

Abstract
The classical view of the structure–function paradigm advanced by Anfinsen in the 1960s is that a protein's function is inextricably linked to its three‐dimensional structure and is encrypted in its amino acid sequence. However, it is now known that a significant fraction of the proteome consists of intrinsically disordered proteins (IDPs). These proteins populate a polymorphic ensemble of conformations rather than a unique structure but are still capable of performing biological functions. At the boundary, between well‐ordered and inherently disordered states are proteins that are on the brink of stability, either weakly stable ordered systems or disordered but on the verge of being stable. In such marginal states, even relatively minor changes can significantly alter the energy landscape, leading to large‐scale conformational remodeling. Some proteins on the edge of stability are metamorphic, with the capacity to switch from one fold topology to another in response to an environmental trigger (e.g., pH, temperature/salt, redox). Many IDPs, on the other hand, are marginally unstable such that small perturbations (e.g., phosphorylation, ligands) tip the balance over to a range of ordered, partially ordered, or even more disordered states. In general, the structural transitions described by metamorphic fold switches and polymorphic IDPs possess a number of common features including low or diminished stability, large‐scale conformational changes, critical disordered regions, latent or attenuated binding sites, and expansion of function. We suggest that these transitions are, therefore, conceptually and mechanistically analogous, representing adjacent regions in the continuum of order/disorder transitions.
Keywords: fold switching, metamorphic proteins, intrinsically disordered proteins, protein malleability
Introduction
It is widely held that cells with identical genomes display identical phenotypes and respond similar to a given stimulation or environmental perturbation. Implicit in this genome centric view, every protein has a well‐defined three‐dimensional structure that is determined by its amino acid sequence encoded in the genome.1 Consequently, a one‐to‐one correlation between the genotype and phenotype is often assumed and there is good evidence to support this thinking. For example, a single point mutation can result in the loss2 or gain3, 4 of function in a given protein. Further research on temperature‐sensitive mutants5 and allostery6, 7 in proteins extended this horizon and revealed that changes in conformation could enable proteins to assume different functions or switch between inactive and active states with high specificity and affinity. The bacterial tryptophan repressor is a good example of how protein conformational dynamics turns a gene on or off.8 However, it should be noted that even in this expanded view it was believed that proteins are highly ordered with characteristic folds.
Contrary to this deterministic interpretation, it is now well established that proteins need not always be folded to remain functional.9 A large fraction of the proteome across all three kingdoms is composed of intrinsically disordered proteins (IDPs) that, by definition, are ensembles of polymorphic conformers lacking rigid three‐dimensional structure.10, 11, 12, 13 IDPs and segments within ordered proteins constituting intrinsically disordered regions (IDRs) are characterized by a combination of low mean hydrophobicity and relatively high net charge, important prerequisites for the absence of three‐dimensional structure in proteins under physiological conditions.14, 15 Therefore, IDPs are marked by a preponderance of polar and charged residues and a paucity of non‐polar residues that include the bulky hydrophobic and aromatic amino acids. However, they are quite rich in the structure‐breaking amino acids proline and glycine.16 IDPs are also more prone to post‐translational modifications (PTMs) such as phosphorylation, and are alternatively spliced more often than ordered proteins.17
At the boundary between well‐folded and random coil polypeptide chains are proteins that are on the brink of thermodynamic stability (Fig. 1). These proteins have shallow energy wells, or no apparent wells at all, that can confer properties generally not seen in more stable folds. Some marginally stable proteins (ΔG unfolding > 0) are metamorphic,18 undergoing large‐scale conformational transitions from one fold topology to another in response to relatively small perturbations.19, 20 In many cases, disordered regions play a role in these metamorphic changes (Fig. 2). On the other side of the stability boundary (ΔG unfolding < 0), certain IDPs appear to be on the verge of being weakly stable folded proteins despite their inherent flexibility. In such cases, binding to a ligand21, 22 or a PTM23 can trigger a significant structural transition from a flexibly disordered state to one with more conformational order. Indeed, while IDPs have different sequence characteristics from metamorphic proteins, it is interesting to note that many IDPs are close to the boundary between disorder and order in plots of mean net charge versus mean hydrophobicity.14 Thus, the examples of fold switching by marginally stable proteins and disorder/order transitions in marginally unstable IDPs are conceptually similar, representing different points on the continuum of order/disorder transitions. Here, we compare and discuss the range of transitions in metamorphic and polymorphic systems further, highlighting features common to both with recent examples.
Figure 1.
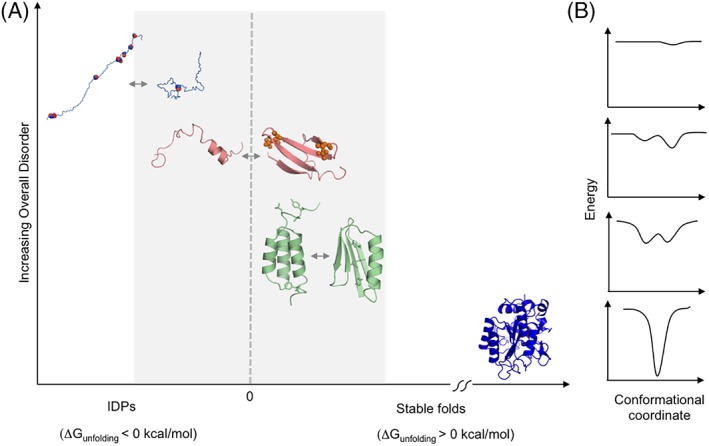
Proteins on the brink of stability can undergo a continuum of order/disorder transitions. (A) Examples of transitions from top left to bottom right: Transition between the extended and collapsed disordered states of prostate associated Gene 4 (PAGE4), modulated by phosphorylation;111 disorder‐to‐order transition of 4E‐BP2 induced by phosphorylation;23 order‐to‐order fold switching between GA98 and GB98, triggered by single amino acid changes or ligand binding.64 In contrast, stable proteins such as subtilisin (shown in dark blue) do not undergo such changes. (B) Approximate energy well diagrams for each protein from PAGE4 (top) to subtilisin (bottom).
Figure 2.
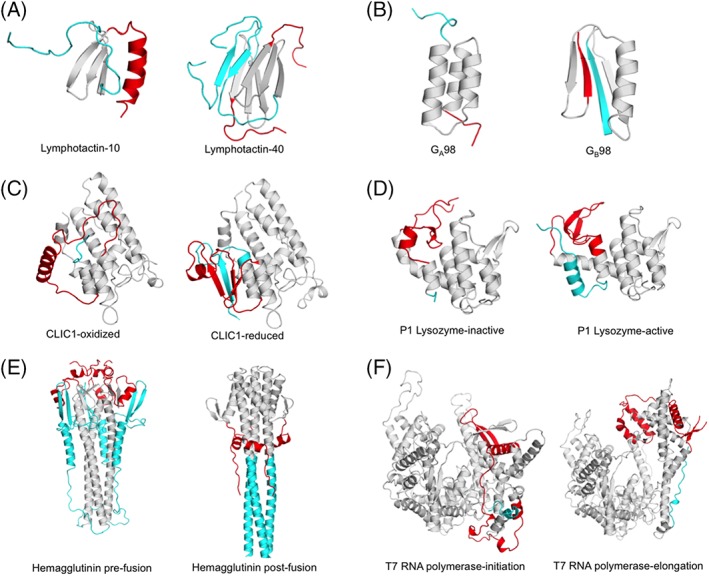
Examples of metamorphic proteins where disordered or partially disordered regions play an important role in remodeling ordered states. N‐ and C‐terminal regions of fold‐switched domains are color coded cyan and red, respectively. All cases are naturally occurring with the exception of (B), which is a designed system. Structures for each panel are identified left to right. (A) Lymphotactin‐10 (PDB 1J8I), Lymphotactin‐40 (PDB 2JP1). (B) GA98 (PDB 2LHC), GB98 (PDB 2LHD). (C) Chloride intracellular channel 1 (CLIC1)‐oxidized (PDB 1RK4), CLIC1‐reduced (PDB 1K0M). (D) P1 lysozyme‐inactive (PDB 1XJU), P1 lysozyme‐active (PDB 1XJT). (E) hemagglutinin pre‐fusion (PDB 5HMG), post‐fusion (PDB 1HTM). (F) T7 RNA polymerase‐initiation state (PDB 1QLN), elongation state (PDB 1MSW).
Large‐scale transitions between folded states
The Anfinsen hypothesis states that the amino acid sequence is sufficient to determine the native fold of a protein and that the three‐dimensional structure is the thermodynamically most stable conformation.1, 24 The latter point implies a unique structure although Anfinsen did not explicitly state this. However, it has been known for some time now that certain polypeptide chains, for example, prions,25, 26, 27 can adopt more than one fold topology. Further examples of naturally occurring fold switches have since accumulated (Table I).28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48 A recent paper showed that there were approximately 100‐fold switches in the Protein Data Bank and further estimated that 0.5–4% of known structures may have fold switched partners.49 These proteins, or regions within these proteins, undergo significant changes in their three‐dimensional structures without any mutations in their amino acid sequences. Environmental triggers such as pH, temperature, salt concentration, redox conditions, ligand binding, proteolytic cleavage, or oligomerization can shift the equilibrium between two different fold topologies, driving conformational switching in these natural examples. Thus, fold switching has been demonstrated to be another post‐translational mechanism, alongside chemical modifications such as phosphorylation, by which a given polypeptide chain can expand its biological function.
Table I.
Examples of Naturally Occurring Protein Fold Switches and Their Triggering Mechanisms
| Naturally occurring fold switches | Trigger | PDB code |
|---|---|---|
| Serpins28 (e.g., antithrombin) | Proteolysis/domain swap | 2ANT (active, latent) |
| Lymphotactin29 | Salt, temperature | 1J8I (monomer) |
| 2JP1 (dimer) | ||
| Chloride ion channel protein30 | Redox | 1K0M (reduced) |
| 1RK4 (oxidized) | ||
| Mad2 spindle checkpoint protein31 | Ligand binding | 1DUJ (inactive) |
| 1S2H (active) | ||
| T7 RNA polymerase32, 33 | Ligand binding | 1QLN (initiation) |
| 1MSW (elongation) | ||
| Viral fusion proteins34, 35 | pH | 5HMG (pre‐fusion) |
| (e.g., influenza virus hemagglutinin) | 1HTM (post‐fusion) | |
| P1 Lysozyme36 | Redox | 1XJU (inactive) |
| 1XJT (active) | ||
| Circadian clock protein KaiB37, 38 | Ligand binding | 2QKE (inactive) |
| 5JWR (active) | ||
| RFaH C‐terminal domain (CTD)39, 40 | Ligand binding | 2OUG (full length) |
| 2LCL (CTD) | ||
| Selecase41 | Concentration | 4QHF (active) |
| 4QHH (inactive) | ||
| Cytolysin A42 | Membrane insertion | 1QOY (monomer) |
| 2WCD (protomer) | ||
| Phytochromes43, 44 | Light | 4O0P (dark) |
| 4O01 (light) | ||
| Retinoic acid receptor45 | Ligand binding | 1DKF (antagonist) |
| 3KMR (agonist) | ||
| TCR ectodomain46 | Unknown | 2VLM (typical) |
| 3MFF (alternative) | ||
| Caspase‐647 | Ligand binding | 2WDB (free) |
| 3OD5 (bound) | ||
| XRCC148 | Redox | 1XNT (reduced) |
| 3LQC (oxidized) |
Concurrent with studies of natural proteins, efforts to design protein fold switches have provided considerable insight into their potential role in fold evolution, suggesting that this phenomenon may be more general and an inherent property of polypeptide chains. Early experiments showed that an 11‐residue region of a longer polypeptide chain could adopt either alpha‐helical or beta‐hairpin conformations, depending on where it was placed in the amino acid sequence.50 Similarly, a 9‐amino acid region in the N‐terminus of the Arc repressor was able to convert between α‐helical and β‐strand conformations with only a single amino acid modification.51 These types of studies were extended to the entire length of polypeptide chains in small proteins. The Paracelsus Challenge52 sought to establish the minimum number of amino acids required to specify one fold over another. This led to a series of investigations demonstrating that it is possible to engineer two proteins to have amino acid sequence similarities/identities of 50–60% while maintaining distinctly folded states.53, 54, 55, 56, 57 Subsequent experiments with the GA 58 and GB 59 domains of streptococcal protein G indicated that different folds with even higher identities were achievable by design.
The GA 3α helical bundle and the GB 4β + α “β‐grasp” structure are two of the most common folds known,60 with the parental GA and GB amino acid sequences having only 16% identity. Designed mutants were co‐evolved to very high identities of 88–98%, with the parent topologies remaining intact.61, 62, 63 A number of mutants at switch points were found to populate both the 3α and 4β + α states.64 The GA 3α‐fold has also been co‐evolved to high identity with other common folds such as the α/β/α‐sandwich and an all‐β structure.65 Together, these results suggest that some folds are likely to have been derived from pre‐existing topologies rather than evolving independently. This idea is further supported by the detection of transitory sequences in fold migration51, 66, 67, 68, 69 as well as by other studies.70, 71, 72
Thus, some structural motifs or even entire domains have the ability to switch from one topologically ordered state to another, exposing a previously hidden binding surface and leading to an expansion of function [Fig. 3(A)]. Recurring features found in both natural and designed fold switches are minimally overlapping cores, latent binding epitopes, disordered regions, and weakened thermodynamic stability.19, 20 Mutual exclusivity in cores allows information for both folded states to co‐exist in a single polypeptide chain. This relatively tight constraint can be loosened by the presence of flexibly disordered regions, which tend to play an important role in the fold switch, often transiting to a more ordered state in the alternative fold (Fig. 2). In particular, the lower stability of switchable systems allows alternative folded states to be more accessible than for stable proteins [Fig. 1(B)]. Proteins with switchable folds can, therefore, be considered metastable and do not fit the traditional thermodynamic hypothesis, which holds for well‐folded stable structures with relatively deep energy wells.
Figure 3.
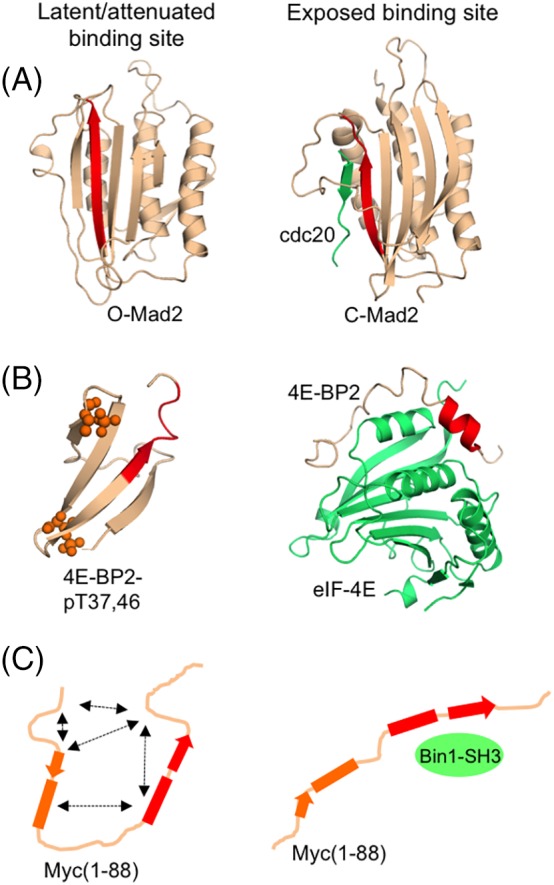
Latent and attenuated binding sites become accessible in a range of order–order, order–disorder, and disorder–disorder transitions involving metamorphic and polymorphic proteins. (A) The metamorphic protein O‐Mad2 (PDB 1DUJ) has a buried binding site (red) that is only accessible to cdc20 (green) upon fold switching to C‐Mad2 (1S2H). (B) Partially disordered 4E‐BP2 binds to eIF4E (green) utilizing an exposed helical motif (red) that is masked in a β‐sheet structure upon phosphorylation. (C) The disordered protein Myc (1‐88) has multiple transient long‐range interactions that attenuate the affinity of its binding epitope (red) for Bin1‐SH3 (green).
Disorder‐to‐order transitions
Similar to the fold switching examples discussed above, many IDPs are known to undergo significant conformational ordering upon binding to a ligand. There are numerous examples of such coupled folding and binding.21, 73, 74, 75, 76, 77, 78, 79 In some cases, where the disordered protein is on the edge of being a globular fold, the binding affinity is related to its thermodynamic stability, suggesting a conformational selection mechanism analogous to the equilibrium shifts inferred in protein fold switches. This is clearly demonstrated in the complex of subtilisin with its N‐terminal prodomain.73 The prodomain is disordered in the absence of subtilisin, but adopts a folded α/β‐plait topology upon binding. When mutations are made to the prodomain that stabilize its bound state conformation, binding affinity to subtilisin increases accordingly and can be linearly related to the extent of prodomain folding. In this particular study, the binding of prodomain to subtilisin was increased by approximately 100‐fold through the use of stabilizing mutations, demonstrating that prodomain binding is highly tunable.80 In many other cases, however, IDP binding is shown to be consistent with an induced fit mechanism where the polypeptide chain folds on the ligand surface.81, 82, 83 Such a mechanism seems to be more prevalent when the bound state of the IDP does not correspond with a globular structure.79
In addition to the use of mutations, IDP binding affinities can also be modulated by PTMs. One of the most common PTMs, phosphorylation, can alter charge distribution and provide new sites for hydrogen bonding interactions. Even relatively small perturbations can modify binding affinity and folding propensity, further emphasizing that IDPs may sometimes be on the verge of being either partially or fully ordered proteins with weak stability. This is illustrated dramatically by a recent example23 where phosphorylation at two threonine residues in the mostly disordered 4E‐BP2 protein leads to folding into a 4‐stranded beta‐domain, even in the absence of a binding partner. The folded state is weakly stable and sequesters a motif that is used to bind the translation initiation factor eIF4E in the less ordered unphosphorylated state. The disorder/order transition, therefore, functions as a switch that controls the binding of 4E‐BP2 to eIF4E, thereby regulating translation. This example serves to highlight another feature of IDPs that has similarity to fold switches, their ability to mask or unmask functional sites through large‐amplitude conformational transitions [Fig. 3(B)]. Further, in analogy with metamorphic proteins, the disorder‐to‐order or order‐to‐disorder transitions in IDPs can involve either structural motifs or entire domains.
IDPs undergo disorder‐to‐order transitions not only in the presence of folded proteins and PTMs but also by interacting with other disordered regions. An example of this type of transition is the DnaE intein, a naturally occurring split intein that has trans‐splicing activity both in vivo and in vitro. This split intein has two subunits, I N and IC, that are both intrinsically disordered by themselves but combine with high affinity (K D 33 nM) to form an ordered complex, which is necessary for the initiation of trans‐splicing.84, 85 Calorimetric analysis indicated that the unfavorable entropy loss in going from disorder to order is outweighed by a favorable enthalpic change likely due to significant interactions between polar groups in the two subunits. This type of enthalpy‐entropy compensation is frequently detected in systems where folding is coupled to binding.22
IDPs often bind their target ligands with high specificity but relatively weak affinities (micromolar or less). These low affinities may be a requirement for correct function, allowing for rapid dissociation and preventing permanent masking of other binding motifs on the polypeptide chain. The dynamic nature and marginal thermodynamic instability of IDPs is therefore advantageous for certain functions (e.g., transcriptional and translational activation, signaling) where further mutations stabilizing more ordered states may actually be deleterious.78 Another characteristic of IDPs is their ability to interact with multiple binding partners.86, 87 They are therefore frequently found to be hub points in protein interaction networks.88 In certain IDPs such as p53, the same amino acid sequence adopts a bound state structure that varies from α‐helical to β‐strand to coil conformations depending on the cognate ligand.89 Other disease‐relevant IDPs such as Aβ and α‐synuclein exhibit similar kinds of polymorphic behavior to p53, undergoing a broad spectrum of disorder‐to‐order transitions that depend on their environment. Both Aβ and α‐synuclein are largely disordered in dilute aqueous solutions,90, 91, 92, 93 helical in a membrane‐like environment,94, 95, 96, 97 and form a wide range of mostly β‐structures depending on the ligand or sample conditions.98, 99, 100, 101, 102, 103, 104, 105 Thus, the structural polymorphism displayed by IDPs that permits them to recognize multiple binding partners is analogous to what is observed in metamorphic fold switches but on a larger scale. While the alternative topology in a fold switch has different ligand binding properties that typically lead to one additional function for the same polypeptide chain, IDPs can adopt a wider array of possible conformers due to their greater inherent malleability, with a corresponding increase in functionality.
Shape‐shifting ensembles
For some IDPs, ligand binding or a modification such as phosphorylation does not lead to ordered or partially ordered states. Rather, the conformational ensemble shifts from one disordered but functional state to another. Underlying these types of cases is the recognition that most IDPs tend not to be true random coils but generally have some weak to moderate conformational preferences. These transient propensities lead to a conformationally biased but still flexible polypeptide, whose ensemble characteristics can change significantly in the presence of a ligand or upon PTMs to the chain. The combination of NMR spectroscopic approaches with other methods such as SAXS, SANS, smFRET, and simulations has provided considerable insight into the mechanism of such transitions between states that are disordered to different extents.106, 107, 108, 109
One recent example of this is the IDP and cancer/testis antigen, prostate‐associated gene 4 (PAGE4), a stress–response protein that is up‐regulated in the fetal and diseased prostate. In prostate cancer cells, PAGE4 is differentially phosphorylated by two kinases, Homeodomain Interacting Protein Kinase 1 (HIPK1) and CDC‐Like Kinase 2 (CLK2).110, 111, 112 Despite both phosphorylated forms being disordered and flexible, biophysical measurements show that HIPK1‐PAGE4 has a less disordered conformational ensemble than CLK2‐PAGE4, primarily due to the presence of acidic and basic motifs that form transient but stabilizing electrostatic interactions. These long‐range effects are disrupted when PAGE4 is hyper‐phosphorylated by CLK2 because phosphorylation occurs at multiple sites in or near the basic motifs, effectively neutralizing the transient electrostatic interactions and leading to a more extended ensemble of conformers (Fig. 4). The large differences in the ensembles are reflected in opposing functions – HIPK1‐PAGE4 binds the transcription factor Jun/Fos more tightly and potentiates c‐Jun, whereas CLK2‐PAGE4 attenuates c‐Jun activity. Thus, multi‐site phosphorylation can promote structure23 or disrupt it.111
Figure 4.
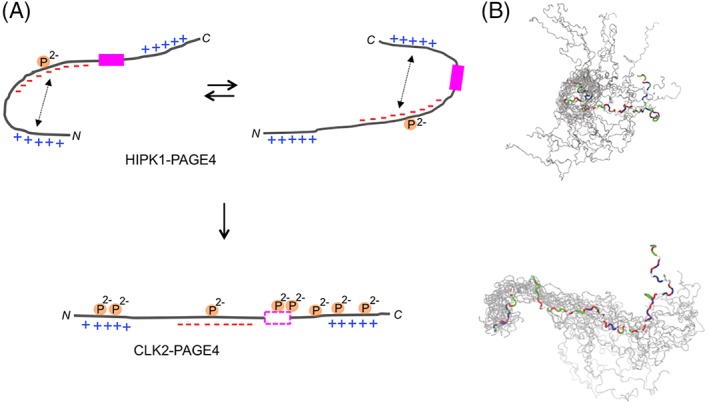
Ensemble switching between relatively closed and open disordered states. (A) Differential phosphorylation remodels the PAGE4 ensemble. Cartoon depiction of the HIPK1‐PAGE4 polypeptide chain (top) showing competing long‐range electrostatic interactions that decrease the radius of gyration of the polypeptide chain. The purple rectangle represents a transient helix. Hyper‐phosphorylation by CLK2 (bottom) weakens these long‐range interactions and decreases the helical propensity, leading to a more extended conformation with larger radius of gyration. (B) Conformational ensembles for HIPK1‐PAGE4 (top) and CLK2‐PAGE4 (bottom) from MD simulations.112
Similar types of transient long‐range interactions have also been observed in other flexible polypeptide chains where they can mask or attenuate the function of a ligand binding site. For example, the high affinity Bin1‐SH3 binding site of Myc (1‐88) consists of an approximately 12‐residue motif which, when incorporated into an isolated short peptide, binds to Bin1‐SH3 with a K D of 4.2 μM.113 In the context of the 88‐amino acid Myc polypeptide, however, Bin1‐SH3 displays significantly weaker and multi‐valent binding to Myc with K D values of 33 μM and 200 μM.114 The transient interactions between different parts of the Myc chain serve to regulate its affinity to Bin‐SH3. Even though these long‐range intramolecular contacts may be weak, the high local concentration of the low affinity sites within the flexible polypeptide chain can lead to significant attenuation and make regulation over an order of magnitude quite feasible. Thus binding (or PTM) to one part of a flexible polypeptide chain can lead to long‐range changes in the conformational ensemble that alter affinity for other ligands by making latent sites more accessible to interactions [Fig. 3(C)]. Such actions at a distance can be considered to be dynamic allosteric effects.115 Other recent examples of these types of transient long‐range interactions are also seen in IDPs such as the human tau protein116 and the disordered region of the glucocorticoid receptor.117 Splice variants of these proteins have disordered regions with varying lengths that can alter the extent of transient intramolecular interactions leading to different binding characteristics and functional outputs.
Thus, while some IDPs transit to more order upon binding to their targets, others remain significantly disordered over extensive regions of the polypeptide chain.118 There are numerous examples of such “fuzzy complexes” where the IDPs involved do not acquire a discernable secondary structure upon interaction. These include the T cell receptor ζ chain‐nef complex,119 the nematode desiccation‐tolerance protein anhydrin in its interaction with DNA,120 tau binding with microtubules,116 and cystic fibrosis transmembrane conductance regulator with a range of partners.121
Functional consequences
Both metamorphic fold switch proteins and IDPs typically carry out more than one function. Naturally occurring fold switches expand their functional capacity by adopting weakly stable alternative topologies with new binding surfaces. For example, lymphotactin can interact with both a chemokine receptor and glycosaminoglycans depending on its folded state, which in turn can be regulated by salt and temperature conditions.29 There are numerous other elegant examples of this phenomenon (Table I) demonstrating the enhanced functional role of metamorphic proteins, where the equilibrium between two distinctly folded states can be shifted by a variety of environmental factors. By comparison, individual IDPs can acquire even more functionality because of their highly flexible and polymorphic nature. Regardless of the type of structural change in an IDP, conformational dynamics has significant effects on function. With multiple states and rapid transitions between them, IDPs can stochastically engage in many interactions and thereby contribute to “conformational noise” in network interactions.122 Therefore, in response to perturbations (e.g., inflammatory stress, drug treatment), myriad network options can be explored and the functionally most advantageous selected. Moreover, the ubiquitous presence of IDPs as transcription factors, and more generally as hubs in protein interaction networks, is indicative of their role in propagating and amplifying transcriptional noise.123 IDPs can thus confer protein interaction networks with remarkable flexibility and resilience.122 One classic example is the utilization of just four IDP transcription factors to reprogram a somatic cell to a pluripotent stem cell.124 Another notable example is the potential role of PAGE4, AP‐1, and the androgen receptor, all IDPs, in the phenotypic switching between androgen‐dependent and androgen‐independent states of prostate cancer cells.111, 112 A recently discovered protein‐based inheritance mechanism was found to be enriched in IDP sequences, providing further support for the conformational noise hypothesis.125
Conclusions
Functional proteins have a wide range of structures and thermodynamic stabilities, varying from well‐ordered folds to highly flexible polypeptide chains. At the boundary between these two extremes are proteins that are on the brink of stability. These are either weakly stable ordered systems or disordered but on the verge of being stable. In such marginal states, where there is a more delicate balance between stabilizing and destabilizing forces, minor changes can have a much larger effect than in well‐stabilized or completely disordered chains. For folded proteins with reduced thermodynamic stability, a relatively small but growing number have been shown to be metamorphic. Such ordered proteins can expand their functional capacity by adopting an alternative fold topology, either through a short mutational path or through environmental factors (e.g., pH, temperature/salt, redox). Many IDPs, on the other hand, are marginally unstable but still highly flexible with no discernible thermodynamic minima. Small perturbations (e.g., phosphorylation, ligands) in these proteins can shift the equilibrium over to a range of ordered, partially ordered, or even more disordered states. Because of their inherent flexibility, IDPs have the potential to adopt a greater number of folded conformations in response to ligand binding than the more constrained metamorphic proteins.
While IDPs and metamorphic proteins have different sequence composition, the order/order transitions seen in metamorphic fold switch proteins and the disorder/order and disorder/disorder transitions observed in polymorphic IDPs have several features in common. First, both fold switches and IDPs have diminished stability. Fold switches tend to be on the margin of thermodynamic stability (0 < ΔG unfolding < 2–3 kcal/mol) whereas IDPs have no detectable energy minima (ΔG unfolding < 0) and are often on the verge of being weakly stable proteins that are either partially or fully folded. Second, while disordered regions in polymorphic IDPs can be remodeled in a wide variety of ways, disordered regions also tend to play an important role in the transition between ordered states in metamorphic fold switching. In both cases, these conversions are sensitive to environmental triggers. Third, both metamorphic transitions and transitions involving IDPs tend to be large‐scale conformational changes where residues in a structural motif or in an entire domain undergo significant alterations in their backbone phi/psi angles. Finally, both metamorphic folded proteins and polymorphic IDPs possess latent or attenuated binding sites that become more exposed upon conformational switching and result in the acquisition of additional function. Such masking and unmasking effects are more typically associated with transitions where at least one of the states is ordered. However, it is becoming increasingly clear that even transitions between two disordered ensembles can lead to increased accessibility to a binding site by virtue of the ability to perturb competing transient interactions within the polypeptide chain. Overall, the parallels drawn here suggest that conformational switches in metamorphic and polymorphic proteins are conceptually and mechanistically similar processes, representing adjacent regions in the continuum of order/disorder transitions.
Acknowledgments
We thank the NIH for supporting our work on protein fold switching (GM062154, J. O. and P. B.) and PAGE4 (CA181730, P. K. and J. O.).
Prakash Kulkarni's current address is Department of Medical Oncology & Therapeutics Research, City of Hope National Medical Center, 1500 East Duarte Road, Duarte, CA 91010. E‐mail: pkulkarni@coh.org.
Contributor Information
Prakash Kulkarni, Email: pkulkar4@ibbr.umd.edu.
Philip N. Bryan, Email: pbryan@umd.edu
John Orban, Email: jorban@umd.edu.
References
- 1. Anfinsen CB, Haber E (1961) Studies on the reduction and re‐formation of protein disulfide bonds. J Biol Chem 236:1361–1363. [PubMed] [Google Scholar]
- 2. Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Molecular cell biology. 4th ed. New York: WH Freeman, 2000. [Google Scholar]
- 3. Bargmann CI, Hung MC, Weinberg RA (1986) Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell 45:649–657. [DOI] [PubMed] [Google Scholar]
- 4. Irby RB, Mao W, Coppola D, Kang J, Loubeau JM, Trudeau W, Karl R, Fujita DJ, Jove R, Yeatman TJ (1999) Activating SRC mutation in a subset of advanced human colon cancers. Nat Genet 21:187–190. [DOI] [PubMed] [Google Scholar]
- 5. Ben‐Aroya S, Pan X, Boeke JD, Hieter P (2010) Making temperature‐sensitive mutants. Methods Enzymol 470:181–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Koshland DE (1959) Enzyme flexibility and enzyme action. J Cell Comp Physiol 54:245–258. [DOI] [PubMed] [Google Scholar]
- 7. Koshland DE (1963) The role of flexibility in enzyme action. Cold Spring Harb Symp Quant Biol 28:473–482. [Google Scholar]
- 8. Somerville R (1992) The Trp repressor, a ligand‐activated regulatory protein. Prog Nucleic Acid Res Mol Biol 42:1–38. [DOI] [PubMed] [Google Scholar]
- 9. Wright PE, Dyson HJ (1999) Intrinsically unstructured proteins: re‐assessing the protein structure–function paradigm. J Mol Biol 293:321–331. [DOI] [PubMed] [Google Scholar]
- 10. Uversky VN. Intrinsically disordered proteins. New York: Springer, 2014. [Google Scholar]
- 11. Bhowmick A, Brookes DH, Yost SR, Dyson HJ, Forman‐Kay JD, Gunter D, Head‐Gordon M, Hura GL, Pande VS, Wemmer DE, Wright PE, Head‐Gordon T (2016) Finding our way in the dark proteome. J Am Chem Soc 138:9730–9742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DeForte S, Uversky VN (2016) Order, disorder, and everything in between. Molecules 21:E1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pauwels K, Lebrun P, Tompa P (2017) To be disordered or not to be disordered: is that still a question for proteins in the cell? Cell Mol Life Sci 74:3185–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Uversky VN (2013) Unusual biophysics of intrinsically disordered proteins. Biochim Biophys Acta 1834:932–951. [DOI] [PubMed] [Google Scholar]
- 15. Huang F, Oldfield CJ, Xue B, Hsu WL, Meng J, Liu X, Shen L, Romero P, Uversky VN, Dunker A (2014) Improving protein order–disorder classification using charge‐hydropathy plots. BMC Bioinformatics 15(Suppl 17):S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Theillet FX, Kalmar L, Tompa P, Han KH, Selenko P, Dunker AK, Daughdrill GW, Uversky VN (2013) The alphabet of intrinsic disorder I. Act like a Pro: on the abundance and roles of proline residues in intrinsically disordered proteins. Intrinsically Disord Proteins 1:e24360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dunker AK, Bondos SE, Huang F, Oldfield CJ (2015) Intrinsically disordered proteins and multicellular organisms. Semin Cell Dev Biol. 37:44–55. [DOI] [PubMed] [Google Scholar]
- 18. Murzin AG (2008) Biochemistry. Metamorphic proteins. Science 320:1725–1726. [DOI] [PubMed] [Google Scholar]
- 19. Bryan PN, Orban J (2010) Proteins that switch folds. Curr Opin Struct Biol 20:482–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bryan PN, Orban J (2013) Implications of protein fold switching. Curr Opin Struct Biol 23:314–316. [DOI] [PubMed] [Google Scholar]
- 21. Radhakrishnan I, Pérez‐Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE (1997) Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator:coactivator interactions. Cell 91:741–752. [DOI] [PubMed] [Google Scholar]
- 22. Dyson HJ, Wright PE (2005) Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 6:197–208. [DOI] [PubMed] [Google Scholar]
- 23. Bah A, Vernon RM, Siddiqui Z, Krzeminski M, Muhandiram R, Zhao C, Sonenberg N, Kay LE, Forman‐Kay JD (2015) Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature 519:106–109. [DOI] [PubMed] [Google Scholar]
- 24. Anfinsen CB (1973) Principles that govern the folding of protein chains. Science 181:223–230. [DOI] [PubMed] [Google Scholar]
- 25. Prusiner SB (1982) Novel proteinaceous infectious particles cause scrapie. Science 216:136–144. [DOI] [PubMed] [Google Scholar]
- 26. Zahn R, Liu A, Lührs T, Riek R, von Schroetter C, López García F, Billeter M, Calzolai L, Wider G, Wüthrich K (2000) NMR solution structure of the human prion protein. Proc Natl Acad Sci USA 97:145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tycko R, Savtchenko R, Ostapchenko VG, Makarava N, Baskakov IV (2010) The α‐helical C‐terminal domain of full‐length recombinant PrP converts to an in‐register parallel β‐sheet structure in PrP fibrils: evidence from solid state nuclear magnetic resonance. Biochemistry 49:9488–9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gettins PGW (2002) Serpin structure, mechanism, and function. Chem Rev 102:4751–4803. [DOI] [PubMed] [Google Scholar]
- 29. Tuinstra RL, Peterson FC, Kutlesa S, Elgin ES, Kron MA, Volkman BF (2008) Interconversion between two unrelated protein folds in the lymphotactin native state. Proc Natl Acad Sci USA 105:5057–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Littler DR, Harrop SJ, Goodchild SC, Phang JM, Mynott AV, Jiang L, Valenzuela SM, Mazzanti M, Brown LJ, Breit SN, Curmi PM (2010) The enigma of the clic proteins: Ion channels, redox proteins, enzymes, scaffolding proteins? FEBS Lett 584:2093–2101. [DOI] [PubMed] [Google Scholar]
- 31. Luo X, Tang Z, Xia G, Wassmann K, Matsumoto T, Rizo J, Yu H (2004) The mad2 spindle checkpoint protein has two distinct natively folded states. Nat Struct Mol Biol 11:338–345. [DOI] [PubMed] [Google Scholar]
- 32. Yin YW, Steitz TA (2002) Structural basis for the transition from initiation to elongation transcription in T7 RNA polymerase. Science 298:1387–1395. [DOI] [PubMed] [Google Scholar]
- 33. Steitz TA (2009) The structural changes of T7 RNA polymerase from transcription initiation to elongation. Curr Opin Struct Biol 19:683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harrison SC (2015) Viral membrane fusion. Virology 479‐480:498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bullough PA, Hughson FM, Skehel JJ, Wiley DC (1994) Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371:37–43. [DOI] [PubMed] [Google Scholar]
- 36. Xu M, Arulandu A, Struck DK, Swanson S, Sacchettini JC, Young R (2005) Disulfide isomerization after membrane release of its SAR domain activates P1 lysozyme. Science 307:113–117. [DOI] [PubMed] [Google Scholar]
- 37. Chang YG, Cohen SE, Phong C, Myers WK, Kim YI, Tseng R, Lin J, Zhang L, Boyd JS, Lee Y, Kang S, Lee D, Li S, Britt RD, Rust MJ, Golden SS, LiWang A (2015) Circadian rhythms. A protein fold switch joins the circadian oscillator to clock output in cyanobacteria. Science 349:324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tseng R, Goularte NF, Chavan A, Luu J, Cohen SE, Chang YG, Heisler J, Li S, Michael AK, Tripathi S, Golden SS, LiWang A, Partch CL (2017) Structural basis of the day‐night transition in a bacterial circadian clock. Science 355:1174–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Burmann BM, Knauer SH, Sevostyanova A, Schweimer K, Mooney RA, Landick R, Artsimovitch I, Rosch P (2012) An alpha helix to beta barrel domain switch transforms the transcription factor rfah into a translation factor. Cell 150:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Belogurov GA, Mooney RA, Svetlov V, Landick R, Artsimovitch I (2009) Functional specialization of transcription elongation factors. EMBO J 28:112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lopez‐Pelegrin M, Cerda‐Costa N, Cintas‐Pedrola A, Herranz‐Trillo F, Bernado P, Peinado JR, Arolas JL, Gomis‐Ruth FX (2014) Multiple stable conformations account for reversible concentration‐dependent oligomerization and autoinhibition of a metamorphic metallopeptidase. Angew Chem Int Ed Engl 53:10624–10630. [DOI] [PubMed] [Google Scholar]
- 42. Mueller M, Grauschopf U, Maier T, Glockshuber R, Ban N (2009) The structure of a cytolytic alpha‐helical toxin pore reveals its assembly mechanism. Nature 459:726–730. [DOI] [PubMed] [Google Scholar]
- 43. Takala H, Bjorling A, Berntsson O, Lehtivuori H, Niebling S, Hoernke M, Kosheleva I, Henning R, Menzel A, Ihalainen JA, Westenhoff S (2014) Signal amplification and transduction in phytochrome photosensors. Nature 509:245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Burgie ES, Zhang J, Vierstra RD (2016) Crystal structure of deinococcus phytochrome in the photoactivated state reveals a cascade of structural rearrangements during photoconversion. Structure 24:448–457. [DOI] [PubMed] [Google Scholar]
- 45. le Maire A, Teyssier C, Erb C, Grimaldi M, Alvarez S, de Lera AR, Balaguer P, Gronemeyer H, Royer CA, Germain P, Bourguet W (2010) A unique secondary‐structure switch controls constitutive gene repression by retinoic acid receptor. Nat Struct Mol Biol 17:801–807. [DOI] [PubMed] [Google Scholar]
- 46. van Boxel GI, Holmes S, Fugger L, Jones EY (2010) An alternative conformation of the T‐cell receptor a constant region. J Mol Biol 400(4):828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dagbay KB, Bolik‐Coulon N, Savinov SN, Hardy JA (2017) Caspase‐6 undergoes a distinct helix‐strand interconversion upon substrate binding. J Biol Chem 292:4885–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cuneo MJ, London RE (2010) Oxidation state of the XRCC1 N‐terminal domain regulates DNA polymerase beta binding affinity. Proc Natl Acad Sci USA 107:6805–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Porter LL, Looger LL (2018) Extant fold‐switching proteins are widespread. Proc Natl Acad Sci USA 115:5968–5973. 10.1073/pnas.1800168115. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Minor DL, Kim PS (1996) Context‐dependent secondary structure formation of a designed protein sequence. Nature 380:730–734. [DOI] [PubMed] [Google Scholar]
- 51. Cordes MH, Burton RE, Walsh NP, McKnight CJ, Sauer RT (2000) An evolutionary bridge to a new protein fold. Nat Struct Biol 7:1129–1132. [DOI] [PubMed] [Google Scholar]
- 52. Rose GD (1997) Protein folding and the Paracelsus challenge. Nat Struct Biol 4:512–514. [DOI] [PubMed] [Google Scholar]
- 53. Jones DT, Moody CM, Uppenbrink J, Viles JH, Doyle PM, Harris CJ, Pearl LH, Sadler PJ, Thornton JM (1996) Towards meeting the Paracelsus challenge: the design, synthesis, and characterization of parcelsin‐43, an alpha‐helical protein with over 50% sequence identity to an all‐beta protein. Proteins: Struct Funct Genet 24:502–513. [DOI] [PubMed] [Google Scholar]
- 54. Dalal S, Balasubramanian S, Regan L (1997) Protein alchemy: changing beta‐sheet into alpha‐helix. Nat Struct Biol 4:548–552. [DOI] [PubMed] [Google Scholar]
- 55. Yuan SM, Clarke ND (1998) A hybrid sequence approach to the Paracelsus challenge. Proteins: Struct Funct Genet 30:136–143. [DOI] [PubMed] [Google Scholar]
- 56. Alexander PA, Rozak DA, Orban J, Bryan PN (2005) Directed evolution of highly homologous proteins with different folds by phage display: implications for the protein folding code. Biochemistry 44:14045–14054. [DOI] [PubMed] [Google Scholar]
- 57. He Y, Yeh DC, Alexander P, Bryan PN, Orban J (2005) Solution NMR structures of IgG binding domains with artificially evolved high levels of sequence identity but different folds. Biochemistry 44:14055–14061. [DOI] [PubMed] [Google Scholar]
- 58. He Y, Rozak DA, Sari N, Chen Y, Bryan P, Orban J (2006) Structure, dynamics, and stability variation in bacterial albumin binding modules: implications for species specificity. Biochemistry 45:10102–10109. [DOI] [PubMed] [Google Scholar]
- 59. Gallagher T, Alexander P, Bryan P, Gilliland GL (1994) Two crystal structures of the B1 immunoglobulin‐binding domain of streptococcal protein G and comparison with NMR. Biochemistry 33:4721–4729. [PubMed] [Google Scholar]
- 60. Day R, Beck DAC, Armen RS, Daggett V (2003) A consensus view of fold space: combining SCOP, CATH, and the Dali domain dictionary. Protein Sci 12:2150–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Alexander PA, He Y, Chen Y, Orban J, Bryan PN (2007) The design and characterization of two proteins with 88% sequence identity but different structure and function. Proc Natl Acad Sci USA 104:11963–11968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. He Y, Chen Y, Alexander P, Bryan PN, Orban J (2008) NMR structures of two designed proteins with high sequence identity but different fold and function. Proc Natl Acad Sci USA 105:14412–14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Alexander PA, He Y, Chen Y, Orban J, Bryan PN (2009) A minimal sequence code for switching protein structure and function. Proc Natl Acad Sci USA 106:21149–21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. He Y, Chen Y, Alexander PA, Bryan PN, Orban J (2012) Mutational tipping points for switching protein folds and functions. Structure 20:283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Porter LL, He Y, Chen Y, Orban J, Bryan PN (2015) Subdomain interactions foster the design of two protein pairs with ~80% sequence identity but different folds. Biophys J 108:154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Newlove T, Konieczka JH, Cordes MH (2004) Secondary structure switching in Cro protein evolution. Structure 12:569–581. [DOI] [PubMed] [Google Scholar]
- 67. Meier S, Jensen PR, David CN, Chapman J, Holstein TW, Grzesiek S, Ozbek S (2007) Continuous molecular evolution of protein‐domain structure by single amino acid changes. Curr Biol 17:173–178. [DOI] [PubMed] [Google Scholar]
- 68. Roessler CG, Hall BM, Anderson WJ, Ingram WM, Roberts SA, Montfort WR, Cordes MH (2008) Transitive homology‐guided structural studies lead to discovery of Cro proteins with 40% sequence identity but different folds. Proc Natl Acad Sci USA 105:2343–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yadid I, Kirshenbaum N, Sharon M, Dym O, Tawfik D (2010) Metamorphic proteins mediate evolutionary transitions of structure. Proc Natl Acad Sci USA 107:7287–7292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Grishin NV (2001) Fold change in evolution of protein structures. J Struct Biol 134:167–185. [DOI] [PubMed] [Google Scholar]
- 71. Meyerguz L, Kleinberg J, Elber R (2007) The network of sequence flow between protein structures. Proc Natl Acad Sci USA 104:11627–11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cao B, Elber R (2010) Computational exploration of the network of sequence flow between protein structures. Proteins: Struct Funct Genet 78:985–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ruvinov S, Wang L, Ruan B, Almog O, Gilliland GL, Eisenstein E, Bryan PN (1997) Engineering the independent folding of the subtilisin BPN’ prodomain: analysis of two‐state folding versus protein stability. Biochemistry 36:10414–10421. [DOI] [PubMed] [Google Scholar]
- 74. Zhou P, Lugovskoy AA, McCarty JS, Li P, Wagner G (2001) Solution structure of DFF40 and DFF45 N‐terminal domain complex and mutual chaperone activity of DFF40 and DFF45. Proc Natl Acad Sci USA 98:6051–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Dames SA, Martinez‐Yamout M, De Guzman RN, Dyson HJ, Wright PE (2002) Structural basis for Hif‐1α/CBP recognition in the cellular hypoxic response. Proc Natl Acad Sci USA 99:5271–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wright PE, Dyson HJ (2009) Linking folding and binding. Curr Opin Struct Biol 19:31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Otieno S, Kriwacki R (2012) Probing the role of nascent helicity in p27 function as a cell cycle regulator. PLoS One 7:e47177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Borcherds W, Theillet FX, Katzer A, Finzel A, Mishall KM, Powell AT, Wu H, Manieri W, Dieterich C, Selenko P, Loewer A, Daughdrill GW (2014) Disorder and residual helicity alter p53‐Mdm2 binding affinity and signaling in cells. Nat Chem Biol 10:1000–1002. [DOI] [PubMed] [Google Scholar]
- 79. Arai M (2018) Unified understanding of folding and binding mechanisms of globular and intrinsically disordered proteins. Biophys Rev 10:163–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ruan B, Hoskins J, Bryan PN (1999) Rapid folding of calcium‐free subtilisin by a stabilized pro‐domain mutant. Biochemistry 38:8562–8571. [DOI] [PubMed] [Google Scholar]
- 81. Narayanan R, Ganesh OK, Edison AS, Hagen SJ (2008) Kinetics of folding and binding of an intrinsically disordered protein: the inhibitor of yeast aspartic proteinase YPrA. J Am Chem Soc 130:11477–11485. [DOI] [PubMed] [Google Scholar]
- 82. Kiefhaber T, Bachmann A, Jensen KS (2012) Dynamics and mechanisms of coupled protein folding and binding reactions. Curr Opin Struct Biol 22:21–29. [DOI] [PubMed] [Google Scholar]
- 83. Schneider R, Maurin D, Communie G, Kragelj J, Hansen DF, Ruigrok RW, Jensen MR, Blackledge M (2015) Visualizing the molecular recognition trajectory of an intrinsically disordered protein using multinuclear relaxation dispersion NMR. J Am Chem Soc 137:1220–1229. [DOI] [PubMed] [Google Scholar]
- 84. Wu H, Hu Z, Liu XQ (1998) Protein trans‐splicing by a split intein encoded in a split DnaE gene of Synechocystis sp. PCC6803. Proc Natl Acad Sci USA 95:9226–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Zheng Y, Wu Q, Wang C, Xu MQ, Liu Y (2012) Mutual synergistic protein folding in split intein. Biosci Rep 32:433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Marcotte EM, Tsechansky M (2009) Disorder, promiscuity, and toxic partnerships. Cell 138:16–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Babu MM, van der Lee R, de Groot NS, Gsponer J (2011) Intrinsically disordered proteins: regulation and disease. Curr Opin Struct Biol 21:432–440. [DOI] [PubMed] [Google Scholar]
- 88. Haynes C, Oldfield CJ, Ji F, Klitgord N, Cusick ME, Radivojac P, Uversky VN, Vidal M, Iakoucheva LM (2006) Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput Biol 2:e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Oldfield CJ, Meng J, Yang JY, Yang MQ, Uversky VN, Dunker AK (2008) Flexible nets: disorder and induced fit in the associations of p53 and 14‐3‐3 with their partners. BMC Genomics 9(Suppl 1):S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Hou LM, Shao HY, Zhang YB, Li H, Menon NK, Neuhaus EB, Brewer JM, Byeon IJL, Ray DG, Vitek MP, Iwashita T, Makula RA, Przybyla AB, Zagorski MG (2004) Solution NMR studies of the Aβ(1‐40) and Aβ(1‐42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J Am Chem Soc 126:1992–2005. [DOI] [PubMed] [Google Scholar]
- 91. Lazo ND, Grant MA, Condron MC, Rigby AC, Teplow DB (2005) On the nucleation of amyloid beta‐protein monomer folding. Protein Sci 14:1581–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Conway KA, Harper JD, Lansbury PT (1998) Accelerated in vitro fibril formation by a mutant α‐synuclein linked to early‐onset Parkinson disease. Nat Med 4:1318–1320. [DOI] [PubMed] [Google Scholar]
- 93. Eliezer D, Kutluay E, Bussell R Jr, Browne G (2001) Conformational properties of alpha‐synuclein in its free and lipid‐associated states. J Mol Biol 307:1061–1073. [DOI] [PubMed] [Google Scholar]
- 94. Coles M, Bicknell W, Watson AA, Fairlie DP, Craik DJ (1998) Solution structure of amyloid beta‐peptide(1‐40) in a water‐micelle environment. Is the membrane‐spanning domain where we think it is? Biochemistry 37:11064–11077. [DOI] [PubMed] [Google Scholar]
- 95. Shao H, Jao SC, Ma K, Zagorski MG (1999) Solution structures of micelle‐bound amyloid β‐(1‐40) and β‐(1‐42) peptides of Alzheimer's disease. J Mol Biol 285:755–773. [DOI] [PubMed] [Google Scholar]
- 96. Ulmer TS, Bax A, Cole NB, Nussbaum RL (2005) Structure and dynamics of micelle‐bound human alpha‐synuclein. J Biol Chem 280:9595–9603. [DOI] [PubMed] [Google Scholar]
- 97. Bodner CR, Dobson CM, Bax A (2009) Multiple tight phospholipid‐binding modes of α‐synuclein revealed by solution NMR spectroscopy. J Mol Biol 390:775–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Luhrs T, Ritter C, Adrian M, Riek‐Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R (2005) 3D structure of Alzheimer's amyloid‐β(1‐42) fibrils. Proc Natl Acad Sci USA 102:17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Paravastu AK, Leapman RD, Yau WM, Tycko R (2008) Molecular structural basis for polymorphism in Alzheimer's β‐amyloid fibrils. Proc Natl Acad Sci USA 105:18349–18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Masters CL, Selkoe DJ (2012) Biochemistry of amyloid β‐protein and amyloid deposits in Alzheimer disease. Cold Spring Harb Perspect Med 2:a006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Crespi GAN, Hermans SJ, Parker MW, Miles LA (2015) Molecular basis for mid‐region amyloid‐β capture by leading Alzheimer's disease immunotherapies. Sci Rep 5:9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Qiang W, Yau WM, Lu JX, Collinge J, Tycko R (2017) Structural variation in amyloid‐β fibrils from Alzheimer's disease clinical subtypes. Nature 541:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Vilar M, Chou HT, Luhrs T, Maji SK, Riek‐Loher D, Verel R, Manning G, Stahlberg H, Riek R (2008) The fold of α‐synuclein fibrils. Proc Natl Acad Sci USA 105:8637–8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Bousset L, Pieri L, Ruiz‐Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Bockmann A, Meier BH, Melki R (2013) Structural and functional characterization of two alpha‐synuclein strains. Nat Commun 4:2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, Wan W, Stubbs G, Schwieters CD, Lee VM, George JM, Rienstra CM (2016) Solid‐state NMR structure of a pathogenic fibril of full‐length human α‐synuclein. Nat Struct Mol Biol 23:409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Sibille N, Bernado P (2012) Structural characterization of intrinsically disordered proteins by the combined use of NMR and SAXS. Biochem Soc Trans 40:955–962. [DOI] [PubMed] [Google Scholar]
- 107. Nath A, Sammalkorpi M, DeWitt DC, Trexler AJ, Elbaum‐Garfinkle S, O'Hern CS, Rhoades E (2012) The conformational ensembles of α‐synuclein and tau: combining single‐molecule FRET and simulations. Biophys J 103:1940–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mazouchi A, Zhang Z, Bahram A, Gomes GN, Lin H, Song J, Chan HS, Forman‐Kay JD, Gradinaru CC (2016) Conformations of a metastable SH3 domain characterized by smFRET and an excluded‐volume polymer model. Biophys J 110:1510–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Aznauryan M, Degado L, Soranno A, Nettels D, Huang JR, Labhardt AM, Grzesiek S, Schuler B (2016) Comprehensive structural and dynamical view of an unfolded protein from the combination of single‐molecule FRET, NMR, and SAXS. Proc Natl Acad Sci USA 113:E5389–E5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. He Y, Chen Y, Mooney SM, Rajagopalan K, Bhargava A, Sacho E, Weninger K, Bryan PN, Kulkarni P, Orban J (2015) Phosphorylation‐induced conformational ensemble switching in an intrinsically disorder cancer/testis antigen. J Biol Chem 290:25090–25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kulkarni P, Jolly MK, Jia D, Mooney SM, Bhargava A, Kagohara LT, Chen Y, Hao P, He Y, Veltri RW, Grishaev A, Weninger K, Levine H, Orban J (2017) Phosphorylation‐induced conformational dynamics in an intrinsically disordered protein and potential role in phenotypic heterogeneity. Proc Natl Acad Sci USA 114:E2644–E2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Lin X, Roy S, Jolly MK, Bocci F, Schafer NP, Tsai MY, Chen Y, He Y, Grishaev A, Weninger K, Orban J, Kulkarni P, Rangarajan G, Levine H, Onuchic JN (2018) PAGE4 and conformational switching: insights from molecular dynamics simulations and implications for prostate cancer. J Mol Biol 430:2422–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Pineda‐Lucena A, Ho CS, Mao DY, Sheng Y, Laister RC, Muhandiram R, Lu Y, Seet BT, Katz S, Szyperski T, Penn LZ, Arrowsmith CH (2005) A structure‐based model of the c‐Myc/Bin1 protein interaction shows alternative splicing of Bin1 and c‐Myc phosphorylation are key binding determinants. J Mol Biol 351:182–194. [DOI] [PubMed] [Google Scholar]
- 114. Andresen C, Helander S, Lemak A, Fares C, Csizmok V, Carlsson J, Penn LZ, Forman‐Kay JD, Arrowsmith CH, Lundstrom P, Sunnerhagen M (2012) Transient structure and dynamics in the disordered c‐Myc transactivation domain affect Bin1 binding. Nucleic Acids Res 40:6353–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Motlagh HN, Wrabl JO, Li J, Hilser VJ (2014) The ensemble nature of allostery. Nature 508:331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Mukrasch MD, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, Mandelkow E, Zweckstetter M (2009) Structural polymorphism of 441‐residue Tau at single residue resolution. PLoS Biol 7:e1000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Li J, White JT, Saavedra H, Wrabl JO, Motlagh HN, Liu K, Sowers J, Schroer TA, Thompson EB, Hilser VJ (2017) Genetically tunable frustration controls allostery in an intrinsically disordered transcription factor. Elife 6:e30688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Fuxreiter M, Tompa P (2012) Fuzzy complexes: a more stochastic view of protein function. Adv Exp Med Biol 725:1–14. [DOI] [PubMed] [Google Scholar]
- 119. Sigalov AB, Kim WM, Saline M, Stern LJ (2008) The intrinsically disordered cytoplasmic domain of the T cell receptor zeta chain binds to the nef protein of simian immunodeficiency virus without a disorder‐to‐order transition. Biochemistry 47:12942–12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Chakrabortee S, Meersman F, Kaminski Schierle GS, Bertoncini CW, McGee B, Kaminski CF, Tunnacliffe A (2010) Catalytic and chaperone‐like functions in an intrinsically disordered protein associated with desiccation tolerance. Proc Natl Acad Sci USA 107:16084–16089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Bozoky Z, Krzeminski M, Muhandiram R, Birtley JR, Al‐Zahrani A, Thomas PJ, Frizzell RA, Ford RC, Forman‐Kay JD (2013) Regulatory R region of the CFTR chloride channel is a dynamic integrator of phosphor‐dependent intra‐ and intermolecular interactions. Proc Natl Acad Sci USA 110:E4427–E4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Mahmoudabadi G, Rajagopalan K, Getzenberg RH, Hannenhalli S, Rangarajan G, Kulkarni P (2013) Intrinsically disordered proteins and conformational noise: implications in cancer. Cell Cycle 12:26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Neems D, Kosak ST (2010) Turning down the volume on transcriptional noise. Nat Cell Biol 12:929–931. [DOI] [PubMed] [Google Scholar]
- 124. Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676. [DOI] [PubMed] [Google Scholar]
- 125. Chakrabortee S, Byers JS, Jones S, Garcia DM, Bhullar B, Chang A, She R, Lee L, Fremin B, Lindquist S, Jarosz DF (2016) Intrinsically disordered proteins drive emergence and inheritance of biological traits. Cell 167:369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]