

Abstract
Background
Spermatogenesis is one of the most complicated cellular differentiation processes in a body. Researchers struggled to find and develop a micro‐environmental condition that can support the process in vitro. Such endeavors can be traced back to a century ago and are yet continuing.
Methods
Reports on in vitro spermatogenesis and related works were selected and classified into four categories based on the method used; organ culture, tubule culture, cell culture, and 3‐dimensional cell culture methods. Each report was critically reviewed from the present point of view by authors who have been working on in vitro spermatogenesis with organ culture method over a decade.
Results
The organ culture method has the longest history and is the most successful method, which produced fertile mouse sperm from spermatogonial stem cells. Formulation of the medium was a key factor, most importantly serum‐derived substances. However, factors in the serum that induce and support spermatogenesis in the cultured tissue remain to be identified. In addition, the success of mouse spermatogenesis is yet to be applied to other animals. On looking into the history of cell culture method, it became clear that Sertoli cells as feeder cells play an important role. Even with Sertoli cells, however, spermatogenic development has been limited to small parts of spermatogenesis, a segmented period of meiotic prophase for instance. Recent developments of organoid or 3‐dimensional culture techniques are promising but they still need further refinements.
Conclusion
The study of in vitro spermatogenesis progressed significantly over the last century. We need more work, however, to establish a culture system that can induce and maintain complete spermatogenesis of many if not all mammalian species.
Keywords: cell culture, in vitro spermatogenesis, male infertility, microfluidics, organ culture
1. INTRODUCTION
Spermatogenesis is a complex cellular process of germ cell differentiation, from spermatogonial stem cells to spermatozoa, taking a long period of time that is species specific. For instance, it takes 74 and 35 days in human and mice, respectively.1, 2 The process could be subdivided into three phases; proliferation of spermatogonia, meiotic division of spermatocytes, and dynamic changes in shape and nuclear contents of haploid spermatids (spermiogenesis). These processes are not germ cell autonomous, but require intimate interaction between germ and somatic cells, particularly Sertoli cells and also peritubular myoid cells. These two cells compose the seminiferous tubule in which the spermatogenesis takes place. Size of each seminiferous tubule of mouse ranges from about 50 μm in diameter at immature stage to about 200 at matured, when containing full repertoire of spermatogenic cells inside. This diameter does not differ much among species, while the total length of seminiferous tubules differs among animal species from centimeters to meters long.3, 4 Both ends of the tubules open to the rete testis, which is connected to efferent ducts that carry sperm to the head of the epididymis. The tubules are folded and packed in the testis, composing most of the testis tissue. Several kinds of somatic cells are located between the tubules, including Leydig cells, peritubular myoid cells, endothelial cells and immune cells, which play important roles in supporting spermatogenesis.5
It is always challenging to reproduce in vivo biological processes, such as spermatogenesis outside of the body, ex vivo or in vitro. Researchers in the field worked hard and yet kept experiencing hardships and disappointments. In this review, we would like to look back such history from our present view point and give some interpretations to each study.
The methods and strategies for in vitro spermatogenesis could be divided into several categories. In this review, we classified them into four groups; organ culture, tubule culture, cell culture, and 3‐dimensional cell culture methods, and discuss each of them independently. We also discuss studies using experimental animals, mostly rodents, and humans separately in each section. Each study is described in a chronological order.
2. ORGAN CULTURE METHOD, CLASSIC PERIOD
2.1. Organ culture of experimental animal samples
The first description on the in vitro spermatogenesis was reported by Champy CH, who used rabbit testis for the organ culture experiment.6 Pieces of rabbit testis were cultured on rabbit plasma for hours or for about a week. The description and drawings appear accurate, precise, and beautiful, even from our present point of view. Somatic and undifferentiated germ cells survived for the period of 1 week, but degeneration of most germ cells occurred rapidly and drastically in a few days. Nonetheless, spermatogonial mitosis in the first week and newly formed leptotene primary spermatocytes after 9 days were described with drawings. So, it was the first report showing male germ cells passing through the transition from spermatogonial phase to the meiotic prophase in vitro.
Later in 1937, Martinovitch reported that pachytene spermatocytes were developed from presumably immature spermatogonia in the newborn mouse testis tissue cultured on a clot composed of equal parts of fowl plasma and fowl embryo extract.7 We assume that this was the first report reliable to judge that spermatogenesis appear to have proceeded up to the mid‐pachytene stage with the organ culture method.
About 20 years later, Trowel developed an organ culture system which he named gas‐liquid interphase method.8 He constructed a culture system in which tissue fragments were placed on a thin layer of agarose covering perforated metal grid. The culture medium was poured in just as the agarose becomes wet, thus soaking the cultured tissue adequately enough to obtain nutrients, yet at the same time securing enough oxygen from the gas surrounding the tissue. This gas‐liquid interphase method became the golden standard of the organ culture thereafter. Trowel cultured various tissues and organs of adult rats. Testis was one of them and was reported that most tubules were degenerated and disintegrated rapidly, only a few tubules survived for 3 days.8 It may appear that Trowel's method might be inferior to that of Martinovitch. We suppose, however, that Trowel had at least three handicaps in the experimental setting against Martinovitch. First, Trowel used testis of adult animal instead of newborn. In general, an adult tissue is much more difficult to culture than an immature tissue. Second, he used rat testis instead of mouse. As we discuss later, rat spermatogenesis is harder to reproduce in vitro than that of mouse (Figure 1). Third, he used synthetic medium. Although we can use various types of synthetic medium, most of which are commercially available today, we still need to add some animal‐derived supplements like serum or albumin in most culture experiments. The totally synthetic medium that Trowel used was likely lacking various essential factors and components. We discuss this aspect of culture more specifically in later sections.
Figure 1.
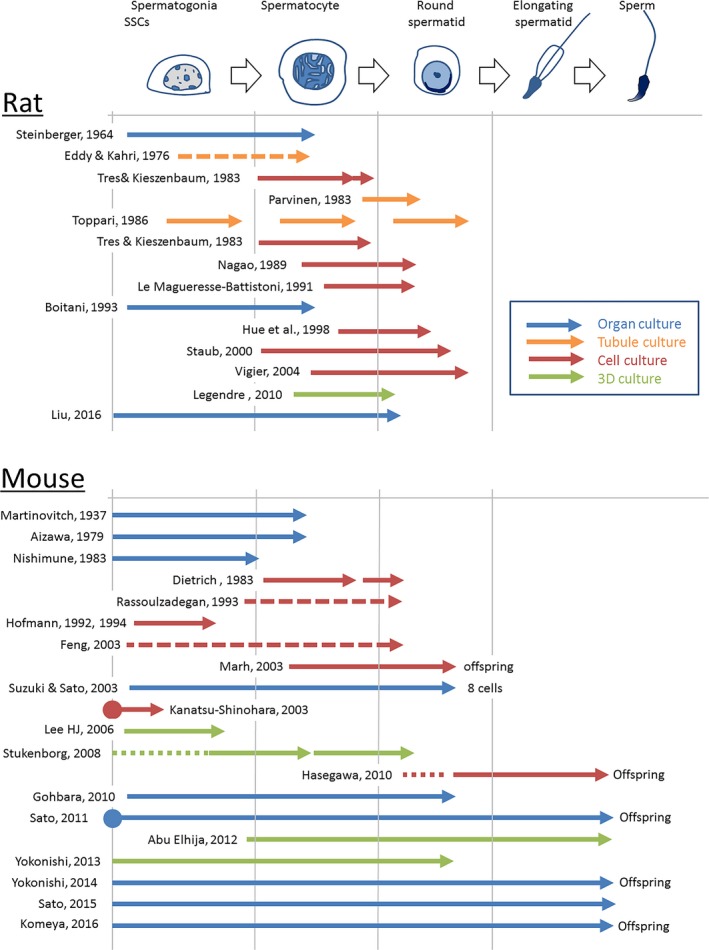
A schematic showing results of in vitro spermatogenesis achieved by each study using rodents
The 1960s was the decade when research on in vitro spermatogenesis advanced significantly. As introduced above, the golden‐standard organ culture method, gas‐liquid interphase method, was established. In addition, understanding of culture medium progressed rapidly and their composition improved significantly. Eagle's medium was pioneered and many others followed to establish the basics of culture medium.9 That was when a team of husband and wife researchers, Emil and Anna Steinberger came into the stage. They adopted Trowel's method and used the Eagle's synthetic medium to tackle in vitro spermatogenesis. In their first publication, which is an abstract of their presentation at a meeting, they reported that fragments of testis tissue of 14‐day‐old rat survived for 4 months. It was written that 10% calf serum was effective for maintaining the viability of the seminiferous epithelium.10 In continuing studies, they observed the progression of spermatogenesis from zygotene to pachytene spermatocytes.11 Then, they examined the effects of hormones and vitamins in their culture condition, using 4‐day‐old rat testes. They found that follicle stimulating hormone (FSH) and human chorionic gonadotropin (hCG) appeared to induce maturation of Sertoli cells, but did not promote spermatogonial differentiation. On the other hand, a combination of vitamins, A, C, and E, promoted the differentiation up to primary spermatocytes, at leptotene and zygotene, and rarely pachytene stages.12 They also examined various culture parameters, including pH, temperature, gas phase, along with composition of the nutrients in the medium.13 In this study, they described that a chemically defined medium based on Eagle's minimum essential medium, supplemented with pyruvate and six non‐essential amino acids, was comparably effective as calf serum‐supplemented medium in inducing spermatogenesis up to spermatocyte. However, when culture period lasted longer than 4 weeks, the chemically defined medium was not able to maintain germ cells, showing their weak point over serum‐added medium. They also examined effect of temperatures, ranging from 31 to 37°C, on organ culture of rat testis to conclude that temperature above 33°C is less optimal for survival and differentiation of germ cells. This certainly corresponds with the scrotal temperature of the animals. They also examined the effect of increased oxygen tension. They reported that the benefit of greater penetration of oxygen into the tissue was counteracted by signs of oxygen poisoning. Higher concentration of oxygen, 5% CO2 + 95% pure oxygen instead of 5% CO2 + 95% air, consistently accompanied diminished number of germ cells and their shorter survival.13 In the next report, they adopted autoradiography technique to unambiguously demonstrate the in vitro differentiation of germ cells.14
In order to define ingredients necessary and effective to promote in vitro spermatogenesis, they abandoned serum and began actively using chemically defined media. First, they tested the effect of vitamins of A, E, and C which they formerly found effective. Through more than 200 independent culture experiments, they confirmed the effect of vitamins to promote spermatogenesis in yielding increased number of spermatocytes. They even found that glutamine at 4 mmol L−1 or higher contributed significantly to induce spermatocyte production than vitamins did.15 Through this experiment, they formulated a chemically defined medium that can promote in vitro spermatogenesis from spermatogonia in 4‐day‐old rat testis to pachytene spermatocyte in 3‐week culture. They confessed yet that those spermatocytes degenerated and disappeared at 5 weeks.
In the original article of 1967, they summarized the results regarding factors in the medium affecting on the spermatogenesis. They presented a summary data obtained using not only rats, but also mice, guinea‐pigs, rabbits, monkeys, and humans. They concluded that glutamine at 4 mmol L−1 or higher was more effective than vitamins to produce spermatocytes. On the other hand, pituitary hormones were not necessary for the initiation of spermatogenesis. Supplements such as sera and tissue extracts neither resulted in improved maintenance or differentiation of the germinal cells. These studies certainly broaden our understanding on the organ culture and spermatogenesis as a whole. Nonetheless, the in vitro spermatogenesis per se showed limited progress, up to pachytene spermatocyte at best. Yet, they demonstrated impressive tissue viability after months in culture. This was performed by grafting testis tissues cultured for 7 weeks into the host testis, which led to regeneration of complete spermatogenesis in the grafted tissues.16 This result certainly proved that the tissue maintained the ability but the culture condition still lacked some environmental milieu necessary for the progression of spermatogenesis.
The Steinbergers made huge contribution to advance in vitro spermatogenesis, which started from the basis of Trowel's method. However, on comparison to the results by Martinovitch, the progress made by Steinberger regarding in vitro progression of spermatogenesis might appear marginal (Figure 1). We consider that rat spermatogenesis, compared to that of mouse, is highly demanding to reproduce in vitro. As we will see below that mouse spermatogenesis was accomplished with organ culture method, while the same method has not worked on rats in similar manner. Although the Steinbergers used many different species of animals in their studies of in vitro spermatogenesis, they mostly used rats instead of mice. We personally wonder if their experimental animal were mice instead of rats, their results might have been different. In any case, their works contributed tremendously to the field and became a basis for the later studies on in vitro spermatogenesis.17
After the Steinbergers’ extensive studies on in vitro spermatogenesis, we speculate that it became a consensus among researchers in those days that in vitro spermatogenesis using an organ culture method has its dead end; it will hardly overcome a stone wall of pachytene arrest. Therefore, researchers turned their attention to other methods. There were, however, groups adhered to the organ culture method. Nishimune's group cultured the matured mouse testis tissues which were pre‐conditioned to be cryptorchid in order to eliminate differentiated germ cells. So, in the testis of those mice, germ cells were solely undifferentiated spermatogonia, as type A spermatogonia. After 15 days of incubation of the tissue in a serum‐supplemented medium, they observed spermatocytes at the leptotene and pachytene stages of meiosis. This was actually reconfirmation of the results by the Steinbergers, using the testis of adult mice. 18 As the Nishimune group found serum to be effective while the Steinbergers did not,16 it was stated in the original article that the discrepancy might arise from the difference between type A spermatogonia in the cryptorchid testis and gonocytes in immature testis. Nishimune et al further speculated that the Steinbergers were observing the differentiation from type B spermatogonia, because serum‐independent differentiation ability of type B spermatogonia was observed by themselves.18
In their subsequent studies, Nishimune's group investigated the specific factors in the serum necessary for differentiation of type A spermatogonia. They found that FSH, but not other hormones like testosterone or factors like insulin and transferrin, had shown stimulating effects on spermatogonial differentiation.19 Later, they further reported that vitamin A and FSH synergistically induced differentiation of type A spermatogonia; FSH stimulated the proliferation of type A spermatogonia, whereas retinoids induced spermatogenesis from type A spermatogonia into intermediate or type B spermatogonia.20 They also paid attention to fetuin, a major serum protein of α‐globulin, or certain other molecules co‐purified from calf serum with fetuin for their effects to promote spermatogonial differentiation.21 Later, they reported that insulin‐like growth factor‐1 (IGF‐1) and transforming growth factor‐α (TGF‐α) had stimulating effects on the differentiation of type A spermatogonia. However, these factors could not promote the differentiation of germ cells through the meiotic stage.22 In a sense, we think that Nishimune's group succeeded the works of Steinberger and tried to find factors in the serum that promote spermatogonial differentiation up to pachytene spermatocytes. They actually found retinol as differentiation‐inducing factor for the spermatogonia, which is now an established notion supported by many studies.23 They also found FSH as stimulant for spermatogonial proliferation, but this is not universally supported by other studies. In addition, the issue whether undifferentiated spermatogonia need serum or factors in the serum to initiate differentiation remains to be elucidated and needs further research.
Boitani and colleagues also repeated the Steinbergers’ method using 9‐day‐old rat testis and observed the appearance of pachytene spermatocytes. The culture medium they used was Eagle's minimum essential medium with Earle's salts supplemented with glutamine, Hepes, and nonessential amino acids. Ovine FSH, ovine luteinizing hormone (LH), testosterone, and vitamins A, C, and E were added in some cases. They emphasized that FSH was essential for the progression of type A spermatogonia up to the stage of pachytene spermatocytes after 3 weeks of culture, while vitamins A, C, and E, LH, and testosterone were not effective.24
Suzuki & Sato used their strong technique of micro‐insemination for the evaluation of in vitro‐produced presumed haploid cells. Five‐day‐old mouse testis tissues were cultured for 2 weeks and round spermatids obtained were injected into the oocytes. The embryos developed up to 8‐cell stage but no offspring was produced.25 To our knowledge, this was the first report that might have overcome the long‐standing pachytene barrier in the organ culture experiment. Culture medium they used was Dulbecco's MEM supplemented with 0.1 mmol L−1 each of alanine, aspartic acid, glutamic acid, glycine, proline and serine, together with 1.0 mmol L−1 sodium pyruvate, 10% fetal bovine serum (FBS), and glutamine. The medium composition they used was quite similar to that of the Steinbergers. Thus, we simply speculate that the quality of culture media and supplements had improved through the decades since the Steinberger's experiments. We also suppose it is because spermatogenesis of mouse is easier to reproduce in vitro than that of rat.
2.2. Organ culture of human testis samples
The research of human in vitro spermatogenesis by the organ culture method was first performed by Steinberger16 (Figure 2). They reported that differentiation of primary spermatocytes took place in 3 weeks from the preleptotene to the pachytene stage which was verified by tritiated thymidine incorporation. In a report in 1971 by another group, the testis tissue obtained by the orchidectomy due to prostate cancer was cultured after the labeling with thymidine in the dish for 15 hours.26 During the culture period of 35 days, labeled spermatogenic cells including meiotic and post‐meiotic cells were observed in supernatant obtained from the medium used for culturing of tissue samples at each observation time points, on 3, 5, 14, 20, 22, 25, 27, 28, 32, and 35 days. It was reported that early spermatids labeled with 3H‐thymidine appeared from 27 days until 35 days. However, the authors mentioned in the discussion that many late spermatids observed after 35 days of culture might be just surviving cells. Three years later, another original article reported a result of in vitro human spermatogenesis. The testes from adult patients were labeled with thymidine in a culture dish and kept cultured for 14 days. The labeled cells showed the chromosomes of prometaphase I, metaphase I, prophase 2, and telophase 2 by autoradiographs.27 Seven years later, Curtis reported a precise culture experiment also using human testis tissue fragment.28 She labeled the tissue with thymidine for 1 or 2 hours just after harvesting and continued the culture for 21 days. It was found that many diakinesis figures, an explicit sign of the end of meiotic prophase I, appeared between 10 and 14 days. It was also described that differentiation beyond diakinesis, however, was not obtained and labeled secondary spermatocytes were not observed. It could be summarized as she wrote in the original article that, under the present culture conditions, spermatogonia are unable to differentiate into meiotically committed cells. Cells already committed to meiosis at culture initiation can proceed through spermatogenesis and were recorded in diakinesis.
Figure 2.
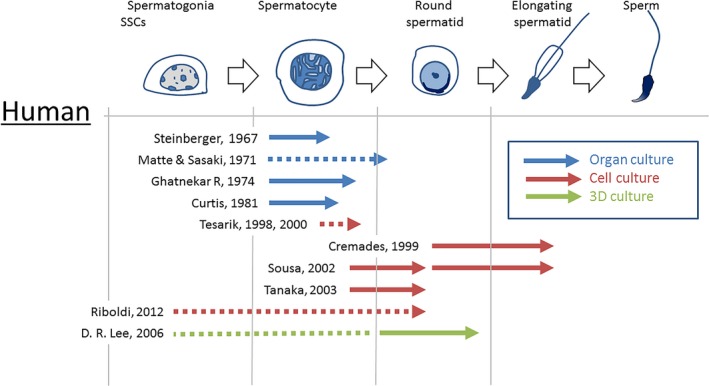
A schematic showing results of human in vitro spermatogenesis
2.3. Hanging drop culture method
Hanging drop culture system is the widely used incubation approach for both embryonic and adult tissues. The system was also applied for the incubation of the testes by Szczepny and colleagues in 2009. They cultured testis tissue of fetal and adult mouse for 48 hours to test the effect of Hedgehog signaling on the testis development.29 The same group applied the method for culturing testis tissue specimens obtained by the orchidectomy of testicular cancer patients. The normal testicular tissues cultured in the culture medium containing Dulbecco's modified Eagle's medium/F12 (DMEM/F12) with either 10% FBS or 0.1% bovine serum albumin (BSA) sustained the proliferation of germ cells for 14 days. However, the progression of spermatogenesis was not recognized.30
3. ORGAN CULTURE METHOD, MODERN DEVELOPMENT
3.1. Serum replacement changed the scene
Over 40 years since the initial report by the Steinbergers, the authors of this review started to reevaluate their results on in vitro spermatogenesis. First of all, we introduced two lines of transgenic mice, harboring Haspin (Gsg2) green fluorescent protein (GFP) and Acr‐GFP, respectively, in order to simplify the evaluation. Because these transgenic mice express GFP at the mid and end stages of meiosis onward, progress in spermatogenesis reaching these stages could be easily detected in each cultured tissue without histological examination. The basic culture method we used was same as that of the Steinbergers or Trowel, namely gas‐liquid interphase method, but we made it simpler by using an agarose gel block, instead of metal grid, as a stage for tissue placement (Figure 3A). The agarose gel block was half‐soaked in the culture media consisting simply of α‐minimum essential medium (α‐MEM), one of the modified versions of Eagle's medium, and 10% FBS. Then, we tried to repeat what the Steinbergers did using mice aged 4‐14 days after birth. Fortunately, we frequently observed the expression of Haspin‐GFP in the cultured tissue. And, although in rare occasions, we found round spermatids in 4 weeks of culture.31 The timing of in vitro differentiation into haploid cells was almost the same as what happens in vivo. Soon thereafter, we found that replacing FBS with knockout serum replacement (KSR) or AlbuMAX (bovine serum albumin purified through chromatography), both of which are commercially available, exhibited stronger effects to induce and maintain spermatogenesis under our organ culture condition. Specifically, mouse testis tissue cultured with medium of α‐MEM supplemented with 10% knockout serum replacement (KSR) or 40 mg/mL AlbuMAX produced haploid cells and sperms. Flow cytometric analyses showed the presence of 1C cells in the cultured tissues. Following micro‐insemination, the live birth of offspring was achieved, demonstrating the in vitro production of the functional sperm for the first time.32 Because KSR was supposed to contain AlbuMAX, the reason of the success was attributable to AlbuMAX. We have further showed that AlbuMAX, but not other popular bovine serum albumin (BSA), has such a strong effect to induce and maintain spermatogenesis.33 In addition, another BSA product, purified by chromatographic method like the preparation of AlbuMAX, also has a similar effect. Thus, we speculate that chromatography purified BSAs contain some important factors effective for in vitro spermatogenesis. This issue certainly needs further studies in the future.
Figure 3.
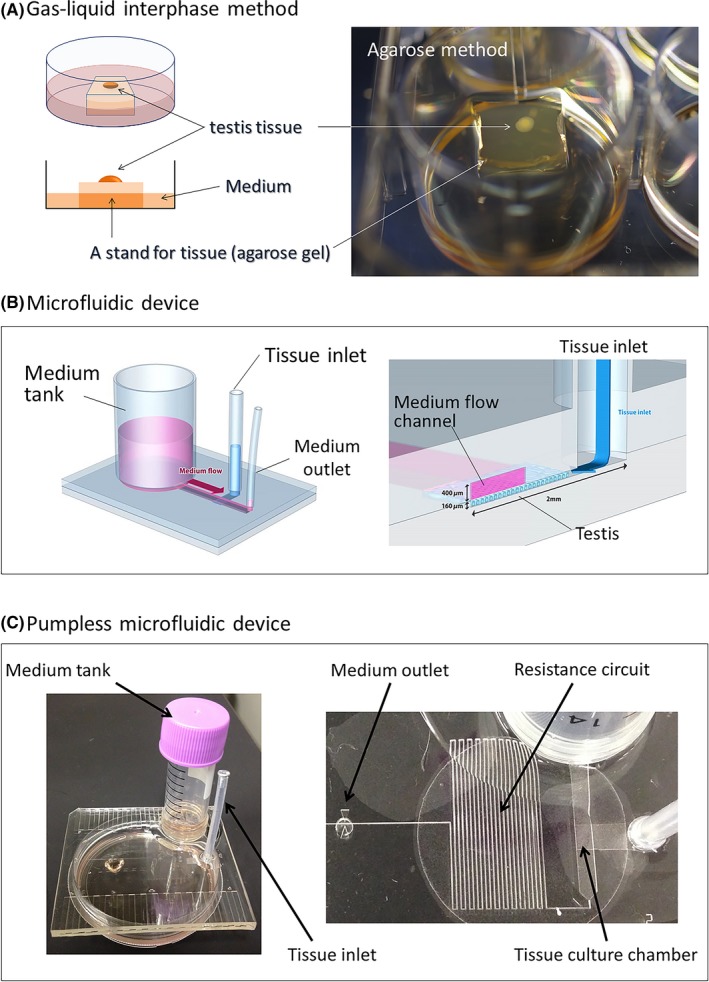
A schematic showing organ culture methods (A) The classical gas‐liquid interphase method is able to support mouse spermatogenesis. (B) In the microfluidic device, mouse testis tissues show efficient spermatogenesis.
The combination of the organ culture method with germ cell transplant provided the derivative technique for research on spermatogonial stem cells (SSCs) and spermatogenesis. As mouse SSCs became possible to culture for their proliferation, we were able to make the cell lines of mouse SSC.34, 35, 36 When cultured or freshly isolated SSCs are injected in the testis of a recipient mouse, they proliferate and differentiate into sperm in vivo. This was an in vivo‐unique phenomenon by that time. Then, we reproduced the same phenomenon in vitro by injecting SSCs to the testis that was excised out of the mouse. The SSC‐injected testis was cut into pieces and cultured on the agarose gel, which resulted in proliferation of SSCs and differentiation up to the sperm formation.37
Subsequently, the agarose gel‐based organ culture system also succeeded in treating mutant infertile mice. Testis tissues of the neonatal mice carrying mutation in the kit‐ligand gene, also named as stem‐cell factor (SCF), were cultured. The addition of recombinant kit‐ligand to the culture medium increased the number of spermatogonia and induced differentiation up to round spermatids. The combination of kit‐ligand and colony‐stimulating factor‐1 (CSF1)‐induced spermatogenesis are more prominent, leading to the production of elongated spermatids, flagellated sperm and offspring produced by micro‐insemination. This study showed the potential to treat spermatogenic impairments by culturing the testis tissue with the supplementation of factors insufficient in the original testis without any genetic manipulations.38
In 2014, the agarose gel organ culture system also achieved complete spermatogenesis in the cryopreserved testis of neonatal mice. After 7 months of cryopreservation, the testis tissues were cultured for producing sperm that were used to fertilize eggs by micro‐insemination and produce healthy offspring that matured to produce progeny of their own through natural mating. The DNA methylation levels at 11 differentially methylated regions of the offspring showed normal state of methylation. Thus, the results showed the potential usage of cryopreserving the biopsied testis tissue fragments to preserve the fertility of male pediatric cancer patients in the future.39
As mentioned above, adult tissue is more difficult to culture than immature tissue in general, and also in the case of testis. Our group challenged this issue again with the agarose gel organ culture method using transgenic mice that express GFP in meiotic cells.40 In order to make the results clear and unambiguous, mice were treated with vitamin A‐deficient (VAD) diet to remove all differentiated germ cells from the testes leaving undifferentiated spermatogonia alone behind. VAD‐treated testis was cultured successfully for spermatogenesis up to sperm formation using our culture technique described above. Thus, it was proven that in vitro spermatogenesis with mature testis tissue is possible. However, the efficiency is far lower than that of using neonatal mice.40 This adult tissue problem is one of the important issues that needs to be addressed in future studies.
Another important issue on testis organ culture experiment is species differences. It became apparent that testis tissues other than the mouse origin are resistant to in vitro spermatogenesis. Even rat testis tissues are reluctant to advance its spermatogenesis under culture condition (Figure 1). Although, rats and mice are both rodents and share many anatomical and physiological properties, there are several different characteristics between them. Our study on in vitro spermatogenesis using rat testis tissues did not achieve complete spermatogenesis like the results obtained with mice. In contrast to our results, Liu and colleagues cultured testis tissue of 7‐day‐old rats and found that spermatogenesis progressed, demonstrating the expression of Sycp3 and Crisp1 genes by RT‐PCR in 4 weeks of culture. Histological findings are not conclusive but possibly spermatids appeared.41 In vitro spermatogenesis of the rat origin remains to be a challenge to be overcome in future studies.
3.2. Microfluidic system
The agarose gel organ culture method became a reliable standard method, repeated by many independent research groups, to induce mouse spermatogenesis.42, 43, 44, 45 However, the efficiency and duration of the spermatogenesis were far from comparable to those observed in vivo.46 To improve the culture condition, replicating the microcirculatory system of the body in the organ culture system could be effective. Based on this concept, we adopted the microfluidic technology into our organ culture system. The culture device consisted of a chamber for a tissue and a channel for medium flow (Figure 3B). The cultured testis tissue was separated from the flowing medium by a thin porous membrane. Thus, substances like nutrients and waste products were exchanged by molecular diffusion mechanism as same as in vivo. In the microfluidic device, spermatogenesis of mouse was efficiently induced and stably maintained up to 6 months. Healthy offspring were produced through micro‐insemination using sperm and spermatids derived from the testis cultured for 6 months.47 In addition, the testosterone production, another major function of the testis, was also exhibited in the microfluidic system. Furthermore, the hormone production was stimulated to a higher level on addition of the luteinizing hormone in the flowing culture medium, that mimicked the in vivo phenomenon. In order to make the system simpler and user‐friendly, we then developed a device that dispensed with the power pump which was used to generate flow of the culture medium in our original microfluidics device. Using the hydrostatic pressure of the medium in a reserve tank, along with the resistance circuit to control the flow‐speed, the pumpless device was able to create the same culture condition for testis tissue in the tissue chamber (Figure 3C). In fact, the device maintained the mouse spermatogenesis as efficiently as the pump‐connected microfluidic device for 3 months.48 Taken together, these microfluidic devices succeeded in creating a novel organ culture system, which radically differs from the classical gas‐liquid interphase method, and will potentially revolutionize the organ culture method as a whole.
4. TUBULE CULTURE, CULTURE OF A SEGMENT OF SEMINIFEROUS TUBULE
As we saw the limitations of organ culture method in 1960s, another approach using the fragments of the seminiferous tubules, instead of testis tissue mass as in organ culture method, was sought in the following decades. Initially, Eddy and Kahri isolated and cultured fragments of seminiferous tubules from 7 to 35‐day‐old rats in the culture dish as submerged style. They carefully observed the migration of cells out of the tubules. Even after 4 weeks of incubation, the presence of spermatocytes, presumed by their round shape, was detectable in both light and electron microscopy, suggesting the possibility that a tubular culture method can support ex vivo spermatogenesis to some extent.49
Parvinen and colleagues elaborated the tubule culture method. They found that spermatogenic cycle was found to be discerned by looking at tubules with transillumination.50, 51 They demonstrated that when segments of rat seminiferous tubules at stages XII and XIII, that is immediately prior to the meiotic reduction divisions in stage XIV, were cultured in a chemically defined medium of Ham's F12/Dulbecco's MEM, the late pachytene spermatocytes and diakinetic spermatocytes completed the meiotic divisions. The spermatids arising from this culture differentiated up to step 5 of spermiogenesis in vitro.52, 53 Toppari et al, then focusing on spermiogenesis, cut out stages II‐III segment of rat seminiferous tubules and cultured them in the same way. In 7 days, the round spermatids at step 2‐3 developed into step 7, mimicking in vivo differentiation. It is reported also that spermatogonia and spermatocytes in that segment developed accordingly as in vivo54 (Figure 1).
The tubule culture method certainly has its own advantages. It can theoretically separate the seminiferous tubule from the interstitial cells and tissues, thereby eliminating the effect from them. It gives clearer and finer view of the sample than the organ culture method. As Parvinen and colleagues showed, a restricted segment contains particular stages of germ cells which can be followed under culture condition. Thus, the effect of culture condition, medium compositions or physical environmental factors, can be more clearly evaluated than the organ culture method. However, the progress of spermatogenesis with the tubule culture method was not comparable to that with the organ culture method. Tubule culture method needs further elaboration to support longer and sustainable fashion of spermatogenesis.
5. CELL CULTURE METHOD, TWO DIMENSIONAL (2D)
As was shown in 1960s‐70s that organ culture method did not promise its success in promoting in vitro spermatogenesis beyond the pachytene stage, researchers turned their attention to cell culture method. However, cell culture methods had two innate disadvantages compared with organ culture methods. First, losing 3D structure of the seminiferous tubule can be detrimental for proper spermatogenesis to take place, which seems to be the case as discussed below. Secondly, it becomes quite difficult to distinguish spermatogenic cells at different stages without original topological relationships in the seminiferous epithelium. In other words, researchers cannot use or rely on the histological evaluation of spermatogenesis. This second point is not a minor issue even today. Instead of histological features, however, researchers are able to use emerging new methods, like flow cytometry, detection of marker genes expression, or immunostaining of marker proteins.
5.1. Primary culture
It should be noted that the Steinbergers tested the cell culture of rat testicular cells.55 Contrary to their organ culture results, they found no evidence for the differentiation of germ cells in the cell culture experiment. Thus, they denied several former studies claiming complete spermatogenesis in cell culture for a number of months. Their observation was quite reasonable and became the start line for succeeding researchers.
It was reported in 1982 that completely dissociated, by collagenase and trypsin, testis cells of mice, 6‐15 weeks old, pretreated with hydroxyurea to remove differentiated germ cells were cultured to survive 12 days and cells at initial prophase reached pachytene and diplotene phases. It was also shown that preexisting diplotene cells went through the two divisions to become spermatocytes.56 Their method contained unique conditions that make it difficult to repeat exactly what was performed. Interestingly, all cultures were said to be performed at 20°C. In the same year, Tres and Kieszenbaum reported that dissociated rat germ cells labeled with [3H] thymidine differentiated into pachytene spermatocytes in a massive way.57 They claimed that coculture with Sertoli cell was essential for survival and differentiation of germ cell based on their meticulous observation. The above two reports were the firsts showing that dissociated male germ cells underwent meiotic divisions under a culture condition.
Another study reported the production of haploid cells from meiotic cells of the rat. The testicular cells from 14‐day postpartum (dpp) rats, enzymatically dispersed, were cultured on the type 1 collagen gels. The culture media was composed of a 1:1 mixture of Ham's F12 medium and Leibovitz's L15 medium containing 10% FBS supplemented with epinephrine or norepinephrine. After 2 weeks of incubation, Giemsa staining and DNA flow cytometry showed the appearance of 1C cells, indicating the differentiation into the haploid cells.58
Jégou's team addressed the question if dissociated pachytene spermatocytes can complete meiosis in vitro under coculture with Sertoli cells. Pachytene primary spermatocytes collected by an elutriation method from testes of adult rats were seeded on a layer of Sertoli cells from 20‐day‐old rats. During the 7‐day experiment, the cells were cultured at 32°C in F12‐Dulbecco modified essential medium (DMEM) supplemented with insulin, transferrin, retinoic acid, transforming growth factor α, FSH, and testosterone. Under this condition, pachytene spermatocytes were able to complete meiosis. A population of haploid cells was identified by flow cytometry within 4 days of culture. They also detected the expression of protamine‐1 gene, specific to postmeiotic germ cells, by Northern blot analysis. They concluded that hormones and other elements added to the culture medium did not seem to improve the rate of cells passing through meiosis, but appeared to have a positive effect on maintaining cell integrity and viability in long‐term cultures.59
In 1993, another new approach for in vitro spermatogenesis was demonstrated. Cuzin's group first established Sertoli cell line, named 15P‐1, from a transgenic mouse which harbored large T antigen of polyoma virus. The 15P‐1 maintained the character of Sertoli cells, such as the expression of specific genes and phagocytotic activity. This group also developed a new assay method to detect haploid cells using LacZ gene. The testicular cells explanted from the Prm‐lacZ transgenic mice expressed the Escherichia coli LacZ gene specifically at the post‐meiotic stage. Germ cells obtained from the testes of this mouse, 10‐18 dpp, were cultured on the feeder of 15P‐1 for 2‐12 days. The culture medium consisted of DMEM and FBS (10%). The progression of spermatogenesis was evaluated by 5‐bromo‐4‐chloro‐3‐indolyl beta‐D‐galactoside (X‐gal) staining to detect beta‐galactosidase activity and also by the flow cytometry method. Generation of round spermatids during the period of 2‐5 days was described. It was also claimed that testicular germ cells from 9‐day‐old mice underwent trans‐meiotic differentiation.60 The innovative methods and resulting in vitro spermatogenesis described by this group fascinated researchers in the field. However, their claims have not yet been independently replicated.
Millán's group immortalized various testicular cells with simian virus 40 (SV40) large tumor antigen (LTAg).61 Actually they effectively established a number of cell lines; peritubular, Leydig, and Sertoli cell lines. When these cell lines were cultured together, they formed a cord‐like structure. In fact, freshly isolated pachytene spermatocytes cocultured with those immortalized cells integrated in the cord. However, differentiation of these cells was not observed. On the other hand, Millan's team took unique strategy to immortalize germ cells by introducing SV40 LTAg gene along with a temperature‐sensitive (ts) mutant of the mouse p53 gene. A resultant cell line, GC‐2spd (ts), was immortalized by active proliferation under LTAg's effect at 39°C, while also being capable of differentiating at 32‐37°C by activated p53 function which neutralized the activity of LTAg. At 17‐30th passages cultured at 37°C, flow cytometric analysis revealed a peak of haploid cells. The cell line also showed the formation of acrosomic granule.62 Nine years later, the same strategy using different immortalizing factor, telomerase, was adopted to produce a spermatogonial cell line.63 In this case, the immortalized cells reportedly showed differentiation ability up to haploid cells. Studies using immortalized germ cells for in vitro spermatogenesis are attractive and potentially useful for elucidating the mechanism of spermatogenesis. However, manipulating the genome of germ cells cannot be used for reproduction of animals, let alone for human. In this sense, this strategy has its own limitation.
5.2. Importance of germ cell‐Sertoli cell interaction
In 1998, Durand's group reported an incubation of the testicular cells from 23‐25 dpp rats labelled with BrdU in vivo, by intraperitoneal injection, 9 days before culture experiment started. In this way, they faithfully evaluated the fate of the pachytene spermatocytes, the most advanced labeled cells at the start of culturing, in culture. They became secondary spermatocyte in 5 days, and round spermatids in 7 days. The duration of the process appears very close to that in vivo. The production of 1C cells was maintained for 3 weeks in the analysis of ploidy. In this study, DMEM F12‐based medium with 0.2% FCS, testosterone, and FSH was used.64 Testis tissues were digested with collagenase and seminiferous tubules were harvested for further dissociation of the cells. Nonetheless, at the same time, researchers tried to maintain the interaction between Sertoli cells and spermatogenic cells as much as possible. This sounds a bit tricky but was written to be an important point to make spermatogenesis occur in vitro.
The same group used the BrdU‐labeling method to extend the results.65 This time, the BrdU was injected to rats aged 20‐28 dpp one day before the culture experiment started. Thus, the labeled cells at the most advanced stage were leptotene spermatocytes and the progress of their differentiation was followed in culture. In 1 week, BrdU‐labeled small pachytene spermatocytes appeared, while in 2 weeks, middle‐to‐late pachytene spermatocytes were observed as labeled cells. After 3 weeks, diakinesis and secondary spermatocytes were observed. In the end, quadruplets of BrdU‐labeled round spermatids in the culture were observed from Day 21 onward. It was described that these spermatids tended to separate from each other thereafter. This report beautifully described the process of meiotic phase progression in vitro.65 It was suggested that in vitro spermatogenesis requires sufficient period of time to pass through the meiotic process like in vivo spermatogenesis does. Then, Durand's group used the culture technique to find the effect of FSH and testosterone on meiosis. They collected pachytene spermatocytes from adult rats by centrifugal elutriation and cocultured with Sertoli cells of 20 dpp rats for 2 weeks. The addition of FSH and testosterone to DMEM/F12 supplemented with 0.2% FCS increased the number of round spermatids produced, confirming the effect of the two hormones.66
Using immature mice, 13‐18 dpp, Marh et al succeeded in in vitro spermatogenesis from pachytene spermatocyte to round spermatid. In this study, they used serum‐free medium, named TKM, which contains various factors like insulin, insulin‐growth factor 1, growth hormone, epidermal growth factor, retinol, testosterone, and dihydrotestosterone. It was also recognized that a Sertoli cell‐feeder layer played an important role for driving the meiotic division forward. After 7‐10 days of incubation, round spermatids appeared and were used for micro‐insemination, which resulted in offspring production.67 This was the first report that in vitro‐produced haploid cells successfully used for offspring generation with micro‐insemination.
The importance of Sertoli cell and hormones of FSH, testosterone, and epinephrine, were also demonstrated in the process of round to elongating spermatids transformation in vitro. Hasegawa et al elegantly cocultured mouse round spermatids with Sertoli cells, which were collected manually with a micromanipulator, in DMEM supplemented with 10% FBS, testosterone, FSH, and epinephrine, for 2 days. Resultant elongating spermatids were capable of inducing embryo development after micro‐insmeination into oocyte to the same extent as that of in vivo‐derived elongating spermatids, while round spermatids were not able to.68
5.3. Human biopsy specimen
As seen above, human spermatogenesis using organ culture method was limited in success compared to that using mice and rats. In late 1990s, biopsy specimens of patients partially digested or mechanically disintegrated were used for culture experiments. Tesarik et al performed human testis cell culture experiment using biopsy specimens of patients with obstructive azoospermia. They incubated the sample for 24 or 48 hours to observe any progression of spermatogenesis. It was reported that post‐meiotic cell differentiation was observed; morphological changes of spermatid nuclei and flagellar growth were noted. In addition, meiotic progression was also detectable. They argued that some of such progression was enhanced by the presence of FSH 69 (Figure 2). This study, however, was too short in observation periods to generate a conclusive statement on the possibility of in vitro human spermatogenesis. The incubation of the cryopreserved testicular biopsy specimen from non‐obstructive azoospermia was also performed by the same group with the same results. They concluded that the progression of spermatogenesis continued only for 24 hours and no additional benefit was observed by prolonging the culture beyond that time point, which appears rather pessimistic.70
Cremades et al collected round spermatids under the microscope with micro‐manipulator from biopsied sample of four infertile patients. They cultured the round spermatids on the Vero cell feeder in a commercially available medium for embryo culture supplemented with 10% synthetic serum substitute for 5 days. They observed that the round spermatids progressed further up to elongated spermatids.71 Vero cell is a cell line derived from kidney epithelial cells of an African green monkey and was reported to support human embryo development in a culture.72 As the culture was done in microdrops, it may be easier to follow the change of cells of interest. It seems, however, that 5 days are not long enough to observe the process of human spermiogenesis. The same group used Vero cell‐conditioned medium, instead of coculturing with Vero cell feeders, in a drop of medium, for testis cells of non‐obstructive azoospermic patients. In this experiment, Sertoli cells, spermatogonia, spermatocytes, and round spermatids were individually collected under microscope for culturing in a drop. During the culture period of 2‐3 weeks, the Vero cell‐conditioned medium along with FSH and testosterone induced the differentiation of round spermatids into elongated spermatids.73
Studies described above all suggested that Vero cell secretes factors effective for advancing spermatogenesis, particularly around end‐meiotic stage and spermiogenesis. Tanaka also used Vero cell as feeder and cultured primary spermatocyte isolated from the biopsy specimens of azoospermic patients. He reported that single primary spermatocyte at pachytene stage underwent differentiation to form four spermatids in 5 days. The culture medium based on minimum essential medium (MEM) with 50% boar rete testicular fluid or human synthetic oviduct fluid and 10% human serum gave the highest production rate of spermatids, reaching at around 10%, meaning that one out of ten primary spermatocyte became spermatids. The number of chromosomes and chromatids in the newly developed spermatids were confirmed to be 23 by a fluorescence in situ hybridization (FISH) analysis. This study clearly showed that end‐meiotic phase, successive cell divisions of late primary spermatocytes, can take place in vitro.74 However, the period of 5 days may be too short to complete that process. In another study using purified SSCs from azoospermic patients, cells were cocultured with Sertoli cells isolated from patients. CD49f (Integrin α6) was used as a marker of SSCs for their isolation with FACS. After a 5‐day incubation, the immunostaining of SCP3 and CREST showed the existence of the meiotic germ cells. FISH analysis confirmed the appearance of the haploid cells.75 In this study again, 5 days are too short a period to complete such a complicated process of differentiation.
5.4. Experiments using bovine testis
Based on the accumulated knowledge on the 2D cell culture system in rodents, research of in vitro spermatogenesis in other species were performed. In many cases, DMEM or DMEM plus F12 medium supplemented with 10% FBS with or without testosterone and FSH was used. Izadyar et al partially purified type A spermatogonia, presumably undifferentiated spermatogonia, from enzymatically dissociated cells of bovine testicle and cultured them under various conditions. The cells differentiated into the c‐kit positive cells, corresponding to differentiating spermatogonia, in 4 weeks of incubation. Although not conclusive, after the long‐term incubation over 3 months, cells appeared like elongated cells with a tail‐like structure containing a condensed nucleus resembling spermatozoa.76 In another report, a mixture of testicular cells derived from 3‐ to 5‐month‐old buffaloes, containing spermatogonia alone as germ cell, was cultured with the medium supplemented with FBS, testosterone, and retinol. It was described that spermatogonia differentiated into spermatid‐like cells with a flagellum after 4 weeks of culture. The expression of the spermatid‐specific marker gene (PRM2) was also identified by RT‐PCR.77
6. CELL CULTURE METHOD, THREE DIMENSIONAL (3D)
Experiencing unsatisfactory results with 2D cell culture methods on in vitro spermatogenesis, researchers naturally turned their strategy toward 3D methods, by artificially constructing three‐dimensional structure in vitro. Such trials have appeared since 2000's. First, Lee et al tested the effect of reconstructing tubular structure with rat testicular cells, using a collagen gel and Matrigel. They reported that a tubular structure was produced in those gel‐assisted spaces and differentiation of germ cells toward meiosis and beyond was possible. The picture of the tubule structure, however, in our view, does not seem to contain germ cells.78 Thus, their results may suggest that simple mixture of germ cells with testicular somatic cells in a 3D environment may favor differentiation of germ cells, through paracrine or cell‐cell attaching effect between germ and somatic cells. This method was also tested with human samples. Authors claimed that spermatocytes could be induced to differentiate into presumptive spermatids, judged by flow cytometry and immunocytochemical analysis, in their collagen gel matrix culture method.79
Stukenborg et al reported a method of 3D culture system, which they introduced from the earlier studies of bone marrow hematopoietic cells. The soft agar culture system (SACS) method had been used as colony‐forming assay to detect different kinds of hematopoietic cells and to support their differentiation. In adopting this concept, Stukenborg et al made their own modification of the system and suggested that a two‐layer agar mimicked the in vivo niche environment of seminiferous tubules supporting for SSCs. The testicular cells obtained from 10‐day‐old mice were mixed with the gel‐agar medium (0.35%) and incubated on a solid‐agar base (0.5%). These agars were mixed with high glucose DMEM solutions. The expression of the markers for meiotic cells, Boule and Crem, were maintained for 3 weeks.80 This group then modified the method by using the methylcellulose as the matrix and adding the hCG and FSH to the culture medium.81 Using the SACS method in 2011, the testicular cells obtained from the 7‐day mouse were cultured in the culture medium containing RPMI plus 20% FBS for 4 weeks. Cultured cells expressed the meiotic (Crem‐1 and LDH) and post‐meiotic stages (Protamine) makers which were detected by RT‐PCR. Immunohistochemistry showed the appearance of the protamine positive cells. Authors claimed that spermatids including spermatozoa were also observed.82 The same method was applied to the monkey. Huleihel et al incubated the testicular cells from juvenile (13‐33 months old) rhesus monkeys in the SACS system. The VASA‐, SALL4‐, and GFR‐α1‐positive cells were constantly observed, suggesting the maintenance of the pre‐meiotic germ cells. After 1‐month incubation, the CREM‐1‐ and acrosin‐positive cells appeared, suggesting the progression of spermatogenesis into the meiotic and post‐meiotic stages.83 Throughout the experiments with SACS method, however, reconstruction of tubular structure or any reminiscents of seminiferous epithelium was not shown. Thus, those results appear to suggest, as mentioned above, that the distribution of testicular cells in a 3D condition per se might have an effect on promoting germ cell differentiation. This raises a long‐lasting fundamental question about spermatogenesis that is, whether the mammalian germ cells in order to become sperms definitely need intra‐seminiferous tubular condition or not. We do not know the answer yet.
Legendre et al developed another type of 3D culture method that uses the bicameral chamber in which a condition similar to the apical and basal compartments of the seminiferous tubule would be replicated. They collected testicular cells from 18‐day‐old rats. Peritubular cells were first cultured on the underside of the insert which was set upside down for cells to adhere. After setting the insert in the original position, a mixture of Sertoli and germ cells were poured in the insert which was coated with an artificial extracellular matrix, mixture of Matrigel matrix and type 1 collagen. The insert was soaked in the culture medium containing DMEM/F‐12 with FSH, testosterone and 5% FBS for 22 days. Under this culture condition, they concluded that haploid cells were produced from early pachytene or zygotene spermatocytes, based on following findings: Giemsa staining showed the cytoplasm and DNA condensation characteristics for the haploid cells. RT‐PCR detected the increase of transition protein 2 mRNA, a post‐meiotic marker.84
Reconstruction of a tubular structure from dissociated testicular cells, if feasible, could be a basis for in vitro spermatogenesis. Our group reported that such a reconstruction of the seminiferous tubule structure is possible in vivo in the subcutaneous space of mice.85 Then, we extended that study by demonstrating the reconstruction of tubular structure with dissociated neonatal testicular cells totally in vitro.86 Specifically, enzymatically dispersed testicular cells from neonatal mice (0.5‐5.5 dpp) were introduced into the well of V‐ bottom plate to let them aggregate. After 2 days of incubation, the formed spheroid was placed on the top of agarose gel block and incubated as regular organ culture experiments. The cells in an aggregate spontaneously formed tubule‐like structures in which germ cells were incorporated inside. Some germ cells differentiated up to round spermatid stage. When SSCs from other sources were intermingled in the aggregate, some of them were also incorporated and they underwent differentiation up to meiotic phase. This the first report showing that reconstructed tubular structure was able to support spermatogenesis from SSCs up to haploid cell formation. However, the efficiency was yet lower than the regular testis tissue organ culture experiments. There is much room to improve culture conditions for more efficient spermatogenesis.
7. CONCLUSION
In this review, we recognized a steady progress of the study of in vitro spermatogenesis over the last century. It was rapid and drastic occasionally but, in most other times, it was rather sluggish. In vitro spermatogenesis has been always a tough project for researchers. It is sure that spermatogenesis is a complex biological process. As a result, the microenvironment in the testis is tuned in each species for maximum productivity which is thus vulnerable for any minor disturbances in that condition. Building up such delicate micro‐environment out of the body is a challenging task and now becoming a prime theme for modern biology. We hope that researchers will conquer this task through applying cutting‐edge technologies and broad knowledge in the related areas. The artificially produced in vitro environment, which is really comparable to that of in vivo, can be used to decipher the relationship between the environment and the quality of the products, as well as the genomic and epigenomic integrity of sperm. Such research will benefit our understanding on the reproduction and our future reproductive medicine.
DISCLOSURE
Conflict of interest: The authors declare that they have no conflict of interest. Human/Animal rights: This article does not contain any studies with human and animal participants performed by any of the authors.
ACKNOWLEDGEMENTS
Authors thank Drs. Makoto Nagano and Kyle Orwig for their critical and constructive comments on the manuscript. This work was supported by grants from a Grant‐in‐Aid for Scientific Research on Innovative Areas, `Mechanisms regulating gamete formation in animals’ 25114007 (to T.O.); Grant‐in‐Aid for Young Scientists (A) 17H05098 (to M.K.); Grant of Yamaguchi Endocrine Research Foundation (to M.K.); Grant for 2016—2018 Strategic Research Promotion of Yokohama City University (SK2811) (to T.O.).
Komeya M, Sato T, Ogawa T. In vitro spermatogenesis: a century‐long research journey, still half way around. Reprod Med Biol. 2018;17:407–420. 10.1002/rmb2.12225
REFERENCES
- 1. Amann RP. The cycle of the seminiferous epithelium in humans: a need to revisit? J Androl. 2008;29:469‐487. [DOI] [PubMed] [Google Scholar]
- 2. Oakberg EF. Duration of spermatogenesis in the mouse and timing of stages of the cycle of the seminiferous epithelium. Am. J. Anat. 1956;99:507‐516. [DOI] [PubMed] [Google Scholar]
- 3. Bascom KF, Osterud HL. Quantitative studies of the testicle. II. Pattern and total tubule length in the testicles of certain common mammals. Anat Rec. 1925;31:159‐169. [Google Scholar]
- 4. Nakata H, Sonomura T, Iseki S. Three‐dimensional analysis of seminiferous tubules and spermatogenic waves in mice. Reproduction. 2017;154:569‐579. [DOI] [PubMed] [Google Scholar]
- 5. Potter SJ, DeFalco T. Role of the testis interstitial compartment in spermatogonial stem cell function. Reproduction. 2017;153:151‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Champy CH. De la méthode de culture des tissus. VI. Le testicule. Arch Zool Exptl Gen. 1920;60:461‐500. [Google Scholar]
- 7. Martinovitch PN. The development in vitro of the mammalian gonad. Ovary and ovogenesis. Proc R Soc B Biol Sci. 1938;125:232‐249. [Google Scholar]
- 8. Trowell OA. The culture of mature organs in a synthetic medium. Exp Cell Res. 1959;16:118‐147. [DOI] [PubMed] [Google Scholar]
- 9. Yao T, Asayama Y. Animal‐cell culture media: history, characteristics, and current issues. Reprod Med Biol. 2017;16:99‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Steinberger A, Steinberger E, Perloff WH. Growth of rat testes fragments in organ culture. Fed Proc. 1963;22:372. [Google Scholar]
- 11. Steinberger E, Steinberger A, Perloff WH. Studies on growth in organ culture of testicular tissue from rats of vaious ages. Anat Rec. 1964;148:581‐589. [DOI] [PubMed] [Google Scholar]
- 12. Steinberger E, Steinberger A, Perloff WH. Initiation of spermatogenesis in vitro. Endocrinology. 1964;74:788‐792. [DOI] [PubMed] [Google Scholar]
- 13. Steinberger A, Steinberger E, Perloff WH. Mammalian testis in organ culture. Exp Cell Res. 1964;36:19‐27. [DOI] [PubMed] [Google Scholar]
- 14. Steinberger A, Steinberger E. Differentiation of rat seminiferous epithelium in organ culture. J Reprod Fertil. 1965;9:243‐248. [DOI] [PubMed] [Google Scholar]
- 15. Steinberger A, Steinberger E. Stimulatory effect of vitamins and glutamine on the differentiation of germ cells in rat testes organ culture grown in chemically defined media. Exp Cell Res. 1966;44:429‐435. [DOI] [PubMed] [Google Scholar]
- 16. Steinberger A, Steinberger E. Factors affecting spermatogenesis in organ cultures of mammalian testes. J Reprod Fertil. 1967;Suppl. 2:117‐124. [Google Scholar]
- 17. Steinberger A, Steinberger E. Tissue culture of male mammalian gonads. In Vitro. 1970;5:17‐27. [DOI] [PubMed] [Google Scholar]
- 18. Aizawa S, Nishimune Y. In‐vitro differentiation of type A spermatogonia in mouse cryptorchid testis. J Reprod Fertil. 1979;56:99‐104. [DOI] [PubMed] [Google Scholar]
- 19. Haneji T, Nishimune Y. Hormones and the differentiation of type A spermatogonia in mouse cryptorchid testes incubated in vitro. J Endocrinol. 1982;94:43‐50. [DOI] [PubMed] [Google Scholar]
- 20. Haneji T, Maekawa M, Nishimune Y. Vitamin A and follicle‐stimulating hormone synergistically induce differentiation of type A spermatogonia in adult mouse cryptorchid testes in vitro. Endocrinology. 1984;114:801‐805. [DOI] [PubMed] [Google Scholar]
- 21. Nishimune YM, Osaka M. In vitro differentiation mouse cryptorchid of type a spermatogonia from testes in serum‐free. Biol Reprod. 1983;28:1217‐1223. [DOI] [PubMed] [Google Scholar]
- 22. Tajima Y, Watanabe D, Koshimizu U, Matsuzawa T, Nishimune Y. Insulin‐like growth factor‐I and transforming growth factor‐α stimulate differentiation of type A spermatogonia in organ culture of adult mouse cryptorchid testes. Int J Androl. 1995;18:8‐12. [DOI] [PubMed] [Google Scholar]
- 23. Busada JT, Geyer CB. The role of retinoic acid (RA) in spermatogonial differentiation. Biol Reprod. 2016;94:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Boitani C, Politi MG, Menna T. Spermatogonial cell proliferation in organ culture of immature rat testis. Biol Reprod. 1993;48:761‐767. [DOI] [PubMed] [Google Scholar]
- 25. Suzuki S, Sato K. The fertilising ability of spermatogenic cells derived from cultured mouse immature testicular tissue. Zygote. 2003;11:307‐316. [DOI] [PubMed] [Google Scholar]
- 26. Matte R, Sasaki M. Autoradiographic evidence of human male germ‐cell differentiation in vitro. Cytologia. 1971;36:298‐303. [DOI] [PubMed] [Google Scholar]
- 27. Ghatnekar R, Lima‐De‐faria A, Rubin S, Menander K. Development of human male meiosis in vitro. Hereditas. 1974;78:265‐271. [DOI] [PubMed] [Google Scholar]
- 28. Curtis D. In vitro differentiation of diakinesis figures in human testis. Hum Genet. 1981;59:406‐411. [DOI] [PubMed] [Google Scholar]
- 29. Szczepny A, Hogarth CA, Young J, Loveland KL. Identification of Hedgehog signaling outcomes in mouse testis development using a hanging drop‐culture system. Biol Reprod. 1999;80:258‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jørgensen A, Young J, Nielsen JE, et al. Hanging drop cultures of human testis and testis cancer samples: a model used to investigate activin treatment effects in a preserved niche. Br J Cancer. 2014;110:2604‐2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gohbara A, Katagiri K, Sato T, et al. In vitro murine spermatogenesis in an organ culture system. Biol Reprod. 2010;83:261‐267. [DOI] [PubMed] [Google Scholar]
- 32. Sato T, Katagiri K, Gohbara A, et al. In vitro production of functional sperm in cultured neonatal mouse testes. Nature. 2011;471:504‐507. [DOI] [PubMed] [Google Scholar]
- 33. Sanjo H, Komeya M, Sato T, et al. In vitro mouse spermatogenesis with an organ culture method in chemically defined medium. PLoS ONE. 2018;13:e0192884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kanatsu‐Shinohara M, Ogonuki N, Inoue K, et al. Long‐term proliferation in culture and germline transmission of mouse male germline stem cells. Biol Reprod. 2003;69:612‐616. [DOI] [PubMed] [Google Scholar]
- 35. Ogawa T, Ohmura M, Tamura Y, et al. Derivation and morphological characterization of mouse spermatogonial stem cell lines. Arch Histol Cytol. 2004;67:297‐306. [DOI] [PubMed] [Google Scholar]
- 36. Kubota H, Avarbock MR, Brinster RL. Growth factors essential for self‐renewal and expansion of mouse spermatogonial stem cells. Proc. Natl. Acad. Sci. U. S. A. 2004;101:16489‐16494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sato T, Katagiri K, Yokonishi T, et al. In vitro production of fertile sperm from murine spermatogonial stem cell lines. Nat Commun. 2011;2:472. [DOI] [PubMed] [Google Scholar]
- 38. Sato T, Yokonishi T, Komeya M, et al. Testis tissue explantation cures spermatogenic failure in c‐Kit ligand mutant mice. Proc. Natl. Acad. Sci. U. S. A. 2012;109:16934‐16938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yokonishi T, Sato T, Komeya M, et al. Offspring production with sperm grown in vitro from cryopreserved testis tissues. Nat Commun. 2014;5:4320. [DOI] [PubMed] [Google Scholar]
- 40. Sato T, Katagiri K, Kojima K, Komeya M, Yao M, Ogawa T. In vitro spermatogenesis in explanted adult mouse testis tissues. PLoS ONE. 2015;10:e0130171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu F, Cai C, Wu X, et al. Effect of KnockOut serum replacement on germ cell development of immature testis tissue culture. Theriogenology. 2016;85:193‐199. [DOI] [PubMed] [Google Scholar]
- 42. Arkoun B, Dumont L, Milazzo JP, et al. Retinol improves in vitro differentiation of pre‐pubertal mouse spermatogonial stem cells into sperm during the first wave of spermatogenesis. PLoS ONE. 2015;10:e0116660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dumont L, Oblette A, Rondanino C, et al. Vitamin A prevents round spermatid nuclear damage and promotes the production of motile sperm during in vitro maturation of vitrified pre‐pubertal mouse testicular tissue. Mol Hum Reprod. 2016;22:819‐832. [DOI] [PubMed] [Google Scholar]
- 44. Isoler‐Alcaraz J, Fernández‐Pérez D, Larriba E, Del Mazo J. Cellular and molecular characterization of gametogenic progression in ex vivo cultured prepuberal mouse testes. Reprod Biol Endocrinol. 2017;15:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nakamura N, Merry GE, Inselman AL, et al. Evaluation of Culture Time and Media in an In Vitro Testis Organ Culture System. Birth Defects Res. 2017;109:465‐474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ogawa T. In vitro spermatogenesis: the dawn of a new era in the study of male infertility. Int J Urol. 2012;19:282‐283. [DOI] [PubMed] [Google Scholar]
- 47. Komeya M, Kimura H, Nakamura H, et al. Long‐term ex vivo maintenance of testis tissues producing fertile sperm in a microfluidic device. Sci Rep. 2016;6:21472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Komeya M, Hayashi K, Nakamura H, et al. Pumpless microfluidic system driven by hydrostatic pressure induces and maintains mouse spermatogenesis in vitro. Sci Rep. 2017;7:15459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Eddy EM, Kahri AI. Cell associations and surface features in cultures of juvenile rat seminiferous tubules. Anat Rec. 1976;185:333‐357. [DOI] [PubMed] [Google Scholar]
- 50. Parvinen M, Vanha‐Perttula T. Identification and enzyme quantitation of the stages of the seminiferous epithelial wave in the rat. Anat Rec. 1972;174:435‐449. [DOI] [PubMed] [Google Scholar]
- 51. Söderström KO, Parvinen M. RNA synthesis in different stages of rat seminiferous epithelial cycle. Mol Cell Endocrinol. 1976;5:181‐199. [DOI] [PubMed] [Google Scholar]
- 52. Parvinen M, Wright WW, Phillips DM, Mather NA, Musto NA, Bardin CW. Spermatogenesis in vitro: completion of meiosis and early spermiogenesis. Endocrinology. 1983;112:1150‐1152. [DOI] [PubMed] [Google Scholar]
- 53. Toppari J, Parvinen M. In vitro differentiation of rat seminiferous tubular segments from defined stages of the epithelial cycle morphologic and immunolocalization analysis. J Androl. 1985;6:334‐343. [DOI] [PubMed] [Google Scholar]
- 54. Toppari J, Mali P, Eerola E. Rat spermatogenesis in vitro traced by quantitative flow cytometry. J Histochem Cytochem. 1986;34:1029‐1035. [DOI] [PubMed] [Google Scholar]
- 55. Steinberger A, Steinberger E. In vitro culture of rat testicular cells. Exp Cell Res. 1966;44:443‐452. [DOI] [PubMed] [Google Scholar]
- 56. Dietrich AJJ, Scholten R, Vink ACG, Oud JL. Testicular cell suspensions of the mouse in vitro. Andrologia. 1983;15:236‐246. [DOI] [PubMed] [Google Scholar]
- 57. Tres LL, Kierszenbaum AL. Viability of rat spermatogenic cells in vitro is facilitated by their coculture with Sertoli cells in serum‐free hormone‐supplemented medium. Proc Natl Acad Sci U S A. 1983;80:3377‐3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nagao Y. Viability of meiotic prophase spermatocytes of rats is facilitated in primary culture of dispersed testicular cells on collagen gel by supplementing epinephrine or norepinephrine: evidence that meiotic prophase spermatocytes complete meiotic divisions in vitro. In Vitro Cell. Dev. Biol. 1989;25:1088‐1098. [DOI] [PubMed] [Google Scholar]
- 59. Le Magueresse‐Battistoni B, Gérard N, Jégou B. Pachytene spermatocytes can achieve meiotic process in vitro. Biochem Biophys Res Commun. 1991;179:1115‐1121. [DOI] [PubMed] [Google Scholar]
- 60. Rassoulzadegan M, Paquis‐Flucklinger V, Bertino B, et al. Transmeiotic differentiation of male germ cells in culture. Cell. 1993;75:997‐1006. [DOI] [PubMed] [Google Scholar]
- 61. Hofmann MC, Narisawa S, Hess RA, Millán JL. Immortalization of germ cells and somatic testicular cells using the SV40 large T antigen. Exp Cell Res. 1992;201:417‐435. [DOI] [PubMed] [Google Scholar]
- 62. Hofmann MC, Hess R a, Goldberg E, Millán JL. Immortalized germ cells undergo meiosis in vitro. Proc. Natl. Acad. Sci. U. S. A. 1994;91:5533‐5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Feng LX, Chen Y, Dettin L, et al. Generation and in vitro differentiation of a spermatogonial cell line. Science. 2002;297:392‐395. [DOI] [PubMed] [Google Scholar]
- 64. Hue D, Staub C, Perrard‐Sapori MH, et al. Meiotic differentiation of germinal cells in three‐week cultures of whole cell population from rat seminiferous tubules. Biol Reprod. 1998;59:379‐387. [DOI] [PubMed] [Google Scholar]
- 65. Staub C, Hue D, Nicolle JC, Perrard‐Sapori MH, Segretain D, Durand P. The whole meiotic process can occur in vitro in untransformed rat spermatogenic cells. Exp Cell Res. 2000;260:85‐95. [DOI] [PubMed] [Google Scholar]
- 66. Vigier M, Weiss M, Perrard MH, Godet M, Durand P. The effects of FSH and of testosterone on the completion of meiosis and the very early steps of spermiogenesis of the rat: an in vitro study. J Mol Endocrinol. 2004;33:729‐742. [DOI] [PubMed] [Google Scholar]
- 67. Marh J, Tres LL, Yamazaki Y, Yanagimachi R, Kierszenbaum AL. Mouse round spermatids developed in vitro from preexisting spermatocytes can produce normal offspring by nuclear injection into in vivo‐developed mature oocytes. Biol Reprod. 2003;69:169‐176. [DOI] [PubMed] [Google Scholar]
- 68. Hasegawa H, Terada Y, Ugajin T, Yaegashi N, Sato K. A novel culture system for mouse spermatid maturation which produces elongating spermatids capable of inducing calcium oscillation during fertilization and embryonic development. J Assist Reprod Genet. 2010;27:565‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tesarik J, Greco E, Rienzi L, et al. Differentiation of spermatogenic cells during in‐vitro culture of testicular biopsy samples from patients with obstructive azoospermia: effect of recombinant follicle stimulating hormone. Hum Reprod. 1998;13:2772‐2781. [DOI] [PubMed] [Google Scholar]
- 70. Tesarik J, Mendoza C, Anniballo R, Greco E. In‐vitro differentiation of germ cells from frozen testicular biopsy specimens. Hum Reprod. 2000;15:1713‐1716. [DOI] [PubMed] [Google Scholar]
- 71. Cremades N, Bernabeu R, Barros A, Sousa M. In‐vitro maturation of round spermatids using co‐culture on Vero cells. Hum Reprod. 1999;14:1287‐1293. [DOI] [PubMed] [Google Scholar]
- 72. Ménézo YJ, Guerin JF, Czyba JC. Improvement of human early embryo development in‐vitro by co‐culture on monolayers of Vero cells. Biol Reprod. 1990;42:301‐306. [DOI] [PubMed] [Google Scholar]
- 73. Sousa M, Cremades N, Alves C, Silva J, Barros A. Developmental potential of human spermatogenic cells co‐cultured with Sertoli cells. Hum Reprod. 2002;17:161‐172. [DOI] [PubMed] [Google Scholar]
- 74. Tanaka A, Nagayoshi M, Awata S, Mawatari Y, Tanaka I, Kusunoki H. Completion of meiosis in human primary spermatocytes through in vitro coculture with Vero cells. Fertil Steril. 2003;79:795‐801. [DOI] [PubMed] [Google Scholar]
- 75. Riboldi M, Rubio C, Pellicer A, Gil‐Salom M, Simón C. In vitro production of haploid cells after coculture of CD49f+ with Sertoli cells from testicular sperm extraction in nonobstructive azoospermic patients. Fertil Steril. 2012;98:580‐590. [DOI] [PubMed] [Google Scholar]
- 76. Izadyar F, Den Ouden K, Creemers LB, Posthuma G, Parvinen M, De Rooij DG. Proliferation and differentiation of bovine type A spermatogonia during long‐term culture. Biol Reprod. 2003;68:272‐281. [DOI] [PubMed] [Google Scholar]
- 77. Xie B, Qin Z, Huang B, et al. In vitro culture and differentiation of buffalo (Bubalus bubalis) Spermatogonia. Reprod Domest Anim. 2010;45:275‐282. [DOI] [PubMed] [Google Scholar]
- 78. Lee JH, Kim HJ, Kim H, Lee SJ, Gye MC. In vitro spermatogenesis by three‐dimensional culture of rat testicular cells in collagen gel matrix. Biomaterials. 2006;27:2845‐253. [DOI] [PubMed] [Google Scholar]
- 79. Lee JH, Gye MC, Choi KW, et al. In vitro differentiation of germ cells from nonobstructive azoospermic patients using three‐dimensional culture in a collagen gel matrix. Fertil Steril. 2007;87:824‐833. [DOI] [PubMed] [Google Scholar]
- 80. Stukenborg JB, Wistuba J, Luetjens CM, et al. Coculture of spermatogonia with somatic cells in a novel three‐dimensional soft‐agar‐culture‐system. J Androl. 2008;29:312‐329. [DOI] [PubMed] [Google Scholar]
- 81. Stukenborg JB, Schlatt S, Simoni M, et al. New horizons for in vitro spermatogenesis? An update on novel three‐dimensional culture systems as tools for meiotic and post‐meiotic differentiation of testicular germ cells. Mol Hum Reprod. 2009;15:521‐529. [DOI] [PubMed] [Google Scholar]
- 82. Abu Elhija M, Lunenfeld E, Schlatt S, Huleihel M. Differentiation of murine male germ cells to spermatozoa in a soft agar culture system. Asian J. Androl. 2012;14:285‐293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Huleihel M, Nourashrafeddin S, Plant TM. Application of three‐dimensional culture systems to study mammalian spermatogenesis, with an emphasis on the rhesus monkey (Macaca mulatta). Asian J. Androl. 2015;17:972‐980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Legendre A, Froment P, Desmots S, Lecomte A, Habert R, Lemazurier E. An engineered 3D blood‐testis barrier model for the assessment of reproductive toxicity potential. Biomaterials. 2010;31:4492‐4505. [DOI] [PubMed] [Google Scholar]
- 85. Kita K, Watanabe T, Ohsaka K, et al. Production of functional spermatids from mouse germline stem cells in ectopically reconstituted seminiferous tubules. Biol Reprod. 2007;76:211‐217. [DOI] [PubMed] [Google Scholar]
- 86. Yokonishi T, Sato T, Katagiri K, Komeya M, Kubota Y, Ogawa T. In vitro reconstruction of mouse seminiferous tubules supporting germ cell differentiation. Biol Reprod. 2013;89:15. [DOI] [PubMed] [Google Scholar]