun G, Denu JM (2008) A combinatorial H4 tail library to explore the histone code. Biochemistry 47:8094–8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Prince VE, Pickett FB (2002) Splitting pairs: the diverging fates of duplicated genes. Nat Rev Genet 3:827–837. [DOI] [PubMed] [Google Scholar]
- 39. Bonsignore LA, Butler JS, Klinge CM, Schaner Tooley CE (2015) Loss of the N‐terminal methyltransferase NRMT1 increases sensitivity to DNA damage and promotes mammary oncogenesis. Oncotarget 6:12248–12263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen T, Brownawell AM, Macara IG (2004) Nucleocytoplasmic shuttling of JAZ, a new cargo protein for exportin‐5. Mol Cell Biol 24:6608–6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McCaffrey LM, Macara IG (2009) The Par3/aPKC interaction is essential for end bud remodeling and progenitor differentiation during mammary gland morphogenesis. Genes Dev 23:1450–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Coutsias EA, Seok C, Dill KA (2004) Using quaternions to calculate RMSD. J Comput Chem 25:1849–1857. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Fig. 1