

Abstract
The catalytically crucial N‐terminal histidine (His1) of fungal lytic polysaccharide monooxygenases (LPMOs) is post‐translationally modified to carry a methylation. The functional role of this methylation remains unknown. We have carried out an in‐depth functional comparison of two variants of a family AA9 LPMO from Thermoascus aurantiacus (TaLPMO9A), one with, and one without the methylation on His1. Various activity assays showed that the two enzyme variants are identical in terms of substrate preferences, cleavage specificities and the ability to activate molecular oxygen. During the course of this work, new functional features of TaLPMO9A were discovered, in particular the ability to cleave xyloglucan, and these features were identical for both variants. Using a variety of techniques, we further found that methylation has minimal effects on the pK a of His1, the affinity for copper and the redox potential of bound copper. The two LPMOs did, however, show clear differences in their resistance against oxidative damage. Studies with added hydrogen peroxide confirmed recent claims that low concentrations of H2O2 boost LPMO activity, whereas excess H2O2 leads to LPMO inactivation. The methylated variant of TaLPMO9A, produced in Aspergillus oryzae, was more resistant to excess H2O2 and showed better process performance when using conditions that promote generation of reactive‐oxygen species. LPMOs need to protect themselves from reactive oxygen species generated in their active sites and this study shows that methylation of the fully conserved N‐terminal histidine provides such protection.
Keywords: lytic polysaccharide monooxygenase, histidine, methylation, hydrogen peroxide, Thermoascus aurantiacus
Abbreviations
- AA
auxiliary activity
- LPMO
lytic polysaccharide monooxygenase
- PASC
phosphoric acid swollen cellulose
Introduction
Copper‐dependent lytic polysaccharide monooxygenases (LPMOs) oxidatively cleave polysaccharides.1, 2, 3, 4, 5 They are classified as Auxiliary Activities in the Carbohydrate‐Active enzyme (CAZy; http://www.cazy.org 6) database and belong to the families AA9, AA10, AA11, AA13, AA14, and AA15.6 The discovery of LPMOs in 20101 has been followed by extensive research related to their structure, function and diversity.4, 5 Since LPMO action renders the crystalline structure of recalcitrant substrates such as cellulose and chitin more susceptible to the action of classical glycoside hydrolases (GHs), LPMOs have become an important ingredient of commercial enzyme cocktails for industrial biomass conversion.7, 8
As of today, around 50 LPMOs have been characterized (according to the CAZy database), of which most are fungal LPMOs belonging to the AA9 family.9 Characterized fungal LPMOs act on various polysaccharides, including cellulose,10, 11 xyloglucan and other (1,4)‐linked β‐glucans,12, 13 starch,14, 15, and xylan grafted onto cellulose.16 Regarding their oxidative regioselectivity, cellulose‐active LPMOs may act on the C1 or the C4 in the scissile glycosidic bond, or on both.2, 17, 18 Structural analyses have shown that the copper ion in the active site is coordinated by a highly conserved “histidine brace” formed by three nitrogen ligands provided by the N‐terminal amino group and the side chains of the N‐terminal histidine (His1) and the side chain of an additional histidine2 (Fig. 1).
Figure 1.
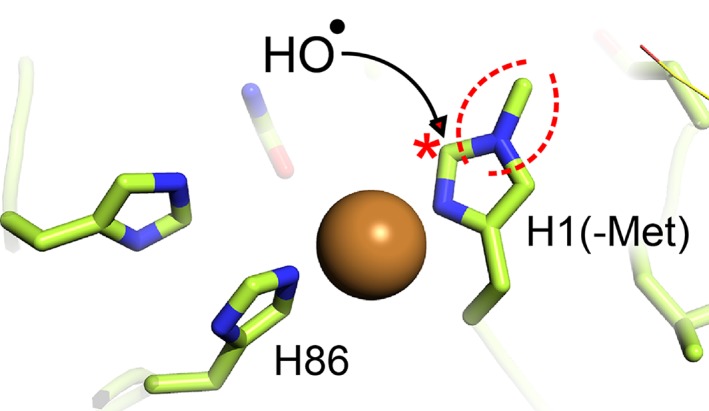
The active site of TaLPMO9A. Two histidine side chains (His1 and His86) contribute to copper coordination in a highly conserved structural arrangement.2 In fungal LPMOs His1 is methylated; the methylgroup is indicated by dotted red lines. Oxidation of histidine, discussed further below, is thought to start at Cε1,41, 53, 54 which is indicated by a red star and which is adjacent to the methylated nitrogen.
Fungal LPMOs have been successfully produced in several fungal species such as Aspergillus oryzae, 2, 19 Neurospora crassa, 17 and Myceliophthora thermophile, 20 and in the yeasts Pichia pastoris (recently renamed to Komagataella phaffii) and Saccharomyces cerevisiae. 18, 21, 22, 23, 24 In nature, fungal LPMOs undergo several post‐translational modifications (PTMs) including signal peptide cleavage, disulfide bond formation and τ‐methylation of the N‐terminal histidine2, 14, 17 (Fig. 1), and may also be subject to O‐ and N‐linked glycosylations.5, 25, 26 Accurate cleavage of the signal peptide is important for activity because the N‐terminal amino group is part of the catalytic center. Remarkably, the role of the τ‐methylation (i.e., methylation of Nε2) of the catalytically crucial His1 (Fig. 1) remains unclear, although some suggestions have been made.4, 5, 27, 28 It is noteworthy that the N‐terminal histidines of bacterial LPMOs are not methylated.4, 5, 29
Fungal LPMOs expressed in filamentous fungi or yeasts will display differences in their PTMs. When expressed in fungi, methylation of the His1 occurs,2, 14, 17, 30 whereas yeasts lack the methyltransferase responsible for this PTM, meaning that His1 remains non‐methylated. Besides this, different expression hosts may yield proteins with different glycosylation patterns, which could affect enzyme properties, although expression in especially P. pastoris is generally considered to result in functional recombinant proteins.25, 31 Expression in P. pastoris will lead to LPMOs lacking their natural PTMs, most notably methylation of the N‐terminus, but LPMOs produced in this manner tend to be active, as illustrated by their ability to boost the overall cellulolytic activity of Celluclast, a commercial LPMO‐poor cellulase cocktail.7, 32 Thus, the functional implications of the methylation remain an enigma.
In this study, we have expressed a cellulose‐active LPMO from Thermoascus aurantiacus (TaLPMO9A, previously known as TaGH61A) in P. pastoris (TaLPMO9A‐Pp), and compared its properties with the properties of the same LPMO expressed in the fungus A. oryzae (TaLPMO9A‐Ao), a host known to carry out PTMs such as methylation.2 The investigated enzyme properties included activity, substrate specificity, substrate binding, the ability to generate H2O2, product profiles, the pK a of His1, redox potential, copper binding, thermal and operational stability, and the boosting effect on the efficiency of Celluclast, an LPMO‐poor cellulase cocktail. These analyses showed that the only major difference between the two enzyme variants concerns their operational stability, in particular their resistance against oxidative damage, which seems to be a major determinant of LPMO performance during biomass conversion.
Results
Heterologous expression of TaLPMO9A in Pichia pastoris and Aspergillus oryzae
TaLPMO9A expressed in P. pastoris (TaLPMO9A‐Pp) or in A. oryzae (TaLPMO9A‐Ao) was purified to homogeneity and the mass of both enzyme variants was between 25 and 30 kDa, based on SDS‐PAGE (Supporting Information Fig. S1). These similar masses were slightly higher than the theoretical mass (24.4 kDa) of the protein calculated from its amino acid sequence. This minor discrepancy is likely due to N‐ and/or O‐glycosylations. The NetNGlyc and NetOGlyc servers (http://www.cbs.dtu.dk/services) predicted one putative N‐glycosylation site, Asn138, and five putative O‐glycosylation sites, Ser35, Thr37, Thr39, Thr47, and Thr144. None of these are located on the catalytic surface of the protein (Supporting Information Fig. S2). The glycosylation sites nearest to the histidine brace, Thr39 and Thr47, are located 13.7 and 15.3 Å away from the catalytic copper, respectively.
First, we analyzed the N‐terminus of the TaLPMO9A preparations to confirm correct processing of the signal peptide and the presence of methylation on His1. The two TaLPMO9A variants were digested with trypsin, which, in case of correct processing, would yield a non‐methylated N‐terminal peptide (HGFVQNIVIDGK) or a methylated N‐terminal peptide (Met‐HGFVQNIVIDGK) with corresponding theoretical masses of 1326.52 or 1340.55 Da, respectively. LC–MS analysis confirmed that the signal peptide was correctly cleaved right before the His1 in 97% or >99% of the cases for TaLPMO9A‐Ao and TaLPMO9A‐Pp, respectively. In addition, methylation of His1 was absent in the TaLPMO9A‐Pp, while in the TaLPMO9A‐Ao sample, the majority of the protein carried the methylation (Supporting Information Fig. S3).
Substrate binding and specificity
TaLPMO9A‐Ao and TaLPMO9A‐Pp showed weak and similar binding to PASC and Avicel. Upon mixing with the substrate, the concentration of free enzyme in solution was lowered by 16% in the presence of PASC (Supporting Information Fig. S4), or ≤5% in case of Avicel (data not shown). The observation of weak binding is congruent with previously reported binding data for LPMOs lacking a CBM domain.33, 34
Both TaLPMO9A‐Ao and TaLPMO9A‐Pp showed activity toward PASC, tamarind xyloglucan (TXG) and steam‐exploded birch (SEB) in the presence of ascorbic acid (Fig. 2 and Supporting Information Fig. S5), while both enzymes were inactive toward cellopentaose (Supporting Information Fig. S6), ivory nut mannan, konjac glucomannan, and xylan from birch wood (data not shown).
Figure 2.
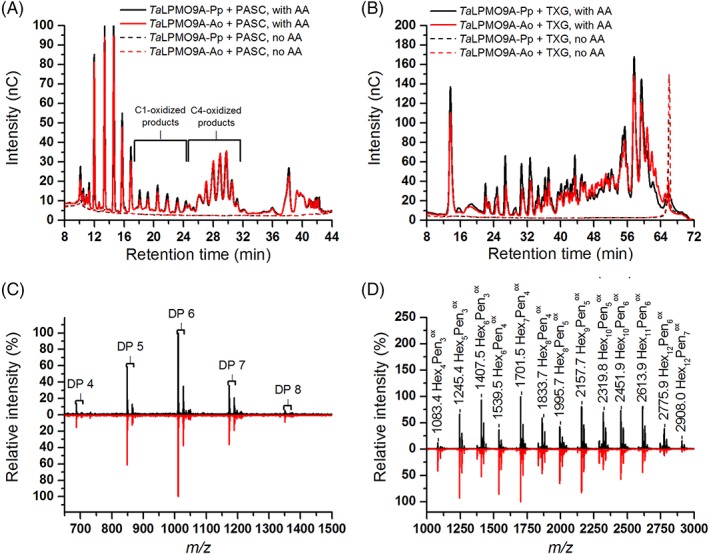
Reaction products generated by TaLPMO9A‐Ao and TaLPMO9A‐Pp from PASC or TXG. (A) HPAEC‐PAD profiles showing products released from PASC. Oxidized products are indicated in the figure and assignments of these peaks were based on the previous work.18, 23 Native products elute prior to the C1‐oxidized products. AA indicates the presence of 1 mM ascorbic acid in the reaction mixture. (B) HPAEC‐PAD profiles showing products released from TXG. (C) MALDI‐ToF MS spectra of products released in reactions containing TaLPMO9A‐Ao (red line) or TaLPMO9A‐Pp (black line), PASC and ascorbic acid, after Na+ saturation. The spectra show the DP4–DP8 region (mass values are provided in Supporting Information Fig. S5). (D) MALDI‐ToF MS spectra of products obtained in reactions containing TaLPMO9A‐Ao (red line) or TaLPMO9A‐Pp (black line), TXG and ascorbic acid, after Na+ saturation. The indicated m/z values refer to sodium adducts and the nature of the products is indicated using the following abbreviations: Hex, hexose; Pen, pentose; ox, oxidized. For more details about detected products, see Supporting Information Figure S7.
In the presence of ascorbic acid, TaLPMO9A‐Ao and TaLPMO9A‐Pp released a mixture of C1‐ and C4‐oxidized oligosaccharides (Fig. 2A). Although product quantification is not straightforward, both the HPAEC‐PAD chromatograms and MALDI‐ToF MS spectra (Fig. 2C and Supporting Information Fig. S7) indicated that C4 oxidation dominates. The MALDI‐TOF MS data showed signal compatible with the formation of double‐oxidized oligosaccharides (Supporting Information Fig. S7).
HPAEC‐PAD analysis of products generated from TXG (Fig. 2B), revealed a broad range of products eluting between 22 and 62 min and the product profiles were similar to those previously observed for the xyloglucan‐active C1/C4‐oxidizing LPMOs FgLPMO9A and GtLPMO9A‐2.23, 35 MALDI‐ToF MS spectra (Fig. 2D and Supporting Information Fig. S7) revealed products with varying combinations of pentoses (Pen) and hexoses (Hex). The appearance of oxidized products with a number of pentoses not equaling a multiple of 3, suggests that TaLPMO9A is able to oxidize TXG at any position in the backbone (the repeating unit in TXG is the Hex4Pen3 heptasaccharide XXXG). Interestingly, a closer look at the MALDI‐ToF MS data for the Hex7Pen4 cluster (Supporting Information Fig. S7) showed a signal that could reflect a double oxidized species, which would mean that the mixed C1/C4 activity also applies to TXG.
TaLPMO9A‐Ao and TaLPMO9A‐Pp also liberated oxidized cello‐oligosaccharides from SEB (Supporting Information Fig. S5) in the absence of ascorbic acid. SEB contains 36% of lignin, which has been shown earlier to drive the LPMO reaction.2, 7, 19, 36 Due to the high background, product identification in the reactions with SEB is not easy, but it is clear form Supporting Information Figure S5, that the two TaLPMO9A variants give identical results.
Importantly, while revealing novel aspects of the functionality of TaLPMO9A, the activity and binding assays described above did not reveal any functional difference between the methylated and the non‐methylated variant of the enzyme.
Variation of the electron donor
Next, we compared product formation by TaLPMO9A‐Ao and TaLPMO9A‐Pp in reactions with PASC using either ascorbic acid or cellobiose dehydrogenase (MtCDH) as electron donor. In the reactions with the enzymatic electron supply, the amount of products increased over 24 h for both LPMOs. After 24 h, product formation was the same for the two versions of TaLPMO9A (114 ± 9 and 126 ± 6 nC × min, for TaLPMO9A‐Ao and TaLPMO9A‐Pp, respectively; Fig. 3A). On the other hand, with the non‐enzymatic electron donor, product formation rapidly reached a plateau, after 2 h for TaLPMO9A‐Pp and after 4 h for TaLPMO9A‐Ao (Fig. 3B). Consequently, the methylated TaLPMO9A‐Ao version gave higher final product yields than the non‐methylated TaLPMO9A‐Pp (192 ± 2 and 153 ± 10 nC × min, respectively). These results indicate that the two enzymes may differ in terms of operational stability, as discussed further, below.
Figure 3.
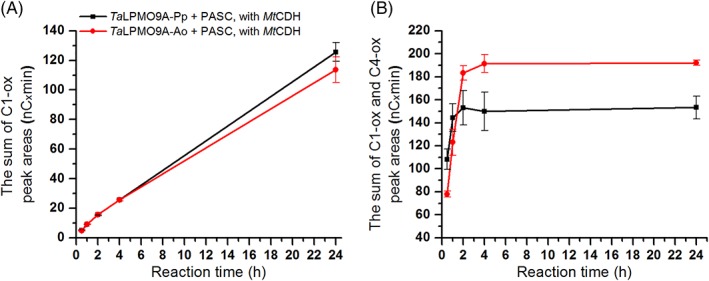
Time course for release of oxidized products during incubation of 1 μM TaLPMO9A‐Ao (red line) or TaLPMO9A‐Pp (black line) with 2 mg/mL PASC and (A) 1 μM MtCDH, or (B) 1 mM ascorbic acid, in 40 mM Bis‐Tris, pH 6.5, at 45°C. The amount of released oxidized products for each sample is expressed as the sum of the integrated peak areas (nC × min) for C1 products only (A; in this case C4 products cannot be detected because they are oxidized by CDH) or C1 and C4 products together (B). Consequently, values on the y‐axis in the graph A and B should not be compared; quantification is only valid for comparing the two enzyme variants within the same panel.
H2O2 production
It has been shown that, in the presence of a suitable reductant and in the absence of substrate, LPMOs accumulate H2O2.21 We tested the ability of both TaLPMO9A‐Ao and TaLPMO9A‐Pp to produce H2O2 in the absence of substrate with ascorbic acid as reductant. The measured H2O2 production rates were the same for both enzymes, 0.93 ± 0.04 and 1.00 ± 0.09 min−1, for TaLPMO9A‐Ao and TaLPMO9A‐Pp, respectively. It is worth noting that, these H2O2 production rates are similar to previously reported LPMO catalytic rates.1, 12, 37 The similarity of the H2O2 production rates of TaLPMO9A‐Ao and TaLPMO9A‐Pp suggests that the methylation on His1 does not affect the LPMO's ability to activate molecular oxygen.
The pKa value of His1
The pK a for methylated His1 in apo‐TaLPMO9A‐Ao was determined to be 7.21 ± 0.16. For non‐methylated apo‐TaLPMO9A‐Pp, the pK a values of the two active site histidines were determined to be 7.36 ± 0.16 and 7.67 ± 0.16 for His1 and His86, respectively (Supporting Information Fig. S8). The pK a value for His1 in apo‐TaLPMO9A‐Pp is thus close to the pK a of 7.21 ± 0.16 for methylated His1 in apo‐TaLPMO9A‐Ao.
Zn2+ is a non‐paramagnetic analogue of Cu2+ that does not cause paramagnetic relaxation enhancement and can thus be used to study the effect of metal binding on the catalytic histidines. pH titrations of both TaLPMO9A forms were carried out in the presence of Zn2+ to investigate the effect of Zn2+ on the pK a of His1. For the Zn2+‐loaded forms of the enzymes, the pK a values for His1 were determined to be 7.43 ± 0.16 and 7.58 ± 0.16 for TaLPMO9A‐Pp and TaLPMO9A‐Ao, respectively. (Nb. Since His86 was not observable in the Zn2+‐loaded form of TaLPMO9A‐Pp, its pKa could not be determined.) The pH titration curves are shown in Supporting Information Figure S8.
Cu2+ binding, redox potential and thermal stability
Intense efforts were made to probe binding of Cu(II) to the apo forms of the LPMOs by isothermal titration calorimetry, using a method that has previously been used successfully for various LPMOs,27, 38 but it was not possible to obtain solid data. At enzyme concentrations of 10 μM and higher and temperatures of 10°C and higher, the stoichiometry of Cu(II) versus LPMO was higher than 1. At lower LPMO concentrations (e.g, 5 μM) and temperature (e.g. 5°C), the heats of binding were too small for the determination of any thermodynamic parameters. Therefore, to get an impression of copper binding, we adopted a stopped‐flow method described previously by Chaplin et al.39 to obtain kinetic information for copper binding.
The apo forms of TaLPMO9A‐Ao or TaLPMO9A‐Pp were mixed with Cu2+ solutions of known concentrations and the quenching of Trp fluorescence was monitored as a function of time in a stopped‐flow spectrophotometer. For both enzymes, the pseudo first‐order rate constants (k obs) were linearly dependent on the Cu2+ concentrations (Supporting Information Fig. S9). The slopes of the k obs versus [Cu2+] plots yielded second‐order rate constants of 2.19 ± 0.06 × 106 and 1.77 ±0.04 × 106 M −1·s−1, for TaLPMO9A‐Ao and TaLPMO9A‐Pp, respectively. Both linear plots cross the axes close to the origin, as previously observed in similar analysis of a family AA10 LPMO from Streptomyces lividans, SliLPMO10E, suggesting that association rates for Cu2+ are high compared with the dissociation rates.39
Redox potentials, assessed through equilibrium reactions with N,N,N′,N′‐tetramethyl‐1,4‐phenylenediamine (TMP), were determined to be 195 ± 7 mV for TaLPMO9A‐Ao and 186 ± 5 mV for TaLPMO9A‐Pp.
The thermal stability of the LPMOs was investigated using DSC analysis. The DSC thermograms showed that TaLPMO9A‐Ao and the majority TaLPMO9A‐Pp are thermostable and unfold irreversibly at an apparent melting temperature (T m,app) of approximately 77.0°C (Supporting Information Fig. S10). The DSC studies showed that the TaLPMO9A‐Pp preparation contained a subfraction with a lower T m,app of approximately 67.0°C. It is worth noting that all enzyme assays in this study were done at the much lower temperature of 45°C.
Boosting effect on cellulase cocktails
Earlier, it has been shown that replacing 15% of a blend of Celluclast and Novozym 188 (C/N188), an LPMO‐poor cellulase preparation, with TaLPMO9A increases saccharification of steam exploded birch.7 Here, we tested the same blend with either of the TaLPMO9A preparations for the saccharification of sulfite‐pulped spruce. As references, we used a mixture of 85% C/N188 blend and 15% BSA as well as Cellic® CTec2, a commercial LPMO‐containing cellulase cocktail.7, 40
As expected on the basis of previous results,32 Fig. 4 shows that addition of the LPMO had an effect on saccharification efficiency and that this effect was much larger (25% increase in glucose yield at 96 h) in the presence of ascorbic acid. Importantly, as opposed to reactions carried out in the absence of ascorbic acid (Fig. 4A), differences between the two enzyme preparations became apparent in the reactions with ascorbic acid (Fig. 4B). During the first 24 h, TaLPMO9A‐Ao and TaLPMO9A‐Pp boosted the C/N188 cellulase blend to the same extent as shown by similar saccharification yields (Fig. 4B). During the next 72 h of incubation, however, saccharification was clearly more efficient in the reactions containing TaLPMO9A‐Ao, compared with reactions containing TaLPMO9A‐Pp. Indeed, after 96 h, the C/N188 blend with TaLPMO9A‐Ao reached 50% saccharification, which is not significantly different from the level that was reached with Cellic CTec2 (Fig. 4B), but higher than the saccharification yield reached with TaLPMO9A‐Pp. While in the case of TaLPMO9A‐Pp, the LPMO effect stopped after 24 h (meaning that after 24 h, the difference between black and blue bars in Fig. 4B is constant), the LPMO effect seemed to continue over the complete incubation period for TaLPMO9A‐Ao (shown as an increasing difference between the red and blue bars between 24 and 96 h). Again, this suggests that the two LPMO variants differ in terms of operational stability.
Figure 4.
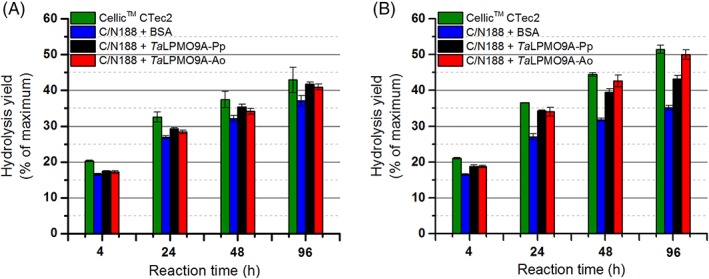
Saccharification of sulfite‐pulped spruce in the (A) absence and (B) presence of ascorbic acid (1 mM), upon substitution of 15% of the proteins in a Celluclast and Novozym 188 (C/N188) blend with the same amount of BSA (blue bar), TaLPMO9A‐Pp (black bar) or TaLPMO9A‐Ao (red bar). For comparison, the results for a reaction with an LPMO‐containing commercial cellulase cocktail, Cellic™ CTec2 (green bar), are included. Note that it has been shown previously that in the absence of an externally added reductant (as in panel A) LPMO activity on this substrate is low.32
The effect of H2O2
It has been shown that LPMOs not only can produce H2O2,21 but that they may also use H2O2 as co‐substrate during polysaccharide cleavage.41, 42, 43 It has further been shown that a surplus of H2O2 leads to oxidative inactivation of LPMOs and that this is due to the action of the LPMO itself (i.e., redox chemistry happening when H2O2 enters a reduced active site in the absence of substrate). Finally, it has been shown that LPMOs differ in terms of their susceptibility to H2O2 and that binding to substrate has a protective effect.41
We tested the effect of different H2O2 concentrations on the activity of TaLPMO9A‐Ao and TaLPMO9A‐Pp in reactions with PASC and ascorbic acid (Fig. 5). Comparison of apparent initial rates, based on the amounts of oxidized oligosaccharides released after 3 min of incubation, revealed that the two enzyme variants responded differently to varying levels of H2O2 (Fig. 6). Consistently with previous observations, a boosting effect of H2O2 was observed (Figs. 5 and 6). However, there is a trade‐off between the boosting effect of H2O2 and H2O2‐mediated enzyme inactivation, which becomes noticeable for the higher H2O2 concentrations and/or after longer incubation times (Figs. 5, 6, 7). When it comes to this trade‐off, the two enzyme variants clearly differ. For example, whilst at 200 μM H2O2 TaLPMO9A‐Ao showed a linear progress curve up to 60 min (Fig. 5C), the progress curve for TaLPMO9A‐Pp was not even linear at 100 μM H2O2 (Fig. 5A). Consequently, at 200 μM H2O2 and after 60 min TaLPMO9A‐Ao yields much more products than TaLPMO9A‐Pp (Fig. 7B). At higher H2O2 concentrations, inactivation becomes more prominent for both enzymes, meaning that the yields after 60 min are reduced. In some cases inactivation is so fast that the apparent initial rates are reduced and in some cases (for TaLPMO9A‐Pp) these (apparent) rates are even lower than those measured in the absence of H2O2 (Figs. 5 and 6).
Figure 5.
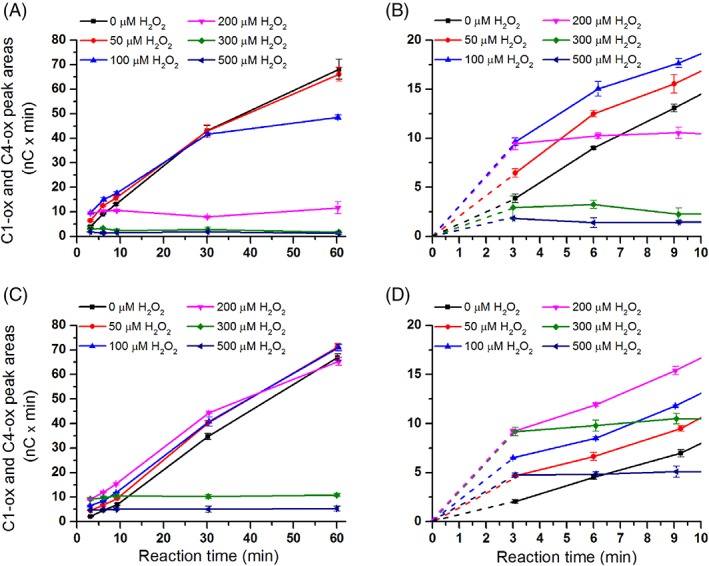
The effect of H2O2 on product generation by TaLPMO9A‐Ao and TaLPMO9A‐Pp over time. The figures show time‐courses for the release of oxidized products in reactions containing 2.5 g/L PASC and 1 μM of TaLPMO9A‐Pp (A; zoom‐in view in B), or TaLPMO9A‐Ao (C; zoom‐in view in D), in the presence of different initial concentrations of exogenous H2O2 (0–500 μM) and 1 mM ascorbic acid. Note that this figure shows that increasing H2O2 concentrations lead to higher initial activity (clearly visible in panels B and D) and to higher rates of LPMO inactivation. The trade‐off between these two phenomena determines the apparent initial rate (measured at 3 min) and the overall shape of the progress curves.
Figure 6.
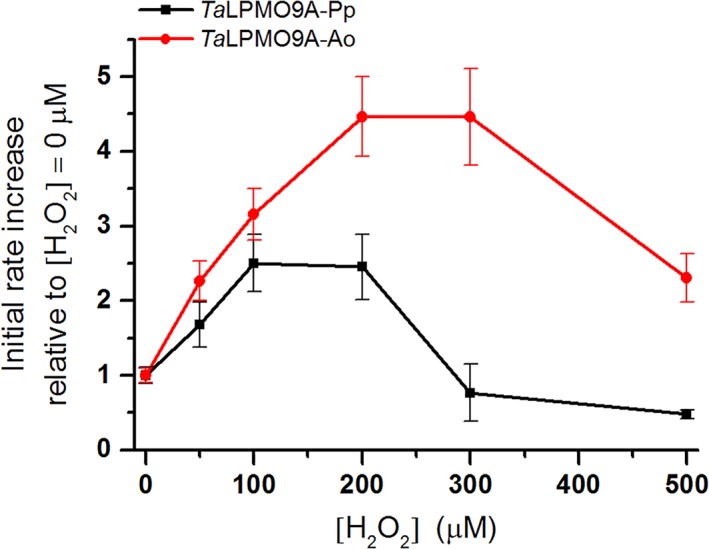
The effect of H2O2 on TaLPMO9A‐Ao and TaLPMO9A‐Pp. (A) Relative increase in apparent initial rate, calculated on the basis of product formation after 3 min, for TaLPMO9A‐Ao (red line) and TaLPMO9A‐Pp (black line) compared with the reference reaction without initial exogenous H2O2 (40 mM Bis‐Tris, pH 6.5, at 45°C). See Figure 7 for the actual product levels and see Figure 5 for the complete progress curves.
Figure 7.
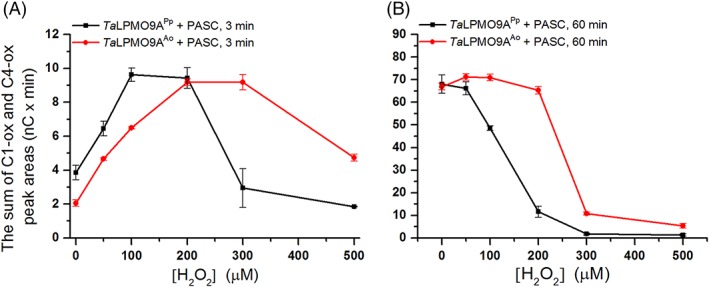
The amount of oxidized products generated during incubation of 1 μM TaLPMO9A‐Ao (red line) or TaLPMO9A‐Pp (black line) with 2 mg/ml PASC, 1 mM ascorbic acid and different initial concentrations of H2O2, after (A) 3 min, and (B) 60 min (40 mM Bis‐Tris, pH 6.5, at 45°C). The amount of released oxidized products is expressed as the sum of the integrated areas (nC × min) of the peaks representing C1 and C4 oxidized products. For complete progress curves, see Figure 5.
It is noteworthy that TaLPMO9A‐Pp was faster than TaLPMO9A‐Ao in the initial phase of the reaction (Fig. 7A). This difference was, however, only observed in the very beginning of the reaction and after 30 min the difference was no longer detectable (Fig. 5). Control reactions showed that oxidation products released from PASC did not originate from Fenton‐type chemistry as shown by the absence of detectable products when the LPMOs were replaced by CuSO4 (Supporting Information Fig. S11) in reactions that contained ascorbic acid and varying amounts of H2O2.
Discussion
Several of the fungal LPMOs characterized so far have been heterologously expressed in fungi or P. pastoris, and harbor different post‐translational modifications (PTMs) depending on the host organism. While minor variations in glycosylation are generally not considered to be of major functional importance, some LPMOs carry another PTM, namely methylation of the catalytically crucial N‐terminal histidine.2 Comparison of the structures of methylated and non‐methylated AA9 LPMOs has not provided obvious clues as to the role of the methylation. We have therefore carried out a functional comparison of TaLPMO9A produced in P. pastoris and TaLPMO9A produced in A. oryzae. Analysis of the N‐terminal peptide confirmed that post‐translational methylation of the N‐terminal histidine occurred only in the protein expressed in A. oryzae.
TaLPMO9A has a limited number of putative glycosylation sites, which reduces the chance of significant differences in glycosylation between TaLPMO9A‐Pp and TaLPMO9A‐Ao. Indeed, the two enzyme variants showed similar mobilities in SDS‐PAGE analysis. Considering the latter observation and the fact that none of the putative glycosylation sites are located near the catalytic center, it is unlikely that a glycosylation, or its absence, would explain functional differences between TaLPMO9A‐Pp and TaLPMO9A‐Ao.
The functional characterization of the two TaLPMO9A variants provided novel information on this LPMO. As expected on the basis of previously published data,2, 7, 44 TaLPMO9A showed a mixed C1/C4‐oxidizing activity on PASC, with the C4‐oxidizing activity being dominating. Importantly, we show that TaLPMO9A is also able to cleave xyloglucan and that cleavage likely can happen independent of the backbone substitution pattern, similar to FgLPMO9A23 and GtLPMO9A‐2.35 Interestingly, the MALDI‐TOF MS data shown in Supporting Information Figure S7B may indicate production of a double‐oxidized xyloglucan fragment, which has also been observed for FgLPMO9A23 and which would unambiguously proof that LPMOs can cleave xyloglucan at both C1 and C4 positions. Further analytical work is needed to finally proof the formation of such fragments. Importantly, the studies with various substrates clearly showed that TaLPMO9A‐Pp and TaLPMO9A‐Ao have identical substrate specificities and yield identical products under the tested conditions.
Reactions with various electron‐donating systems (ascorbic acid, CDH, or natural compounds in steam‐exploded birch) did not reveal differences in terms of the LPMO's ability to recruit electrons from varying sources. While progress curves with CDH showed that product formation increased during most of the monitored incubation period, the reactions with ascorbic acid, expectedly,45, 46 terminated before the monitoring period was over (Fig. 3). Termination of the reaction could be due to depletion of ascorbic acid, which in the presence of transition metals and O2 will engage in a variety of redox side reactions, and/or to damage done by the reactive oxygen species generated in such side reactions, including H2O2 produced by non‐substrate bound LPMOs.21, 41 The experiments with ascorbic acid revealed a difference between the two enzyme variants: the activity of TaLPMO9A‐Pp terminated more rapidly than the activity of TaLPMO9A‐Ao, suggesting that the latter is operationally more stable.
Previous studies have shown that τ‐methylation increases the pK a of histidine in solution by 0.46.47 Our NMR‐based determinations of the pK a values of His1 in TaLPMO9A showed that methylation hardly affects the pK a value, regardless of whether a metal ion (Zn2+) is bound. The minimal effect of Zn2+ on the pK a contrasts to earlier observations for SmLPMO10A, showing strong binding of Zn2+ leading to an expected reduction of the pK a of the catalytic histidines.27 The present observations suggest that LPMOs belonging to the AA9 family (such as TaLPMO9A) interact with Zn2+ more weakly than LPMOs belonging to the AA10 family (such as SmLPMO10A), as has indeed been observed by isothermal titration calorimetry.2
Our measurements of the kinetics of copper binding indicate strong binding and showed that copper binding may be slightly more effective in the methylated enzyme. It is conceivable that methylation locks the histidine side chain in a single tautomeric conformation that is beneficial for copper binding, analogous to what has been suggested for copper binding by amyloid‐β peptides.48
The observed apparent melting temperatures for both enzymes were similar, although the DSC experiments did show differences between the two enzyme variants. While proteins forms showed a main unfolding peak at 77°C, the unfolding curve for TaLPMO9A‐Pp showed a minor peak at 67°C. The extra peak observed TaLPMO9A‐Pp indicates a change in unfolding mechanism or a heterogeneity in the TaLPMO9A‐Pp preparation. A change in unfolding mechanism and sample heterogeneity could both be caused by variation in glycosylation. Notably, such variation cannot have been large because the two enzyme forms have very similar masses (Supporting Information Fig. S1). Importantly, the apparent melting temperatures (67°C–77°C) are far above the temperature that was used in all performed experiments (45°C). Therefore, we do not expect experimental artifacts due to differences in the thermal stability of the two enzyme variants.
Looking further into features that affect and/or reflect catalytic efficiency, we found that the two enzyme variants have almost identical redox potentials as well as almost identical abilities to generate H2O2. These experimental observations are in accordance with a theoretical study of LPMOs by Kim et al.,49 who concluded that the methylation of His1 has only minor effects on the structure and functionality of the catalytic center.
The experiments discussed above all lead to the conclusion that the two LPMO forms are functionally very similar, in accordance with conclusions based on theoretical studies.49 However, in more application oriented experiments, the two TaLPMO9A preparations showed clear differences. Although both the methylated and non‐methylated versions boosted cellulose saccharification by an LPMO‐poor cellulase cocktail, the methylated TaLPMO9A‐Ao outperformed the non‐methylated version, TaLPMO9A‐Pp in longer reactions. This difference is likely caused by inactivation of the TaLPMO9A‐Pp alone and not by inactivation of other enzymes in the reaction, since after 24 h, when TaLPMO9A‐Pp no longer had a boosting effect, cellulose hydrolysis continued (Fig. 4B, black and blue bars).
Recently, it has been shown that small amounts of H2O2 boost LPMO activity, while higher amounts lead to inactivation of the enzyme.41, 42, 43, 50 The presence of ascorbic acid in the reactions described above will lead to generation of H2O2,51, 52 which perhaps could explain the observed apparent enzyme inactivation. It was also shown41 that inactivation of LPMOs by excess H2O2 is accompanied by oxidative damage in the catalytic center only, with the N‐terminal histidine being especially vulnerable. Thus, it may seem that H2O2‐driven inactivation is an autocatalytic process.
The studies with H2O2 showed a clear boosting effect of H2O2 on LPMO activity and H2O2‐driven enzyme inactivation. Importantly, these studies revealed that TaLPMO9A‐Ao is more H2O2 resistant than TaLPMO9A‐Pp, suggesting that methylation of His1 protects the enzyme against autoxidation of the LPMO's catalytic center. Thus, methylation of His1 increases the operational stability of the LPMO. In this respect, it is interesting to note the relatively stable kinetics obtained with CDH (Fig. 3A), which is known to form H2O2 in a controlled manner at rates comparable to observed catalytic rates of LPMOs.21, 45 In line with the recent claims by Bissaro et al.,41 it is tempting to speculate that in these reactions the produced H2O2 is consumed immediately by the LPMO, avoiding H2O2 accumulation and thus oxidative inactivation.
Oxidation of histidines tends to generate 2‐oxo‐histidine, which may undergo ring opening and further degradation.41, 53, 54 It would be of interest to study the inactivation process of LPMOs in more detail, and to explore how τ‐methylation, that is, methylation of Nε2, reduces the vulnerability of the Cε for oxygenation. Of note, auto‐oxidative inactivation is a well‐known phenomenon in the field of peroxidases55 and is usually the result of a surplus of H2O2 that leads to either heme destruction or protein modification, via molecular mechanisms that remain to be fully resolved.55, 56, 57 Interestingly, it has recently been shown that Nδ‐methylation of the imidazole ring of a proximal histidine in a heme‐iron peroxidase led to an increase in total turnover numbers.58 The authors of this study attributed this positive effect to the fixing of the His in a single tautomeric form, which could reduce chances of entering off‐pathway processes that lead to enzyme inactivation. Non‐methylated LPMOs show variation in their vulnerability for inactivation indicating that other structural factors than methylation co‐determine operational stability. The effect of the methyl group in fungal LPMOs may thus be co‐determined by interactions that involve specific structural features of these proteins. Another aspect that warrants further studies is that, despite being more prone to oxidative damage, TaLPMO9A‐Pp showed higher initial rates than methylated TaLPMO9A‐Ao (Figs. 5 and 7A). There is no straightforward explanation for this observation, which is of particular interest in light of the notion that the N‐terminal histidines of bacterial LPMOs are not methylated.
In conclusion, we have compared the catalytic properties of TaLPMO9A expressed in two commonly used expression hosts, P. pastoris and A. oryzae. The most important physical difference between the two enzyme versions concerns the methylation of their N‐terminal histidine. We show that this methylation plays a role in protecting the LPMO from oxidative damage, which is most likely autocatalytic. It is interesting to note that our data suggest that the protection against auto‐oxidation comes at a cost, since methylation seems to reduce the initial activity. Interestingly, bacterial LPMOs lack the N‐terminal methylation and one may wonder whether this has to do with the degree of “oxidative pressure” that the various microbes meet, including, perhaps levels of available hydrogen peroxide. From an applied point of view, it is important to note that the methylation of His1, and thus the selection of the expression host for production of recombinant LPMOs, is important for the robustness of this redox enzyme.
Materials and Methods
Cloning, expression, and purification of TaLPMO9A in Pichia pastoris
TaLPMO9A from Thermoascus aurantiacus [UniProt: G3XAP7] was cloned, expressed in P. pastoris, and purified as described before.32 The enzyme was saturated with Cu(II) by incubating with an excess of Cu(II)SO4 (at ~3:1 molar ratio of copper:enzyme) for 90 min at room temperature as described previously.59 Subsequently, the sample was subjected to size‐exclusion chromatography using a HiLoad 16/60 Superdex 75 column (GE Healthcare, Uppsala, Sweden) equilibrated with 50 mM Bis‐Tris/HCl buffer (pH 6.5) containing 150 mM NaCl, using a flow rate of 0.75 mL/min. Fractions containing the pure protein were identified using SDS‐PAGE and subsequently pooled, concentrated and buffer exchanged to 50 mM Bis‐Tris/HCl buffer, pH 6.5, using Amicon Ultra centrifugal filters (MWCO 3 kDa, Merck Millipore, NJ). The resulting solution with purified protein was filtered through a 0.22 μm Millex®‐GV filter (Merck Millipore, NJ) and stored at 4°C.
Expression and purification of TaLPMO9A in Aspergillus oryzae
TaLPMO9A was expressed and partially purified at Novozymes (Bagsvaerd, Denmark) as described before.60 The enzyme was saturated with Cu(II) followed by additional purification using the HiLoad 16/60 Superdex 75 size exclusion column (GE Healthcare, Uppsala, Sweden), and subsequent buffer exchange to 50 mM Bis‐Tris/HCl buffer, pH 6.5, as described above for P. pastoris expressed TaLPMO9A.
The protein concentrations of both final TaLPMO9A preparations were determined by measuring absorbance at 280 nm and using a molar extinction coefficient of 45,630 M −1·cm−1 determined using ExPASy's ProtParam tool.61
Apo‐enzymes (for the kinetics of Cu2+ binding) were generated as described before62 by 30 min incubation with EDTA in a 1:4 molar ratio (LPMO:EDTA), at room temperature.
Production of 13C, 15N‐labeled TaLPMO9A in Pichia pastoris
The production of the isotopically labeled TaLPMO9A‐Pp was done as described before, using shake flask cultures.63, 64 Briefly, P. pastoris harboring the pPink‐GAP plasmid, carrying the Ta‐lpmo9a gene, was grown in 50 mL of 13C, 15N‐labeled buffered minimal glucose medium (13C, 15N‐BMD), in a 250‐mL shake flask at 29°C and 200 rpm for 24 h. This culture was used to inoculate 450 mL 13C, 15N‐BMD medium in 2‐L shake flasks followed by incubation at 29°C and 200 rpm for 48 h. After the first 24 h, the medium was re‐supplemented with 1% (w/v) 13C‐labeled glucose. Cells were removed by centrifugation and the supernatant was concentrated to 100 mL using a VivaFlow 50 tangential crossflow concentrator (MWCO 10 kDa, Sartorius Stedim Biotech Gmbh, Goettingen, Germany).
The concentrated supernatant was supplemented with ammonium sulfate to a final concentration of 1.42 M and loaded onto a 5‐mL HiTrap Phenyl FF column (GE Healthcare, Uppsala, Sweden), equilibrated with 50 mM Bis‐Tris/HCl buffer (pH 6.5) containing 1.42 M ammonium sulfate, using a flow rate of 1 mL/min. Proteins bound to the column were eluted using a 25‐mL linear gradient from 1.42 to 0 M ammonium sulfate in 50 mM Bis‐Tris/HCl buffer (pH 6.5). Fractions containing the pure protein were identified using SDS‐PAGE and subsequently pooled, concentrated and buffer exchanged to 50 mM Bis‐Tris/HCl buffer, pH 6.5.
For NMR experiments, the apo‐forms of TaLPMO9A‐Ao and 13C, 15N‐labeled TaLPMO9A‐Pp were obtained by incubating the protein samples with 10 mM Na‐EDTA for 45 min at room temperature, followed by buffer exchange to 20 mM MES buffer, pH 5.5.
Analysis of N‐terminal methylation
Protein samples were prepared for liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis as described before.65 Briefly, dried protein samples were dissolved in 4% SDS, 10 mM dithiothreitol. After incubation for 25 min at 95°C, iodoacetamide was added to a final concentration of 50 mM. The protein solution was then transferred to an STrap tip that was packed with two Empore™ C18 extraction disks (3M, St. Paul, MN) and 11 MK360 quartz filters (Ahlstrom Munktell, Falun, Sweden), and that was prefilled with 90% (v/v) methanol, 100 mM Tris, pH 7.1 (STrapping solution). The samples in the STrap tips were washed first with the STrapping solution and then with 50 mM ammonium bicarbonate. Subsequently, trypsin was added to the STrap tips, which were then incubated for 45 min at 37°C. The digestion was stopped by washing the STrap tips with 0.5% (v/v) trifluoroacetic acid and peptides were eluted with a solution containing 80% (v/v) acetonitrile and 0.1% (v/v) TFA.
The tryptic peptides (~1 μg) were separated on an Acclaim™ PepMap™ 100 C18 column, 50 cm × 75 μm (Thermo Fisher Scientific, Bremen, Germany), using a 40 min gradient running from 12% to 44% acetonitrile. Chromatography was conducted using an Ultimate3000 RSLCnano/QExactive (Thermo Fisher Scientific, Bremen, Germany) system, set up with a Nanospray Flex ion source. MS and MS/MS data were recorded using a standard data dependent acquisition method: m/z range 300–1,500; automatic gain control targets of 3 × 106 (MS) and 5 × 104 (MS/MS); resolution of 70,000 (MS) and 35,000 (MS/MS), dynamic exclusion set to 20 s, and normalized collision energy set to 28.
Thermo's Xcalibur software (v3.0) was used to evaluate raw data, and to generate extracted ion chromatograms. The raw data were converted to mgf format using the msconvert module of Proteowizard (v 3.0.9016). The mgf files were used in Mascot searches against a database constructed by appending N‐terminal variants of the target protein sequence to the reference proteome of Komagataella phaffii/Pichia pastoris (UniProt proteomes ID: UP000000314).
Substrates and electron donors
The following substrates were used in this study: phosphoric acid swollen cellulose (PASC), prepared from Avicel as described before66; xyloglucan from tamarind seed (TXG), ivory nut mannan, konjac glucomannan and cellopentaose, all purchased from Megazyme (Bray, Ireland); xylan from birch wood obtained from Sigma‐Aldrich (St. Louis, MO); steam exploded birch (Betula pubescens) wood (SEB) was prepared at 210°C, with 10 min residence time, as described earlier.67
Electron donors for the LPMOs were L‐ascorbic acid (≥99.0%) purchased from Sigma‐Aldrich (St. Louis, MO) and cellobiose dehydrogenase from Myriococcum thermophilum (MtCDH) expressed in P. pastoris and purified as previously reported.68
LPMO reactions
Unless otherwise stated, reaction mixtures contained 2 mg/mL substrate, 1 μM LPMO and, as electron donor, 1 mM ascorbic acid or 1 μM MtCDH in 40 mM Bis‐Tris/HCl buffer (pH 6.5). Reactions were performed in 2 mL Eppendorf tubes containing 100 μL total reaction volume, which were incubated at 45°C with shaking at 1,000 rpm in an Eppendorf Thermomixer (Eppendorf, Hamburg, Germany). Reactions were stopped by filtration using a 96‐well plate equipped with a 0.45 μm filter (Merck Millipore, Billerica, MA) that was operated with a vacuum manifold. Control experiments were performed in the absence of reductant.
Substrate specificity of the two TaLPMO9A preparations was characterized using ascorbic acid as electron donor, and the reactions were incubated for 16 h in a total volume of 100 μL.
Product accumulation from PASC over time was followed using ascorbic acid or MtCDH as electron donor. The total reaction volume was 500 μL, from which 100 μL aliquots were taken after 0.5, 1, 2, 4, and 24 h of incubation. Each reaction was done in triplicates.
Detection of oxidized products
Oxidized products were analyzed using high‐performance anion exchange chromatography with pulsed amperometric detection (HPAEC‐PAD) and by matrix‐assisted laser desorption ionization–time of flight mass spectrometry (MALDI‐ToF MS).
HPAEC was performed on a Dionex ICS5000 system, equipped with a CarboPac PA1 analytical column (2 × 250 mm) and a CarboPac PA1 guard column (2 × 50 mm), using a 50‐min gradient69 for cellulosic and a 75‐min gradient12 for hemicellulosic substrates. Chromatograms were recorded with Chromeleon and analyzed using Origin 9.1 software (OriginLab, Northampton, MA).
MALDI‐ToF MS was performed on an Ultraflex MALDI‐ToF/ToF instrument (Bruker Daltonik GmbH, Bremen, Germany) equipped with a Nitrogen 337 nm laser, as described earlier.12 The data were analyzed using mMass software.70 Baseline correction and Gaussian smoothing (window size 0.3 m/z) were applied to all spectra.
Binding of TaLPMO9A to insoluble substrates
Binding studies with PASC were performed as described before.33 The reaction mixture contained 2 mg/mL substrate and 5 μM of the enzyme in 50 mM Bis‐Tris/HCl buffer, pH 6.5, and was carried out at 45°C with shaking at 1,000 rpm in an Eppendorf Thermomixer (Eppendorf, Hamburg, Germany).
Determination of the pKa of His1
NMR pH titrations of the apo forms of TaLPMO9A were performed on approximately 0.3 mM non‐isotopic labeled (natural abundance) methylated TaLPMO9A‐Ao and on approximately 0.6 mM 13C and 15N labeled non‐methylated TaLPMO9A‐Pp, using 20 mM sodium phosphate buffers with 10 mM NaCl and 10% D2O in the pH range 5.0–8.5. pH titrations were also carried out it in the presence of Zn2+ (a non‐paramagnetic analogue of Cu2+ that has been previously used for NMR investigations of LPMOs27), using 20 mM ammonium acetate buffers with 10 mM NaCl, 2 mM ZnCl2, and 10% D2O in the pH range 3.5–8.5. Buffers were exchanged using Amicon Ultra centrifugal filters (MWCO 3 kDa, Merck Millipore, NJ). Equation (1), which is a rearrangement of the Henderson–Hasselbach equation, was used to estimate the pK a by using a least squares minimization algorithm to fit the pK a in Excel, where δ BH is the chemical shift of the fully protonated histidine, δ B is the chemical shift of the fully deprotonated histidine, and δ is the measured chemical shift at a certain pH.
| (1) |
In the case of methylated TaLPMO9A‐Ao, a 13C Heteronuclear Single Quantum Coherence (HSQC) spectrum for the aliphatic region was recorded at each titration point to monitor the changes in chemical shifts of the methyl group of His1. This methyl group has a unique chemical shift71 usually not observed in proteins, which was assigned at pH 6.5: δ1H/δ13C {3.8 ppm; 35.2 ppm}.
For 13C and 15N labeled non‐methylated TaLPMO9A‐Pp, a 13C HSQC spectrum for the aromatic region was recorded at each titration point to monitor the changes in chemical shifts of the epsilon atoms (Cε1/Hε1) of His1, which was assigned at pH 6.5: δ1H/δ13C {8.3 ppm; 133.5 ppm}.
Saccharification experiments
Saccharification of sulfite‐pulped Norway spruce prepared using the BALI™ process72 was conducted in 60‐mL rubber sealed glass bottles (Wheaton, Millville, NJ) with 10‐mL working volume and 50 g/L total solids loading in 50 mM sodium acetate buffer, pH 5.0, in the presence or absence of 1 mM ascorbic acid. The reactions contained 4 mg of enzyme per g of glucan, and the enzyme mixture was composed of 85% (w/w) Celluclast 1.5 L:Novozym 188 blend (in a 5:1 [w/w] ratio), and 15% (w/w) TaLPMO9A‐Pp, TaLPMO9A‐Ao or bovine serum albumin (BSA). Earlier, this C/N188 and LPMO ratio had been found optimal for obtaining the highest sugar recovery from steam‐exploded birch.7 Reactions were incubated at 50°C, with mixing at 38 rpm in a Multi RS‐60 programmable rotator (Biosan, Riga, Latvia). About 100 μL aliquots were taken after 4, 24, 48, and 96 h of incubation, followed by heat inactivation at 100°C for 15 min. As a reference, Cellic® CTec2 was used. All three commercial preparations, Celluclast 1.5 L, Novozym 188, and Cellic® CTec2, were kindly provided by Novozymes A/S (Bagsværd, Denmark) and their protein concentration was determined with the Bio‐Rad Protein Assay (Bio‐Rad, Hercules, CA), based on the Bradford method,73 using BSA as a standard.
Glucose and cellobiose released during enzymatic hydrolysis were quantified with High‐Performance Liquid Chromatography (HPLC) using a Dionex Ultimate 3000 system (Dionex, Sunnyvale, CA) coupled to a refractive index (RI) detector 101 (Shodex, Tokyo, Japan), as described earlier.32 Hydrolysis yields were calculated based on detected glucose and cellobiose (typically <1% of the total).
Determination of the Cell Potential (E°)
The cell potentials for the redox couples TaLPMO9A‐Ao(Cu2+)/TaLPMO9A‐Ao(Cu+) and TaLPMO9A‐Pp(Cu2+)/TaLPMO9A‐Pp(Cu+) were determined as described by Aachmann et al.27 Solutions (50 μL) of oxygen‐free N,N,N′,N′‐tetramethyl‐1,4 phenylenediamine (TMPred) in its reduced form (200 μM) and Cu2+‐charged TaLPMO9A‐Ao or TaLPMO9A‐Pp (70 μM) in 20 mM MES buffer (pH 5.5, t = 25°C) were mixed and the extent of reaction was determined by measuring absorbance from the formed TMP radical cation (TMPox) at λ = 610 nm. The concentrations of TMPox, which equal concentrations of LPMO‐Cu+, were calculated by using an extinction coefficient of 14.0 mM −1 cm−1.74 From the determined concentrations of TMPox and LPMO‐Cu+, the equilibrium constant (K) was calculated. The cell potential for the LPMO‐Cu2+/LPMO‐Cu+ redox couple was determined by adding the known cell potential of 273 mV for TMPred/TMPox 75 to the cell potential of the equilibrium reaction of TMPred and LPMO‐Cu2+.27
Stopped‐flow kinetics
Kinetic experiments to measure changes in fluorescence emission upon binding of Cu2+ were carried out using a SFM4000 stopped‐flow spectrophotometer (BioLogic Science Instruments, Grenoble, France). Apo‐LPMO samples (2.0 μM, final concentration) were prepared in 20 mM MES buffer, pH 5.5, and mixed with samples of Cu2+ (in the form of CuSO4 · 5H2O) in the same buffer with concentrations ranging from 6 to 125 μM after mixing. We used a single mixing set‐up, which means that for each experiment a new syringe with the appropriate concentration of CuSO4 was prepared. Tryptophan residues were excited at 295 nm and emitted light was detected after passage through a cut‐off filter permitting light with wavelengths above 320 nm to pass, using a photomultiplier tube (670 V) positioned at a 90 degrees angle relative to the exciting beam. Kinetic data were analyzed using the Bio‐Kine software (BioLogic Science Instruments, Grenoble, France).
Differential scanning calorimetry
Differential scanning calorimetry (DSC) to analyze the thermal stabilities of TaLPMO9A‐Ao and TaLPMO9A‐Pp was carried out with a Nano‐Differential Scanning Calorimeter III instrument (TA Instruments, New Castle, DE). TaLPMO9A‐Ao and TaLPMO9A‐Pp (15 and 10 μM, respectively) were saturated with Cu(II) by incubating with an excess of Cu(II)SO4 (at 3:1 molar ratio of copper:enzyme) for 90 min at room temperature. Subsequently, the enzymes were dialyzed against 2 × 1,000 volumes of 50 mM Na phosphate buffer, pH 6.5, for 24 h at 4°C. Prior to loading into sample cell, the enzyme solutions were filtered through a 0.22 μm filter (Merck Millipore, Billerica, MA) and degassed. The dialysis buffer was also filtered, degassed and loaded into reference cell. Scanning was performed in the range from 30°C to 90°C, with a scan rate of 1°C/min. The experiments were carried out in duplicate, using freshly dialyzed enzyme for each scan. The data were analyzed using NanoAnalyze software (TA Instruments, New Castle, DE) whereby baseline scans, collected with buffer in both, reference and sample cells, were subtracted from the sample scans. Transition temperatures, T m (defined as the temperature of maximum apparent heat capacity) were estimated from the baseline‐corrected DSC thermograms.
H2O2 production
The method for measuring H2O2 production was adapted from a previously reported protocol.21 A reaction mixture (180 μL) containing 1 μM TaLPMO9A, 5 U/mL horseradish peroxidase (HRP) and 100 μM Amplex® Red (Thermo Fisher Scientific, Bremen, Germany) in 50 mM Bis‐Tris/HCl buffer pH 6.5 was incubated for 5 min at 40°C in a 96‐well microtiter plate in a plate reader (Multiskan™ FC Microplate Photometer (Thermo Fisher Scientific, Bremen Germany). The reaction was initiated by the addition of 20 μL of 500 μM ascorbic acid (50 μM final concentration) in each well and the production of resorufin was monitored at 540 nm. Control reactions in the absence of TaLPMO9A were carried out to obtain the LPMO‐independent resorufin production rate. This control reaction provided a background signal equal to 0.3% of the LPMO‐catalyzed reaction and was subtracted from the latter. A H2O2 standard curve was prepared using the same conditions (without ascorbic acid and LPMO). The reactions were monitored for 45 min and H2O2 production rates were derived from data points in the linear region, between 2 and 20 min.
Sensitivity toward H2O2
To assess the impact of H2O2 on TaLPMO9A, 500 μL reaction mixtures were prepared as described above with the addition of H2O2 corresponding to 0, 50, 100, 200, 300, and 500 μM final concentrations. H2O2 was added to the reaction mixture just before initiation of the reaction by addition of ascorbic acid (1 mM final concentration). At regular intervals of 3, 6, 9, 30, and 60 min about 55 μL samples were taken from the reaction mixtures, and LPMO activity was immediately stopped by filtration using a 96‐well filter plate (Merck Millipore, Billerica, MA) operated with a vacuum manifold. Filtered samples were frozen (−20°C) prior to further analysis.
Conflicts of Interest
The authors declare that they have no conflicts of interest with the contents of this article.
Supporting information
Supplementary material: The supplementary material includes eleven figures.
Figure S1: SDS‐PAGE of purified TaLPMO9A‐Ao and TaLPMO9A‐Pp.
Figure S2: 3D structure of TaLPMO9A (PDB ID: 3ZUD).
Figure S3: LC–MS chromatograms of N‐terminal peptides.
Figure S4: Binding to PASC.
Figure S5: Reaction products generated from steam exploded birch.
Figure S6: HPAEC‐PAD chromatograms for the reactions with cellopentaose.
Figure S7: Close‐up of MALDI‐ToF MS spectra for the reactions with PASC or xyloglucan.
Figure S8: NMR pH titration curves.
Figure S9: Kinetic information for copper binding obtained by stopped‐flow spectrophotometry.
Figure S10: Differential scanning calorimetry analysis.
Figure S11: Control reactions to check for occurrence of copper‐catalyzed Fenton‐type chemistry.
Acknowledgments
This work was supported by the Research Council of Norway through grants 226244 and 243663. We thank Novozymes for supply of TaLPMO9A produced in Aspergillus oryzae.
Lytic polysaccharide monooxygenases (LPMOs) are abundant mono‐copper enzymes that are important in industrial biomass processing and the global carbon cycle. We have studied the role of a relatively rare post‐translational modification found in fungal LPMOs, namely methylation of the copper coordinating N‐terminal histidine. We show that methylation has surprisingly little effect on the properties of His1 and the LPMO as a whole, with one important exception: methylation reduces oxidative self‐inactivation, thus improving overall enzyme performance.
Filename: Petrovic_Role_of_histidine_methylation_in_fungal_LPMOs_Supplementary_material.
References
- 1. Vaaje‐Kolstad G, Westereng B, Horn SJ, Liu Z, Zhai H, Sorlie M, Eijsink VGH (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330:219–222. [DOI] [PubMed] [Google Scholar]
- 2. Quinlan RJ, Sweeney MD, Lo Leggio L, Otten H, Poulsen JCN, Johansen KS, Krogh KBRM, Jorgensen CI, Tovborg M, Anthonsen A, Tryfona T, Walter CP, Dupree P, Xu F, Davies GJ, Walton PH (2011) Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc Natl Acad Sci U S A 108:15079–15084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Horn SJ, Vaaje‐Kolstad G, Westereng B, Eijsink VGH (2012) Novel enzymes for the degradation of cellulose. Biotechnol Biofuels 5:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beeson WT, Vu VV, Span EA, Phillips CM, Marletta MA (2015) Cellulose degradation by polysaccharide monooxygenases. Annu Rev Biochem 84:923–946. [DOI] [PubMed] [Google Scholar]
- 5. Hemsworth GR, Johnston EM, Davies GJ, Walton PH (2015) Lytic polysaccharide monooxygenases in biomass conversion. Trends Biotechnol 33:747–761. [DOI] [PubMed] [Google Scholar]
- 6. Levasseur A, Drula E, Lombard V, Coutinho PM, Henrissat B (2013) Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes. Biotechnol Biofuels 6:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muller G, Varnai A, Johansen KS, Eijsink VGH, Horn SJ (2015) Harnessing the potential of LPMO‐containing cellulase cocktails poses new demands on processing conditions. Biotechnol Biofuels 8:187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu JG, Chandra R, Arantes V, Gourlay K, van Dyk JS, Saddler JN (2015) The addition of accessory enzymes enhances the hydrolytic performance of cellulase enzymes at high solid loadings. Bioresour Technol 186:149–153. [DOI] [PubMed] [Google Scholar]
- 9. Lombard V, Ramulu HG, Drula E, Coutinho PM, Henrissat B (2014) The carbohydrate‐active enzymes database (CAZy) in 2013. Nucleic Acids Res 42:490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Phillips CM, Beeson WT, Cate JH, Marletta MA (2011) Cellobiose dehydrogenase and a copper‐dependent polysaccharide monooxygenase potentiate cellulose degradation by Neurospora crassa. ACS Chem Biol 6:1399–1406. [DOI] [PubMed] [Google Scholar]
- 11. Westereng B, Ishida T, Vaaje‐Kolstad G, Wu M, Eijsink VGH, Igarashi K, Samejima M, Stahlberg J, Horn SJ, Sandgren M (2011) The putative endoglucanase PcGH61D from Phanerochaete chrysosporium is a metal‐dependent oxidative enzyme that cleaves cellulose. PLoS One 6:e27807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Agger JW, Isaksen T, Varnai A, Vidal‐Melgosa S, Willats WGT, Ludwig R, Horn SJ, Eijsink VGH, Westereng B (2014) Discovery of LPMO activity on hemicelluloses shows the importance of oxidative processes in plant cell wall degradation. Proc Natl Acad Sci U S A 111:6287–6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bennati‐Granier C, Garajova S, Champion C, Grisel S, Haon M, Zhou S, Fanuel M, Ropartz D, Rogniaux H, Gimbert I, Record E, Berrin JG (2015) Substrate specificity and regioselectivity of fungal AA9 lytic polysaccharide monooxygenases secreted by Podospora anserina. Biotechnol Biofuels 8:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lo Leggio L, Simmons TJ, Poulsen JCN, Frandsen KEH, Hemsworth GR, Stringer MA, von Freiesleben P, Tovborg M, Johansen KS, De Maria L, Harris PV, Soong CL, Dupree P, Tryfona T, Lenfant N, Henrissat B, Davies GJ, Walton PH (2015) Structure and boosting activity of a starch‐degrading lytic polysaccharide monooxygenase. Nat Commun 6:5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vu VV, Beeson WT, Span EA, Farquhar ER, Marletta MA (2014) A family of starch‐active polysaccharide monooxygenases. Proc Natl Acad Sci U S A 111:13822–13827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Frommhagen M, Sforza S, Westphal AH, Visser J, Hinz SW, Koetsier MJ, van Berkel WJ, Gruppen H, Kabel MA (2015) Discovery of the combined oxidative cleavage of plant xylan and cellulose by a new fungal polysaccharide monooxygenase. Biotechnol Biofuels 8:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vu VV, Beeson WT, Phillips CM, Cate JH, Marletta MA (2014) Determinants of regioselective hydroxylation in the fungal polysaccharide monooxygenases. J Am Chem Soc 136:562–565. [DOI] [PubMed] [Google Scholar]
- 18. Isaksen T, Westereng B, Aachmann FL, Agger JW, Kracher D, Kittl R, Ludwig R, Haltrich D, Eijsink VGH, Horn SJ (2014) A C4‐oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello‐oligosaccharides. J Biol Chem 289:2632–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harris PV, Welner D, McFarland KC, Re E, Poulsen JCN, Brown K, Salbo R, Ding HS, Vlasenko E, Merino S, Xu F, Cherry J, Larsen S, Lo Leggio L (2010) Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: structure and function of a large, enigmatic family. Biochemistry 49:3305–3316. [DOI] [PubMed] [Google Scholar]
- 20. Frommhagen M, Koetsier MJ, Westphal AH, Visser J, Hinz SWA, Vincken JP, van Berkel WJH, Kabel MA, Gruppen H (2016) Lytic polysaccharide monooxygenases from Myceliophthora thermophila C1 differ in substrate preference and reducing agent specificity. Biotechnol Biofuels 9:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kittl R, Kracher D, Burgstaller D, Haltrich D, Ludwig R (2012) Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol Biofuels 5:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu M, Beckham GT, Larsson AM, Ishida T, Kim S, Payne CM, Himmel ME, Crowley MF, Horn SJ, Westereng B, Igarashi K, Samejima M, Ståhlberg J, Eijsink VG, Sandgren M (2013) Crystal structure and computational characterization of the lytic polysaccharide monooxygenase GH61D from the basidiomycota fungus Phanerochaete chrysosporium. J Biol Chem 288:12828–12839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nekiunaite L, Petrovic DM, Westereng B, Vaaje‐Kolstad G, Hachem MA, Varnai A, Eijsink VGH (2016) FgLPMO9A from Fusarium graminearum cleaves xyloglucan independently of the backbone substitution pattern. FEBS Lett 590:3346–3356. [DOI] [PubMed] [Google Scholar]
- 24. Saloheimo M, Nakari‐Setala T, Tenkanen M, Penttila M (1997) cDNA cloning of a Trichoderma reesei cellulase and demonstration of endoglucanase activity by expression in yeast. Eur J Biochem 249:584–591. [DOI] [PubMed] [Google Scholar]
- 25. Demain AL, Vaishnav P (2009) Production of recombinant proteins by microbes and higher organisms. Biotechnol Adv 27:297–306. [DOI] [PubMed] [Google Scholar]
- 26. Nevalainen H, Peterson R (2014) Making recombinant proteins in filamentous fungi‐ are we expecting too much? Front Microbiol 5:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aachmann FL, Sørlie M, Skjåk‐Bræk G, Eijsink VGH, Vaaje‐Kolstad G (2012) NMR structure of a lytic polysaccharide monooxygenase provides insight into copper binding, protein dynamics, and substrate interactions. Proc Natl Acad Sci U S A 109:18779–18784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hemsworth GR, Henrissat B, Davies GJ, Walton PH (2014) Discovery and characterization of a new family of lytic polysaccharide rnonooxygenases. Nat Chem Biol 10:122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vaaje‐Kolstad G, Forsberg Z, Loose JS, Bissaro B, Eijsink VGH (2017) Structural diversity of lytic polysaccharide monooxygenases. Curr Opin Struct Biol 44:67–76. [DOI] [PubMed] [Google Scholar]
- 30. Li X, Beeson WT, Phillips CM, Marletta MA, Cate JHD (2012) Structural basis for substrate targeting and catalysis by fungal polysaccharide monooxygenases. Structure 20:1051–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lambertz C, Garvey M, Klinger J, Heesel D, Klose H, Fischer R, Commandeur U (2014) Challenges and advances in the heterologous expression of cellulolytic enzymes: a review. Biotechnol Biofuels 7:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chylenski P, Petrović DM, Müller G, Dahlström M, Bengtsson O, Lersch M, Siika‐aho M, Horn SJ, Eijsink VGH (2017) Enzymatic degradation of sulfite‐pulped softwoods and the role of LPMOs. Biotechnol Biofuels 10:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Forsberg Z, Nelson CE, Dalhus B, Mekasha S, Loose JSM, Crouch LI, Rohr AK, Gardner JG, Eijsink VGH, Vaaje‐Kolstad G (2016) Structural and functional analysis of a lytic polysaccharide monooxygenase important for efficient utilization of chitin in Cellvibrio japonicus. J Biol Chem 291:7300–7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Crouch LI, Labourel A, Walton PH, Davies GJ, Gilbert HJ (2016) The contribution of non‐catalytic carbohydrate binding modules to the activity of lytic polysaccharide monooxygenases. J Biol Chem 291:7439–7449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kojima Y, Varnai A, Ishida T, Sunagawa N, Petrovic DM, Igarashi K, Jellison J, Goodell B, Alfredsen G, Westereng B, Eijsink VGH, Yoshida M (2016) Characterization of an LPMO from the brown‐rot fungus Gloeophyllum trabeum with broad xyloglucan specificity, and its action on cellulose‐xyloglucan complexes. Appl Environ Microbiol 82:6557–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Westereng B, Cannella D, Agger JW, Jorgensen H, Andersen ML, Eijsink VGH, Felby C (2015) Enzymatic cellulose oxidation is linked to lignin by long‐range electron transfer. Sci Rep 5:18561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Frandsen KEH, Simmons TJ, Dupree P, Poulsen JCN, Hemsworth GR, Ciano L, Johnston EM, Tovborg M, Johansen KS, von Freiesleben P, Marmuse L, Fort S, Cottaz S, Driguez H, Henrissat B, Lenfant N, Tuna F, Baldansuren A, Davies GJ, Lo Leggio L, Walton PH (2016) The molecular basis of polysaccharide cleavage by lytic polysaccharide monooxygenases. Nat Chem Biol 12:298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Borisova AS, Isaksen T, Dimarogona M, Kognole AA, Mathiesen G, Varnai A, Rohr AK, Payne CM, Sorlie M, Sandgren M, Eijsink VGH (2015) Structural and functional characterization of a lytic polysaccharide monooxygenase with broad substrate specificity. J Biol Chem 290:22955–22969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chaplin AK, Wilson MT, Hough MA, Svistunenko DA, Hemsworth GR, Walton PH, Vijgenboom E, Worrall JA (2016) Heterogeneity in the histidine‐brace copper coordination sphere in auxiliary activity family 10 (AA10) lytic polysaccharide monooxygenases. J Biol Chem 291:12838–12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cannella D, Hsieh CWC, Felby C, Jorgensen H (2012) Production and effect of aldonic acids during enzymatic hydrolysis of lignocellulose at high dry matter content. Biotechnol Biofuels 5:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bissaro B, Rohr AK, Muller G, Chylenski P, Skaugen M, Forsberg Z, Horn SJ, Vaaje‐Kolstad G, Eijsink VGH (2017) Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2. Nat Chem Biol 13:1123–1128. [DOI] [PubMed] [Google Scholar]
- 42. Bissaro B, Rohr AK, Skaugen M, Forsberg Z, Horn SJ, Vaaje‐Kolstad G, Eijsink V (2016) Fenton‐type chemistry by a copper enzyme: molecular mechanism of polysaccharide oxidative cleavage. bioRxiv 097022: 10.1101/097022. [DOI]
- 43. Hangasky JA, Iavarone AT, Marletta MA (2018) Reactivity of O2 versus H2O2 with polysaccharide monooxygenases. Proc Natl Acad Sci U S A 115:4915–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim IJ, Seo N, An HJ, Kim JH, Harris PV, Kim KH (2017) Type‐dependent action modes of TtAA9E and TaAA9A acting on cellulose and differently pretreated lignocellulosic substrates. Biotechnol Biofuels 10:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Loose JS, Forsberg Z, Kracher D, Scheiblbrandner S, Ludwig R, Eijsink VGH, Vaaje‐Kolstad G (2016) Activation of bacterial lytic polysaccharide monooxygenases with cellobiose dehydrogenase. Prot Sci 25:2175–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kracher D, Scheiblbrandner S, Felice AK, Breslmayr E, Preims M, Ludwicka K, Haltrich D, Eijsink VGH, Ludwig R (2016) Extracellular electron transfer systems fuel cellulose oxidative degradation. Science 352:1098–1101. [DOI] [PubMed] [Google Scholar]
- 47. Paiva AC, Juliano L, Boschcov P (1976) Ionization of methyl derivatives of imidazole, histidine, thyreotropin releasing factor, and related compounds. J Am Chem Soc 98:7645–7648. [DOI] [PubMed] [Google Scholar]
- 48. Tickler AK, Smith DG, Ciccotosto GD, Tew DJ, Curtain CC, Carrington D, Masters CL, Bush AI, Cherny RA, Cappai R, Wade JD, Barnham KJ (2005) Methylation of the imidazole side chains of the Alzheimer disease amyloid‐beta peptide results in abolition of superoxide dismutase‐like structures and inhibition of neurotoxicity. J Biol Chem 280:13355–13363. [DOI] [PubMed] [Google Scholar]
- 49. Kim S, Stahlberg J, Sandgren M, Paton RS, Beckham GT (2014) Quantum mechanical calculations suggest that lytic polysaccharide monooxygenases use a copper‐oxyl, oxygen‐rebound mechanism. Proc Natl Acad Sci U S A 111:149–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kuusk S, Bissaro B, Kuusk P, Forsberg Z, Eijsink VGH, Sorlie M, Valjamae P (2018) Kinetics of H2O2‐driven degradation of chitin by a bacterial lytic polysaccharide monooxygenase. J Biol Chem 293:523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peterson RW, Walton JH (1943) The autoxidation of ascorbic acid. J Am Chem Soc 65:1212–1217. [Google Scholar]
- 52. Boatright WL (2016) Oxygen dependency of one‐electron reactions generating ascorbate radicals and hydrogen peroxide from ascorbic acid. Food Chem 196:1361–1367. [DOI] [PubMed] [Google Scholar]
- 53. Uchida K, Kawakishi S (1993) 2‐Oxo‐histidine as a novel biological marker for oxidatively modified proteins. FEBS Lett 332:208–210. [DOI] [PubMed] [Google Scholar]
- 54. Uchida K, Kawakishi S (1989) Ascorbate‐mediated specific oxidation of the imidazole ring in a histidine derivative. Bioorg Chem 17:330–343. [Google Scholar]
- 55. Valderrama B, Ayala M, Vazquez‐Duhalt R (2002) Suicide inactivation of peroxidases and the challenge of engineering more robust enzymes. Chem Biol 9:555–565. [DOI] [PubMed] [Google Scholar]
- 56. Mao L, Luo S, Huang Q, Lu J (2013) Horseradish peroxidase inactivation: heme destruction and influence of polyethylene glycol. Sci Rep 3:3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gonzalez‐Perez D, Garcia‐Ruiz E, Ruiz‐Duenas FJ, Martinez AT, Alcalde M (2014) Structural determinants of oxidative stabilization in an evolved versatile peroxidase. ACS Catal 4:3891–3901. [Google Scholar]
- 58. Green AP, Hayashi T, Mittl PRE, Hilvert D (2016) A Chemically programmed proximal ligand enhances the catalytic properties of a heme enzyme. J Am Chem Soc 138:11344–11352. [DOI] [PubMed] [Google Scholar]
- 59. Loose JSM, Forsberg Z, Fraaije MW, Eijsink VGH, Vaaje‐Kolstad G (2014) A rapid quantitative activity assay shows that the Vibrio cholerae colonization factor GbpA is an active lytic polysaccharide monooxygenase. FEBS Lett 588:3435–3440. [DOI] [PubMed] [Google Scholar]
- 60. Dotson WD, Greenier J, Ding H (2006) Polypeptides having cellulolytic enhancing activity and polynucleotides encoding same. US20060005279A1.
- 61. Gasteiger EHC, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server In: Walker JM, Ed., 2005. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press; p. 571–607. [Google Scholar]
- 62. Forsberg Z, Mackenzie AK, Sorlie M, Rohr AK, Helland R, Arvai AS, Vaaje‐Kolstad G, Eijsink VGH (2014) Structural and functional characterization of a conserved pair of bacterial cellulose‐oxidizing lytic polysaccharide monooxygenases. Proc Natl Acad Sci U S A 111:8446–8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pickford AR, O'Leary JM. Isotopic labeling of recombinant proteins from the methylotrophic yeast Pichia pastoris In: Downing AK, Ed., 2004. Protein NMR Techniques. Totowa, NJ: Humana Press; p. 17–33. [DOI] [PubMed] [Google Scholar]
- 64. Courtade G, Wimmer R, Rohr AK, Preims M, Felice AK, Dimarogona M, Vaaje‐Kolstad G, Sorlie M, Sandgren M, Ludwig R, Eijsink VGH, Aachmann FL (2016) Interactions of a fungal lytic polysaccharide monooxygenase with beta‐glucan substrates and cellobiose dehydrogenase. Proc Natl Acad Sci U S A 113:5922–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zougman A, Selby PJ, Banks RE (2014) Suspension trapping (STrap) sample preparation method for bottom‐ up proteomics analysis. Proteomics 14:1006–1010. [DOI] [PubMed] [Google Scholar]
- 66. Wood TM (1988) Preparation of crystalline, amorphous and dyed cellulase substrates. Method Enzymol 160:19–25. [Google Scholar]
- 67. Vivekanand V, Olsen EF, Eijsink VGH, Horn SJ (2013) Effect of different steam explosion conditions on methane potential and enzymatic saccharification of birch. Bioresour Technol 127:343–349. [DOI] [PubMed] [Google Scholar]
- 68. Zamocky M, Schumann C, Sygmund C, O'Callaghan J, Dobson ADW, Ludwig R, Haltrich D, Peterbauer CK (2008) Cloning, sequence analysis and heterologous expression in Pichia pastoris of a gene encoding a thermostable cellobiose dehydrogenase from Myriococcum thermophilum. Prot Expr Purif 59:258–265. [DOI] [PubMed] [Google Scholar]
- 69. Westereng B, Agger JW, Horn SJ, Vaaje‐Kolstad G, Aachmann FL, Stenstrom YH, Eijsink VGH (2013) Efficient separation of oxidized cello‐oligosaccharides generated by cellulose degrading lytic polysaccharide monooxygenases. J Chromatogr A 1271:144–152. [DOI] [PubMed] [Google Scholar]
- 70. Strohalm M, Kavan D, Novak P, Volny M, Havlicek V (2010) mMass 3: a cross‐platform software environment for precise analysis of mass spectrometric data. Anal Chem 82:4648–4651. [DOI] [PubMed] [Google Scholar]
- 71. Henderson WW, Shepherd RE, Abola J (1986) Proton and carbon‐13 NMR spectra of a series of methyl‐substituted imidazole complexes of pentaamminecobalt(III) and crystal structure of the remote isomer of [(4‐methylimidazole)pentaamminecobalt(III)] trichloride dihydrate. Inorg Chem 25:3157–3163. [Google Scholar]
- 72. Rodsrud G, Lersch M, Sjode A (2012) History and future of world's most advanced biorefinery in operation. Biomass Bioenergy 46:46–59. [Google Scholar]
- 73. Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein‐dye binding. Anal Biochem 72:248–254. [DOI] [PubMed] [Google Scholar]
- 74. Sorlie M, Seefeldt LC, Parker VD (2000) Use of stopped‐flow spectrophotometry to establish midpoint potentials for redox proteins. Anal Biochem 287:118–125. [DOI] [PubMed] [Google Scholar]
- 75. Liu Y, Seefeldt LC, Parker VD (1997) Entropies of redox reactions between proteins and mediators: the temperature dependence of reversible electrode potentials in aqueous buffers. Anal Biochem 250:196–202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material: The supplementary material includes eleven figures.
Figure S1: SDS‐PAGE of purified TaLPMO9A‐Ao and TaLPMO9A‐Pp.
Figure S2: 3D structure of TaLPMO9A (PDB ID: 3ZUD).
Figure S3: LC–MS chromatograms of N‐terminal peptides.
Figure S4: Binding to PASC.
Figure S5: Reaction products generated from steam exploded birch.
Figure S6: HPAEC‐PAD chromatograms for the reactions with cellopentaose.
Figure S7: Close‐up of MALDI‐ToF MS spectra for the reactions with PASC or xyloglucan.
Figure S8: NMR pH titration curves.
Figure S9: Kinetic information for copper binding obtained by stopped‐flow spectrophotometry.
Figure S10: Differential scanning calorimetry analysis.
Figure S11: Control reactions to check for occurrence of copper‐catalyzed Fenton‐type chemistry.