

Abstract
Background
One recent study reports cancer driver mutations in deep endometriosis, but its biological/clinical significance remains unclear. Since the natural history of endometriosis is essentially gradual progression toward fibrosis, it is thus hypothesized that the six driver genes reported to be mutated in endometriosis (the RP set) may play important roles in fibrogenesis but not necessarily malignant transformation.
Methods
Extensive PubMed search to see whether RP and another set of driver genes not yet reported (NR) to be mutated in endometriosis have any roles in fibrogenesis. All studies reporting on the role of fibrogenesis of the genes in both RP and NR sets were retrieved and evaluated in this review.
Results
All six RP genes were involved in various aspects of fibrogenesis as compared with only three NR genes. These nine genes can be anchored in networks linking with their upstream and downstream genes that are known to be aberrantly expressed in endometriosis, piecing together seemingly unrelated findings.
Conclusions
Given that somatic driver mutations can and do occur frequently in physiologically normal tissues, it is argued that these mutations in endometriosis are not necessarily synonymous with malignancy or premalignancy, but the result of enormous pressure for fibrogenesis.
Keywords: cancer driver mutation, endometriosis, fibrogenesis, natural history, repeated tissue injury and repair
1. INTRODUCTION
Endometriosis, defined to be the deposition and growth of endometrial‐like tissues outside of uterine cavity, is a benign and a major contributor to pelvic pain and subfertility affecting 6%‐10% of women of reproductive age. Despite extensive research, our knowledge on its etiology, pathogenesis and pathophysiology is still fragmentary. Consequently, its effective treatment still remains a challenge.1 So far the quest for novel nonhormonal therapeutics has not been successful,2 and there is no single biomarker that has unequivocally been shown to be clinically useful in diagnosing endometriosis.3, 4, 5
Given its high prevalence, its negative impact on quality of life in afflicted women,6, 7 and its heavy socioeconomic burden,8, 9, 10 there has been an ever burgeoning interest in finding its pathogenesis, pathophysiology, and its optimal management, as evidenced by the exponential growth in the number of articles on endometriosis.2 It is well accepted that endometriosis is first and foremost an estrogen‐dependent disease, characterized by the estrogen‐dependent growth and maintenance of ectopic endometrium and the increased local production of estrogens.11 It is also considered as an inflammatory condition, featuring increased production of proinflammatory cytokines and chemokines.11 Recent evidence is accumulating that it is also a procoagulant disease.12, 13
It has been well‐recognized that endometriosis sometimes behaves like a tumor14 and that it exhibits many features reminiscent of malignancy, such as invasion,15 neoangiogenesis,16 and recurrence.17 As with most neoplasms, which are monoclonal in origin,18, 19 each focus of endometriotic lesions is also monoclonal.20, 21 Similar to cancers,22 endometriosis also exhibits various epigenetic aberrations.23 These similarities prompted numerous studies in search of germline mutations or polymorphisms that predispose women to endometriosis or of lesion‐bearing mutations or genomic aberrations that can facilitate their malignant potential. Cytogenetic studies demonstrate that there are aberrant somatic genetic alterations, such as loss of heterozygosity (LOH),24 chromosome aneuploidy,25, 26, 27 and copy number changes,28, 29, 30, 31 which are also hallmarks of cancer,22, 32 even though earlier such attempts found nothing.33, 34 The endeavor to search for genetic predisposing variants all started in the 1990s,35, 36 and took a full swing in the era of genome‐wide association studies (GWASs) when high‐throughput genotyping methods became available and more affordable.37, 38 With the demand for larger sample sizes with detailed phenotypic and clinical information and thus more resources, the GWAS advocates promise to provide new insights into disease risk, classification, and comorbidity.39
Although endometriotic lesions can be found throughout the abdominal‐pelvic peritoneum and visceral organs, there are three major subtypes of endometriosis based primarily on their anatomical locations: ovarian endometrioma (OMA) which is reported to be the most common,40, 41 superficial peritoneal endometriosis (SPE), and deep endometriosis (DE).42 While all subtypes of endometriosis are benign, patients with OMA are well‐documented to have elevated risk of developing two particular histotypes of ovarian carcinomas—the endometrioid and the clear cell carcinoma,43, 44 although the magnitude of risk is rather moderate.45 Given the neoplastic potential of OMA, a slew of studies have been published on the expression and genomic alteration of ARID1A in endometriotic lesions46, 47, 48, 49 following the heels of the report that the gene is frequently mutated in endometriosis‐associated ovarian carcinomas.50
In contrast, extraovarian DE lesions have not been reported to transform into malignancy. Consequently, it came as a complete surprise when a recent high‐profile study reports that the majority (79%) of DE lesions harbor somatic mutations and a sizeable portion of them (26%) contain known cancer driver mutations at ARID1A, PIK3CA, KRAS, or PPP2R1A in the epithelial component.51
In contrast to those genomic alterations termed “passenger” mutations,52 “driver” mutations are implicated in pathways that are critical to the propensity of tumor cells for growth, survival, and metastasis.53 As such, driver mutations are supposedly rare in benign conditions such as endometriosis, but nonetheless are present in premalignancy and most frequent in metastatic cancer or those with a metastatic potential.53 While the authors of the driver mutation article were cautious by not sounding any alarm, the heightened vigilance, though guarded and veiled, expressed in the article is nonetheless unmistakable: “cancer‐associated mutations” of “driver genes” occur in DE, and “should a more complete genomic or epigenomic analysis be applied, additional drive mutations may be uncovered”.51
Granted, the paucity of report that DE lesions can be transformed into malignancy may be simply attributable to the rarity or to the lack of attention to this issue. Indeed, other fibrotic diseases, such as cirrhosis, have been reported to have increased potential for malignant transformation.54 Since the identification of tumor‐specific mutations in endometriosis has enormous potential to transform cancer diagnostics, monitoring, and screening if validated, there is a pressing need to evaluate the significance of the reported cancer driver mutations in endometriosis.
Similar to Darwinian evolution, all cells in endometriotic lesions are under selection pressure. Cell clones with driver mutations must have some distinctively selective advantages over those without. As one single most powerful driving force in shaping the natural history of lesions is to progress to fibrosis through ReTIAR, it can be hypothesized that these driver mutations in endometriosis may have more to do with fibrogenesis than from tumorigenesis.
In this article, after providing a brief overview on the natural history of endometriotic lesions, I shall provide an overview on the driver mutations in endometriosis and try to address the following questions from the vista of lesional natural history: What kind of role, if any, do these genes play in fibrogenesis? Do endometriotic lesions harboring these driver mutations truly drive neoplastic transformation? Do patients with DE have an increased risk of developing into cancer? Why these mutations are seen mostly in endometriotic epithelial cells as opposed to the stromal counterpart? What clinical implications, if any, does the finding of drive mutations in lesions have? Can driver mutations in endometriotic lesions reported thus far tell us anything about or shed new light into the pathophysiology of endometriosis?
As the major focus of this review is strictly on somatic cancer driver mutations in endometriotic lesions, I shall not touch upon germline mutations, genetic polymorphisms, genomic alterations, copy number changes, or passenger mutations. The driver genes reviewed here are comprised of two sets: the RP set, which includes TP53, PTEN, ARID1A, PIK3CA, KRAS, and PPP2R1A that have been reported to be mutated in endometriosis, and the NR set, which includes ALK, BRAF, CDKN2A, FGFR3, GNAQ, NF1, NF2, NOTCH1, and NRAS, that have not been reported to be mutated in endometriosis. All genes in the NR set were listed in a recent review article on driver mutations in benign diseases,53 and they were chosen mainly as a contrast without any prejudice. If the hypothesis is true, then we should expect to see the higher proportion of genes in the RP set than that from the NR set to be documented to play fibrogenic roles, or vice versa. All these genes and their related proteins, along with their upstream regulators and downstream target genes, will be reviewed for their possible roles in endometriosis and fibrogenesis.
2. METHODS
The PubMed database was searched for all original and review articles published in English until July 18, 2017. Search terms included ‘mutation and endometriosis’, ‘genomic alteration and endometriosis’, ‘aneuploidy and endometriosis’, ‘p53 and endometriosis’, ‘PTEN and endometriosis’, ‘ARID1A and endometriosis’, ‘PIK3CA and endometriosis’, ‘KRAS and endometriosis’, ‘protein phosphatase 2A and endometriosis’, ‘PI3K and endometriosis’, ‘anaplastic lymphoma kinase and endometriosis’, ‘BRAF and endometriosis’, ‘CDKN2A and endometriosis’, ‘p16 and endometriosis’, ‘FGFR3 and endometriosis’, ‘GNAQ and endometriosis’, ‘PTEN and endometriosis’, ‘NF1 and endometriosis’, ‘NF2 and endometriosis’, ‘NOTCH1 and endometriosis’, ‘NRAS and endometriosis’, ‘p53 and fibrosis’, ‘PTEN and fibrosis’, ‘ARID1A and fibrosis’, ‘PIK3CA and fibrosis’, ‘KRAS and fibrosis’, ‘PPP2R1A and fibrosis’, ‘protein phosphatase 2A and fibrosis’, ‘anaplastic lymphoma kinase and fibrosis’, ‘BRAF and fibrosis’, ‘CDKN2A and fibrosis’, ‘p16 and fibrosis’, ‘FGFR3 and fibrosis’, ‘GNAQ and fibrosis’, ‘NF1 and fibrosis’, ‘NF2 and fibrosis’, ‘NOTCH1 and fibrosis’, ‘NRAS and fibrosis’, and ‘oxidative stress and endometriosis’. The resultantly retrieved articles were reviewed and manually curated, and the gene/protein was considered to be of relevance and included when there was a least one article presented any mechanistic link between the gene/protein of interest and fibrosis.
Fisher's exact test was used to evaluate the statistical significance when comparing the proportions between two groups. Wilcoxon's test was used when comparing the distributions of continuous variables between two groups. P values of <0.05 were considered statistically significant. All computations were made with R 3.4.1.55
3. A PRIMER ON THE NATURAL HISTORY OF ENDOMETRIOSIS
While the pathogenesis of endometriosis is still unclear, recent research has provided sufficient details of the natural history of endometriotic lesions. One single defining hallmark of endometriotic lesions is their cyclic bleeding and subsequent tissue repair, just like the eutopic endometrium.56 Because of bleeding—an indication for tissue injury, platelets are involved and indeed have recently been shown to play important roles in the development of endometriosis.57 In particular, platelet‐derived TGF‐β1 drives smooth muscle metaplasia (SMM) and fibrosis through the induction of epithelial‐mesenchymal transition (EMT) and fibroblast‐to‐myofibroblast transdifferentiation (FMT) in endometriotic lesions.13 Activated platelets impair natural killer cell reactivity and function in endometriosis through multiple mechanisms.58, 59 They also secrete many bioactive factors, including thromboxane A2 (TXA2), which may also act as a neutrophin, leading to hyperinnervation within or surround endometriotic lesions.60 Moreover, platelets may also induce epigenetic changes, facilitating the gradual but progressive development of endometriosis, leading ultimately to tissue fibrosis.61 Both animal and human data lend support for the notion that endometriotic lesions are fundamentally wounds undergoing repeated tissue injury and repair (ReTIAR).12, 62, 63 Similar processes also occur in adenomyosis, due to the shared commonality of cyclic bleeding.64, 65
Note that this natural history may not be necessarily equivalent to the natural history of endometriosis at the organismic level. Women with identical lesions at the same locations may not display identical symptoms or the same severity. This is primarily due to the difference in genetic background and life history, neuronal wiring and coping strategy since endometriosis‐associated pelvic pain involves central sensitization66, 67 and is associated with altered brain chemistry and function in the pain matrix.68, 69 Nonetheless, the lesional natural history should constitute the basis for the natural history of endometriosis at the organismic level.
It has long been regarded that OMA, SPE, and DE are three different disease entities and, as such, may have different pathogenesis and pathophysiology. Indeed, gene profiling studies depict different transcriptional signatures in OMA and non‐OMA lesions.70 However, our histological and immunohistochemistry analyses indicate that both OMA and DE share essentially the same features of ReTIAR, and the two conditions differ only by the extent or thoroughness of EMT, FMT, SMM, and the extent of fibrosis, along with different epigenetic aberrations.71 It turned out that the key factors responsible for the differences between these two subtypes of endometriosis lie in the difference in the microenvironment that OMA and DE lesions are situated: the proximity to sensory nerve plexuses in DE but not in OMA. Sensory nerve‐secreted substance P and calcitonin gene‐related peptide (CGRP) accelerate the fibrogenic progression, making DE lesions more fibrotic than OMA lesions72 (Yan et al, unpublished data).
Based on the above discussion, the natural history of endometriotic lesions can be depicted in Figure 1, which also indicates that fibrogenesis of endometriotic lesions also induces cancer driver mutations.
Figure 1.
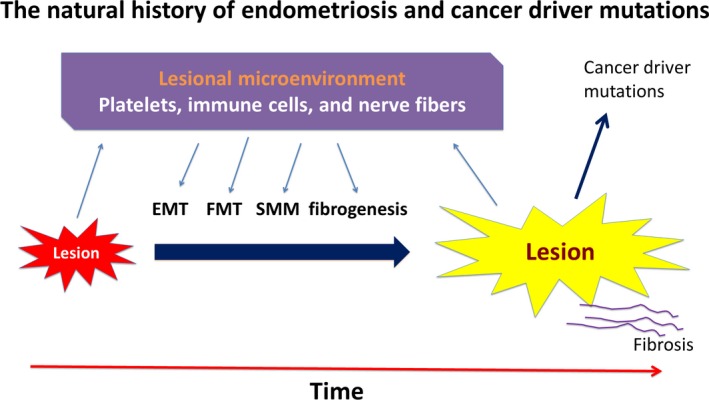
The natural history of endometriosis and cancer driver mutations (adapted from Ref. 401). This diagram sketches, in broad strokes, the progression of endometriotic lesions, which interact with various players in their microenvironment, through epithelial‐mesenchymal transition (EMT), fibroblast‐to‐myofibroblast transdifferentiation (FMT), and smooth muscle metaplasia (SMM), leading ultimately to fibrosis. In addition, it depicts cancer driver mutations are likely induced by the pressure of fibrogenesis of endometriosis. EMT, epithelial‐mesenchymal transition; FMT, fibroblast‐to‐myofibroblast transdifferentiation; SMM, smooth muscle metaplasia
4. WHY FIBROGENESIS?
As of writing, there are 24 328 published articles on endometriosis (Accessed on September 25, 2017) that are indexed in PubMed. Hundreds, if not thousands, of genes and proteins have been reported to be aberrantly expressed in endometriosis, and many of them have been shown, by painstaking experimentations and to various degrees, to be involved in many aspects of endometriosis development. Despite these discoveries, there is a real issue as how these seemingly unrelated results can be pieced together to have a whole picture as how endometriotic lesions progress and cause symptoms.
Granted, many, if not all, genes are involved in multiple cellular processes. In endometriosis, for example, tissue factor (TF) was first reported in the context of angiogenesis,73 but now with the elucidation of the role of platelets in particular and coagulation in general in the development of endometriosis,57, 74 there is reason to believe that the major role of TF in endometriosis is the activation of the coagulation, and its role in angiogenesis may well be its sideline job. This can be understood once we understand that TF plays a vital role in the activation of the coagulation pathways.
Through the prism of ReTAIR, it is easy to see that fibrogenesis starts after lesions are initially formed and becomes an integral part of the lesional development, as well as an end result. Thus, fibrogenesis should be a major theme throughout the progression of endometriotic lesions, and cancer driver mutations are likely to be variations on the theme.
By definition, fibrogenesis is a pathological process characterized by excessive production and deposition of extracellular matrix (ECM) products in response to uncontrolled tissue repair.75 Despite a wide diversity in different fibrotic diseases, all of them share several commonalities, ie, parenchyma injury, accumulation of fibrillar ECM, accumulation of fibroblasts, rarefication of the microvasculature, and a mononuclear infiltrate.75 In endometriotic lesions, similar features can be seen: injury to glandular epithelium due to cyclic bleeding, accumulation (due to EMT and perhaps also proliferation) and activation of fibroblasts (due to FMT), reduced vascularity in DE,71 and infiltration of macrophages and dendritic cells.76, 77
In addition, endometriotic lesions are very similar to organs that undergo fibrogenesis. In fact, they behave very much like an organ, having all the physiological processes that a normal organ does, such as angiogenesis, lymphoangenesis, and neurogenesis,78 which provide means for lesions to access to nutrients and oxygen, waste disposal, and communication with the outside just like an organ.
Further thinking along this line would reveal that one can weave many, if not all, known results together to come up with a more or less complete tapestry on the physiopathology of endometriosis. Indeed, without a framework, many of us would be groping, often seeing a leaf very clearly but having no idea as what the forest likes like. Once we have an even skeletal framework, we can piece together all the rest of puzzles more quickly. More importantly, it can help us understand why there are such aberrations and guide us to discover new things.
5. MUTATIONS OF CANCER DRIVER GENES IN ENDOMETRIOSIS AND THEIR POSSIBLE ROLES IN FIBROGENESIS
The numbers of articles retrieved from the search of PubMed using different search phrases are listed in Table 1. The average number of papers that were found to be more or less related with fibrosis for the genes in the RP is 232.2 (±259.4), which is 7.2 folds higher than that for the genes in the NR set (32.3 ± 34.5). This difference is nearly statistically significant (P = 0.065, by Wilcoxon's test), indicating that the genes/proteins in the RP set had an enrichment of articles on their link with fibrosis. Consistent with the division of the RP and NR sets, the genes/proteins in the RP set had significantly more articles that are related with endometriosis (58.8 ± 44.4 vs 6.1 ± 8.2; P = 0.006).
Table 1.
Number of articles retrieved from PubMed on the cancer driver genes and their roles in fibrogenesis and endometriosis (accessed on July 18, 2017)
| Gene/protein name | Possible roles in fibrosis | Aberration in endometriosis |
|---|---|---|
| p53 | 619 | 128 |
| PTEN | 165 | 86 |
| ARID1A | 2 | 66 |
| PI3K | 493 | 37 |
| KRAS | 59 | 33 |
| Protein phosphatase 2A | 55 | 3 |
| Anaplastic lymphoma kinase | 16 | 0 |
| BRAF | 38 | 13 |
| CDKN2A | 43 | 14 |
| p16 | 110 | 24 |
| FGFR3 | 9 | 1 |
| GNAQ | 1 | 0 |
| NF1 | 30 | 2 |
| NF2 | 7 | 0 |
| NOTCH1 | 67 | 6 |
| NRAS | 2 | 1 |
5.1. Mutations in ARID1A, PIK3CA, KRAS, and PPP2R1A in endometriosis and their roles in fibrogenesis
So far six cancer driver genes, TP53, PTEN, ARID1A, PIK3CA, KRAS, and PPP2R1A have ever been reported to be mutated in endometriosis. In particular, TP53, PTEN, and ARID1A mutations are inactivating, and that of PIK3CA, KRAS, and PPP2R1A are of activating in nature.51 Before attempting to understand why and how these mutations occur, it may be helpful to evaluate their possible roles in fibrogenesis, which is the end result of the natural history of endometriosis.
5.1.1. TP53
Tumor protein p53 (TP53), also known as p53, is a well‐recognized tumor suppressor gene (TSG) that regulates cellular proliferation and apoptosis.79 It is hailed as “the guardian of the genome” due to its role in conserving stability by preventing genome mutation.80 It is frequently mutated in various cancers,81 especially in serous endometrial carcinomas (89% of patients) and serous ovarian carcinomas (95%).82
Since the first report of TP53 mutation in an endometriotic lesion (out of 14) adjacent to ovarian carcinoma in 1998,83 TP53 loss was soon reported,84 followed by a report of LOH.85 Later studies reported somewhat mixed results. Several studies reported no LOH at the TP53 locus in OMA (0/16),86 no hot spot mutation at TP53 in endometriosis (0/23),87 no mutation but only focal expression of TP53,88 and no mutation in endometriotic lesions associated with ovarian cancer (0/12).89 However, since the mutation rate is presumably low, a large sample size is typically needed to detect it. For example, even if the mutation rate is 10%, the probability of observing none among 16 and 23 samples would be 0.19 and 0.09, respectively, certainly not a small‐probability event. Therefore, the negative reports that evaluated just small or moderate samples of endometriotic lesions should be viewed as suggestive, in need for independent confirmation. Assuming a mutation rate of 5% (or 10%), one needs to examine at least 59 (or 29) samples in order to have the 95% probability of observing at least one mutation in the samples. A mutation rate lower than 5% would require even more samples to evaluate.
A recent study demonstrated that conditional deletion of p53 coupled with the activating K‐ras mutation led to development of endometrioid ovarian carcinosarcomas.90 In addition, the combined mutations resulted in simple endometrioid glandular morphology and peritoneal endometriotic lesions as early as 4 weeks after AdCre injection through the ovarian,90 indicating that TP53 loss, in conjunction with KRAS activation, may transform OMA into malignancy.
The published results on TP53 expression in endometriotic lesions are also conflicting. TP53 overexpression in the epithelial component of lesions,88, 91 in atypical endometriosis,92 and in OMA 93 has been reported, but this is at direct odds with a later gene profiling study that reported its downregulation.94 Another study found the TP53 expression to be the highest in OMA, followed by colorectal endometriosis (presumably DE) and then SPE—all in the epithelial, but not stromal, component.95 One study found no difference in TP53 expression between ectopic and control endometrium,96 the others found no TP53 expression in endometriosis,97 OMA,98, 99 and DE.100 One recent study reported downregulation of TP53 in OMA, which is concomitant with increased expression of genes involved in autophagy and elevated protein expression of heme oxygenase‐1 (HO‐1), a sign of oxidative stress.101
Based on the findings published so far and reviewed above, there are reasons to believe that TP53 is likely to be downregulated or even silenced in endometriotic lesions. As such, especially considering the central role of TP53 in tissue repair102 and the risk of malignant transformation of OMA, the possibility of inactivating TP53 mutation or epigenetic silencing in endometriosis cannot be ruled out. This leaves out the question as what biological consequence, if any, it would entail if TP53 is inactive.
Depending on cell/tissue type and disease models, TP53 has been reported to have different roles in the pathogenesis of fibrosis. The deletion of p53 in proximal tubule cells is reported to prevent interstitial fibrogenesis after acute kidney injury (AKI) in mice.103 However, other studies reported that p53 inhibition/knockout promoted renal fibrosis.104, 105
One important mechanism that TP53 loss is responsible for fibrogenesis is due to its role in regulating cellular senescence.106 In responding to DNA damage, oncogene activation, hypoxia, and telomere shortening, TP53 becomes transcriptionally activated, leading to cell‐cycle arrest, DNA repair, and apoptosis.107 Yet cellular senescence is simply a stable form of cell‐cycle arrest that limits the cellular proliferative potential.108 Indeed, activation of endogenous p53 in liver cancer induced senescence and tumor regression in mouse.109 In normal wounds, activated fibroblasts initially proliferate in response to tissue damage and secrete extracellular matrix (ECM) products, then senesce, and are eventually removed from the wounding sites. In pathological conditions such as chronic tissue injury, however, repeated tissue injury, followed by fibroblast proliferation, results in the production of senescent cells outpacing their removal, leading to persistent inflammation and progressive fibrosis.110 TP53 deletion in fibroblasts is found to diminish senescence and to increase TGF‐β1 expression, leading to increased activated fibroblasts, more ECM deposition, diminished immune surveillance, and fibrosis.110 In a nutshell, the senescence program curtails the fibrogenic response to tissue damage, but TP53 loss impairs this program, exacerbating the fibrogenic response.
Consistent with the potential role of TP53 in fibrogenesis in endometriosis, endometriotic cells are reported to have longer telomeres and higher telomerase expression.111, 112, 113 In addition, miR‐125b is found to be critical for FMT and fibrosis, and to promote fibroblast proliferation through suppression of TP53.114 Remarkably, miR‐125b upregulation has been reported in endometriosis.115, 116 Moreover, PPARγ is expressed in normal endometrium117 but is downregulated in ectopic endometrium.118, 119 This appears to mirror what has been reported in liver fibrogenesis: PPARγ is highly expressed in quiescent hepatic stellate cells (HSCs, ie, fibroblasts) but downregulated in activated HSCs.120, 121 PPARγ promotes cellular senescence in fibroblasts,122 and PPARγ agonists induce apoptosis and cell‐cycle arrest in cancer cells.123
In endometriosis, the PPARγ staining levels correlated negatively with the extent of fibrosis,71 similar to the PPARγ inactivation that has been reported to be involved in fibrosis in various organs.124, 125, 126 In particular, MeCP2‐mediated enhancer of zeste homolog 2 (EZH2) activation, trimethylation of histone 3 lysine 27 (H3K27me3) and PPARγ suppression have been reported during myofibroblast activation,125 and in both OMA and DE lesions PPARγ suppression, EZH2 activation and increased H3K27me3 expression have been reported.71 In fact, EZH2 is found to induce EMT and thus fibrogenesis in endometriosis, and there are signs to indicate that platelet aggregation in lesions may be responsible for EZH2 activation.61 Importantly, EZH2 may mediate the repression of GSK‐3β and TP53 and promote the activation of the Wnt/β‐catenin signaling pathway.127 The profibrogenic role of the Wnt/β‐catenin signaling pathway in endometriosis has recently been reported.128, 129 EZH2 can also suppress Dkk1, a negative regulator of the Wnt/β‐catenin signaling, inducing fibrosis.130 Again, Dkk1 expression is reported to be reduced in endometriosis.131
TP53 is recently found to restrict the expression of de novo DNA methyltransferases DNMT3A and DNMT3B while upregulating TET1 and TET2 that promote demethylation.132 TP53 loss results in augmented overall DNA methylation and increased methylation landscape heterogeneity. Incidentally or not, DNMT3A and DNMT3B are both reported to be elevated in endometriotic lesions.133, 134 In addition, TET1, TET2, and TET3 expression is reported to be decreased.135
Taken together, there are reasons to believe that TP53 is downregulated or even inactivated in endometriotic stromal cells. This inactivation may be due to inactivating mutation, but could also be attributable to its hypermethylation136 or transcriptional suppression by, say, miR‐125b overexpression. The loss of TP53 may retard the senescence of fibroblasts in endometriotic lesions, facilitating fibrogenesis. This assertion may be bolstered by an integrative analysis of gene expression database that identified TP53 as one of 26 transcription factors involved in the regulatory programs associated with differential gene expression in endometriosis.137
Note that inactivating TP53 mutation also has been reported in rheumatoid arthritis, which does not confer increased cancer risk.138, 139 This seems to indicate that TP53 mutations can and do occur in nonmalignant tissues which are not premalignant. Thus, inactivating TP53 mutation alone in lesions may not be equated with premalignancy.
5.1.2. PTEN
PTEN, or phosphatase and tensin homolog deleted on chromosome 10, is a powerful tumor suppressor through regulation of proliferation and survival and has been dubbed as “a new guardian of the genome”.140 It is the second most frequently mutated gene in human cancers after TP53, but the mutation spectrums of PTEN and TP53 are different.140 In addition, p53‐null mice are viable, develop normally and exhibit spontaneous tumors,141 but homozygous deletion of Pten is embryonically lethal.142 It inhibits PI3K/AKT signaling by converting PIP3 to PIP2.140 Of particular interest to endometriosis is its ability in maintaining genomic stability.143, 144, 145
In endometriosis, the first attempt to evaluate PTEN mutation yielded negative results,146 but the other study reported a mutation frequency of 21% (7/34) in OMA.147 One study found no mutation at PTEN (0/23),87 but the negative finding may be simply attributable to lack of adequate statistical power due to moderate sample size. Another recent study detected 17 mutations in 32 (53%) rASRM III/IV lesions.148 PTEN loss and LOH have been reported in endometriosis malignant transformation.89, 147, 149 Conditional deletion of Pten is found to induce endometriosis in mouse.150
Reduced PTEN expression has been reported in endometriosis.151, 152, 153 17β‐estradiol promotes cell proliferation through activating PI3K/AKT and MAPK/ERK signaling pathways via an NF‐κB/PTEN‐dependent pathway in endometriotic epithelial cells.152 IL‐8 also is reported to enhance proliferation, reduce apoptosis in endometrial stromal cells through the upregulation of survivin and Bcl‐2, inhibition of PTEN and activation of AKT.154 Consistently, AKT activation has been shown to promote the establishment of endometriosis.155 PTEN suppression inhibits the proliferation and angiogenesis, increases apoptosis and cell‐cycle arrest in endometriotic epithelial cells but forced PTEN expression reverses these changes.156
During normal tissue repair, excessive fibroblasts are removed by apoptosis, thus limiting fibrosis.157 Specifically, fibroblasts transdifferentiate into myofibroblasts in response to injury,158 as seen also in endometriosis.62 Myofibroblasts produce and then deposit type I collagen into the provisional wound matrix.159 They also contract the type I collagen matrix, and, in conjunction with reepithelialization, facilitate wound closure.157, 159 In response to collagen matrix contraction, fibroblasts incorporated into type I collagen matrices undergo apoptosis.159 The contraction of collagen matrix induces PTEN expression, resulting in reduced AKT activation and subsequent apoptosis.160 Conversely, PTEN inactivation augments AKT activity, suppresses the apoptosis of fibroblasts, and enhances their proliferation and invasiveness.160, 161, 162, 163 PTEN loss in renal injury also initiates SMAD3‐ and TP53‐dependent fibrotic response.164 Overexpression of PTEN reduces fibroblast activation, viability, caspase‐3 activity, cell‐cycle arrest in the G0/G1 and G2/M phases and suppression of PI3K/AKT and FAK/ERK signaling pathways.165 PTEN also regulates M2 macrophage polarization through activation of PI3K/AKT/STAT6, with its loss yielding more profibrotic M2 macrophages.166
There is a wealth of literature documenting the critical role of PTEN loss in fibrogenesis. In many fibrotic diseases, myofibroblasts have diminished PTEN expression161 but overexpression of PTEN or its reconstitution results in suppression of AKT and consequent fibrogenesis.167, 168, 169 In many fibrotic diseases, PTEN expression is reduced or simply absent.170, 171, 172, 173, 174, 175 Aberrant PTEN/AKT signaling inactivates FOXO3a, a proapoptotic factor, promoting fibrosis.176, 177 Several putative regulators of PTEN have been reported, including caveolin‐1,178, 179 DJ‐1,180 and miR‐21.181, 182, 183, 184, 185, 186, 187, 188 Remarkably, lower expression of caveolin‐1 has been reported in adenomyosis,189 and higher expression of DJ‐1153, 190 and miR‐21191, 192 has been reported in endometriosis as well. Inactivation of FOXO3a by SGK1 and ERβ also has been reported in endometriosis.193 Fibrosis caused by the PTEN loss has been shown to be critically dependent on CCN2/CTGF,194, 195 which is reported to be upregulated in endometriosis.62, 196, 197 Evidence also shows that one mechanism for PTEN loss or silencing is its DNMT1‐mediated promoter hypermethylation.198 Inhibition of EZH2, a histone methyltransferase catalyzing trimethylation of H3K27 and of H3K9 which serve as anchorage points for the recruitment of additional PRC2 proteins, has been shown to attenuate fibrosis through induction of PTEN expression.199 Interestingly, EZH2, along with its PRC2 partners EED and SUZ12, is found to induce EMT in endometriosis61 and its expression is elevated in endometriosis, especially in DE lesions that exhibit higher fibrotic content.71
In light of the above discussion, it can be concluded that as endometriotic lesions undergo ReTIAR and progress to fibrosis, PTEN expression is reduced or perhaps even silenced, leading to the activation of PI3K/AKT/mTOR and also FAK/ERK signaling pathways, and ultimately to fibrosis. The inactivating PTEN mutation may be simply due to the strong pressure of fibrogenesis.
5.1.3. ARID1A
ARID1A (the AT‐rich interactive domain 1A) encodes the protein BAF250a that participates in forming Switch/Sucrose nonfermentable (SWI/SNF) chromatin remodeling complexes, which are crucial for regulating temporal and spatial gene expression during development.200 In particular, the SWI/SNF subunit BAF250a has been reported to affect self‐renewal and differentiation in many tissues and embryonic stem cells.201, 202 As a TSG, ARID1A is frequently mutated in ovarian clear cell and endometrioid carcinomas as well as in uterine endometrioid carcinomas.50, 203, 204 One recent in vitro study reports that ARID1A knockdown is sufficient to initiate neoplastic transformation in conjunction with epigenetic reprogramming in nontumorigenic endometriotic cells.205
Complete absence of ARID1A staining has been reported in 15% (3/20) of OMA, 5% (1/22) of DE, but in none of SPE (0/16) lesions and eutopic endometrium samples (0/30).46 Partial loss of ARID1A staining (ie, in one tissue section some cells are stained positive while others are negative) has been reported to be in 36% (9/25) of rectovaginal DE samples.48 ARID1A gene expression levels in endometriosis are significantly lower than that of control endometrium, and oxidative stress downregulates ARID1A.206 Incidentally or not, loss of chromosome 1p36.12, where the ARID1A locus is located, also was reported in both ectopic and eutopic endometrium from a woman with SPE.31
Tissue regeneration or repair has been reported to resemble the embryonic developmental process since both processes undergo reorganization and rearrangement of tissue architecture concomitant with distinct transcriptional patterns.207 Thus, it comes with little surprise that the loss of ARID1A is reported to promote liver and ear hole wound regeneration but its forced expression impairs regeneration in mouse models.208 More specifically, ARID1A knockdown remodels chromatin through reduced H3K4me2 marks, disengaging the transcriptional access by C/EBPα, which enforces differentiation, and E2F4, which suppresses proliferation and regeneration through cell‐cycle reentry.208 It also reduces the expression of FOXA2,208 a key transcription factor that regulates cell differentiation and tissue regeneration,209 and HNF4A, a transcription factor. ARID1A expression is suppressed in regenerating tissues, and genetic deletion of ARID1A enhances tissue repair.208 Remarkably, ARID1A is suppressed after tissue injury and the enhancement of tissue repair by ARID1A loss is not tissue‐specific.208 ARID1A is reported to play a critical role in modulating epithelial proliferation in endometrium and to be essential for endometrial function during early pregnancy.210
As endometriotic lesions are fundamentally wounds undergoing ReTAIR, the loss of ARID1A following cyclic bleeding, especially in the epithelial component, may be more common than previously thought. Chronic inflammation211, 212 and prolonged transcriptional suppression213, 214 could lead to promoter hypermethylation and eventually to inactivating mutation at ARID1A when exposed constantly in a hostile microenvironment. Interestingly, LSD1 is reported to be overexpressed in endometriosis, concomitant with reduced H3K4me marks as well.215, 216 In addition, C/EBPα is reported to be epigenetically silenced in endometriotic epithelial cells, which can be reactivated by valproic acid, an HDAC inhibitor.217 Activation of C/EBPα induces apoptosis and activation of caspase‐3 and caspase‐7, and its suppression leads to downregulation of PPARγ, p53, Bax, caspase‐8, and caspase‐10, p16INK4a, p21Waf1/Cip1, CDK2, and CDK4.217 FOXA2 is found to be downregulated in endometriosis.137 Along with TP53, ER‐β, Smad3, β‐catenin, c‐Myc and others, ARID1A regulated FOXA2, HNF4A, and E2F4 are among the 26 transcription factors identified to be involved in the regulatory programs associated with differential gene expression in endometriosis.137
In light of the above discussion, there are reasons to believe that AIRD1A is under constant pressure for its suppression in endometriotic epithelial cells because of ReTIAR. Prolonged transcriptional suppression may lead to epigenetic suppression, which, in some cases, could further lead to its inactivating mutation. Epigenetic suppression through promoter hypermethylation is likely since both de novo and maintenance DNMTs are all upregulated in endometriosis.133
5.1.4. PIK3CA
The phosphatidylinositol‐4,5‐bisphosphate 3‐kinase (PI3K)/AKT signaling pathway is one of the most frequently mutated in human cancers.218PI3K/AKT pathway is the primary physiological target of PTEN.219 Somatic alterations in this pathway include mutation and/or amplification of the genes encoding the PI3K catalytic subunits p110α (PIK3CA) and p110β (PIK3CB), the PI3K regulatory subunit p85 (PIK3R1), the PI3K effectors AKT1, AKT2, and PDK1, and the loss of the lipid phosphatases PTEN and INPP4B.220 PIK3CA uses ATP to phosphorylate phosphatidylinositols and its mutations are the most common genetic alteration of this pathway, conferring enhanced growth and survival propensity.221 About 40% of ER‐positive breast cancers harbor activating PIK3CA mutations.218 Tumors carrying PIK3CA mutations may respond to PI3K/AKT/mTOR inhibitors.222
In endometriosis, PIK3CA mutation is reported in the context of its link with ovarian cancer 223, 224 but none was found among 23 patients with only endometriosis,87 possibly due to the lack of adequate statistical power. However, PI3K/AKT/mTOR signaling pathway has been strongly implicated in the development of endometriosis.190, 225, 226, 227, 228, 229 PDK1 overexpression also has been reported in endometriosis.230
Activating PIK3CA mutation has been implicated in several fibrotic diseases. It is reported to be found in 83% of radial scars with epithelial atypia231 and in 90% of patients with fibroadipose hyperplasia,232 a nonmalignant progressive segmental overgrowth of fibrous and adipose tissues.
In hepatocellular carcinoma, the accelerated tumorigenesis is reported to be attributable to increased injury and inflammation, unrestricted oxidative stress, fibrosis, and compensatory increase in hepatocyte proliferation secondary to PDGFRα/PIK3CA/AKT activation and c‐Myc overexpression.233 In lung epithelial cells, IL‐17A inhibits autophagy through activation of PIK3CA to interrupt the GSK‐3β‐mediated degradation of BCL2, which is be primarily responsible for the development and progression of IL‐17A‐induced pulmonary fibrosis.234
Given the above discussion, it seems that PIK3CA expression is likely to be upregulated in endometriosis, which is consistent with the activation of the PI3K signaling pathway. In addition, activating PIK3CA mutation in endometriosis is credible, and there is also a likelihood that PIK3CA upregulation might be attributable to hypomethylation.
5.1.5. KRAS
RAS proteins, which include H‐, K‐ and N‐RAS, the three closely related members with a molecular mass of ~21 kDa, are small GTPases with two conformational states, a guanosine diphosphate (GDP)‐bound “off” state and a guanosine triphosphate (GTP)‐bound “on” state.235 Exchange of GDP for GTP results in a conformational change, turning the molecular switch from the “off” to the “on” position. Ras proteins activate a wide array of downstream signaling pathways with a multitude of effector proteins, including Raf/ERK and PI3K/AKT.236 ERK1/2 have several substrates, including EGF and estrogen receptors (ERs).236 They function as intracellular switches in signal transduction cascades that regulate many biological functions including proliferation, apoptosis, and differentiation,237 which all play important roles in fibrogenesis.
Oncogenic KRAS is encoded by KRAS‐2 gene,235 is downstream of EGFR and an essential component of the EGFR signaling cascade.238 It is frequently mutated in many malignancies, such as colorectal cancer (~40%), lung cancer (~25%), and pancreatic cancer (~90%).239, 240 The activating mutation of KRAS isolates the EGFR pathway from the effect of EGFR2, rendering EGFR inhibitors ineffective.241 KRAS also can be activated by TGF‐β1,242 angiotensin II,243 EGF,244 endothelin‐1,245 PDGF,246 and thrombin.247 Importantly, these genes/molecules or their receptors (eg, angiotensin II receptors AT‐1 and AT‐2) are all reported to be overexpressed/elevated in endometriosis.74, 248, 249, 250, 251
KRAS mutation attracted much attention after the report that the activation of a Kras allele resulted in peritoneal endometriosis in mice.150 Since in this model the onset of endometriosis appeared to be quite late (~8 months after conditional induction of K‐ras),150 there is question as whether this mouse model of endometriosis truly recapitulates the human counterpart. Indeed, KRAS activating mutation is reported to be rare in presumably OMA,87 even though elevated KRAS expression in eutopic endometrium in women with endometriosis has been reported.252, 253, 254 The somewhat prolonged latency period in inducing endometriosis seems to suggest that the KRAS mutation alone may not be sufficient to induce endometriosis. Indeed, well over a decade has been passed since the report on the K‐ras induced endometriosis in mouse, but the model does not seem to gain any traction in the mainstream research, even though the transplantation of steroid‐manipulated, menstrual like endometrium from conditionally activated K‐ras (K‐rasG12V/+/Ah‐Cre+/+/ROSA26R‐LacZ+/+) mice into gonad‐intact immune‐competent wild‐type mice also induced endometriosis.255This mouse model of endometriosis also shows that the growth of lesions is ER‐dependent since estrogen antagonism suppresses the lesion growth.255 In addition, the lesions exhibit fibrosis as seen by marked collagen deposition.255
KRAS mutations have been identified in 12 (29%) of 42 endometriosis‐associated ovarian low‐grade endometrioid adenocarcinomas.256 Inactivating mutation has recently reported in DE lesions.51
ERK activation has been reported to be a necessary step in the induction of EMT257 which plays a critical role in fibrogenesis. While TGF‐β1 is well‐documented to induce EMT,258 the intracellular signaling responsible for this induction includes activation of ERK1/2,259, 260 p38 MAPK,257 and JNK.261 Again, ERK1/2, p38 MAPK, and JNK have all been reported to be involved in endometriosis.262, 263, 264, 265, 266, 267
Activated KRAS can cooperate with Snail to promote fibrosis.268 One important mechanism underlying this promotion is through the induction of Stem‐Cell Factor (SCF) and the enhancement of mast cell infiltration.268 SCF neutralization can block Snail‐induced migration of mast cells.268 In addition, MT1‐MMP can cooperate with KRAS to promote pancreatic fibrosis through enhanced TGF‐β1 signaling.269 Importantly, SCF levels are found to be elevated in the peritoneal fluid from women with endometriosis270 and the expression levels of c‐Kit, the receptor for SCF, are reported to be elevated in ectopic endometrium.271 And increased mast cell infiltration in lesions also has been well‐documented,272, 273 especially in DE.274 MT1‐MMP expression has been consistently to be documented to be elevated in ectopic endometrium275 and also reported to be elevated in peritoneal fluid276 and eutopic endometrium277 from women with endometriosis.
Thus, there is reason to believe that KRAS mutation in endometriosis is possible but may be rare. It is very likely to be activated in endometriosis.
5.1.6. PPP2R1A
The phosphorylation/dephosphorylation of proteins is controlled by protein kinases and protein phosphatases (PP), which plays a critical role in regulating a variety of cellular processes. PPP2R1A encodes the enzyme serine/threonine‐protein phosphatase 2A 65 kDa regulatory subunit A alpha isoform, and is implicated in the negative control of cell growth and division.278 Its mutation is reported to be common in the serous type of endometrial cancer.279 All subunits of PP2A can and have been be mutated in cancer, but PPP2R1A, has the highest mutation rate.280 Activating PPP2R1A mutations have been reported in 7.1% of patients with ovarian clear cell carcinoma in 2010.203 Since then, there has been a moderate interest in PPP2R1A mutations in endometriosis‐associated ovarian cancer but its mutation is found to be rare.281, 282
There are scanty reports on the role of PPP2R1A in fibrogenesis. However, one study reports that forced miR‐16 expression suppressed the activation of HSCs, which is the critical event in liver fibrogenesis, likely through negative cell‐cycle regulation, enhanced fibrolysis, and increased apoptosis.283 Subsequent integrative analysis identified the regulatory network of miR‐16 and found that PPP2R1A is a target of miR‐16 and that PP2R1A constitutes a key regulatory network node that mediates the miR‐16 action in proliferation, ECM deposition, and survival.283 Interestingly, treatment of endometriotic cells with peritoneal fluid from women with endometriosis reduced the expression of miR‐16.284, 285 Thus, there is a possibility that miR‐16 downregulation may induce PPP2R1A activation, facilitating the progression of fibrogenesis in endometriosis.
Note that PPP2R1A is part of the protein phosphatase 2 (PP2 or PP2A), an enzyme coded by the PPP2CA gene.286 PP2A is a major cellular phosphatase that regulates many protein targets. Its substrate includes cellular proteins, viral proteins, and protein kinases. It functions as a heterotrimeric complex consisting of a catalytic subunit C and two regulatory subunits, A and B.287 Its specificity, (sub)cellular localization, and catalytic activity are determined by the unique combination of regulatory subunits associated with the catalytic subunit.287 Moreover, the catalytic subunit is subject to two types of posttranslational modification, phosphorylation and methylation, which can also be important regulatory devices. The A regulatory subunit is encoded by one of two genes α (PPP2R1A) and β (PPP2R1B), which are 86% identical.
The phosphatase activity of PP2A is present in the subunit C and it can dephosphorylate various transcription factors and protein kinases, including MEK, ERK1/2, AKT, and sphingosine kinase (SK).288 PPP2R1B is reported to be downregulated in cultured dermal fibroblasts from patients with systemic sclerosis, a fibrotic disorder.289 The reported PPP2R1A activating mutation (p.S256F)51 is shown recently in endometrial cancer cells to behave in a dominant‐negative manner due to gain‐of‐function interactions with the PP2A inhibitor TIPRL1, resulting in hyperphosphorylation of AKT, GSK3β, and mTOR signaling pathways.290 That is, the activating mutation leads to reduced PP2A activity and consequent increased AKT activity. PP2A negatively regulates Wnt/β‐catenin and ERK.291
Reduced PP2A activity has been shown in many cancers and Alzheimer's disease.292, 293, 294, 295. Hence, PP2A is considered as a tumor suppressor.296
Yet PP2A inactivation is involved in fibrogenesis. Both trichostatin A (TSA), a pan‐HDAC inhibitor, and HDAC4 knockdown are reported to be sufficient to decrease phosphorylation of AKT and block TGF‐β1‐stimulated α‐SMA expression and thus fibroblast activation, and the pharmacological inhibition of PP1 and PP2A rescues the α‐SMA expression in response to TGF‐β1.297 In other words, suppression of PP2A and PP1 is sufficient to facilitate TGF‐β1‐induced FMT.
In contrast to normal fibroblasts that undergo apoptosis upon collagen matrix, the lung fibroblasts from patients with idiopathic pulmonary fibrosis (IPF) are reported to have low expression of α2β1 integrin, the major receptor for collagen, resulting in a failure to activate PP2A, which, in turn, leads to aberrant activation of β‐catenin pathway and subsequent increased proliferation of fibroblasts.298 Similarly, myofibroblasts on fibrillar type I collagen had reduced PTEN and α2β1 integrin expression as well as reduced PP2A activity, leading to increased AKT, but not ERK, activity, and enhanced proliferation.299 Overexpression of the catalytic subunit of PP2A or just PP2A is found to impair cardiac function and to promote fibrosis.300, 301
Taken together, the above discussion strongly suggests that PPP2R1A mutation is likely to reduce PP2A activity, resulting in enhanced fibrogenesis. Increased PPP2R1A expression or reduced PP2A activity has not been reported, but this might be due to the fact that downregulated genes are less likely to be reported unless the genes play critical roles in pathogenesis or pathophysiology. It is likely that PPP2R1A expression is increased in endometriosis. Alternatively, PP2A activity might be reduced.
5.2. Mutations in ALK, BRAF, FGFR3, GNAQ, NF1, NF2, NOTCH1, and NRAS in endometriosis and their roles in fibrogenesis
5.2.1. ALK
ALK encodes for anaplastic lymphoma kinase, also known as ALK tyrosine kinase receptor or CD246, which is an enzyme that plays an important role in brain development.302 Its fusion with various genomic partners is reported to result in constitutively activation and drive tumorigenesis.302, 303 Its expression or mutation has never been reported in endometriosis. There is no documentation of its direct role in fibrogenesis.
5.2.2. BRAF
BRAF is a well‐known protooncogene that encodes for B‐Raf, which belongs to the RAF kinase family, and plays a role in regulating the MAPK/ERKs signaling pathway that regulating cell division, differentiation, and secretion.304 Its activating mutations have been reported in many cancers.305 BRAF mutation is not found in endometriosis87 and is found to be rare even in ovarian cancer or endometrial cancer‐related endometriosis.256, 306 Despite these negative findings, one study found that BRAF is overexpressed in endometriosis.307 Another study reports that treatment with PLX4032, a potent‐specific BRAF inhibitor, decreased ERK1/2 activity in endometriotic epithelial and stromal cells, with subsequent decreased proliferation, suggesting that the BRAF pathway may be functional in endometriotic lesions.308
BRAF is found to be overexpressed in tissue samples from IPF,309 but a recent study on lung cancer associated with IPF reports that BRAF mutation is found only in a patient with drug‐induced pulmonary fibrosis.310 There is no documentation of its role in fibrogenesis.
5.2.3. CDKN2A
Cyclin‐dependent kinase inhibitor 2A (CDKN2A) is a TSG which encodes for two proteins, p16INK4a, a CDK inhibitor, and p14arf, a p53 stablizer, from two independent promoters. Inactivation of CDKN2A due to either mutation or hypermethylation can result in alteration in the CDK pathway and tumorigenesis. Somatic mutations of CDKN2A are common in many cancers.
LOH at p16INK4a in endometriosis is reported at the turn of this century.85 Aberrant methylation at p16INK4a also was reported in 2002 151 but a more recent study failed to find such an aberration.87 Expression of p16INK4a was reported to be reduced in endometriosis.311 Treatment of endometriotic stromal cells with HDAC inhibitors is reported to induce an accumulation of acetylated histones and in the promoter regions of p16INK4a, suggesting that the p16INK4a expression is likely to be low in endometriosis.312 Indeed, suppression of C/EBPα downregulates p16INK4a and CDK4.217
Because of its role in regulating cell cycles, p16INK4a is often used, along with p21CIP1/WAF1 and senescence‐associated ß‐galactosidase (SA‐β‐gal), as a marker for cellular senescence. Since senescence limits fibrogenesis,110 activation of p16INK4a may signal cellular senescence and thus restrict fibrotic progression. Consistent with this view, it is reported that the expressions of senescence‐associated genes p21, p16 , and p27 are upregulated in keloids fibroblasts exposed to X‐ray radiation, which may explain as why X‐ray radiation is part of the effective therapy for keloids since the radiation may prevent the recurrence of keloids by suppressing fibroblast proliferation, and arresting the cell cycle through inducing premature cellular senescence.313 Similarly, expression of p21CIP1/WAF1, SA‐β‐gal, and p16INK4a is found to be increased in perivascular fibrotic areas after transverse aortic constriction, and these senescent cells are found to be predominantly myofibroblasts, indicating that some myofibroblasts are undergoing premature senescence in the heart.314 Inactivation of the premature senescence program by genetic ablation of both p53 and p16INK4 leads to aggravated fibrosis in mice after transverse aortic constriction.314 More interestingly, cardiac‐specific expression of CCN1 (CYR61), a potent inducer of premature senescence,315 resulted in substantial reduction of perivascular fibrosis after transverse aortic constriction.314 While CCN1 has been consistently reported to be upregulated in endometriotic lesions,196, 316, 317 it should be noted that “older” lesions, which have higher fibrotic content than “younger” ones,62 appear to have diminished CCN1 expression,316 suggesting that reduced cellular senescence in older lesions permits fibrogenesis.110 Of course, CCN1 may be important in the establishment of lesions.316, 318
Given the above discussion, there are reasons to believe that CDKN2A may be downregulated, hypermethylated or even inactively mutated in older lesions that have a high fibrotic content, resulting in attenuated cellular senescence in myofibroblasts in lesions.
5.2.4. FGFR3
FGFR3 encodes for fibroblast growth factor receptor (FGFR) 3 and is a member of the FGFR family. It is considered as an oncogene and is frequently mutated in cancers.319 FGFR3 overexpression is reported in IPF.320 But one recent study on lung cancer associated with IPF reports that no FGFR3 mutation is found in patients with IPF.310 FGFR3 has no documented role in fibrogenesis.
FGFR3 mutation has not been reported in endometriosis,87 but its ligands FGF1 and FGF2 have been reported to be overexpressed in endometriosis.321 Given the lack of any direct role in fibrogenesis, it is likely that FGFR3 is not mutated in endometriosis.
5.2.5. GNAQ
GNAQ gene encodes for guanine nucleotide‐binding protein G(q) subunit α, and its activating somatic mutations result in increased MAPK signaling pathway and is found in Sturge‐Weber syndrome that increases the malignancy potential.322 GNAQ mutation or expression has never been reported in endometriosis as of writing, nor has it been documented to play a direct role in fibrogenesis.
5.2.6. NF1
NF1 encodes for neurofibromin 1 and its mutations are associated with neurofibromatosis type I and Watson syndrome.323 It is considered as a TSG, and its inactivating mutations or loss leads to RAS hyperactivation, resulting in increased activity of its downstream effectors, such as PI3K/AKT/mTOR and MAPK.240 Interestingly, NF1 mutation is found only in endometriosis‐associated ovarian cancer,282 but its expression is found to be reduced in OMA.96
Decreased expression of NF1 is reported to contribute to EMT in neurofibroma specimens and NF1‐derived Schwann cells.324 In a murine fracture model, increased fibrosis is found in Nf1 (null) mouse.325 Another study reports that forced miR‐16 expression suppressed the activation of HSCs (fibroblasts), and NF1 is identified to be one of the key nodes in the functional layer of a regulatory network that mediates the miR‐16 action in proliferation, ECM deposition, and survival.283 Aside from these reports, the role of NF1 in fibrogenesis appears to be limited.
5.2.7. NF2
NF2 also is a tumor suppressor and encodes for neurofibromin 2, also known as Merlin, which suppresses mTORC1 and the complex formation of SRC/FAK, hence regulating PI3K and RAS/MAPK signaling pathways.326, 327 Nearly 75% of malignant mesothelioma has inactivating NF2 mutations.328 Merlin also is known to negatively regulate Yes‐associated protein (YAP), an effector of the Hippo signaling pathway.329 Merlin can also inhibit Wnt/β‐catenin signaling through inhibiting phosphorylation of β‐catenin, thus blocking the translocation of β‐catenin from membrane to nucleus by inhibiting dissociation of β‐catenin from adherens junction.330, 331
While NF2 expression or mutation has never been reported in endometriosis, YAP overexpression has.332 The involvement of Wnt/β‐catenin signaling pathway in endometriosis,70, 333 especially in the context of fibrogenesis,128 also has been reported. Incidentally or not, YAP acts as critical regulators of HSC activation upon chronic injury, and pharmacological inhibition of YAP prevents HSC activation in vitro and fibrogenesis in vivo.334 In IPF, both YAP and TAZ expression levels are elevated and display a predominantly nuclear localization, which indicates increased transcriptional activity.335 The elevated YAP and TAZ expression also corresponds to the increased levels of nuclear β‐catenin and phosphorylated R‐Smads found in fibrotic tissues,330 suggesting that three signaling pathways, ie, TGF‐β/Smad, Wnt/β‐catenin, and the Hippo, converge to regulate fibrogenic processes.336
In light of these discussions, it seems likely that NF2 expression is likely to be reduced in endometriotic lesions. One might also see NF2 inactivating mutations in lesions.
5.2.8. NOTCH1
NOTCH1 encodes a member of the Notch family of receptors. Notch is an evolutionarily conserved intercellular signaling pathway that regulates interactions between physically adjacent cells and plays many roles in regulation of cell‐fate acquisition,337 such as survival or apoptosis, proliferation, differentiation, and maintenance of stem‐cell quiescence and identity.338 In mammalians, there are four Notch receptors (NOTCH1‐4) and five ligands (Jagged1, Jagged2, Delta‐like ligand (Dll)1, Dll3, and Dll4).339The NOTCH1 protein has such diverse functions that the gene is considered both an oncogene and a tumor suppressor.340 NOTCH1 inactivating mutations have been found in cutaneous and lung squamous cell carcinoma,341 and also in sun‐exposed physiologically normal skin342
NOTCH1 overexpression has been shown to facilitate the myofibroblast differentiation from lung fibroblasts343 and to induce COL1A1 and COL1A2 expression in airway fibroblasts,344 suggesting a possible role of Notch1 in fibrogenesis. Activation of Notch1 signaling has also been reported to induce EMT, while Notch1 suppression reverses the EMT process both in vitro and in vivo.345, 346 NOTCH1, along with its ligand Jag1, has been shown to be regulated by TGF‐β1, induce EMT, proliferation, and renal fibrosis in mouse and humans.347 Jagged1‐NOTCH1 signaling also has been shown to be activated in keloid tissues, a fibrotic disease,348 and in hypertrophic scar formation. Activation of NOTCH1 with Dll4 causes a premature cellular senescence and maladaptive repair, facilitating renal fibrogenesis.349 NOTCH1 and TGF‐β1 are shown to be responsible for the induction of Hes1, an important gene for HSC activation.350 In contrast, conditional ablation of NOTCH1 attenuates pulmonary fibrosis through reduced myofibroblast activation and expression of α‐SMA and collagen I,351 and inhibition of Notch signaling attenuates Schistosomiasis‐induced hepatic fibrosis via suppression of macrophage M2 polarization.347
In endometriosis, Musashi‐1, a positive regulator of NOTCH1 through suppression of NOTCH1 inhibitor NUMB, has been shown to be elevated.353 NOTCH1 expression has been reported to be increased in peritoneum adjacent to endometriotic lesions,354 although it is found to be decreased in eutopic endometrium in women with endometriosis, along with Jagged2 and Dll4.355 The expression of c‐Myc, one NOTCH1 downstream gene, has been well‐documented to be increased in endometriosis.97, 356, 357, 358 Suppression of NOTCH1 results in reduced lesion size in a murine model of endometriosis.354 NOTCH1 mutation has been reported in endometriosis‐associated ovarian cancer.282 Recently, the profibrotic role of ADAM17/NOTCH signaling induced by oxidative stress in endometriosis has been reported.360
To summarize, it seems that NOTCH1 signaling pathway is likely to be activated in endometriosis. It is likely that NOTCH1 expression may be increased
5.2.9. NRAS
NRAS is a member of the RAS family, and is mutated in 18%‐28% of melanoma.239, 240 Direct sequencing did not find NRAS mutation in 23 lesion samples,87 but NRAS upregulation is reported in endometriosis.307 However, there is no documentation of its role in fibrogenesis.
5.3. Variations on a major theme
Admittedly, there are more TSGs and oncogenes (eg, GSTP1, SOCS1, RASSF1, APC, and YAP, just to name a few) that are not included for this review, but the choice of the 6 + 9 = 15 driver genes is based on the considerations that (a) whether it has ever been reported to be mutated in endometriosis; and (b) it may be pointless, and perhaps impossible, to review all cancer drive genes—one has to step somewhere, somehow. In this sense, the list in Kato et al53 provides a good compromise for the choice of a “control” set that is not prohibitively too large or inherently biased. Thus, there is no a priori selection bias to choose the NR set. Nonetheless, one overwhelming message gleaned from this overview is that all six cancer driver genes in the RP set are all involved in fibrogenesis. This is in stark contrast to only three (CDKN2A, NF2, and NOTCH1) out of nine driver genes in the NR set that have been credibly documented in fibrogenesis (Table 2), the difference is statistically significant (P = 0.028, by Fisher's exact test). Even if NF1 is counted to have some profibrotic capability, the difference is still statistically significant (P = 0.044, by Fisher's exact test).
Table 2.
Summary of compiled findings regarding mutation and expression of the cancer driver genes in endometriosis and fibrogenesis
| Gene | Chromosomal location | Role in tumorigenesis | Mutations reported in benign conditions | Mutation in endometriosis | Expression in endometriosis | Possible roles in fibrogenesis |
|---|---|---|---|---|---|---|
| TP53 | 17p13.1 | Tumor suppressor | Inactivating mutation in rheumatoid arthritis138, 139 | Found in adjacent to ovarian cancer,83 also loss.84Negative reports also. | Mixed results.Likely to be downregulated | TP53 loss suppress senescence of activated fibroblasts, promoting fibrogenesis |
| PTEN | 10q23.3 | Tumor suppressor | Inactivating,51One reports 21% in OMA147; Another reports 53% 148 | Reduced expression in endometriosis.151, 152, 153 | Activates the PI3K/AKT signaling pathway, suppresses the apoptosis of fibroblasts, and enhances their proliferation and invasiveness. Initiates SMAD3‐ and TP53‐dependent fibrotic response | |
| ARID1A | 1p36.11 | Tumor suppressor | Inactivating,51Partial or complete loss of expression,46 , 48 , 206 | Loss of ARID1A promotes wound regeneration | ||
| PIK3CA | 3q26.32 | Oncogene | Soborrheickeatosis: ~16%Fibroadipose hyperplasia: 90% | Activating, Also reported in the context of its link with ovarian cancer223, 224 | Activated PI3K/AKT/mTOR signaling pathway has been implicated190, 225, 226, 227, 228, 229 | Not documented. |
| KRAS | 12p12.1 | Oncogene | Activating51 | Upregulated in eutopic endometrium252, 253, 254 | Induction of ERK1/2 and in cooperation with Snail. | |
| PPP2AR1 | 19q13.41 | Oncogene | Activating 51 | NR | Impair PP2A activity | |
| ALK | 2p23.1 | Oncogene | Inflammatory myofibroblastic tumor: ~50%53 | NR | NR | Not well documented |
| BRAF | 7q34 | Protooncogene | Melanocytic nevi: 70‐88% 53 | NR | Overexpressed307 | Not well documented |
| CDKN2A | 9p21.3 | Tumor suppressor | LOH.85 Aberrant methylation151 |
Reduced expression of p16311; Likely to be low312 |
A marker for cellular senescence, which can restrict fibrogenesis | |
| FGFR3 | 4p16.3 | Oncogene | NR | NR | Not documented | |
| GNAQ | 9q21.2 | Oncogene | Sturge‐Weber syndrome: 88%53 | NR | NR | Not documented |
| NF1 | 17q11.2 | Tumor suppressor | Neurofibromas and pilocyticastrocytomas53 | only in endometriosis‐associated ovarian cancer282 | Reduced expression in OMA96 | Not well documented |
| NF2 | 22q12.2 | Tumor suppressor | Schwannomas, meningioma, glioma and ependymomaastrocytomas53 | NR | NR | Its suppression is likely to promote fibrogenesis through interaction with Hippo, TGFβ and Wnt‐β‐catenin pathways |
| NOTCH1 | 9q34.4 | Tumor‐suppressive and Oncogenic | Sun‐exposed skin53 | NR | Increased expression in peritoneum adjacent to endometriotic lesions354 | Facilitates FMT and fibrogenesis |
| NRAS | 1p13.2 | Oncogene | Melanocytic nevi: 6%‐14%53 | NR | Overexpressed307 | Not documented |
ALK, anaplastic lymphoma kinase; ARID1A, the AT‐rich interactive domain 1A; FGFR3, Fibroblast growth factor receptor 3; PTEN, phosphatase and tensin homolog deleted on chromosome 10; NF1, Neurofibromin 1; NF2, Neurofibromin 2; OMA, ovarian endometrioma; NR, not reported.
Figure 2 summarizes the above discussion by anchoring the nine cancer driver genes (in solid background) in a vast sea of discoveries reported in endometriosis. Among the nine genes, six are from the RP set and the remaining three, the NR set. It can be seen from the figure that the diagram pieces together many genes/proteins reported to be aberrantly expressed in endometriosis that are otherwise scattered in the ever‐growing literature. It highlights fibrosis as the final destiny of all endometriotic lesions if unimpeded, and provides a more coherent account for many culprits, aiders and abettors involved in EMT and fibrogenesis in endometriosis. In addition, it seems that several pathways lead to the activation of the PI3K/AKT signaling pathway, underscoring its vital importance in fibrogenesis of endometriosis. In fact, this is consistent with the published studies demonstrating its involvement in endometriosis development.155, 229, 361, 362
Figure 2.
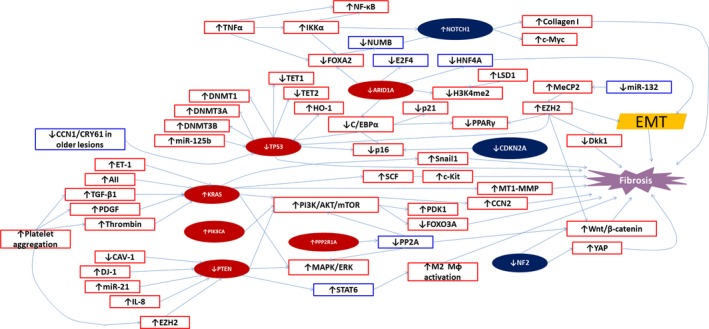
Waiving a tapestry of possible gene network involved in fibrogenesis that anchors the cancer driver genes reported or unreported to be mutated in endometriosis. The genes/proteins in solid maroon oval are those genes reported to be mutated in endometriosis, while those in solid dark blue oval are those that have not been reported to be mutated. The genes/proteins within the red rectangles are those that have been reported in the literature, while those within the blue rectangles are those that have not been reported. ↑ means activating mutation or overexpressed gene/protein, while ↓ indicates an inactivating mutation or overexpressed gene/protein. → means “leads to”, “results in”, or “induces”. AII, Angiotensin II; ARID1A, the AT‐rich interactive domain 1A; CAV‐1, caveolin‐1; C/EBPα, CCAAT‐enhancer‐binding protein α; CCN1/CRY61, CCN family member 1/cysteine‐rich angiogenic inducer 61; DNMT, DNA methyltransferase; E2F4, E2F transcription factor 4; ERK, extracellular signal‐regulated kinase; ET‐1, endothelin 1; EMT, epithelial‐mesenchymal transition; Dkk1, dickkopf homolog 1; FOXO3A, forkhead box O3A or FOXO3; H3K4me2, dimethylated histone 3 lysine 4; HNF4A, hepatocyte nuclear factor 4α; LSD1, lysine‐specific demethylase 1; PDGF, platelet‐derived growth factor; EZH2, enhancer of zeste homolog 2; FOXA2, forkhead box A2; HO‐1, hemeoxygenase 1; IKKα, inhibitor of nuclear factor kappa‐B kinase subunit α; mTOR, mammalian target of rapamycin; MeCP2, methyl CpG‐binding protein 2; MAPK, mitogen‐activated protein kinase; Mɸ, macrophage; MT1‐MMP, membrane‐type 1 matrix metalloproteinase, also called matrix metalloproteinase‐14 or MMP‐14; NF1, Neurofibromin 1; NF2, Neurofibromin 2; NF‐κB, nuclear factor kappa‐light‐chain‐enhancer of activated B cells; PDK1, Pyruvate dehydrogenase lipoamide kinase isozyme 1; PI3K, phosphatidylinositol‐4,5‐bisphosphate 3‐kinase; PP2A, protein phosphatase 2; PPARγ, peroxisome proliferator‐activated receptorγ; PTEN, phosphatase and tensin homolog deleted on chromosome 10; SCF, stem cell factor; STAT6, Signal transducer and activator of transcription 6; TET, ten‐eleven translocation methylcytosine dioxygenase; TGF‐β1, transforming growth factor β1; TNFα, tumor necrosis factor α; YAP, Yes‐associated protein
Moreover, it also lists several suspects, such as miR‐132, HNF4A, STAT6, and E2F4, that so far have not ever been reported to be involved in endometriosis but are very likely to play important roles in the development of endometriosis.
In fact, it has been reported that IKKα, induced by TNFα, can suppress NUMB and thus activate NOTCH1 through suppression of FOXA2.363 HNF4A activation has been reported to attenuate liver fibrosis through suppression of EMT.364 Combined activation of HNF4A, HNF1A, FOXA3, and GATA4 has been reported to reprogram hepatic myofibroblasts into hepatocytes in vivo, suppressing liver fibrosis,365 raising the possibility of reversing fibrosis through transcriptional reprogramming via activation of select transcription factors. Importantly, increased IKKα expression has been reported in endometriosis366 and so have TN pronounced in mice that had lesions mor pronounced in mice that had lesions mor Fα367 and Musashi‐1, a negative regulator of NUMB, which suppresses NOTCH1.353 Incidentally, ovarian‐like differentiation in endometriotic lesions is found to be associated with increased expression of GATA4/6.368
It should be noted that this diagram is by no means all‐inclusive and complete. As we understand more about the pathogenesis and pathophysiology of both endometriosis and fibrogenesis, there is no doubt that we should have a better grasp of the complexity of endometriosis.
5.4. Do driver mutations occur before or after lesions are formed?
Aside from reporting mutations in four cancer driver genes, Anglesio et al also reported a patient with DE who harbored an identical c.35G→A (p.G12D) KRAS mutation in three distinct DE lesions and also in normal sampling of eutopic endometrial and endocervical epithelium.51 Since the mutation occurred in normal endometrium and the endocervix, the finding raises the issue as whether or not the mutation is an early event occurred before any lesion is formed.
This issue is probably better framed in a bigger context. One most widely accepted theory for the pathogenesis of endometriosis is Sampson's retrograde menstruation theory. However, retrograde menstruation has been reported to be nearly universal for women with patent fallopian tubes.369 Given the moderate prevalence of endometriosis,1 several theories have been proposed to explain the vast gap between the moderate prevalence and the ubiquity of retrograde menstruation. One view, expressed by many papers, is that endometriosis may be an endometrial disease, in the sense that the eutopic endometrium may have already harbored molecular/cellular aberrations that predispose their host to endometriosis (see, for example370). However, a close inspection of all articles documenting the aberrations indicates that these data are all based on patients who have already been diagnosed with endometriosis. Hence, it is unclear whether these aberrations are the cause or merely the consequence of endometriosis. Unfortunately, experimental data that support this hypothesis are lacking.
In contrast, there are experimental data that actually provide the evidence that all these endometrial aberrations may well be the consequence of endometriosis. Lee et al reports that, 14 weeks after mice received surgically placed endometrial fragments from donors, their eutopic endometrium showed aberrant gene expression as well as aberrant DNA methylation.371 Similar data have also been reported by baboon models of endometriosis.372 A recent study even showed that 12 weeks after surgical induction of endometriosis, the eutopic endometrium showed altered gene expression which became more pronounced in mice that had lesions more proximal to their uteri than their counterpart that had distal lesions.368
A close examination of the finding by Anglesio et al actually shows that the mutation is unlikely an early event. As figure 4 in Anglesio et al shows, the patient harbored identical KRAS mutation in three distinct DE lesions and also in normal sampling of eutopic endometrial and endocervical epithelium.51 Importantly, the allelic frequency of the mutation in the three DE lesions and normal sampling of eutopic endometrial and endocervical epithelium were 38.57%, 31.22%, 9.37%, 0.28%, and 0.05%, respectively. In other words, the allelic frequency is one to two orders of magnitude higher in DE lesions than that in eutopic endometrium and endocervix.
Since all current cells in an organism descended from a single cell (fertilized egg) through successive cell divisions, there is reason to believe that the mutation that the three lesions, the eutopic endometrium, and the endocervix shared came from the same common ancestor cell, as shown in Figure 3. It is well known in phylogenic analysis that clones with higher mutation frequency harbored the mutation longer than those with lower frequencies, simply because the former had more chances than the latter to propagate.374 In other words, the mutations harbored by DE lesions existed in longer time than those in eutopic endometrium and the endocervix (Figure 3), and the mutation occurred in normal tissues resulted from clonal expansion and/or diffusion originating from the DE lesions due to increased invasiveness of endometriotic epithelial cells resulting from EMT. As argued in the above, mutations occur because of selection pressure, and now given the evidence that DE lesions harbor mutations longer than that of normal tissues, it can be concluded that driver mutations must occur after lesions are formed. Otherwise, a nearly 100% mutation frequency in lesions or higher mutation frequency in normal tissues adjacent to the DE lesions would have been observed.
Figure 3.
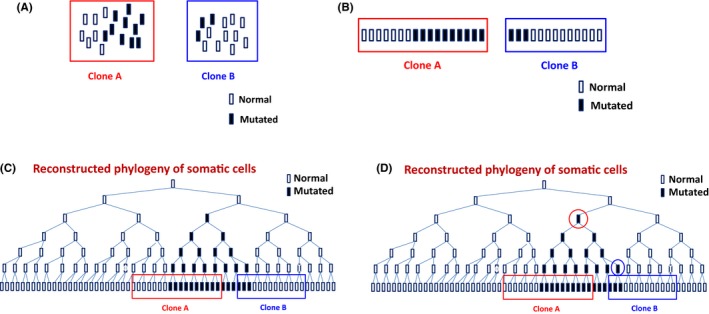
A cartoon illustration showing that clones with higher mutation frequencies harbored the mutation longer than clones with lower mutation frequencies. A, Two clones or tissue samples, A and B, with different frequencies of the mutant allele. In this case, clone A has a higher frequency of the mutant allele than that of clone B. B and C, Since all somatic cells are descended from their parental cells (ie, by mitosis, one cell gives rise to two daughter cells), all cells in clones A and B, mutated or not, can be arranged based on their genetic similarity—ie, two cells are genetically closer if they share more similarity in terms of DNA sequences. D, For clone A, all cells carrying the mutant allele are descended from one common ancestor cell (circled in red) while all cells with the mutation are descended from the common ancestor cell that is circled in blue. However, this mutated cell, circled in blue, actually descended from the cell, circled in red, that is the common ancestor for all cells with the mutatnt allele in clone A. Therefore, clone A harbored the mutation earlier than clone B. The exact inference of this phylogenetic relationship would require statistical calculations with some very elaborate models. See, for example 374
It is a pity that the study by Anglesio et al did not capitalize the opportunity to further sequence these samples. If they did, they could have been able to use the sequence data to reconstruct a precise phylogenic relationship among these tissues, possibly elucidating the lineage relationship among these tissues.
6. CHANGING PRESSURE FOR GENOMIC ALTERATION DUE TO OXIDATIVE STRESS
Oxidative stress has been well‐documented to be involved in the pathophysiology of endometriosis.375 DNA is particularly vulnerable to the damaging effect of reactive oxygen species (ROS), as are other biomolecules. The presence or absence of oxidative stress lies in the imbalance between the production of free radicals and their elimination by antioxidants.376 As ectopic endometrium experiences cyclic bleeding, cell‐free hemoglobin and its highly toxic by‐products heme and iron are released from erythrocytes in and around lesions. Lysis of erythrocytes by macrophages results in iron overload, inducing iron‐mediated damage, oxidative injury and inflammation and oxidative stress.375 Consistently, granulosa cells from patients with infertility and endometriosis exhibit more signs of severe oxidative DNA damage when compared with cells from other patients with infertility.377 Expression levels of 8‐hydroxydeoxyguanosine (8‐Oh‐dG), a sensitive indicator of DNA damage resulting from oxidative stress, are reported to be significantly higher in samples of normal ovarian cortex surrounding endometriotic cysts.378 Interestingly, however, the staining of γ‐H2AX, a marker for DNA damage, is found to be decreased as endometriotic lesions aged.113
In endometriotic lesions, repeated bleeding and the DNA‐damaging molecules secreted by lesions themselves should result in severe oxidative stress and thus impose a strong pressure for mutation on endometroitic cells. However, as DNA damage, oxidative stress, hypoxia, and oncogene (such as KRAS and YAP) activation induce cellular senescence 107 and senescence would curtail fibrosis, it is likely that in older and thus more fibrotic lesions may experience less hypoxia and less oxidative stress (due to increased fibrotic content and thus reduced cellularity, less hemorrhage and thus iron overload) and less DNA damage (as seen by reduced γ‐H2AX expression 113), the pressure for cellular senescence may be reduced, especially in the stromal compartment. This would be consistent with reduced Caveolin‐1 expression in adenomyosis,189 which can be highly fibrotic,65 especially as lesions get older.64 This also would be consistent with seemingly progressive decrease in CCN1 expression in older lesions.316 This dynamic view could also explain the conflicting reports published so far since most, if not all, published studies habitually treat endometriotic cells as if static or even immutable. Obviously, when epithelial cells are differentiated into mesenchymal cells or even smooth muscle‐like cells, their phenotypes and functionality change accordingly.
6.1. Mismatch repair genes in endometriosis
The preservation of genomic integrity is of vital importance to the perpetuation of life. On the other hand, somatic genomic alterations are bound to occur since DNA is susceptible to chemical modifications by endogenous and exogenous agents, although it is less susceptible to replication errors. Thanks to evolution, all cells are equipped with intricate and sophisticated defense systems, such as DNA repair, damage tolerance, cell‐cycle checkpoints, and apoptosis that collectively function to safeguard against DNA damage. Through institution of several mechanisms, including base excision repair, nucleotide excision repair, mismatch repair (MMR), homologous recombination, and nonhomologous end joining, a robust DNA damage response (DDR) is established in different stages of the cell cycle to repair any DNA damage.379 Interestingly, DDR is activated through the inhibition of PP2A.380
Somatic point mutation can conceivably occur due to the failure in base excision repair, nucleotide excision repair, or MMR. In endometriosis, however, only the aspect of MMR has been investigated so far, mostly in the context of potential for malignant transformation.
Higher DNA repair capability in women with endometriosis has been reported.381 Higher DNA damage and lower DNA repair activity have been reported to be related to endometriosis progression, ie, rASRM stage,382 although the use of rASRM stage as a measurement for disease progression is debatable.
Significant loss of MMR proteins, including MLH1, MSH2, MSH6, and PMS2, that are known to be associated with microsatellite instability (MSI),383 in the stromal component of ectopic endometrium has been reported.384 In endometriotic glandular epithelial cells, cytoplasmic staining for aurora A kinase is found to have higher MMR protein expression, suggesting an increased activity of the MMR system.384
MLH1 is a member of the MMR system in humans which rectifies errors in DNA replication during mitosis to ensure genomic fidelity. LOH at MLH1 and MSH2 in adenomyosis also has been reported.385 Transcriptional inactivation of the MLH1 gene through promoter hypermethylation is often associated with MSI,383 which is reported to be associated with the malignant transformation of endometriosis.382, 383, 384, 385, 386
Promoter hypermethylation of MLH1 has been found in ~4% (2/46) of endometriotic lesions.151 Loss of MMR protein expression is reported in 10.1% of endometriosis‐associated ovarian carcinomas.389
Taken together, there is evidence to suggest that the DDR system appears to be activated but somehow its activity seems to be lowered in endometriosis, raising the prospect of genomic alteration.
6.2. Endometriotic epithelial cells are more vulnerable than stromal cells to mutational pressure
In endometrium, the coiling and maturation of the spiral arterioles and growth of the subepithelial capillary plexus take place in the secretory phase.390 After progesterone withdrawal, the luminal portion of the endometrium is shed and the menstruation occurs. This is followed by endometrial repair, which seems to be analogous to classic wound healing and include inflammation, its resolution, angiogenesis, tissue formation, and tissue remodeling or reepithelialization.390 As in eutopic endometrium, the ectopic endometrium starts in each menstrual cycle with the shedding of glandular epithelial cells, but much less so in the endometriotic stromal cells. This relatively more rapid cellular turnover in epithelium than the stroma may explain, at least in part, as why the mutations are seen only in the epithelial cells but not in stromal cells of DE lesions as reported in Anglesio et al.51 This seems to be supported by the recent report that of all 19 mutations detected in 24 DE lesions, all were significantly enriched in epithelial but not in stromal components of every lesion examined.391 The only exception is TP53, since its inactivation, which prevents cellular senescence, is expected to occur in the stromal component of endometriotic lesions, which promotes fibrogenesis.
The presence of mutations in the epithelium, but not in the stroma, of endometriotic lesions suggests that the two cell components do not share the same lineage. This is simply due to the fact that, should they share the same lineage, it is unlikely that the all the driver mutations seen in the endometriotic epithelial cells just occurred after one or more cell divisions, nor could they reverted back to wild types.
Given the most, if not all, studies reporting genomic alterations in endometriosis used careful tissue microdissection techniques31, 51 to evaluate mutations in epithelial and stromal cells separately, one possibility that some studies that failed to find any mutation is because the mixed use of different cell types may have obscured the true signal.
6.3. Spontaneous somatic mutations in apparently normal tissues are not rare
As noted Anglesio et al51 and others, spontaneous somatic mutations or genomic alterations in apparently normal tissues and benign conditions are not rare.53, 352, 392, 393 While the theory of clonal selection of driver mutations drive the progression from benign lesions to premalignancy and then to malignancy appears to hold true, exceptions do exist.53 In addition, while ARID1A acts as tumor suppressor and KRAS and PIK3CA often behave as oncogenic, their mutation frequencies vary wildly among different cancers, attesting to the notion that cellular context also is important, and that perhaps multiple gene mutations or deregulations may be required for tumorigenesis.
Animal models have shown that endometriosis‐like lesions can be induced in mice by activation of KRAS or deletion of PTEN,150 and a combination of these abnormalities, such as KRAS activation plus TP53 loss,90 concurrent inactivation of TP53 and Rb1,394 PTEN loss plus PIK3CA activation,395 and KRAS activation plus PTEN loss150 leads to the development of malignant ovarian tumors that resemble endometrial cancer in humans. Thus, these data seem to suggest that more than one driver mutation is needed for malignant transformation.
7. SOME PREDICTIONS
Given the above discussion, several predictions can be made. First, as lesions become older or more fibrotic, the activated myofibroblasts may become less senescent. Alternatively, lesions exposed to higher concentration of profibrotic milieu should be less senescent, as manifested by decreased expression of TP53, p16, and SA‐β‐gal, and also reduced expression of CCN1 and caveolin‐1.
Second, given the natural history of endometriotic lesions, it seems that the extent of fibrosis should be proportional to the age of the lesion, which should, in turn, be proportional to the increased frequency of mutations. Consequently, these driver mutations as well as those passenger mutations should be observed primarily in DE tissues with higher fibrotic content. It is unfortunate that the extent of fibrosis in tissue samples used in Anglesio et al51 was not evaluated—in fact the word “fibrosis” was not even mentioned once in the paper. However, Figure 3A in Anglesio et al51 seems to show the increased fibromuscular content characteristically DE,396 very likely a manifestation of SMM.62, 74 Note that this assertion can be easily proven or refuted thorough careful evaluation of the tissue fibrotic content and the mutation status in both driver and passenger mutation genes.
As a corollary, we should expect to see much less frequent mutations in both driver and passenger mutation genes in OMA lesions than that of DE since the extent of fibrosis in OMA is less than that of DE.71 One possible exception is perhaps ARID1A, since OMA lesions may also be under strong pressure for tissue repair.
Third, reduced expression or inactivation of TP53 and PTEN, due to either somatic mutation or hypermethylation, should be seen mostly in the stromal component of endometriotic lesions. This is mainly due to the former's role in releasing the break on fibrogenesis when inactivated110 and the latter's role in activating the PI3K/AKT signaling pathway that drives fibrogenesis.
Fourth, since sensory nerve‐derived substance P and CGRP accelerate fibrogenesis in endometriotic lesions, and, in particular, the close correlation between the extent of fibrosis and the immunostaining of NK1R, an SP receptor, we should expect to see that DE lesions harboring either passenger or driver mutations also express NK1R. Figure 4 in Anglesio et al51 is consistent with the locations and the abundance of sensory nerve plexuses seems to correlate with the frequency of activating KRAS mutations in different DE and adenomyotic lesions.
Fifth, those endometriotic lesions showing diminished E‐cadherin but increased vimentin or even α‐SMA staining in the glandular epithelium may be at increased risk of harboring driver mutations. We have shown recently that DE lesions seem to have undergone more complete and thorough EMT, FMT, and SMM as compared with OMA lesions.71 Hence, stromal endometriosis, ie, lesions showing the absence of glandular epithelial cells,397, 398 is frequently seen in DE lesions.399, 400 As most driver mutations are reported to occur only in the lesional epithelial cells in DE,51 these cells may also exhibit lower or even the absence of E‐cadherin expression but elevated expression of mesenchymal markers such as vimentin or even markers of smooth muscle cells such as α‐SMA.71 Due to the increased cellular mobility and invasiveness resulting from EMT, patients with these types of DE lesions may be at elevated risk of dissimilating or spreading of endometriotic cells in other locations, and thus risk of recurrence.
Sixth, since CDKN2A, NF2, and NOTCH1 also appear to play critical roles in fibrogenesis of endometriosis (Figure 2), their mutations in endometriosis are also likely. In addition, for the 6 + 3 driver genes play important roles in fibrogenesis (Figure 2), it is likely that we may see their aberrant methylation even if no mutation is found.
Lastly, aside from the use of more complete genomic and epigenomic analyses, studies with larger sample sizes, the employment of tissue microdissection techniques, and the focus on lesions with higher fibrotic content should have increased chance to detect driver mutations in lesions. Smaller sample sizes will compromise statistical power, the use of mixed endometriotic stromal and epithelial cells would obscure the true signals since in many lesions the stromal component comprises ~70% or higher proportion of the entire lesions, especially when the extent of fibrosis is higher.
8. CLINICAL SIGNIFICANCE
In light of the above discussion, the clinical significance of driver mutations in endometriosis, in particular in DE,51 can be further illuminated as follows.
First, the driver mutations in endometriotic lesions may not necessarily be synonymous with malignancy or premalignancy. The labeling may incite unnecessary, and perhaps ungrounded, fear or even panic in the patient. As discussed above, the driver mutations may be both the cause and consequence of fibrogenesis. As such, evidence that the presence of these driver mutations would increase the risk of malignant transformation needs to be gathered and carefully analyzed.
Second, mutations suggest permanent, irreversible genomic changes. This could mean that treatment of patients with these genomic alterations would be more challenging. This is especially true for hormonal therapy, which is unlikely to rectify the genomic aberrations.
For those patients whose lesions harbor driver mutations, because of their increased fibrotic content, the vascularity is concomitantly reduced and the PR‐B expression levels are also reduced.71 For these patients, hormonal drug treatment is expected to be ineffective, due to the difficulty in delivering the drug to the intended target tissues and the reduced PR‐B expression.
Lastly, in view of the seemingly central role of the PI3K/AKT/mTOR signaling pathway in fibrogenesis in endometriosis (Figure 2), naturally this pathway can be considered as a potential target for therapeutics. Indeed, inhibition of the AKT/mTOR has been reported to be effective in preventing DE in mouse.401 In addition, Figure 2 also underscores the potential roles of epigenetic modification in intervention, which has shown some promising results in treating adenomyosis.402, 403 Future research is warranted to further explore these potentially profitable avenues.
9. CONCLUSIONS
As growing evidence indicates, fibrosis is the ultimate fate of all endometriotic lesions without intervention, simply because they are fundamentally wounds undergoing ReTIAR.12, 62, 63 Although fibrogenesis takes time, one needs to keep in mind that there is a well‐documented and significant diagnostic delay in endometriosis, typically a mean or median of 7 or more years from the time a patient first experiences symptoms until she receives a definitive diagnosis.6, 404, 405, 406 In baboon models of endometriosis, lesions of 1 year or older after induction demonstrate substantial fibrosis.62 This may explain as why one recurring theme is fibrosis when reviewing the six driver genes in the RP set, while only a portion of the other nine driver genes in the NR set are ever documented to be involved in fibrogenesis.
This review should help to shed more light on the pathophysiology of endometriosis by weaving a tapestry of fibrogenesis network, piecing together many genes/proteins known to have aberrations that are otherwise strewed in the literature. This tapestry, albeit still incomplete, provides more clues to the pathophysiology of endometriosis and represents potential therapeutic targets. For example, we can now appreciate the roles of TP53 inactivation in suppression of senescence in fibroblasts and in promoting fibrosis in endometriosis, as most, if not all, previous studies have focused exclusively on its role in malignant transformation. In fact, through the lens of the ReTIAR and fibrogenesis, we can now understand as why many clinical trials on endometriosis have failed.407We can also appreciate the physiological roles of ARID1A downregulation in facilitating tissue repair, which endometriotic lesions encounter almost all the time. In addition, several genes/proteins, such as E2F4 and PP2A, so far have not been reported to be involved in endometriosis, and should be closely scrutinized in future research. Moreover, this review highlights the point made previously that endometriotic lesions are not a static entity; instead, they evolve gradually but progressively to SMM and fibrosis if unimpeded.74
While the finding that DE lesions harbor cancer driver mutations is rather surprising, the recently unveiled natural history of endometriotic lesions, coupled with the reported roles in fibrogenesis of these driver gene mutations, tells us that these mutations are likely not the harbingers of imminent malignant transformation. Rather, they may be the result of combined fibrogenesis, mutational pressure due to oxidative stress, and more cell turnover. Consequently, these mutations should not sound undue alarm unless proven otherwise.
Admittedly, the inference drawn above is based on observations made in other cell types or organs. However, as the God does roll dices (hence more cell turnover increases the chance of genomic alterations) and often acts as a tinkerer who makes with existing machinery and tools at hand, there are strong reasons to believe that similar processes occur in endometriotic lesions.
Cell clones with driver mutations have selective advantages over those without, be it faster or more proliferation, or less requirement for oxygen. In many cancers, this manifests as an enrichment of protein‐altering mutations in cancer genes compared to that expected for the background mutational rate. However, since the frequency of driver mutations in sun‐exposed but physiologically normal skin cells can be high,342 it may not be so surprising that endometriotic lesions, which are under constant assault of hypoxia, proinflammatory cytokines, and oxidative stress, harbor somehow low‐grade driver mutations. As shown elegantly by that article, even the size of clonal expansion induced by a somatic mutation need not to correlate with its potential to induce malignant transformation.342
Despite new lights shed on the pathophysiology of endometriosis through this review, there are still many stones that are left unturned. Except knowing that endometriotic cells are under pressure for mutations to gain certain selective advantages, it is unclear as which factor(s) are mainly responsible for causing the mutation. In addition, KRAS and PIK3CA are oncogenes, and their activating mutations, once occur, should activate these oncogenes. However, oncogene activation in a normal cell does not necessarily lead to cell transformation but, instead, induces cellular senescence.108, 408 Repeated cell division and strong mitogenic signals are also inducers of senescence.409 Yet senescence restricts fibrosis.106 So does this mean that the oncogene activating mutations as reported would lead to the restricted fibrosis? Or endometriotic cells have developed some mechanisms that elude senescence? Moreover, consistent with the view that fibrogenesis entails epigenetic aberrations,410 growing evidence indicates that in endometriosis fibrogenesis also involves epigenetic changes.61, 71, 411, 412 This raises the question as whether hormonal therapeutics can actually modify the epigenetic aberration, resulting in a gradual reverse of fibrogenesis, especially in highly fibrotic lesions such as DE. A recent study reports that, dienogest, a synthetic progestin, is effective in alleviating symptoms but does not reduce nodular size in DE,413 suggesting that hormonal therapeutics cannot modify the epigenetic aberration that is already in place. This would mean the necessity of taking these drugs for a long period in order to alleviate the symptoms without changing the existing epigenetic aberrations. This, of course, presents an issue for the management of endometriosis, but it also presents an opportunity to explore new avenues for rectifying epigenetic aberrations to achieve better treatment results. Again, future research is needed to clarify these issues.
In summary, cancer driver mutations arise in endometriotic lesions very likely from strong pressure for progressive fibrogenesis. The network anchored by the nine driver genes should help to gain new insight into the pathophysiology of endometriosis. It highlights the powerful pressure of fibrogenesis that endometriotic lesions are experiencing, especially in DE lesions. Given that somatic driver mutations can and do occur frequently in physiologically normal tissues, it is argued that these mutations in endometriosis are not necessarily synonymous with malignancy or premalignancy. The fibrogenesis networks linking all genes of driver mutations and others should point to new therapeutic targets for endometriosis.
DISCLOSURES
The author declares no conflict of interest. Human/Animal rights: This article does not contain any studies with human and animal subjects performed by any of the authors.
ACKNOWLEDGMENT
The author would like to thank Professor Fellice Petraglia for his helpful comments on an earlier version of this manuscript.
Guo S‐W. Cancer driver mutations in endometriosis: Variations on the major theme of fibrogenesis. Reprod Med Biol. 2018;17:369–397. 10.1002/rmb2.12221
Funding information
This work was supported by the grants (81471434, 81530040 and 81771553) from the National Science Foundation of China.
REFERENCES
- 1. Giudice LC. Clinical practice. Endometriosis. N Engl J Med. 2010;362:2389‐2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guo SW. An overview of the current status of clinical trials on endometriosis: issues and concerns. Fertil Steril. 2014;101:183‐190; e4. [DOI] [PubMed] [Google Scholar]
- 3. Nisenblat V, Bossuyt PM, Farquhar C, Johnson N, Hull ML. Imaging modalities for the non‐invasive diagnosis of endometriosis. Cochrane Database Syst Rev. 2016;2:CD009591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nisenblat V, Bossuyt PM, Shaikh R, et al. Blood biomarkers for the non‐invasive diagnosis of endometriosis. Cochrane Database Syst Rev. 2016;5:CD012179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nisenblat V, Prentice L, Bossuyt PM, Farquhar C, Hull ML, Johnson N. Combination of the non‐invasive tests for the diagnosis of endometriosis. Cochrane Database Syst Rev. 2016;7:CD012281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nnoaham KE, Hummelshoj L, Webster P, et al. Impact of endometriosis on quality of life and work productivity: a multicenter study across ten countries. Fertil Steril. 2011;96:366‐373;e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gao X, Yeh YC, Outley J, Simon J, Botteman M, Spalding J. Health‐related quality of life burden of women with endometriosis: a literature review. Curr Med Res Opin. 2006;22:1787‐1797. [DOI] [PubMed] [Google Scholar]
- 8. Gao X, Outley J, Botteman M, Spalding J, Simon JA, Pashos CL. Economic burden of endometriosis. Fertil Steril. 2006;86:1561‐1572. [DOI] [PubMed] [Google Scholar]
- 9. Simoens S, Hummelshoj L, Dunselman G, et al. Endometriosis cost assessment (the EndoCost study): a cost‐of‐illness study protocol. Gynecol Obstet Invest. 2011;71:170‐176. [DOI] [PubMed] [Google Scholar]
- 10. Klein S, D'Hooghe T, Meuleman C, Dirksen C, Dunselman G, Simoens S. What is the societal burden of endometriosis‐associated symptoms? a prospective Belgian study. Reprod Biomed Online. 2014;28:116‐124. [DOI] [PubMed] [Google Scholar]
- 11. Bulun SE. Endometriosis. N Engl J Med. 2009;360:268‐279. [DOI] [PubMed] [Google Scholar]
- 12. Wu Q, Ding D, Liu X, Guo SW. Evidence for a Hypercoagulable State in Women With Ovarian Endometriomas. Reprod Sci. 2015;22:1107‐1114. [DOI] [PubMed] [Google Scholar]
- 13. Zhang Q, Duan J, Liu X, Guo SW. Platelets drive smooth muscle metaplasia and fibrogenesis in endometriosis through epithelial‐mesenchymal transition and fibroblast‐to‐myofibroblast transdifferentiation. Mol Cell Endocrinol. 2016;428:1‐16. [DOI] [PubMed] [Google Scholar]
- 14. Serov SF, Scully RE, Sobin LH. Histological Typing of Ovarian Tumours. International Histological Classification of Tumours. No. 9. Geneva, Switzerland: World Health Organization, 1973. [Google Scholar]
- 15. Gaetje R, Kotzian S, Herrmann G, Baumann R, Starzinski‐Powitz A. Invasiveness of endometriotic cells in vitro. Lancet. 1995;346:1463‐1464. [DOI] [PubMed] [Google Scholar]
- 16. Donnez J, Smoes P, Gillerot S, Casanas‐Roux F, Nisolle M. Vascular endothelial growth factor (VEGF) in endometriosis. Hum Reprod. 1998;13:1686‐1690. [DOI] [PubMed] [Google Scholar]
- 17. Guo SW. Recurrence of endometriosis and its control. Hum Reprod Update. 2009;15:441‐461. [DOI] [PubMed] [Google Scholar]
- 18. Fialkow PJ. Clonal origin of human tumors. Biochim Biophys Acta. 1976;458:283‐321. [DOI] [PubMed] [Google Scholar]
- 19. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23‐28. [DOI] [PubMed] [Google Scholar]
- 20. Nilbert M, Pejovic T, Mandahl N, Iosif S, Willen H, Mitelman F. Monoclonal origin of endometriotic cysts. Int J Gynecol Cancer. 1995;5:61‐63. [DOI] [PubMed] [Google Scholar]
- 21. Wu Y, Basir Z, Kajdacsy‐Balla A, et al. Resolution of clonal origins for endometriotic lesions using laser capture microdissection and the human androgen receptor (HUMARA) assay. Fertil Steril. 2003;79(Suppl 1):710‐717. [DOI] [PubMed] [Google Scholar]
- 22. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 23. Guo SW. Epigenetics of endometriosis. Mol Hum Reprod. 2009;15:587‐607. [DOI] [PubMed] [Google Scholar]
- 24. Jiang X, Hitchcock A, Bryan EJ, et al. Microsatellite analysis of endometriosis reveals loss of heterozygosity at candidate ovarian tumor suppressor gene loci. Cancer Res. 1996;56:3534‐3539. [PubMed] [Google Scholar]
- 25. Ballouk F, Ross JS, Wolf BC. Ovarian endometriotic cysts. An analysis of cytologic atypia and DNA ploidy patterns. Am J Clin Pathol. 1994;102:415‐419. [DOI] [PubMed] [Google Scholar]
- 26. Shin JC, Ross HL, Elias S, et al. Detection of chromosomal aneuploidy in endometriosis by multi‐color fluorescence in situ hybridization (FISH). Hum Genet. 1997;100:401‐406. [DOI] [PubMed] [Google Scholar]
- 27. Kosugi Y, Elias S, Malinak LR, et al. Increased heterogeneity of chromosome 17 aneuploidy in endometriosis. Am J Obstet Gynecol. 1999;180:792‐797. [DOI] [PubMed] [Google Scholar]
- 28. Gogusev J, Bouquet de Joliniere J, Telvi L, et al. Detection of DNA copy number changes in human endometriosis by comparative genomic hybridization. Hum Genet 1999;105:444‐451. [DOI] [PubMed] [Google Scholar]
- 29. Gogusev J, Bouquet de Joliniere J, Telvi L, et al. Genetic abnormalities detected by comparative genomic hybridization in a human endometriosis‐derived cell line. Mol Hum Reprod 2000;6:821‐827. [DOI] [PubMed] [Google Scholar]
- 30. Guo SW, Wu Y, Strawn E, et al. Genomic alterations in the endometrium may be a proximate cause for endometriosis. Eur J Obstet Gynecol Reprod Biol. 2004;116:89‐99. [DOI] [PubMed] [Google Scholar]
- 31. Wu Y, Strawn E, Basir Z, et al. Genomic alterations in ectopic and eutopic endometria of women with endometriosis. Gynecol Obstet Invest. 2006;62:148‐159. [DOI] [PubMed] [Google Scholar]
- 32. Duesberg P, Rausch C, Rasnick D, Hehlmann R. Genetic instability of cancer cells is proportional to their degree of aneuploidy. Proc Natl Acad Sci U S A. 1998;95:13692‐13697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dangel A, Medchill MT, Davis G, Meloni AM, Sandberg AA. Cytogenetic studies in endometriosis tissue. Cancer Genet Cytogenet. 1994;78:172‐174. [DOI] [PubMed] [Google Scholar]
- 34. Vercellini P, Trecca D, Oldani S, Fracchiolla NS, Neri A, Crosignani PG. Analysis of p53 and ras gene mutations in endometriosis. Gynecol Obstet Invest. 1994;38:70‐71. [DOI] [PubMed] [Google Scholar]
- 35. Baranov VS, Ivaschenko T, Bakay B, et al. Proportion of the GSTM1 0/0 genotype in some Slavic populations and its correlation with cystic fibrosis and some multifactorial diseases. Hum Genet. 1996;97:516‐520. [DOI] [PubMed] [Google Scholar]
- 36. Baranova H, Bothorishvilli R, Canis M, et al. Glutathione S‐transferase M1 gene polymorphism and susceptibility to endometriosis in a French population. Mol Hum Reprod. 1997;3:775‐780. [DOI] [PubMed] [Google Scholar]
- 37. Uno S, Zembutsu H, Hirasawa A, et al. A genome‐wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat Genet. 2010;42:707‐710. [DOI] [PubMed] [Google Scholar]
- 38. Rahmioglu N, Macgregor S, Drong AW, et al. Genome‐wide enrichment analysis between endometriosis and obesity‐related traits reveals novel susceptibility loci. Hum Mol Genet. 2015;24:1185‐1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Montgomery GW, Zondervan KT, Nyholt DR. The future for genetic studies in reproduction. Mol Hum Reprod. 2014;20:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jenkins S, Olive DL, Haney AF. Endometriosis: pathogenetic implications of the anatomic distribution. Obstet Gynecol. 1986;67:335‐338. [PubMed] [Google Scholar]
- 41. Gylfason JT, Kristjansson KA, Sverrisdottir G, Jonsdottir K, Rafnsson V, Geirsson RT. Pelvic endometriosis diagnosed in an entire nation over 20 years. Am J Epidemiol. 2010;172:237‐243. [DOI] [PubMed] [Google Scholar]
- 42. Koninckx PR, Martin D. Treatment of deeply infiltrating endometriosis. Curr Opin Obstet Gynecol. 1994;6:231‐241. [PubMed] [Google Scholar]
- 43. Brinton LA, Gridley G, Persson I, Baron J, Bergqvist A. Cancer risk after a hospital discharge diagnosis of endometriosis. Am J Obstet Gynecol. 1997;176:572‐579. [DOI] [PubMed] [Google Scholar]
- 44. Pearce CL, Templeman C, Rossing MA, et al. Association between endometriosis and risk of histological subtypes of ovarian cancer: a pooled analysis of case‐control studies. Lancet Oncol. 2012;13:385‐394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guo SW. Endometriosis and ovarian cancer: potential benefits and harms of screening and risk‐reducing surgery. Fertil Steril. 2015;104:813‐830. [DOI] [PubMed] [Google Scholar]
- 46. Samartzis EP, Samartzis N, Noske A, et al. Loss of ARID1A/BAF250a‐expression in endometriosis: a biomarker for risk of carcinogenic transformation? Mod Pathol. 2012;25:885‐892. [DOI] [PubMed] [Google Scholar]
- 47. Chene G, Ouellet V, Rahimi K, Barres V, Provencher D, Mes‐Masson AM. The ARID1A pathway in ovarian clear cell and endometrioid carcinoma, contiguous endometriosis, and benign endometriosis. Int J Gynaecol Obstet. 2015;130:27‐30. [DOI] [PubMed] [Google Scholar]
- 48. Borrelli GM, Abrao MS, Taube ET, et al. (Partial) Loss of BAF250a (ARID1A) in rectovaginal deep‐infiltrating endometriosis, endometriomas and involved pelvic sentinel lymph nodes. Mol Hum Reprod. 2016;22:329‐337. [DOI] [PubMed] [Google Scholar]
- 49. Stamp JP, Gilks CB, Wesseling M, et al. BAF250a Expression in Atypical Endometriosis and Endometriosis‐Associated Ovarian Cancer. Int J Gynecol Cancer. 2016;26:825‐832. [DOI] [PubMed] [Google Scholar]
- 50. Wiegand KC, Shah SP, Al‐Agha OM, et al. ARID1A mutations in endometriosis‐associated ovarian carcinomas. N Engl J Med. 2010;363:1532‐1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Anglesio MS, Papadopoulos N, Ayhan A, et al. Cancer‐Associated Mutations in Endometriosis without Cancer. N Engl J Med. 2017;376:1835‐1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Muller FL, Colla S, Aquilanti E, et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature. 2012;488:337‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kato S, Lippman SM, Flaherty KT, Kurzrock R. The conundrum of genetic “Drivers” in benign conditions. J Natl Cancer Inst 2016;108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A. 2001;98:12072‐12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. R Development Core Team . R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- 56. Brosens IA. Endometriosis–a disease because it is characterized by bleeding. Am J Obstet Gynecol. 1997;176:263‐267. [DOI] [PubMed] [Google Scholar]
- 57. Ding D, Liu X, Duan J, Guo SW. Platelets are an unindicted culprit in the development of endometriosis: clinical and experimental evidence. Hum Reprod. 2015;30:812‐832. [DOI] [PubMed] [Google Scholar]
- 58. Guo SW, Du Y, Liu X. Platelet‐derived TGF‐beta1 mediates the down‐modulation of NKG2D expression and may be responsible for impaired natural killer (NK) cytotoxicity in women with endometriosis. Hum Reprod. 2016;31:1462‐1474. [DOI] [PubMed] [Google Scholar]
- 59. Du Y, Liu X, Guo SW. Platelets impair natural killer cell reactivity and function in endometriosis through multiple mechanisms. Hum Reprod 2017;32:1‐17. [DOI] [PubMed] [Google Scholar]
- 60. Yan D, Liu X, Guo SW. Endometriosis‐derived thromboxane A2 induces neurite outgrowth. Reprod Sci. 2016;7:6084. [DOI] [PubMed] [Google Scholar]
- 61. Zhang Q, Dong P, Liu X, Sagkuragi N, Guo S‐W. Enhancer of Zeste homolog 2 (EZH2) induces epithelial‐mesenchymal transition in endometriosis. Sci Rep. 2017;7:6084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang Q, Duan J, Olson M, Fazleabas A, Guo SW. Cellular changes consistent with epithelial‐mesenchymal transition and fibroblast‐to‐myofibroblast transdifferentiation in the progression of experimental endometriosis in baboons. Reprod Sci 2016;23:1309‐1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhang Q, Liu X, Guo SW. Progressive development of endometriosis and its hindrance by anti‐platelet treatment in mice with induced endometriosis. Reprod Biomed Online. 2017;34:124‐136. [DOI] [PubMed] [Google Scholar]
- 64. Shen M, Liu X, Zhang H, Guo SW. Transforming growth factor β1 signaling coincides with ‐mediated epithelial‐mesenchymal transition and fibroblast‐to‐myofibroblast transdifferentiation in drive the development of adenomyosis in mice. Hum Reprod 2016;31:355‐369. [DOI] [PubMed] [Google Scholar]
- 65. Liu X, Shen S, Qi Q, Zhang H, Guo S‐W. Corroborating evidence for platelet‐induced epithelial‐mesenchymal transition and fibroblast‐to‐myofibroblast transdifferentiation in the development of adenomyosis. Hum Reprod. 2016;31:734‐749. [DOI] [PubMed] [Google Scholar]
- 66. Bajaj P, Bajaj P, Madsen H, Arendt‐Nielsen L. Endometriosis is associated with central sensitization: a psychophysical controlled study. J Pain. 2003;4:372‐380. [DOI] [PubMed] [Google Scholar]
- 67. He W, Liu X, Zhang Y, Guo SW. Generalized hyperalgesia in women with endometriosis and its resolution following a successful surgery. Reprod Sci. 2010;17:1099‐1111. [DOI] [PubMed] [Google Scholar]
- 68. As‐Sanie S, Harris RE, Napadow V, et al. Changes in regional gray matter volume in women with chronic pelvic pain: a voxel‐based morphometry study. Pain. 2012;153:1006‐1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. As‐Sanie S, Kim J, Schmidt‐Wilcke T, et al. Functional connectivity is associated with altered brain chemistry in women with endometriosis‐associated chronic pelvic pain. J Pain. 2016;17:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wu Y, Kajdacsy‐Balla A, Strawn E, et al. Transcriptional characterizations of differences between eutopic and ectopic endometrium. Endocrinology. 2006;147:232‐246. [DOI] [PubMed] [Google Scholar]
- 71. Liu X, Zhang Q, Guo S‐W. Histological and immunohistochemical characterization of the similarity and difference between ovarian endometriomas and deep infiltrating endometriosis. Reprod Sci. 2018;25:329‐340. [DOI] [PubMed] [Google Scholar]
- 72. Yan D, Liu X, Guo SW. Nerve fibers and endometriotic lesions: partners in crime in inflicting pains in women with endometriosis. Eur J Obstet Gynecol Reprod Biol. 2017;209:14‐24. [DOI] [PubMed] [Google Scholar]
- 73. Krikun G. Endometriosis, angiogenesis and tissue factor. Scientifica (Cairo). 2012;2012:306830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang Q, Duan J, Liu X, Guo SW. Platelets drive smooth muscle metaplasia and fibrogenesis in endometriosis through epithelial‐mesenchymal transition and fibroblast‐to‐myofibroblast transdifferentiation. Mol Cell Endocrinol 2016;428:1‐16. In press. [DOI] [PubMed] [Google Scholar]
- 75. Zeisberg M, Kalluri R. Cellular mechanisms of tissue fibrosis. 1. Common and organ‐specific mechanisms associated with tissue fibrosis. Am J Physiol Cell Physiol. 2013;304:C216‐C225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Khan KN, Masuzaki H, Fujishita A, Kitajima M, Sekine I, Ishimaru T. Differential macrophage infiltration in early and advanced endometriosis and adjacent peritoneum. Fertil Steril. 2004;81:652‐661. [DOI] [PubMed] [Google Scholar]
- 77. Schulke L, Berbic M, Manconi F, Tokushige N, Markham R, Fraser IS. Dendritic cell populations in the eutopic and ectopic endometrium of women with endometriosis. Hum Reprod. 2009;24:1695‐1703. [DOI] [PubMed] [Google Scholar]
- 78. Sheveleva T, Bejenar V, Komlichenko E, Dedul A, Malushko A. Innovative approach in assessing the role of neurogenesis, angiogenesis, and lymphangiogenesis in the pathogenesis of external genital endometriosis. Gynecol Endocrinol. 2016;32:75‐79. [DOI] [PubMed] [Google Scholar]
- 79. Surget S, Khoury MP, Bourdon JC. Uncovering the role of p53 splice variants in human malignancy: a clinical perspective. Onco Targets Ther. 2013;7:57‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15‐16. [DOI] [PubMed] [Google Scholar]
- 81. Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25:304‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Petitjean A, Mathe E, Kato S, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28:622‐629. [DOI] [PubMed] [Google Scholar]
- 83. Jiang X, Morland SJ, Hitchcock A, Thomas EJ, Campbell IG. Allelotyping of endometriosis with adjacent ovarian carcinoma reveals evidence of a common lineage. Cancer Res. 1998;58:1707‐1712. [PubMed] [Google Scholar]
- 84. Bischoff FZ, Heard M, Simpson JL. Somatic DNA alterations in endometriosis: high frequency of chromosome 17 and p53 loss in late‐stage endometriosis. J Reprod Immunol. 2002;55:49‐64. [DOI] [PubMed] [Google Scholar]
- 85. Goumenou AG, Arvanitis DA, Matalliotakis IM, Koumantakis EE, Spandidos DA. Microsatellite DNA assays reveal an allelic imbalance in p16(Ink4), GALT, p53, and APOA2 loci in patients with endometriosis. Fertil Steril. 2001;75:160‐165. [DOI] [PubMed] [Google Scholar]
- 86. Prowse AH, Fakis G, Manek S, et al. Allelic loss studies do not provide evidence for the “endometriosis‐as‐tumor” theory. Fertil Steril. 2005;83(Suppl 1):1134‐1143. [DOI] [PubMed] [Google Scholar]
- 87. Vestergaard AL, Thorup K, Knudsen UB, et al. Oncogenic events associated with endometrial and ovarian cancers are rare in endometriosis. Mol Hum Reprod. 2011;17:758‐761. [DOI] [PubMed] [Google Scholar]
- 88. Nakayama K, Toki T, Zhai YL, et al. Demonstration of focal p53 expression without genetic alterations in endometriotic lesions. Int J Gynecol Pathol. 2001;20:227‐231. [DOI] [PubMed] [Google Scholar]
- 89. Xu B, Hamada S, Kusuki I, Itoh R, Kitawaki J. Possible involvement of loss of heterozygosity in malignant transformation of ovarian endometriosis. Gynecol Oncol. 2011;120:239‐246. [DOI] [PubMed] [Google Scholar]
- 90. Tang FH, Hsieh TH, Hsu CY, et al. KRAS mutation coupled with p53 loss is sufficient to induce ovarian carcinosarcomas in mice. Int J Cancer. 2017;140:1860‐1869. [DOI] [PubMed] [Google Scholar]
- 91. Toki T, Nakayama K. Proliferative activity and genetic alterations in TP53 in endometriosis. Gynecol Obstet Invest. 2000;50(Suppl 1):33‐38. [DOI] [PubMed] [Google Scholar]
- 92. Sainz de la Cuesta R, Izquierdo M, Canamero M, Granizo JJ, Manzarbeitia F. Increased prevalence of p53 overexpression from typical endometriosis to atypical endometriosis and ovarian cancer associated with endometriosis. Eur J Obstet Gynecol Reprod Biol. 2004;113:87‐93. [DOI] [PubMed] [Google Scholar]
- 93. Goumenou A, Panayiotides I, Mahutte NG, Matalliotakis I, Fragouli Y, Arici A. Immunohistochemical expression of p53, MDM2, and p21Waf1 oncoproteins in endometriomas but not adenomyosis. J Soc Gynecol Investig. 2005;12:263‐266. [DOI] [PubMed] [Google Scholar]
- 94. Arimoto T, Katagiri T, Oda K, et al. Genome‐wide cDNA microarray analysis of gene‐expression profiles involved in ovarian endometriosis. Int J Oncol. 2003;22:551‐560. [PubMed] [Google Scholar]
- 95. Dufournet C, Uzan C, Fauvet R, Cortez A, Siffroi JP, Darai E. Expression of apoptosis‐related proteins in peritoneal, ovarian and colorectal endometriosis. J Reprod Immunol. 2006;70:151‐162. [DOI] [PubMed] [Google Scholar]
- 96. Laudanski P, Szamatowicz J, Kowalczuk O, Kuzmicki M, Grabowicz M, Chyczewski L. Expression of selected tumor suppressor and oncogenes in endometrium of women with endometriosis. Hum Reprod. 2009;24:1880‐1890. [DOI] [PubMed] [Google Scholar]
- 97. Schneider J, Jimenez E, Rodriguez F, del Tanago JG. c‐myc, c‐erb‐B2, nm23 and p53 expression in human endometriosis. Oncol Rep. 1998;5:49‐52. [DOI] [PubMed] [Google Scholar]
- 98. Bayramoglu H, Duzcan E. Atypical epithelial changes and mutant p53 gene expression in ovarian endometriosis. Pathol Oncol Res. 2001;7:33‐38. [DOI] [PubMed] [Google Scholar]
- 99. Nezhat F, Cohen C, Rahaman J, Gretz H, Cole P, Kalir T. Comparative immunohistochemical studies of bcl‐2 and p53 proteins in benign and malignant ovarian endometriotic cysts. Cancer. 2002;94:2935‐2940. [DOI] [PubMed] [Google Scholar]
- 100. Bassi MA, Arias V, Filho ND, Gueuvoghlanian‐Silva BY, Abrao MS, Podgaec S. Deep invasive endometriosis lesions of the rectosigmoid may be related to alterations in cell kinetics. Reprod Sci 2015;22:1122‐1128. [DOI] [PubMed] [Google Scholar]
- 101. Allavena G, Carrarelli P, Del Bello B, Luisi S, Petraglia F, Maellaro E. Autophagy is upregulated in ovarian endometriosis: a possible interplay with p53 and heme oxygenase‐1. Fertil Steril. 2015;103:1244‐1251;e1. [DOI] [PubMed] [Google Scholar]
- 102. Charni M, Aloni‐Grinstein R, Molchadsky A, Rotter V. p53 on the crossroad between regeneration and cancer. Cell Death Differ. 2017;24:8‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ying Y, Kim J, Westphal SN, Long KE, Padanilam BJ. Targeted deletion of p53 in the proximal tubule prevents ischemic renal injury. J Am Soc Nephrol. 2014;25:2707‐2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Dagher PC, Mai EM, Hato T, et al. The p53 inhibitor pifithrin‐alpha can stimulate fibrosis in a rat model of ischemic acute kidney injury. Am J Physiol Renal Physiol. 2012;302:F284‐F291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Fukuda R, Suico MA, Kai Y, et al. Podocyte p53 limits the severity of experimental alport syndrome. J Am Soc Nephrol. 2016;27:144‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Tyner SD, Venkatachalam S, Choi J, et al. p53 mutant mice that display early ageing‐associated phenotypes. Nature. 2002;415:45‐53. [DOI] [PubMed] [Google Scholar]
- 107. Adamovich Y, Adler J, Meltser V, Reuven N, Shaul Y. AMPK couples p73 with p53 in cell fate decision. Cell Death Differ. 2014;21:1451‐1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729‐740. [DOI] [PubMed] [Google Scholar]
- 109. Xue W, Zender L, Miething C, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Krizhanovsky V, Yon M, Dickins RA, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Dracxler RC, Oh C, Kalmbach K, et al. Peripheral blood telomere content is greater in patients with endometriosis than in controls. Reprod Sci. 2014;21:1465‐1471. [DOI] [PubMed] [Google Scholar]
- 112. Valentijn AJ, Saretzki G, Tempest N, Critchley HO, Hapangama DK. Human endometrial epithelial telomerase is important for epithelial proliferation and glandular formation with potential implications in endometriosis. Hum Reprod. 2015;30:2816‐2828. [DOI] [PubMed] [Google Scholar]
- 113. Hapangama DK, Turner MA, Drury J, et al. Aberrant expression of regulators of cell‐fate found in eutopic endometrium is found in matched ectopic endometrium among women and in a baboon model of endometriosis. Hum Reprod. 2010;25:2840‐2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Nagpal V, Rai R, Place AT, et al. MiR‐125b is critical for fibroblast‐to‐myofibroblast transition and cardiac fibrosis. Circulation. 2016;133:291‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Ohlsson Teague EM, Van der Hoek KH, Van der Hoek MB, et al. MicroRNA‐regulated pathways associated with endometriosis. Mol Endocrinol. 2009;23:265‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Cosar E, Mamillapalli R, Ersoy GS, Cho S, Seifer B, Taylor HS. Serum microRNAs as diagnostic markers of endometriosis: a comprehensive array‐based analysis. Fertil Steril. 2016;106:402‐409. [DOI] [PubMed] [Google Scholar]
- 117. Wanichkul T, Han S, Huang RP, Sidell N. Cytokine regulation by peroxisome proliferator‐activated receptor gamma in human endometrial cells. Fertil Steril. 2003;79(Suppl 1):763‐769. [DOI] [PubMed] [Google Scholar]
- 118. Hornung D, Klingel K, Dohrn K, Kandolf R, Wallwiener D, Taylor RN. Regulated on activation, normal T‐cell‐expressed and ‐secreted mRNA expression in normal endometrium and endometriotic implants: assessment of autocrine/paracrine regulation by in situ hybridization. Am J Pathol. 2001;158:1949‐1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Lebovic DI, Kavoussi SK, Lee J, Banu SK, Arosh JA. PPARgamma activation inhibits growth and survival of human endometriotic cells by suppressing estrogen biosynthesis and PGE2 signaling. Endocrinology. 2013;154:4803‐4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Marra F, Efsen E, Romanelli RG, et al. Ligands of peroxisome proliferator‐activated receptor gamma modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology. 2000;119:466‐478. [DOI] [PubMed] [Google Scholar]
- 121. Zheng S, Chen A. Activation of PPARgamma is required for curcumin to induce apoptosis and to inhibit the expression of extracellular matrix genes in hepatic stellate cells in vitro. Biochem J. 2004;384:149‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Gan Q, Huang J, Zhou R, et al. PPAR{gamma} accelerates cellular senescence by inducing p16INK4{alpha} expression in human diploid fibroblasts. J Cell Sci. 2008;121:2235‐2245. [DOI] [PubMed] [Google Scholar]
- 123. Lu M, Kwan T, Yu C, et al. Peroxisome proliferator‐activated receptor gamma agonists promote TRAIL‐induced apoptosis by reducing survivin levels via cyclin D3 repression and cell cycle arrest. J Biol Chem. 2005;280:6742‐6751. [DOI] [PubMed] [Google Scholar]
- 124. Kon K, Ikejima K, Hirose M, et al. Pioglitazone prevents early‐phase hepatic fibrogenesis caused by carbon tetrachloride. Biochem Biophys Res Commun. 2002;291:55‐61. [DOI] [PubMed] [Google Scholar]
- 125. Mann J, Chu DC, Maxwell A, et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 2010;138:705‐714, 14; e1‐4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Dantas AT, Pereira MC, de Melo Rego MJ, et al. The role of PPAR gamma in systemic sclerosis. PPAR Res. 2015;2015:124624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Chen Q, Zheng PS, Yang WT. EZH2‐mediated repression of GSK‐3beta and TP53 promotes Wnt/beta‐catenin signaling‐dependent cell expansion in cervical carcinoma. Oncotarget. 2016;7:36115‐36129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Matsuzaki S, Darcha C. Involvement of the Wnt/beta‐catenin signaling pathway in the cellular and molecular mechanisms of fibrosis in endometriosis. PLoS ONE. 2013;8:e76808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Li J, Dai Y, Zhu H, Jiang Y, Zhang S. Endometriotic mesenchymal stem cells significantly promote fibrogenesis in ovarian endometrioma through the Wnt/beta‐catenin pathway by paracrine production of TGF‐beta1 and Wnt1. Hum Reprod. 2016;31:1224‐1235. [DOI] [PubMed] [Google Scholar]
- 130. Yang Y, Chen XX, Li WX, et al. EZH2‐mediated repression of Dkk1 promotes hepatic stellate cell activation and hepatic fibrosis. J Cell Mol Med 2017;21:2317‐2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Pabona JM, Simmen FA, Nikiforov MA, et al. Kruppel‐like factor 9 and progesterone receptor coregulation of decidualizing endometrial stromal cells: implications for the pathogenesis of endometriosis. J Clin Endocrinol Metab. 2012;97:E376‐E392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Tovy A, Spiro A, McCarthy R, et al. p53 is essential for DNA methylation homeostasis in naive embryonic stem cells, and its loss promotes clonal heterogeneity. Genes Dev. 2017;31:959‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Wu Y, Strawn E, Basir Z, Halverson G, Guo SW. Aberrant expression of deoxyribonucleic acid methyltransferases DNMT1, DNMT3A, and DNMT3B in women with endometriosis. Fertil Steril. 2007;87:24‐32. [DOI] [PubMed] [Google Scholar]
- 134. Dyson MT, Kakinuma T, Pavone ME, et al. Aberrant expression and localization of deoxyribonucleic acid methyltransferase 3B in endometriotic stromal cells. Fertil Steril. 2015;104:953‐963; e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Roca FJ, Loomans HA, Wittman AT, Creighton CJ, Hawkins SM. Ten‐eleven translocation genes are downregulated in endometriosis. Curr Mol Med. 2016;16:288‐298. [DOI] [PubMed] [Google Scholar]
- 136. Geraldes C, Goncalves AC, Cortesao E, et al. Aberrant p15, p16, p53, and DAPK gene methylation in myelomagenesis: clinical and prognostic implications. Clin Lymphoma Myeloma Leuk. 2016;16:713‐720;e2. [DOI] [PubMed] [Google Scholar]
- 137. Yang H, Kang K, Cheng C, Mamillapalli R, Taylor HS. Integrative analysis reveals regulatory programs in endometriosis. Reprod Sci 2015;22:1060‐1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc Natl Acad Sci U S A. 1997;94:10895‐10900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Reme T, Travaglio A, Gueydon E, Adla L, Jorgensen C, Sany J. Mutations of the p53 tumour suppressor gene in erosive rheumatoid synovial tissue. Clin Exp Immunol. 1998;111:353‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Yin Y, Shen WH. PTEN: a new guardian of the genome. Oncogene. 2008;27:5443‐5453. [DOI] [PubMed] [Google Scholar]
- 141. Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215‐221. [DOI] [PubMed] [Google Scholar]
- 142. Di Cristofano A, Pesce B, Cordon‐Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet 1998;19:348‐355. [DOI] [PubMed] [Google Scholar]
- 143. Abreu JP, Rebelatto CLK, Savari CA, et al. The effect of mesenchymal stem cells on fertility in experimental retrocervical endometriosis. Rev Bras Ginecol Obstet. 2017;39:217‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Puc J, Keniry M, Li HS, et al. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell. 2005;7:193‐204. [DOI] [PubMed] [Google Scholar]
- 145. Puc J, Parsons R. PTEN loss inhibits CHK1 to cause double stranded‐DNA breaks in cells. Cell Cycle. 2005;4:927‐929. [DOI] [PubMed] [Google Scholar]
- 146. Obata K, Hoshiai H. Common genetic changes between endometriosis and ovarian cancer. Gynecol Obstet Invest. 2000;50(Suppl 1):39‐43. [DOI] [PubMed] [Google Scholar]
- 147. Sato N, Tsunoda H, Nishida M, et al. Loss of heterozygosity on 10q23.3 and mutation of the tumor suppressor gene PTEN in benign endometrial cyst of the ovary: possible sequence progression from benign endometrial cyst to endometrioid carcinoma and clear cell carcinoma of the ovary. Cancer Res. 2000;60:7052‐7056. [PubMed] [Google Scholar]
- 148. Govatati S, Kodati VL, Deenadayal M, Chakravarty B, Shivaji S, Bhanoori M. Mutations in the PTEN tumor gene and risk of endometriosis: a case‐control study. Hum Reprod. 2014;29:324‐336. [DOI] [PubMed] [Google Scholar]
- 149. Obata K, Morland SJ, Watson RH, et al. Frequent PTEN/MMAC mutations in endometrioid but not serous or mucinous epithelial ovarian tumors. Cancer Res. 1998;58:2095‐2097. [PubMed] [Google Scholar]
- 150. Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K‐ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med. 2005;11:63‐70. [DOI] [PubMed] [Google Scholar]
- 151. Martini M, Ciccarone M, Garganese G, et al. Possible involvement of hMLH1, p16(INK4a) and PTEN in the malignant transformation of endometriosis. Int J Cancer. 2002;102:398‐406. [DOI] [PubMed] [Google Scholar]
- 152. Zhang H, Zhao X, Liu S, Li J, Wen Z, Li M. 17betaE2 promotes cell proliferation in endometriosis by decreasing PTEN via NFkappaB‐dependent pathway. Mol Cell Endocrinol. 2010;317:31‐43. [DOI] [PubMed] [Google Scholar]
- 153. Rai P, Shivaji S. The role of DJ‐1 in the pathogenesis of endometriosis. PLoS ONE. 2011;6:e18074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Li MQ, Luo XZ, Meng YH, et al. CXCL8 enhances proliferation and growth and reduces apoptosis in endometrial stromal cells in an autocrine manner via a CXCR1‐triggered PTEN/AKT signal pathway. Hum Reprod. 2012;27:2107‐2116. [DOI] [PubMed] [Google Scholar]
- 155. Kim TH, Yu Y, Luo L, Lydon JP, Jeong JW, Kim JJ. Activated AKT pathway promotes establishment of endometriosis. Endocrinology. 2014;155:1921‐1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Lv J, Zhu Q, Jia X, Yu N, Li Q. In vitro and in vivo effects of tumor suppressor gene PTEN on endometriosis: an experimental study. Med Sci Monit. 2016;22:3727‐3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Shaw TJ, Martin P. Wound repair at a glance. J Cell Sci. 2009;122:3209‐3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C. Alpha‐smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001;12:2730‐2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005;13:7‐12. [DOI] [PubMed] [Google Scholar]
- 160. Nho RS, Xia H, Diebold D, et al. PTEN regulates fibroblast elimination during collagen matrix contraction. J Biol Chem. 2006;281:33291‐33301. [DOI] [PubMed] [Google Scholar]
- 161. White ES, Atrasz RG, Hu B, et al. Negative regulation of myofibroblast differentiation by PTEN (Phosphatase and Tensin Homolog Deleted on chromosome 10). Am J Respir Crit Care Med. 2006;173:112‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Geng J, Huang X, Li Y, et al. Phosphatase and tensin homolog deleted on chromosome 10 contributes to phenotype transformation of fibroblasts in idiopathic pulmonary fibrosis via multiple pathways. Exp Biol Med (Maywood). 2016;241:157‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Kimura M, Hashimoto N, Kusunose M, et al. Exogenous induction of unphosphorylated PTEN reduces TGFbeta‐induced extracellular matrix expressions in lung fibroblasts. Wound Repair Regen. 2017;25:86‐97. [DOI] [PubMed] [Google Scholar]
- 164. Samarakoon R, Helo S, Dobberfuhl AD, et al. Loss of tumour suppressor PTEN expression in renal injury initiates SMAD3‐ and p53‐dependent fibrotic responses. J Pathol. 2015;236:421‐432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. An J, Zheng L, Xie S, et al. Regulatory effects and mechanism of adenovirus‐mediated PTEN gene on hepatic stellate cells. Dig Dis Sci. 2016;61:1107‐1120. [DOI] [PubMed] [Google Scholar]
- 166. Cheng Y, Tian Y, Xia J, et al. The role of PTEN in regulation of hepatic macrophages activation and function in progression and reversal of liver fibrosis. Toxicol Appl Pharmacol. 2017;317:51‐62. [DOI] [PubMed] [Google Scholar]
- 167. Kattla JJ, Carew RM, Heljic M, Godson C, Brazil DP. Protein kinase B/Akt activity is involved in renal TGF‐beta1‐driven epithelial‐mesenchymal transition in vitro and in vivo. Am J Physiol Renal Physiol. 2008;295:F215‐F225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Takashima M, Parsons CJ, Ikejima K, Watanabe S, White ES, Rippe RA. The tumor suppressor protein PTEN inhibits rat hepatic stellate cell activation. J Gastroenterol. 2009;44:847‐855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Chung EJ, Lee HK, Jung SA, et al. Transduction of PTEN proteins using the tat domain modulates TGF‐beta1‐mediated signaling pathways and transdifferentiation in subconjunctival fibroblasts. Invest Ophthalmol Vis Sci. 2012;53:379‐386. [DOI] [PubMed] [Google Scholar]
- 170. Xia H, Diebold D, Nho R, et al. Pathological integrin signaling enhances proliferation of primary lung fibroblasts from patients with idiopathic pulmonary fibrosis. J Exp Med. 2008;205:1659‐1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Hao LS, Zhang XL, An JY, et al. PTEN expression is down‐regulated in liver tissues of rats with hepatic fibrosis induced by biliary stenosis. APMIS. 2009;117:681‐691. [DOI] [PubMed] [Google Scholar]
- 172. Parapuram SK, Shi‐wen X, Elliott C, et al. Loss of PTEN expression by dermal fibroblasts causes skin fibrosis. J Invest Dermatol. 2011;131:1996‐2003. [DOI] [PubMed] [Google Scholar]
- 173. Guo L, Chen L, Bi S, et al. PTEN inhibits proliferation and functions of hypertrophic scar fibroblasts. Mol Cell Biochem. 2012;361:161‐168. [DOI] [PubMed] [Google Scholar]
- 174. Lan R, Geng H, Polichnowski AJ, et al. PTEN loss defines a TGF‐beta‐induced tubule phenotype of failed differentiation and JNK signaling during renal fibrosis. Am J Physiol Renal Physiol. 2012;302:F1210‐F1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Angadi PV, Krishnapillai R. Evaluation of PTEN immunoexpression in oral submucous fibrosis: role in pathogenesis and malignant transformation. Head Neck Pathol. 2012;6:314‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Nho RS, Hergert P, Kahm J, Jessurun J, Henke C. Pathological alteration of FoxO3a activity promotes idiopathic pulmonary fibrosis fibroblast proliferation on type i collagen matrix. Am J Pathol. 2011;179:2420‐2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Nho RS, Peterson M, Hergert P, Henke CA. FoxO3a (Forkhead Box O3a) deficiency protects Idiopathic Pulmonary Fibrosis (IPF) fibroblasts from type I polymerized collagen matrix‐induced apoptosis via caveolin‐1 (cav‐1) and Fas. PLoS ONE. 2013;8:e61017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Xia H, Khalil W, Kahm J, Jessurun J, Kleidon J, Henke CA. Pathologic caveolin‐1 regulation of PTEN in idiopathic pulmonary fibrosis. Am J Pathol. 2010;176:2626‐2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Gao Y, Chu M, Hong J, Shang J, Xu D. Hypoxia induces cardiac fibroblast proliferation and phenotypic switch: a role for caveolae and caveolin‐1/PTEN mediated pathway. J Thorac Dis. 2014;6:1458‐1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Yao Y, Wei H, Liu L, et al. Upregulated DJ‐1 promotes renal tubular EMT by suppressing cytoplasmic PTEN expression and Akt activation. J Huazhong Univ Sci Technolog Med Sci. 2011;31:469‐475. [DOI] [PubMed] [Google Scholar]
- 181. Kumarswamy R, Volkmann I, Jazbutyte V, Dangwal S, Park DH, Thum T. Transforming growth factor‐beta‐induced endothelial‐to‐mesenchymal transition is partly mediated by microRNA‐21. Arterioscler Thromb Vasc Biol. 2012;32:361‐369. [DOI] [PubMed] [Google Scholar]
- 182. Roy S, Khanna S, Hussain SR, et al. MicroRNA expression in response to murine myocardial infarction: miR‐21 regulates fibroblast metalloprotease‐2 via phosphatase and tensin homologue. Cardiovasc Res. 2009;82:21‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Dey N, Ghosh‐Choudhury N, Kasinath BS, Choudhury GG. TGFbeta‐stimulated microRNA‐21 utilizes PTEN to orchestrate AKT/mTORC1 signaling for mesangial cell hypertrophy and matrix expansion. PLoS ONE. 2012;7:e42316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Wei J, Feng L, Li Z, Xu G, Fan X. MicroRNA‐21 activates hepatic stellate cells via PTEN/Akt signaling. Biomed Pharmacother. 2013;67:387‐392. [DOI] [PubMed] [Google Scholar]
- 185. Zhou Y, Xiong M, Fang L, et al. miR‐21‐containing microvesicles from injured tubular epithelial cells promote tubular phenotype transition by targeting PTEN protein. Am J Pathol. 2013;183:1183‐1196. [DOI] [PubMed] [Google Scholar]
- 186. Liu Y, Wang X, Yang D, Xiao Z, Chen X. MicroRNA‐21 affects proliferation and apoptosis by regulating expression of PTEN in human keloid fibroblasts. Plast Reconstr Surg. 2014;134:561e‐573e. [DOI] [PubMed] [Google Scholar]
- 187. Shen W, Chen G, Dong R, Zhao R, Zheng S. MicroRNA‐21/PTEN/Akt axis in the fibrogenesis of biliary atresia. J Pediatr Surg. 2014;49:1738‐1741. [DOI] [PubMed] [Google Scholar]
- 188. Liu Z, Wang J, Guo C, Fan X. microRNA‐21 mediates epithelial‐mesenchymal transition of human hepatocytes via PTEN/Akt pathway. Biomed Pharmacother. 2015;69:24‐28. [DOI] [PubMed] [Google Scholar]
- 189. Zhao L, Zhou S, Zou L, Zhao X. The expression and functionality of stromal caveolin 1 in human adenomyosis. Hum Reprod. 2013;28:1324‐1338. [DOI] [PubMed] [Google Scholar]
- 190. Guo J, Gao J, Yu X, Luo H, Xiong X, Huang O. Expression of DJ‐1 and mTOR in eutopic and ectopic endometria of patients with endometriosis and adenomyosis. Gynecol Obstet Invest. 2015;79:195‐200. [DOI] [PubMed] [Google Scholar]
- 191. Braza‐Boils A, Salloum‐Asfar S, Mari‐Alexandre J, et al. Peritoneal fluid modifies the microRNA expression profile in endometrial and endometriotic cells from women with endometriosis. Hum Reprod. 2015;30:2292‐2302. [DOI] [PubMed] [Google Scholar]
- 192. Park JH, Lee SK, Kim MK, et al. Saponin extracts induced apoptosis of endometrial cells from women with endometriosis through modulation of miR‐21‐5p. Reprod Sci. 2018;25:292‐301. [DOI] [PubMed] [Google Scholar]
- 193. Monsivais D, Dyson MT, Yin P, et al. Estrogen receptor beta regulates endometriotic cell survival through serum and glucocorticoid‐regulated kinase activation. Fertil Steril 2016;105:1266‐1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Liu S, Parapuram SK, Leask A. Fibrosis caused by loss of PTEN expression in mouse fibroblasts is crucially dependent on CCN2. Arthritis Rheum. 2013;65:2940‐2944. [DOI] [PubMed] [Google Scholar]
- 195. Parapuram SK, Thompson K, Tsang M, et al. Loss of PTEN expression by mouse fibroblasts results in lung fibrosis through a CCN2‐dependent mechanism. Matrix Biol. 2015;43:35‐41. [DOI] [PubMed] [Google Scholar]
- 196. Eyster KM, Hansen KA, Winterton E, Klinkova O, Drappeau D, Mark‐Kappeler CJ. Reciprocal communication between endometrial stromal cells and macrophages. Reprod Sci. 2010;17:809‐822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Matsuzaki S, Darcha C. Antifibrotic properties of epigallocatechin‐3‐gallate in endometriosis. Hum Reprod. 2014;29:1677‐1687. [DOI] [PubMed] [Google Scholar]
- 198. Bian EB, Huang C, Ma TT, et al. DNMT1‐mediated PTEN hypermethylation confers hepatic stellate cell activation and liver fibrogenesis in rats. Toxicol Appl Pharmacol. 2012;264:13‐22. [DOI] [PubMed] [Google Scholar]
- 199. Zhou X, Zang X, Ponnusamy M, et al. Enhancer of zeste homolog 2 inhibition attenuates renal fibrosis by maintaining smad7 and phosphatase and tensin homolog expression. J Am Soc Nephrol. 2016;27:2092‐2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Lei I, Gao X, Sham MH, Wang Z. SWI/SNF protein component BAF250a regulates cardiac progenitor cell differentiation by modulating chromatin accessibility during second heart field development. J Biol Chem. 2012;287:24255‐24262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Lei I, West J, Yan Z, et al. BAF250a protein regulates nucleosome occupancy and histone modifications in priming embryonic stem cell differentiation. J Biol Chem. 2015;290:19343‐19352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Jones S, Wang TL, Shih Ie M, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Wiegand KC, Lee AF, Al‐Agha OM, et al. Loss of BAF250a (ARID1A) is frequent in high‐grade endometrial carcinomas. J Pathol. 2011;224:328‐333. [DOI] [PubMed] [Google Scholar]
- 205. Lakshminarasimhan R, Andreu‐Vieyra C, Lawrenson K, et al. Down‐regulation of ARID1A is sufficient to initiate neoplastic transformation along with epigenetic reprogramming in non‐tumorigenic endometriotic cells. Cancer Lett. 2017;401:11‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Winarto H, Tan MI, Sadikin M, Wanandi SI. ARID1A expression is down‐regulated by oxidative stress in endometriosis and endometriosis‐associated ovarian cancer. Transl Oncogenomics. 2017;9:1177272716689818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Levin M. Endogenous bioelectrical networks store non‐genetic patterning information during development and regeneration. J Physiol. 2014;592:2295‐2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Sun X, Chuang JC, Kanchwala M, et al. Suppression of the SWI/SNF component arid1a promotes mammalian regeneration. Cell Stem Cell. 2016;18:456‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Christodoulou C, Longmire TA, Shen SS, et al. Mouse ES and iPS cells can form similar definitive endoderm despite differences in imprinted genes. J Clin Invest. 2011;121:2313‐2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Kim TH, Yoo JY, Wang Z, et al. ARID1A Is Essential for Endometrial Function during Early Pregnancy. PLoS Genet. 2015;11:e1005537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Hsieh CJ, Klump B, Holzmann K, Borchard F, Gregor M, Porschen R. Hypermethylation of the p16INK4a promoter in colectomy specimens of patients with long‐standing and extensive ulcerative colitis. Cancer Res. 1998;58:3942‐3945. [PubMed] [Google Scholar]
- 212. Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA. Accelerated age‐related CpG island methylation in ulcerative colitis. Cancer Res. 2001;61:3573‐3577. [PubMed] [Google Scholar]
- 213. Song JZ, Stirzaker C, Harrison J, Melki JR, Clark SJ. Hypermethylation trigger of the glutathione‐S‐transferase gene (GSTP1) in prostate cancer cells. Oncogene. 2002;21:1048‐1061. [DOI] [PubMed] [Google Scholar]
- 214. Stirzaker C, Song JZ, Davidson B, Clark SJ. Transcriptional gene silencing promotes DNA hypermethylation through a sequential change in chromatin modifications in cancer cells. Cancer Res. 2004;64:3871‐3877. [DOI] [PubMed] [Google Scholar]
- 215. Ding D, Liu X, Guo SW. Overexpression of lysine‐specific demethylase 1 in ovarian endometriomas and its inhibition reduces cellular proliferation, cell cycle progression, and invasiveness. Fertil Steril. 2014;101:740‐749. [DOI] [PubMed] [Google Scholar]
- 216. Sun Q, Ding D, Liu X, Guo SW. Tranylcypromine, a lysine‐specific demethylase 1 (LSD1) inhibitor, suppresses lesion growth and improves generalized hyperalgesia in mouse with induced endometriosis. Reprod Biol Endocrinol. 2016;14:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Kawano Y, Nasu K, Hijiya N, et al. CCAAT/enhancer‐binding protein alpha is epigenetically silenced by histone deacetylation in endometriosis and promotes the pathogenesis of endometriosis. J Clin Endocrinol Metab. 2013;98:E1474‐E1482. [DOI] [PubMed] [Google Scholar]
- 218. Karakas B, Bachman KE, Park BH. Mutation of the PIK3CA oncogene in human cancers. Br J Cancer. 2006;94:455‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Sulis ML, Parsons R. PTEN: from pathology to biology. Trends Cell Biol. 2003;13:478‐483. [DOI] [PubMed] [Google Scholar]
- 220. Mayer IA, Arteaga CL. PIK3CA activating mutations: a discordant role in early versus advanced hormone‐dependent estrogen receptor‐positive breast cancer? J Clin Oncol. 2014;32:2932‐2934. [DOI] [PubMed] [Google Scholar]
- 221. Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988‐1004. [DOI] [PubMed] [Google Scholar]
- 222. Janku F, Hong DS, Fu S, et al. Assessing PIK3CA and PTEN in early‐phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep. 2014;6:377‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Yamamoto S, Tsuda H, Takano M, Iwaya K, Tamai S, Matsubara O. PIK3CA mutation is an early event in the development of endometriosis‐associated ovarian clear cell adenocarcinoma. J Pathol. 2011;225:189‐194. [DOI] [PubMed] [Google Scholar]
- 224. Matsumoto T, Yamazaki M, Takahashi H, et al. Distinct beta‐catenin and PIK3CA mutation profiles in endometriosis‐associated ovarian endometrioid and clear cell carcinomas. Am J Clin Pathol. 2015;144:452‐463. [DOI] [PubMed] [Google Scholar]
- 225. Grund EM, Kagan D, Tran CA, et al. Tumor necrosis factor‐alpha regulates inflammatory and mesenchymal responses via mitogen‐activated protein kinase kinase, p38, and nuclear factor kappaB in human endometriotic epithelial cells. Mol Pharmacol. 2008;73:1394‐1404. [DOI] [PubMed] [Google Scholar]
- 226. Honda H, Barrueto FF, Gogusev J, Im DD, Morin PJ. Serial analysis of gene expression reveals differential expression between endometriosis and normal endometrium. Possible roles for AXL and SHC1 in the pathogenesis of endometriosis. Reprod Biol Endocrinol. 2008;6:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Yin X, Pavone ME, Lu Z, Wei J, Kim JJ. Increased activation of the PI3K/AKT pathway compromises decidualization of stromal cells from endometriosis. J Clin Endocrinol Metab. 2012;97:E35‐E43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Choi J, Jo M, Lee E, Lee DY, Choi D. Dienogest enhances autophagy induction in endometriotic cells by impairing activation of AKT, ERK1/2, and mTOR. Fertil Steril 2015;104:655‐664.e1. [DOI] [PubMed] [Google Scholar]
- 229. Zhou Y, Zeng C, Li X, et al. IGF‐I stimulates ERbeta and aromatase expression via IGF1R/PI3K/AKT‐mediated transcriptional activation in endometriosis. J Mol Med (Berl). 2016;94:887‐897. [DOI] [PubMed] [Google Scholar]
- 230. Young VJ, Brown JK, Maybin J, Saunders PT, Duncan WC, Horne AW. Transforming growth factor‐beta induced Warburg‐like metabolic reprogramming may underpin the development of peritoneal endometriosis. J Clin Endocrinol Metab. 2014;99:3450‐3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Wolters KL, Ang D, Warrick A, Beadling C, Corless CL, Troxell ML. Frequent PIK3CA mutations in radial scars. Diagn Mol Pathol. 2013;22:210‐214. [DOI] [PubMed] [Google Scholar]
- 232. Lindhurst MJ, Parker VE, Payne F, et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet. 2012;44:928‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Zhang XF, Tan X, Zeng G, et al. Conditional beta‐catenin loss in mice promotes chemical hepatocarcinogenesis: role of oxidative stress and platelet‐derived growth factor receptor alpha/phosphoinositide 3‐kinase signaling. Hepatology. 2010;52:954‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Liu H, Mi S, Li Z, Hua F, Hu ZW. Interleukin 17A inhibits autophagy through activation of PIK3CA to interrupt the GSK3B‐mediated degradation of BCL2 in lung epithelial cells. Autophagy. 2013;9:730‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Takai Y, Sasaki T, Matozaki T. Small GTP‐binding proteins. Physiol Rev. 2001;81:153‐208. [DOI] [PubMed] [Google Scholar]
- 236. Martinez‐Salgado C, Rodriguez‐Pena AB, Lopez‐Novoa JM. Involvement of small Ras GTPases and their effectors in chronic renal disease. Cell Mol Life Sci. 2008;65:477‐492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Rojas JM, Santos E. Ras genes and human cancer: Different implications and different roles. Curr Genomics. 2002;3:295‐311. [Google Scholar]
- 238. Baselga J. The EGFR as a target for anticancer therapy–focus on cetuximab. Eur J Cancer. 2001;37(Suppl 4):S16‐S22. [DOI] [PubMed] [Google Scholar]
- 239. Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25:272‐281. [DOI] [PubMed] [Google Scholar]
- 240. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014;13:828‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Karapetis CS, Khambata‐Ford S, Jonker DJ, et al. K‐ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med. 2008;359:1757‐1765. [DOI] [PubMed] [Google Scholar]
- 242. Santibanez JF, Guerrero J, Quintanilla M, Fabra A, Martinez J. Transforming growth factor‐beta1 modulates matrix metalloproteinase‐9 production through the Ras/MAPK signaling pathway in transformed keratinocytes. Biochem Biophys Res Commun. 2002;296:267‐273. [DOI] [PubMed] [Google Scholar]
- 243. Guo DF, Sun YL, Hamet P, Inagami T. The angiotensin II type 1 receptor and receptor‐associated proteins. Cell Res. 2001;11:165‐180. [DOI] [PubMed] [Google Scholar]
- 244. Robin P, Boulven I, Bole‐Feysot C, Tanfin Z, Leiber D. Contribution of PKC‐dependent and ‐independent processes in temporal ERK regulation by ET‐1, PDGF, and EGF in rat myometrial cells. Am J Physiol Cell Physiol. 2004;286:C798‐C806. [DOI] [PubMed] [Google Scholar]
- 245. Wang L, Proud CG. Regulation of the phosphorylation of elongation factor 2 by MEK‐dependent signalling in adult rat cardiomyocytes. FEBS Lett. 2002;531:285‐289. [DOI] [PubMed] [Google Scholar]
- 246. Che W, Abe J, Yoshizumi M, et al. p160 Bcr mediates platelet‐derived growth factor activation of extracellular signal‐regulated kinase in vascular smooth muscle cells. Circulation. 2001;104:1399‐1406. [DOI] [PubMed] [Google Scholar]
- 247. Lin CC, Shyr MH, Chien CS, et al. Thrombin‐stimulated cell proliferation mediated through activation of Ras/Raf/MEK/MAPK pathway in canine cultured tracheal smooth muscle cells. Cell Signal. 2002;14:265‐275. [DOI] [PubMed] [Google Scholar]
- 248. Loverro G, Maiorano E, Napoli A, Selvaggi L, Marra E, Perlino E. Transforming growth factor‐beta 1 and insulin‐like growth factor‐1 expression in ovarian endometriotic cysts: a preliminary study. Int J Mol Med. 2001;7:423‐429. [DOI] [PubMed] [Google Scholar]
- 249. Nakao T, Chishima F, Sugitani M, Tsujimura R, Hayashi C, Yamamoto T. Expression of angiotensin II types 1 and 2 receptors in endometriotic lesions. Gynecol Obstet Invest. 2017;82:294‐302. [DOI] [PubMed] [Google Scholar]
- 250. Rakhila H, Al‐Akoum M, Bergeron ME, et al. Promotion of angiogenesis and proliferation cytokines patterns in peritoneal fluid from women with endometriosis. J Reprod Immunol. 2016;116:1‐6. [DOI] [PubMed] [Google Scholar]
- 251. Guo SW, Du Y, Liu X. Endometriosis‐derived stromal cells secrete thrombin and thromboxane A2, inducing platelet activation. Reprod Sci. 2016;23:1044‐1052. [DOI] [PubMed] [Google Scholar]
- 252. Yotova IY, Quan P, Leditznig N, Beer U, Wenzl R, Tschugguel W. Abnormal activation of Ras/Raf/MAPK and RhoA/ROCKII signalling pathways in eutopic endometrial stromal cells of patients with endometriosis. Hum Reprod. 2011;26:885‐897. [DOI] [PubMed] [Google Scholar]
- 253. Farahani MS, Shahbazi S, Moghaddam SA, Mahdian R. Evaluation of KRAS Gene Expression and LCS6 Variant in Genomic and Cell‐Free DNA of Iranian Women With Endometriosis. Reprod Sci. 2015;22:679‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Shahrabi‐Farahani M, Shahbazi S, Mahdian R, Amini‐Moghaddam S. K‐Ras 4A Transcript variant is up‐regulated in eutopic endometrium of endometriosis patients during proliferative phase of menstrual cycle. Arch Gynecol Obstet. 2015;292:225‐229. [DOI] [PubMed] [Google Scholar]
- 255. Cheng CW, Licence D, Cook E, et al. Activation of mutated K‐ras in donor endometrial epithelium and stroma promotes lesion growth in an intact immunocompetent murine model of endometriosis. J Pathol. 2011;224:261‐269. [DOI] [PubMed] [Google Scholar]
- 256. Stewart CJ, Leung Y, Walsh MD, Walters RJ, Young JP, Buchanan DD. KRAS mutations in ovarian low‐grade endometrioid adenocarcinoma: association with concurrent endometriosis. Hum Pathol. 2012;43:1177‐1183. [DOI] [PubMed] [Google Scholar]
- 257. Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the Erk pathway is required for TGF‐beta1‐induced EMT in vitro. Neoplasia. 2004;6:603‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial‐mesenchymal transitions in development and disease. Cell. 2009;139:871‐890. [DOI] [PubMed] [Google Scholar]
- 259. Janda E, Lehmann K, Killisch I, et al. Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signaling pathways. J Cell Biol. 2002;156:299‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Rhyu DY, Yang Y, Ha H, et al. Role of reactive oxygen species in TGF‐beta1‐induced mitogen‐activated protein kinase activation and epithelial‐mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol. 2005;16:667‐675. [DOI] [PubMed] [Google Scholar]
- 261. Hocevar BA, Brown TL, Howe PH. TGF‐beta induces fibronectin synthesis through a c‐Jun N‐terminal kinase‐dependent, Smad4‐independent pathway. EMBO J. 1999;18:1345‐1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Yoshino O, Osuga Y, Hirota Y, et al. Possible pathophysiological roles of mitogen‐activated protein kinases (MAPKs) in endometriosis. Am J Reprod Immunol. 2004;52:306‐311. [DOI] [PubMed] [Google Scholar]
- 263. Ngo C, Nicco C, Leconte M, et al. Protein kinase inhibitors can control the progression of endometriosis in vitro and in vivo. J Pathol. 2010;222:148‐157. [DOI] [PubMed] [Google Scholar]
- 264. Lee DH, Kim SC, Joo JK, et al. Effects of 17beta‐estradiol on the release of monocyte chemotactic protein‐1 and MAPK activity in monocytes stimulated with peritoneal fluid from endometriosis patients. J Obstet Gynaecol Res. 2012;38:516‐525. [DOI] [PubMed] [Google Scholar]
- 265. Oh HK, Choi YS, Yang YI, Kim JH, Leung PC, Choi JH. Leptin receptor is induced in endometriosis and leptin stimulates the growth of endometriotic epithelial cells through the JAK2/STAT3 and ERK pathways. Mol Hum Reprod. 2013;19:160‐168. [DOI] [PubMed] [Google Scholar]
- 266. Sui C, Mecha E, Omwandho CO, et al. PAI‐1 secretion of endometrial and endometriotic cells is Smad2/3‐ and ERK1/2‐dependent and influences cell adhesion. Am J Transl Res. 2016;8:2394‐2402. [PMC free article] [PubMed] [Google Scholar]
- 267. McKinnon BD, Kocbek V, Nirgianakis K, Bersinger NA, Mueller MD. Kinase signalling pathways in endometriosis: potential targets for non‐hormonal therapeutics. Hum Reprod Update 2016;22:382‐403. [DOI] [PubMed] [Google Scholar]
- 268. Shields MA, Ebine K, Sahai V, et al. Snail cooperates with KrasG12D to promote pancreatic fibrosis. Mol Cancer Res. 2013;11:1078‐1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Krantz SB, Shields MA, Dangi‐Garimella S, et al. MT1‐MMP cooperates with Kras(G12D) to promote pancreatic fibrosis through increased TGF‐beta signaling. Mol Cancer Res. 2011;9:1294‐1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Osuga Y, Koga K, Tsutsumi O, et al. Stem cell factor (SCF) concentrations in peritoneal fluid of women with or without endometriosis. Am J Reprod Immunol. 2000;44:231‐235. [DOI] [PubMed] [Google Scholar]
- 271. Pacchiarotti A, Caserta D, Sbracia M, Moscarini M. Expression of oct‐4 and c‐kit antigens in endometriosis. Fertil Steril. 2011;95:1171‐1173. [DOI] [PubMed] [Google Scholar]
- 272. Konno R, Yamada‐Okabe H, Fujiwara H, et al. Role of immunoreactions and mast cells in pathogenesis of human endometriosis–morphologic study and gene expression analysis. Hum Cell. 2003;16:141‐149. [DOI] [PubMed] [Google Scholar]
- 273. Sugamata M, Ihara T, Uchiide I. Increase of activated mast cells in human endometriosis. Am J Reprod Immunol. 2005;53:120‐125. [DOI] [PubMed] [Google Scholar]
- 274. Anaf V, Chapron C, El Nakadi I, De Moor V, Simonart T, Noel JC. Pain, mast cells, and nerves in peritoneal, ovarian, and deep infiltrating endometriosis. Fertil Steril. 2006;86:1336‐1343. [DOI] [PubMed] [Google Scholar]
- 275. Ueda M, Yamashita Y, Takehara M, et al. Survivin gene expression in endometriosis. J Clin Endocrinol Metab. 2002;87:3452‐3459. [DOI] [PubMed] [Google Scholar]
- 276. Laudanski P, Szamatowicz J, Ramel P. Matrix metalloproteinase‐13 and membrane type‐1 matrix metalloproteinase in peritoneal fluid of women with endometriosis. Gynecol Endocrinol. 2005;21:106‐110. [DOI] [PubMed] [Google Scholar]
- 277. Chung HW, Lee JY, Moon HS, et al. Matrix metalloproteinase‐2, membranous type 1 matrix metalloproteinase, and tissue inhibitor of metalloproteinase‐2 expression in ectopic and eutopic endometrium. Fertil Steril. 2002;78:787‐795. [DOI] [PubMed] [Google Scholar]
- 278. Everett AD, Xue C, Stoops T. Developmental expression of protein phosphatase 2A in the kidney. J Am Soc Nephrol. 1999;10:1737‐1745. [DOI] [PubMed] [Google Scholar]
- 279. Nagendra DC, Burke J 3rd, Maxwell GL, Risinger JI. PPP2R1A mutations are common in the serous type of endometrial cancer. Mol Carcinog. 2012;51:826‐831. [DOI] [PubMed] [Google Scholar]
- 280. Sangodkar J, Farrington CC, McClinch K, Galsky MD, Kastrinsky DB, Narla G. All roads lead to PP2A: exploiting the therapeutic potential of this phosphatase. FEBS J. 2016;283:1004‐1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. McConechy MK, Ding J, Senz J, et al. Ovarian and endometrial endometrioid carcinomas have distinct CTNNB1 and PTEN mutation profiles. Mod Pathol. 2014;27:128‐134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Er TK, Su YF, Wu CC, et al. Targeted next‐generation sequencing for molecular diagnosis of endometriosis‐associated ovarian cancer. J Mol Med (Berl). 2016;94:835‐847. [DOI] [PubMed] [Google Scholar]
- 283. Pan Q, Guo C, Sun C, Fan J, Fang C. Integrative analysis of the transcriptome and targetome identifies the regulatory network of miR‐16: an inhibitory role against the activation of hepatic stellate cells. Bio‐Med Mater Eng. 2014;24:3863‐3871. [DOI] [PubMed] [Google Scholar]
- 284. Braza‐Boils A, Gilabert‐Estelles J, Ramon LA, et al. Peritoneal fluid reduces angiogenesis‐related microRNA expression in cell cultures of endometrial and endometriotic tissues from women with endometriosis. PLoS ONE. 2013;8:e62370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285. Braza‐Boils A, Mari‐Alexandre J, Gilabert J, et al. MicroRNA expression profile in endometriosis: its relation to angiogenesis and fibrinolytic factors. Hum Reprod. 2014;29:978‐988. [DOI] [PubMed] [Google Scholar]
- 286. Jones TA, Barker HM, da Cruz e Silva EF, et al. Localization of the genes encoding the catalytic subunits of protein phosphatase 2A to human chromosome bands 5q23–>q31 and 8p12–>p11.2, respectively. Cytogenet Cell Genet. 1993;63:35‐41. [DOI] [PubMed] [Google Scholar]
- 287. Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417‐439.11171037 [Google Scholar]
- 288. Nematullah M, Hoda MN, Khan F. Protein phosphatase 2A: a double‐faced phosphatase of cellular system and its role in neurodegenerative disorders. Mol Neurobiol 2018;55:1750‐1761. [DOI] [PubMed] [Google Scholar]
- 289. Tan FK, Hildebrand BA, Lester MS, et al. Classification analysis of the transcriptosome of nonlesional cultured dermal fibroblasts from systemic sclerosis patients with early disease. Arthritis Rheum. 2005;52:865‐876. [DOI] [PubMed] [Google Scholar]
- 290. Haesen D, Abbasi Asbagh L, Derua R, et al. Recurrent PPP2R1A mutations in uterine cancer act through a dominant‐negative mechanism to promote malignant cell growth. Cancer Res. 2016;76:5719‐5731. [DOI] [PubMed] [Google Scholar]
- 291. Wlodarchak N, Xing Y. PP2A as a master regulator of the cell cycle. Crit Rev Biochem Mol Biol. 2016;51:162‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Cristobal I, Manso R, Rincon R, Carames C, Zazo S, Del Pulgar TG, et al. Phosphorylated protein phosphatase 2A determine poor outcome in patients with metastatic colorectal cancer. Br J Cancer. 2014;111:756‐762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends Mol Med. 2008;14:152‐160. [DOI] [PubMed] [Google Scholar]
- 294. Xiong Y, Jing XP, Zhou XW, Wang XL, Yang Y, Sun XY, et al. Zinc induces protein phosphatase 2A inactivation and tau hyperphosphorylation through Src dependent PP2A (tyrosine 307) phosphorylation. Neurobiol Again. 2013;34:745‐756. [DOI] [PubMed] [Google Scholar]
- 295. Yu UY, Yoo BC, Ahn JH. Regulatory B subunits of protein phosphatase 2A are involved in site‐specific regulation of tau protein phosphorylation. Korean J Physiol Pharmacol. 2014;18:155‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta. 2009;1795:1‐16. [DOI] [PubMed] [Google Scholar]
- 297. Guo W, Shan B, Klingsberg RC, Qin X, Lasky JA. Abrogation of TGF‐beta1‐induced fibroblast‐myofibroblast differentiation by histone deacetylase inhibition. Am J Physiol Lung Cell Mol Physiol. 2009;297:L864‐L870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Xia H, Seeman J, Hong J, et al. Low alpha(2)beta(1) integrin function enhances the proliferation of fibroblasts from patients with idiopathic pulmonary fibrosis by activation of the beta‐catenin pathway. Am J Pathol. 2012;181:222‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Hong J, Chu M, Qian L, Wang J, Guo Y, Xu D. Fibrillar type I collagen enhances the differentiation and proliferation of myofibroblasts by lowering alpha2beta1 integrin expression in cardiac fibrosis. Biomed Res Int. 2017;2017:1790808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Gergs U, Boknik P, Buchwalow I, et al. Overexpression of the catalytic subunit of protein phosphatase 2A impairs cardiac function. J Biol Chem. 2004;279:40827‐40834. [DOI] [PubMed] [Google Scholar]
- 301. Hoehn M, Zhang Y, Xu J, et al. Overexpression of protein phosphatase 2A in a murine model of chronic myocardial infarction leads to increased adverse remodeling but restores the regulation of beta‐catenin by glycogen synthase kinase 3beta. Int J Cardiol. 2015;183:39‐46. [DOI] [PubMed] [Google Scholar]
- 302. Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non‐Hodgkin's lymphoma. Science. 1994;263:1281‐1284. [DOI] [PubMed] [Google Scholar]
- 303. Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer. 2013;13:685‐700. [DOI] [PubMed] [Google Scholar]
- 304. Sullivan RJ, Flaherty K. MAP kinase signaling and inhibition in melanoma. Oncogene. 2013;32:2373‐2379. [DOI] [PubMed] [Google Scholar]
- 305. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949‐954. [DOI] [PubMed] [Google Scholar]
- 306. Zannoni GF, Improta G, Chiarello G, et al. Mutational status of KRAS, NRAS, and BRAF in primary clear cell ovarian carcinoma. Virchows Arch. 2014;465:193‐198. [DOI] [PubMed] [Google Scholar]
- 307. Chen Q, Zhang C, Chen Y, Lou J, Wang D. Identification of endometriosis‐related genes by representational difference analysis of cDNA. Aust N Z J Obstet Gynaecol. 2012;52:140‐145. [DOI] [PubMed] [Google Scholar]
- 308. Santulli P, Marcellin L, Chouzenoux S, et al. Role of the protein kinase BRAF in the pathogenesis of endometriosis. Expert Opin Ther Targets. 2016;20:1017‐1029. [DOI] [PubMed] [Google Scholar]
- 309. Antoniou KM, Margaritopoulos GA, Soufla G, et al. Expression analysis of Akt and MAPK signaling pathways in lung tissue of patients with idiopathic pulmonary fibrosis (IPF). J Recept Signal Transduct Res. 2010;30:262‐269. [DOI] [PubMed] [Google Scholar]
- 310. Guyard A, Danel C, Theou‐Anton N, et al. Morphologic and molecular study of lung cancers associated with idiopathic pulmonary fibrosis and other pulmonary fibroses. Respir Res. 2017;18:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Goumenou AG, Matalliotakis IM, Tzardi M, Fragouli IG, Mahutte NG, Arici A. p16, retinoblastoma (pRb), and cyclin D1 protein expression in human endometriotic and adenomyotic lesions. Fertil Steril. 2006;85(Suppl 1):1204‐1207. [DOI] [PubMed] [Google Scholar]
- 312. Kawano Y, Nasu K, Li H, et al. Application of the histone deacetylase inhibitors for the treatment of endometriosis: histone modifications as pathogenesis and novel therapeutic target. Hum Reprod. 2011;26:2486‐2498. [DOI] [PubMed] [Google Scholar]
- 313. Ji J, Tian Y, Zhu YQ, et al. Ionizing irradiation inhibits keloid fibroblast cell proliferation and induces premature cellular senescence. J Dermatol. 2015;42:56‐63. [DOI] [PubMed] [Google Scholar]
- 314. Meyer K, Hodwin B, Ramanujam D, Engelhardt S, Sarikas A. Essential Role for Premature Senescence of Myofibroblasts in Myocardial Fibrosis. J Am Coll Cardiol. 2016;67:2018‐2028. [DOI] [PubMed] [Google Scholar]
- 315. Chen CC, Lau LF. Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol. 2009;41:771‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Gashaw I, Hastings JM, Jackson KS, Winterhager E, Fazleabas AT. Induced endometriosis in the baboon (Papio anubis) increases the expression of the proangiogenic factor CYR61 (CCN1) in eutopic and ectopic endometria. Biol Reprod. 2006;74:1060‐1066. [DOI] [PubMed] [Google Scholar]
- 317. Absenger Y, Hess‐Stumpp H, Kreft B, et al. Cyr61, a deregulated gene in endometriosis. Mol Hum Reprod. 2004;10:399‐407. [DOI] [PubMed] [Google Scholar]
- 318. Zhao Y, Li Q, Katzenellenbogen BS, et al. Estrogen‐induced CCN1 is critical for establishment of endometriosis‐like lesions in mice. Mol Endocrinol. 2014;28:1934‐1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Shaw AT, Hsu PP, Awad MM, Engelman JA. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat Rev Cancer. 2013;13:772‐787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. McKenzie KJ, Newport R. Increased somatic sensations are associated with reduced limb ownership. J Psychosom Res. 2015;78:88‐90. [DOI] [PubMed] [Google Scholar]
- 321. Ferriani RA, Charnock‐Jones DS, Prentice A, Thomas EJ, Smith SK. Immunohistochemical localization of acidic and basic fibroblast growth factors in normal human endometrium and endometriosis and the detection of their mRNA by polymerase chain reaction. Hum Reprod. 1993;8:11‐16. [DOI] [PubMed] [Google Scholar]
- 322. Shirley MD, Tang H, Gallione CJ, et al. Sturge‐Weber syndrome and port‐wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971‐1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol. 2000;151:33‐40. [DOI] [PubMed] [Google Scholar]
- 324. Arima Y, Hayashi H, Kamata K, et al. Decreased expression of neurofibromin contributes to epithelial‐mesenchymal transition in neurofibromatosis type 1. Exp Dermatol. 2010;19:e136‐e141. [DOI] [PubMed] [Google Scholar]
- 325. El‐Hoss J, Sullivan K, Cheng T, et al. A murine model of neurofibromatosis type 1 tibial pseudarthrosis featuring proliferative fibrous tissue and osteoclast‐like cells. J Bone Miner Res. 2012;27:68‐78. [DOI] [PubMed] [Google Scholar]
- 326. Xiao GH, Gallagher R, Shetler J, et al. The NF2 tumor suppressor gene product, merlin, inhibits cell proliferation and cell cycle progression by repressing cyclin D1 expression. Mol Cell Biol. 2005;25:2384‐2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Schroeder RD, Angelo LS, Kurzrock R. NF2/merlin in hereditary neurofibromatosis 2 versus cancer: biologic mechanisms and clinical associations. Oncotarget. 2014;5:67‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Murakami H, Mizuno T, Taniguchi T, et al. LATS2 is a tumor suppressor gene of malignant mesothelioma. Cancer Res. 2011;71:873‐883. [DOI] [PubMed] [Google Scholar]
- 329. Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Bosco EE, Nakai Y, Hennigan RF, Ratner N, Zheng Y. NF2‐deficient cells depend on the Rac1‐canonical Wnt signaling pathway to promote the loss of contact inhibition of proliferation. Oncogene. 2010;29:2540‐2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Zhou L, Ercolano E, Ammoun S, Schmid MC, Barczyk MA, Hanemann CO. Merlin‐deficient human tumors show loss of contact inhibition and activation of Wnt/beta‐catenin signaling linked to the PDGFR/Src and Rac/PAK pathways. Neoplasia. 2011;13:1101‐1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Song Y, Fu J, Zhou M, et al. Activated Hippo/Yes‐Associated Protein Pathway Promotes Cell Proliferation and Anti‐apoptosis in Endometrial Stromal Cells of Endometriosis. J Clin Endocrinol Metab. 2016;101:1552‐1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Sanchez AM, Vigano P, Quattrone F, et al. The WNT/beta‐catenin signaling pathway and expression of survival promoting genes in luteinized granulosa cells: endometriosis as a paradigm for a dysregulated apoptosis pathway. Fertil Steril. 2014;101:1688‐1696. [DOI] [PubMed] [Google Scholar]
- 334. Mannaerts I, Leite SB, Verhulst S, et al. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol. 2015;63:679‐688. [DOI] [PubMed] [Google Scholar]
- 335. Liu F, Lagares D, Choi KM, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2015;308:L344‐L357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Piersma B, Bank RA, Boersema M. Signaling in fibrosis: TGF‐beta, WNT, and YAP/TAZ converge. Front Med (Lausanne). 2015;2:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Bi P, Kuang S. Notch signaling as a novel regulator of metabolism. Trends Endocrinol Metab. 2015;26:248‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Fleming RJ. Structural conservation of Notch receptors and ligands. Semin Cell Dev Biol. 1998;9:599‐607. [DOI] [PubMed] [Google Scholar]
- 340. Ntziachristos P, Lim JS, Sage J, Aifantis I. From fly wings to targeted cancer therapies: a centennial for notch signaling. Cancer Cell. 2014;25:318‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Wang NJ, Sanborn Z, Arnett KL, et al. Loss‐of‐function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci U S A. 2011;108:17761‐17766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Martincorena I, Roshan A, Gerstung M, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880‐886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343. Liu T, Hu B, Choi YY, et al. Notch1 signaling in FIZZ1 induction of myofibroblast differentiation. Am J Pathol. 2009;174:1745‐1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Hu M, Ou‐Yang HF, Wu CG, Qu SY, Xu XT, Wang P. Notch signaling regulates col1alpha1 and col1alpha2 expression in airway fibroblasts. Exp Biol Med (Maywood). 2014;239:1589‐1596. [DOI] [PubMed] [Google Scholar]
- 345. Namba T, Tanaka KI, Ito Y, et al. Induction of EMT‐like phenotypes by an active metabolite of leflunomide and its contribution to pulmonary fibrosis. Cell Death Differ. 2010;17:1882‐1895. [DOI] [PubMed] [Google Scholar]
- 346. Shao S, Zhao X, Zhang X, et al. Notch1 signaling regulates the epithelial‐mesenchymal transition and invasion of breast cancer in a Slug‐dependent manner. Mol Cancer. 2015;14:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Bielesz B, Sirin Y, Si H, et al. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest. 2010;120:4040‐4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Syed F, Bayat A. Notch signaling pathway in keloid disease: enhanced fibroblast activity in a Jagged‐1 peptide‐dependent manner in lesional vs. extralesional fibroblasts. Wound Repair Regen. 2012;20:688‐706. [DOI] [PubMed] [Google Scholar]
- 349. Sorensen‐Zender I, Rong S, Susnik N, et al. Renal tubular Notch signaling triggers a prosenescent state after acute kidney injury. Am J Physiol Renal Physiol. 2014;306:F907‐F915. [DOI] [PubMed] [Google Scholar]
- 350. Zhang K, Zhang YQ, Ai WB, et al. Hes1, an important gene for activation of hepatic stellate cells, is regulated by Notch1 and TGF‐beta/BMP signaling. World J Gastroenterol. 2015;21:878‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Hu B, Wu Z, Bai D, Liu T, Ullenbruch MR, Phan SH. Mesenchymal deficiency of Notch1 attenuates bleomycin‐induced pulmonary fibrosis. Am J Pathol. 2015;185:3066‐3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Zheng S, Zhang P, Chen Y, Zheng S, Zheng L, Weng Z. Inhibition of Notch Signaling Attenuates Schistosomiasis Hepatic Fibrosis via Blocking Macrophage M2 Polarization. PLoS ONE. 2016;11:e0166808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Gotte M, Wolf M, Staebler A, et al. Increased expression of the adult stem cell marker Musashi‐1 in endometriosis and endometrial carcinoma. J Pathol. 2008;215:317‐329. [DOI] [PubMed] [Google Scholar]
- 354. Young VJ, Brown JK, Saunders PT, Duncan WC, Horne AW. The peritoneum is both a source and target of TGF‐beta in women with endometriosis. PLoS ONE. 2014;9:e106773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Su RW, Strug MR, Joshi NR, et al. Decreased Notch pathway signaling in the endometrium of women with endometriosis impairs decidualization. J Clin Endocrinol Metab. 2015;100:E433‐E442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Schenken RS, Johnson JV, Riehl RM. c‐myc protooncogene polypeptide expression in endometriosis. Am J Obstet Gynecol. 1991;164:1031‐1036; discussion 6‐7. [DOI] [PubMed] [Google Scholar]
- 357. Pellegrini C, Gori I, Achtari C, et al. The expression of estrogen receptors as well as GREB1, c‐MYC, and cyclin D1, estrogen‐regulated genes implicated in proliferation, is increased in peritoneal endometriosis. Fertil Steril. 2012;98:1200‐1208. [DOI] [PubMed] [Google Scholar]
- 358. Proestling K, Birner P, Gamperl S, et al. Enhanced epithelial to mesenchymal transition (EMT) and upregulated MYC in ectopic lesions contribute independently to endometriosis. Reprod Biol Endocrinol. 2015;13:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359. He H, Liu R, Xiong W, Pu D, Wang S, Li T. Lentiviral vector‐mediated down‐regulation of Notch1 in endometrial stem cells results in proliferation and migration in endometriosis. Mol Cell Endocrinol. 2016;434:210‐218. [DOI] [PubMed] [Google Scholar]
- 360. Gonzalez‐Foruria I, Santulli P, Chouzenoux S, Carmona F, Chapron C, Batteux F. Dysregulation of the ADAM17/Notch signalling pathways in endometriosis: from oxidative stress to fibrosis. Mol Hum Reprod 2017;23:488‐499. [DOI] [PubMed] [Google Scholar]
- 361. Matsuzaki S, Darcha C. Co‐operation between the AKT and ERK signaling pathways may support growth of deep endometriosis in a fibrotic microenvironment in vitrodagger. Hum Reprod. 2015;30:1606‐1616. [DOI] [PubMed] [Google Scholar]
- 362. Jana S, Chatterjee K, Ray AK, DasMahapatra P, Swarnakar S. Regulation of Matrix Metalloproteinase‐2 Activity by COX‐2‐PGE2‐pAKT Axis Promotes Angiogenesis in Endometriosis. PLoS ONE. 2016;11:e0163540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363. Liu M, Lee DF, Chen CT, et al. IKKalpha activation of NOTCH links tumorigenesis via FOXA2 suppression. Mol Cell. 2012;45:171‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Yue HY, Yin C, Hou JL, et al. Hepatocyte nuclear factor 4alpha attenuates hepatic fibrosis in rats. Gut. 2010;59:236‐246. [DOI] [PubMed] [Google Scholar]
- 365. Song G, Pacher M, Balakrishnan A, et al. Direct Reprogramming of Hepatic Myofibroblasts into Hepatocytes In Vivo Attenuates Liver Fibrosis. Cell Stem Cell. 2016;18:797‐808. [DOI] [PubMed] [Google Scholar]
- 366. Ilad RS, Fleming SD, Murphy CR, Fazleabas AT. Immunohistochemical study of the ubiquitin‐nuclear factor‐kB pathway in the endometrium of the baboon (Papio anubis) with and without endometriosis. Reprod Fertil Dev. 2010;22:1118‐1130. [DOI] [PubMed] [Google Scholar]
- 367. Cakmak H, Guzeloglu‐Kayisli O, Kayisli UA, Arici A. Immune‐endocrine interactions in endometriosis. Front Biosci (Elite Ed). 2009;1:429‐443. [DOI] [PubMed] [Google Scholar]
- 368. Fouquet B, Santulli P, Noel JC, Misrahi M. Ovarian‐like differentiation in eutopic and ectopic endometrioses with aberrant FSH receptor, INSL3 and GATA4/6 expression. BBA Clin. 2016;6:143‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369. Halme J, Hammond MG, Hulka JF, Raj SG, Talbert LM. Retrograde menstruation in healthy women and in patients with endometriosis. Obstet Gynecol. 1984;64:151‐154. [PubMed] [Google Scholar]
- 370. Vinatier D, Cosson M, Dufour P. Is endometriosis an endometrial disease? Eur J Obstet Gynecol Reprod Biol. 2000;91:113‐125. [DOI] [PubMed] [Google Scholar]
- 371. Lee B, Du H, Taylor HS. Experimental murine endometriosis induces DNA methylation and altered gene expression in eutopic endometrium. Biol Reprod. 2009;80:79‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372. Kim JJ, Taylor HS, Lu Z, et al. Altered expression of HOXA10 in endometriosis: potential role in decidualization. Mol Hum Reprod. 2007;13:323‐332. [DOI] [PubMed] [Google Scholar]
- 373. Naqvi H, Ilagan Y, Krikun G, Taylor HS. Altered genome‐wide methylation in endometriosis. Reprod Sci. 2014;21:1237‐1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374. Felsenstein J. Inferring Phylogenies, 2nd edn Sunderland, MA, USA: Sinauer Associates; 2003. [Google Scholar]
- 375. Donnez J, Binda MM, Donnez O, Dolmans MM. Oxidative stress in the pelvic cavity and its role in the pathogenesis of endometriosis. Fertil Steril. 2016;106:1011‐1017. [DOI] [PubMed] [Google Scholar]
- 376. Aon MA, Cortassa S, O'Rourke B. Redox‐optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta. 2010;1797:865‐877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Seino T, Saito H, Kaneko T, Takahashi T, Kawachiya S, Kurachi H. Eight‐hydroxy‐2'‐deoxyguanosine in granulosa cells is correlated with the quality of oocytes and embryos in an in vitro fertilization‐embryo transfer program. Fertil Steril. 2002;77:1184‐1190. [DOI] [PubMed] [Google Scholar]
- 378. Matsuzaki S, Schubert B. Oxidative stress status in normal ovarian cortex surrounding ovarian endometriosis. Fertil Steril. 2010;93:2431‐2432. [DOI] [PubMed] [Google Scholar]
- 379. Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58:235‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Ferrari E, Bruhn C, Peretti M, Cassani C, Carotenuto WV, Elgendy M, et al. PP2A controls genome integrity by integrating nutrient‐sensing and metabolic pathways with the DNA damage response. Mol Cell. 2017;67(266–281):e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381. Matta JL, Flores I, Morales LM, Monteiro J, Alvarez‐Garriga C, Bayona M. Women with endometriosis have a higher DNA repair capacity and diminished breast cancer risk. Mol Cancer Biol 2013;1(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382. Carvalho LF, Abrao MS, Biscotti C, Sharma R, Nutter B, Falcone T. Oxidative cell injury as a predictor of endometriosis progression. Reprod Sci. 2013;20:688‐698. [DOI] [PubMed] [Google Scholar]
- 383. Leung SY, Yuen ST, Chung LP, Chu KM, Chan AS, Ho JC. hMLH1 promoter methylation and lack of hMLH1 expression in sporadic gastric carcinomas with high‐frequency microsatellite instability. Cancer Res. 1999;59:159‐164. [PubMed] [Google Scholar]
- 384. Grassi T, Calcagno A, Marzinotto S, et al. Mismatch repair system in endometriotic tissue and eutopic endometrium of unaffected women. Int J Clin Exp Pathol. 2015;8:1867‐1877. [PMC free article] [PubMed] [Google Scholar]
- 385. Goumenou AG, Arvanitis DA, Matalliotakis IM, Koumantakis EE, Spandidos DA. Loss of heterozygosity in adenomyosis on hMSH2, hMLH1, p16Ink4 and GALT loci. Int J Mol Med. 2000;6:667‐671. [DOI] [PubMed] [Google Scholar]
- 386. Amemiya S, Sekizawa A, Otsuka J, Tachikawa T, Saito H, Okai T. Malignant transformation of endometriosis and genetic alterations of K‐ras and microsatellite instability. Int J Gynaecol Obstet. 2004;86:371‐376. [DOI] [PubMed] [Google Scholar]
- 387. Ali‐Fehmi R, Khalifeh I, Bandyopadhyay S, et al. Patterns of loss of heterozygosity at 10q23.3 and microsatellite instability in endometriosis, atypical endometriosis, and ovarian carcinoma arising in association with endometriosis. Int J Gynecol Pathol. 2006;25:223‐229. [DOI] [PubMed] [Google Scholar]
- 388. Ren F, Wang D, Jiang Y, Ren F. Epigenetic inactivation of hMLH1 in the malignant transformation of ovarian endometriosis. Arch Gynecol Obstet. 2012;285:215‐221. [DOI] [PubMed] [Google Scholar]
- 389. Lu FI, Gilks CB, Mulligan AM, et al. Prevalence of loss of expression of DNA mismatch repair proteins in primary epithelial ovarian tumors. Int J Gynecol Pathol. 2012;31:524‐531. [DOI] [PubMed] [Google Scholar]
- 390. Maybin JA, Critchley HO. Menstrual physiology: implications for endometrial pathology and beyond. Hum Reprod Update. 2015;21:748‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391. Noe M, Ayhan A, Wang TL, Shih IM. Independent development of endometrial epithelium and stroma within the same endometriosis. J Pathol 2018;245:265‐269. [DOI] [PubMed] [Google Scholar]
- 392. McConnell MJ, Lindberg MR, Brennand KJ, et al. Mosaic copy number variation in human neurons. Science. 2013;342:632‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Krimmel JD, Schmitt MW, Harrell MI, et al. Ultra‐deep sequencing detects ovarian cancer cells in peritoneal fluid and reveals somatic TP53 mutations in noncancerous tissues. Proc Natl Acad Sci U S A. 2016;113:6005‐6010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Flesken‐Nikitin A, Choi KC, Eng JP, Shmidt EN, Nikitin AY. Induction of carcinogenesis by concurrent inactivation of p53 and Rb1 in the mouse ovarian surface epithelium. Cancer Res. 2003;63:3459‐3463. [PubMed] [Google Scholar]
- 395. Kinross KM, Montgomery KG, Kleinschmidt M, et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J Clin Invest. 2012;122:553‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396. van Kaam KJ, Schouten JP, Nap AW, Dunselman GA, Groothuis PG. Fibromuscular differentiation in deeply infiltrating endometriosis is a reaction of resident fibroblasts to the presence of ectopic endometrium. Hum Reprod. 2008;23:2692‐2700. [DOI] [PubMed] [Google Scholar]
- 397. Clement PB. The pathology of endometriosis: a survey of the many faces of a common disease emphasizing diagnostic pitfalls and unusual and newly appreciated aspects. Adv Anat Pathol. 2007;14:241‐260. [DOI] [PubMed] [Google Scholar]
- 398. Mai KT, Yazdi HM, Perkins DG, Parks W. Pathogenetic role of the stromal cells in endometriosis and adenomyosis. Histopathology. 1997;30:430‐442. [DOI] [PubMed] [Google Scholar]
- 399. Abrao MS, Neme RM, Carvalho FM, Aldrighi JM, Pinotti JA. Histological classification of endometriosis as a predictor of response to treatment. Int J Gynaecol Obstet. 2003;82:31‐40. [DOI] [PubMed] [Google Scholar]
- 400. Kamergorodsky G, Ribeiro PA, Galvao MA, et al. Histologic classification of specimens from women affected by superficial endometriosis, deeply infiltrating endometriosis, and ovarian endometriomas. Fertil Steril. 2009;92:2074‐2077. [DOI] [PubMed] [Google Scholar]
- 401. Leconte M, Nicco C, Ngo C, et al. The mTOR/AKT inhibitor temsirolimus prevents deep infiltrating endometriosis in mice. Am J Pathol. 2011;179:880‐889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Liu X, Guo SW. A pilot study on the off‐label use of valproic acid to treat adenomyosis. Fertil Steril. 2008;89:246‐250. [DOI] [PubMed] [Google Scholar]
- 403. Xishi L, Lei Y, Guo SW. Valproic acid as a therapy for adenomyosis: a comparative case series. Reprod Sci. 2010;17:904‐912. [DOI] [PubMed] [Google Scholar]
- 404. Hadfield R, Mardon H, Barlow D, Kennedy S. Delay in the diagnosis of endometriosis: a survey of women from the USA and the UK. Hum Reprod. 1996;11:878‐880. [DOI] [PubMed] [Google Scholar]
- 405. Hudelist G, Fritzer N, Thomas A, et al. Diagnostic delay for endometriosis in Austria and Germany: causes and possible consequences. Hum Reprod. 2012;27:3412‐3416. [DOI] [PubMed] [Google Scholar]
- 406. Staal AH, van der Zanden M, Nap AW. Diagnostic Delay of Endometriosis in the Netherlands. Gynecol Obstet Invest. 2016;81:321‐324. [DOI] [PubMed] [Google Scholar]
- 407. Guo S.W., Groothuis P. Is it time for a paradigm shift in drug research and development in endometriosis/adenomyosis? Hum Reprod Update 2018; [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 408. Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol. 2008;20:150‐155. [DOI] [PubMed] [Google Scholar]
- 409. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123:966‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Moran‐Salvador E, Mann J. Epigenetics and liver fibrosis. Cell Mol Gastroenterol Hepatol. 2017;4:125‐134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Delaney AA, Khan Z, Zheng Y, et al. KLF10 Mediated Epigenetic Dysregulation of Epithelial CD40/CD154 Promotes Endometriosis. Biol Reprod. 2016;95:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412. Zheng Y, Khan Z, Zanfagnin V, Correa LF, Delaney AA, Daftary GS. Epigenetic modulation of collagen 1A1: therapeutic implications in fibrosis and endometriosis. Biol Reprod. 2016;94:87. [DOI] [PubMed] [Google Scholar]
- 413. Leonardo‐Pinto JP, Benetti‐Pinto CL, Cursino K, Yela DA. Dienogest and deep infiltrating endometriosis: The remission of symptoms is not related to endometriosis nodule remission. Eur J Obstet Gynecol Reprod Biol. 2017;211:108‐111. [DOI] [PubMed] [Google Scholar]