

Summary
The purpose of this study was to investigate whether growth factors produced by activated human lung mast cells (HLMCs) impair β2‐adrenoceptor (β2‐AR) function in human airway smooth muscle (ASM) cells. Protein array analysis confirmed the presence of various growth factors, including transforming growth factor (TGF)‐β1, in the supernatants of high‐affinity IgE receptor (FcεRI)‐activated HLMCs which, when applied to ASM cells, impaired albuterol‐induced cyclic adenosine monophosphate (cAMP) production, an effect that was prevented following neutralization of TGF‐β1. This blunted β2‐AR response was reproduced by treating ASM cells with TGF‐β1 or fibroblast growth factor (FGF)‐2, which induced β2‐AR phosphorylation at tyrosine residues Tyr141 and Tyr350, and significantly reduced the maximal bronchorelaxant responses to isoproterenol in human precision cut lung slices (PCLS). Finally, ASM cells isolated from severe asthmatics displayed constitutive elevated β2‐AR phosphorylation at both Tyr141 and Tyr350 and a reduced relaxant response to albuterol. This study shows for the first time that abnormal β2‐AR phosphorylation/function in ASM cells that is induced rapidly by HLMC‐derived growth factors, is present constitutively in cells from severe asthmatics.
Keywords: allergy, inflammation, mast cells, signal transduction
Introduction
β2‐adrenoceptor (β2‐AR) agonists represent the most commonly used drugs in the management of pulmonary diseases, including asthma 1. These drugs provide relief from acute bronchoconstriction by promoting bronchodilation through direct activation of β2‐AR on airway smooth muscle (ASM) and promoting its relaxation. Despite their clinical benefits, the use of β2‐AR agonists in asthma has been associated with loss of β2‐AR function, deleterious effects and/or worsening of the symptoms and asthma deaths 2. Understanding the potential mechanisms that result in the loss of function and/or deleterious clinical effects in patients treated with β2‐agonists could offer novel therapeutic alternatives.
Studies performed in different cell types have reported that some, but not all, growth factors can impair cell responsiveness to β2‐agonists by phosphorylating β2‐AR on several serine (ser345/346) or tyrosine residues (Tyr350/354/141), resulting in receptor uncoupling and internalization 3, 4, 5, 6. In human lung mast cells (HLMCs), we showed that stem cell factor (SCF) can similarly uncouple β2‐AR following receptor phosphorylation at similar Tyr350 residues 7. More recently, we observed that β2‐AR responses were suppressed in both mast cells (suppression of FcεR1‐induced activation) and ASM cells (inhibition of ASM contraction) following cell–cell contact via β2‐AR phosphorylation, on Tyr350 residues 8. Whether mast cells also blunt β2‐AR responses in ASM cells via the action of secreted different growth factors remains unknown.
The present study provides the first evidence that abnormal β2‐AR phosphorylation/function can be induced experimentally by exposing healthy ASM cells to growth factors released from activated mast cells, a feature that was constitutively present in ASM cells from severe asthmatics. Abnormal tyrosine phosphorylation of β2‐AR represents a novel mechanism that could explain the poor clinical efficacy of β2‐AR therapy seen in severe asthma.
Materials and methods
Culture of human airway smooth muscle cells
Primary human ASM cells were obtained from consented healthy subjects and asthmatic patients isolated from endobronchial biopsies as described previously 9. The demographics of subjects used in the study are shown in Fig3d.
Figure 3.
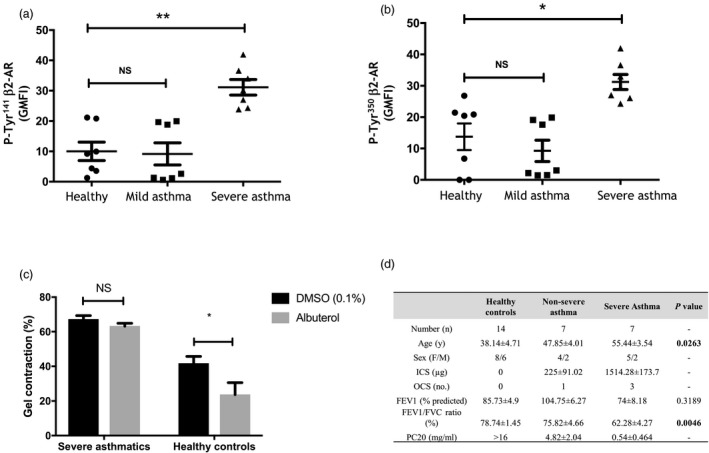
Airway smooth muscle (ASM) cells from patients with severe asthma displayed increased β2‐adrenoceptor (β2‐AR) phosphorylation and reduced bronchorelaxant responses. (a,b) Phosphorylation of β2‐AR at Tyr350 or Tyr141 was assessed by flow cytometry and expressed as the fold increase in geometric mean fluorescence intensity (GMFI) over the isotype control antibody. Data are presented as means ± standard error of the mean (s.e.m.) of experiments performed in ASM cells derived from seven subjects. *P < 0·05 and **P < 0·01 versus untreated cells. (c) Effect of albuterol on the spontaneous contraction of ASM cells embedded in collagen gel matrices in three healthy and severe asthmatic subjects. *P < 0·01 versus dimethylsulphoxide (DMSO) controls. Statistical analysis was performed using one‐way (a/b) or two‐way analysis of variance (anova) (c). (d) Demographics of subjects used in the study. Bold indicates signficance compared to healhty subjects.
Mast cell isolation, culture and stimulation
Isolation and stimulation of HLMC were performed as described previously 10.
Flow cytometry
Flow cytometry assays were performed as described previously using the Becton Dickinson FACScan (Oxford, UK) 9. The antibodies used were rabbit anti‐human β2‐AR (Tyr141) antibody, rabbit anti‐human β2‐AR (Tyr350) antibody (2 μg/ml; Santa Cruz Biotechnology, Santa Cruz, CA, USA), isotype‐matched control (rabbit immunoglobulin (Ig)G; Imminostep, Salamanca, Spain) and secondary anti‐rabbit fluorescein isothiocyanate (FITC; Cell Signaling Technology, Danvers, MA, USA).
Measurement of cyclic adenosine monophosphate (cAMP) production
ASM cells were stimulated with 10 µM albuterol for 10 min in the presence or absence of FcεR1‐activated HLMC supernatants (1 : 4 dilution, added for different time‐points or 10 min, as indicated) before cell extracts were used for the determination of intracellular cAMP concentration using the cAMP EIA kit from Amersham Biosciences (Little Chalfont, UK) (detection limit curve range > 25 fmol/well) following the manufacturer's protocol.
Collagen gel contraction assay
Spontaneous ASM cells contraction was examined in collagen gels, as described previously 8.
Growth factor array
Supernatants were prepared from FcεR1‐activated HLMCs, as described previously 10, and were used to perform the growth factor array analysis which allows the simultaneous detection of 41 different human growth factors, following the manufacturer's protocol (Abcam, Cambridge, UK).
Human lung slice preparation
The bronchorelaxant responses using the six precision cut lung slices (PCLS) were performed as described previously 11. The slices were incubated with transforming growth factor (TGF)‐®1 or fibroblast growth factor (FGF)‐2 at 100 ng/ml for 30 min before responses to isoproterenol were assessed on PCLS precontracted with 10–4 M carbachol. The airway dilation data were expressed as percentage dilation of the maximum carbachol constricted airway luminal area with the baseline value prior to contraction considered as 100%, as described previously 11.
Statistical analysis
All data are presented as mean ± standard error of the mean (s.e.m.). Statistical analysis was performed by two‐ or one‐way analysis of variance (anova) with Bonferroni's correction for multiple comparisons. Differences were considered significant when P < 0·05. Statistical analysis was performed using GraphPad Prism version 6 (GraphPad Software, San Diego, CA, USA).
Results
FcεR1‐activated HLMCsproduced growth factors and attenuated the response to albuterol in human ASMcells
Representative protein array membrane showing growth factors) produced in the supernatants of resting and FcεR1‐activated HLMCs [IgE (2·5 µg/ml)‐anti‐IgE (1 : 1000 dilution)] (Fig1a). Densitometric analyses of the membrane arrays revealed that levels of FGF‐2, epidermal growth factor (EGF), heparin‐binding (HB)‐EGF, hepatocyte growth factor (HGF), granulocyte colony‐stimulating factor (G‐CSF), granulocyte–macrophage (GM)‐CSF, insulin‐like growth factor binding protein IGF)‐BP2, IGF‐BP6, IGF‐II, SCF, SCF‐R, macrophage CSF (M‐CSF), vascular endothelial growth factor (VEGF)‐A, VEGF‐D, VEGF‐R3 and TGF‐β1 were produced by FcεR1‐activated HLMCs, although only four growth factors (GM‐CSF, HGF, IGF‐II and TGF‐β1, P < 0·01) reached statistical significance. ASM cells first incubated with supernatants from FcεR1‐activated HLMCs (compared to that of resting HLMCs) for different time‐points (10–30 min) displayed a complete inhibition of cAMP accumulation measured in ASM cells treated with 10 µM albuterol for 10 min (n = 3 ASM cell lines, P < 0·01) (Fig1b). The inhibitory effect of a 10‐min incubation with FcεR1‐activated HLMC supernatants on albuterol responses was almost completely prevented by incubating supernatants of FcεR1‐activated HLMCs with 10 µg/ml neutralizing TGF‐β1 antibody (Fig1c).
Figure 1.
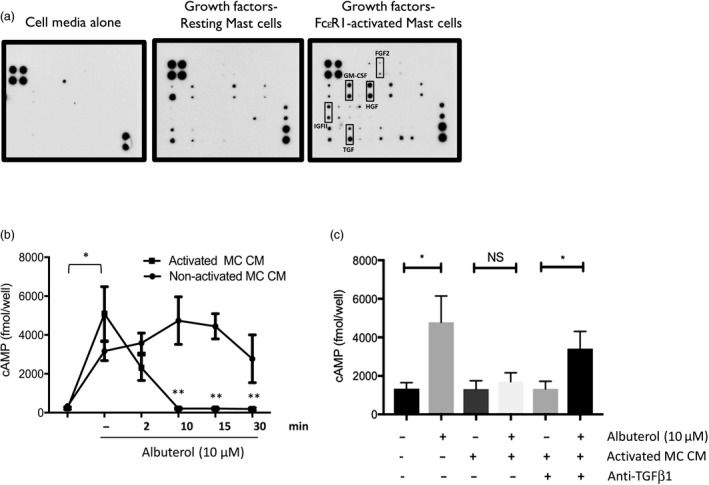
Supernatants of high‐affinity immunoglobulin (Ig)E receptor (FcεR1)‐activated human lung mast cells (HLMCs), which contained different growth factors, suppressed albuterol‐induced cyclic adenosine monophosphate (cAMP) production in airway smooth muscle (ASM) cells. (a) Representative blot showing the growth factor membrane arrays incubated with HLMC media alone, supernatants from resting HLMCs or FcεR1‐activated HLMCs. Representative of three individual donors. (b) ASM cells were treated with supernatants of FcεR1‐activated HLMCs (1 : 4 dilution, 30 min) or non‐activated HLMCs for 2, 10, 15 and 30 min before cAMP production (fmol/well) in response to a 10‐min stimulation with 10 µM albuterol was investigated using enzyme immunoassay (n = 3 donors). *P < 0·05 versus basal, **P < 0·01 versus cells treated with non‐activated HLMCs. (c) Incubating ASM cells for 10 min with the conditioned medium (CM) of FcεRI‐activated HLMCs (MC) containing neutralizing anti‐transforming growth factor (TGF)‐β1 antibody (10 µg/ml) restored the albuterol (10 µM)‐dependent cAMP response. *P = 0·033 (n = 4). Statistical analysis was performed using two‐way analysis of variance (anova).
TGF‐β1 and FGF‐2 induced β2‐AR phosphorylation on key tyrosine residues and attenuated β2‐AR responsiveness
Interestingly, only pretreatment of human ASM cells with either TGF‐β1 or FGF‐2 at 10 ng/ml for 10 min increased β2‐AR phosphorylation significantly on both Tyr141 (Fig2a) and Tyr350 (Fig2b), while VEGF or nerve growth factor (NGF) had no effect, despite having functional receptors in ASM cells 12, 13, 14, 15. Figure2c shows that TGF‐β1 and FGF‐2 also reduced cAMP responses to albuterol by more than 85 and 72%, respectively (n = 3). Figure 2d shows that isoproterenol‐evoked bronchodilatation assessed using human PCLS was inhibited by TGF‐β1 or FGF‐2 (30 min, 100 ng/ml), with a reduction of the maximum % bronchodilation from 57·44 ± 4·11 to 34·29 ± 5·37 and 32·4 ± 2·39 (n = 6 slices, P < 0·05), respectively. Both growth factors had no effect on isoproterenol EC50 value (logEC50 = –6·27 ± 0·2099).
Figure 2.
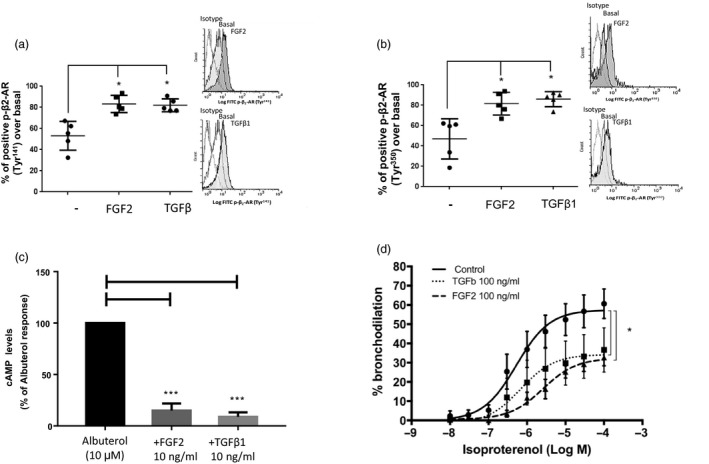
Transforming growth factor (TGF)‐β1 and fibroblast growth factor (FGF)‐2 increased β2‐adrenoceptor (β2‐AR) phosphorylation on tyrosine residues and reduced β2‐AR responsiveness. (a,b) Means ± standard error of the mean (s.e.m.) of the % positive p‐β2‐AR on Tyr141 and Tyr350 staining over basal assessed by flow cytometry (with representative histograms) in human healthy airway smooth muscle (ASM) cells (n = 5) treated with 10 ng/ml FGF‐2 or TGF‐β for 10 min, *P < 0·05 versus resting conditions. (c). ASM cells pretreated with TGF‐β or FGF‐2 (10 min) were incubated with albuterol (10 μM) for 15 min before accumulation of cyclic adenosine monophosphate (cAMP) was assessed by enzyme immunoassay. Data are expressed as % of albuterol‐induced cAMP response performed in three healthy subjects and analysed using the one‐sample t‐test. ***P < 0·001 versus albuterol. (d) Human PCLS precontracted with 10–4 M carbachol were incubated with control diluent, FGF‐2 or TGF‐β for 30 min prior isoproterenol (10−8 to 10−4 M). Each concentration–response curve is expressed as means ± s.e.m. performed in six precision cut lung slices (PCLS) isolated from two donors. *P < 0·01 versus control PCLS. Statistical analysis was performed using one‐way analysis of variance (anova) on the maximum bronchodilatory responses.
ASM cells from severe asthmatics display an increased basal β2‐AR phosphorylation at tyrosine residues and an impaired relaxant response to albuterol
Our flow cytometry data show that ASM cells derived from severe asthmatics, but not from non‐severe asthmatics, displayed a constitutive increased phosphorylation of β2‐AR at both Tyr141 (Fig3a, geometric mean fluorescence intensity (GMFI) = 31·13 ± 2·54, P = 0·0045, n = 7) and Tyr350 (Fig. 3b, GMFI = 31·22 ± 2·40, P = 0·0224, n = 7) when compared to levels seen in healthy subjects (GMFI = 10·03 ± 3·05 and 13·76 ± 4·23, n = 7). Gel contraction assays also revealed that albuterol failed to abrogate the spontaneous contraction of severe asthmatic ASM cells while reducing by more than 47% the response seen in healthy ASM cells (Fig3c, P < 0·05, n = 3). Figure 3d shows the demographics of the subjects used in the studies. Data are shown as means ± s.e.m.
Discussion
The possibility that cell–cell interactions between mast cells and ASM cells may play a role in the pathogenesis of asthma is supported by multiple in‐vitro reports showing the ability of mast cell mediators to induce various pro‐asthmatic responses in ASM cells/tissues 2. We found that β2‐AR dysfunction in human ASM cells could be induced experimentally by exposing human ASM cells with supernatants from activated HLMCs, which produced a number of growth factors, although only four (GM‐CSF, HGF, IGF‐II and TGF‐β1) reached statistical significance. The observation that HLMC effect was largely prevented by the presence of neutralizing TGF‐β1 antibody, or mimicked by the pretreatment of ASM cells with exogenous TGF‐β1 (or FGF‐2), identified TGF‐β1 as a novel modulator of β2‐AR function. We believe that the central mechanism by which TGF‐β1 impaired β2‐AR function in ASM cells results from its capacity to phosphorylate the receptor rapidly at two key tyrosine residues, Tyr141 and Tyr350, which was seen within 10 min. This hypothesis is supported by our recent finding that β2‐AR phosphorylation at Tyr350 in HLMCs was essential in driving the loss of β2‐AR responses when exposed to another mast cell mediator SCF 7 or following adhesion of HLMCs to ASM cells 8. Whether TGF‐β1 interferes with β2‐AR responses by acting at a level downstream to β2‐AR remains a possibility, although a previous study also performed in human ASM cells showed that TGF‐β1 inhibited isoproterenol‐induced cAMP production by decreasing receptor number without altering receptor coupling (Gs/adenylyl cyclase) 16. Our combined observations suggest that the inhibition of β2‐AR responses by TGF‐β1 results from different changes occurring mainly at the receptor level. Interestingly, while FGF‐2 also induced β2‐AR phosphorylation, we found that VEGF and NGF failed to have any effect (data not shown), despite previous studies showing the existence of functional receptors in human ASM cells 12, 13, 14, 17, This supports the interesting concept that β2‐AR phosphorylation by RTK ligands is highly stimuli‐ and cell‐specific. Indeed, depending on the cell types, β2‐AR can be phosphorylated at Tyr350/354, Tyr364, Ser346 or Ser346 by insulin or Tyr132 and Tyr141 by insulin‐like growth factor‐1 4, 6, 18. It is noteworthy to mention that, irrespective of the type of growth factors, β2‐AR phosphorylation on tyrosine residues is a key trigger of receptor dysfunction 6, 18. Our PCLS model, used widely to study ASM contractile/relaxant responses 19, confirmed that either TGF‐β1 and FGF‐2 (although used at much higher concentrations, due possibly to tissue complexity of the PCLS) reduced β2‐AR‐evoked bronchorelaxant responses dramatically. Although a different agonist (isoproterenol) was used in PCLS, both albuterol and isoproterenol were shown to have a similar bronchodilatory profile in patients 20. Our study shows that in addition to tumour necrosis factor (TNF)‐α, interleukin (IL)‐1β, IL‐13 or IL‐5, both TGF‐β1 or FGF‐2 can also drive β2‐AR dysfunction in human ASM cells (reviewed in 21). We also made the novel finding that ASM cells from severe asthmatics displayed a constitutive tyrosine phosphorylation of β2‐ARs and blunted response to albuterol in the collagen gel assays. Because ASM cells from severe asthmatics also have a reduced sensitivity to corticosteroids 22, 23, 24, our study further supports the existence of the factors in severe asthma that blunt the response to the two main anti‐asthma drugs.
Our study shows that β2‐AR dysfunction in ASM in asthma could result from the transient (induced by growth factors) or permanent (unknown mechanisms) phosphorylation of the receptor at both Tyr141 and Tyr350 residues. We propose that targeting the mechanisms leading to abnormal β2‐AR phosphorylation have the potential to enhance β2‐agonist activity in patients with asthma, and perhaps reduce the unwanted adverse effects sometimes evident with their chronic use 21.
Author contributions
L. C. performed most of the experiments, generated, analysed and interpreted the data. A. Z., R. B. and M. B. helped with the experiments and data analysis. R. A. P. and C. K.‐W. performed the P. C. L. S. studies and contributed to data analysis/interpretation. Y. A. and P. B. conceived the project, designed the experiments and analysed the data and Y. A. wrote the paper.
Disclosures
None.
Acknowledgements
This study was funded by Wellcome Trust Project Grant (094058) and supported by the National Institute for Health Research Leicester Biomedical Research Center Respiratory. 2Funded in part by NIH PO1‐HL114471 (RAP, CKW). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR and the Department of Health.
References
- 1. Papi A, Caramori G, Adcock IM, Barnes PJ. Rescue treatment in asthma. More than as‐needed bronchodilation. Chest 2009; 135:1628–33. [DOI] [PubMed] [Google Scholar]
- 2. Bradding P, Arthur G. Mast cells in asthma – state of the art. Clin Exp Allergy 2016; 46:194–263. [DOI] [PubMed] [Google Scholar]
- 3. Karoor V, Wang L, Wang HY, Malbon CC. Insulin stimulates sequestration of beta‐adrenergic receptors and enhanced association of beta‐adrenergic receptors with Grb2 via tyrosine 350. J Biol Chem 1998; 273:33035–41. [DOI] [PubMed] [Google Scholar]
- 4. Karoor V, Malbon CC. Insulin‐like growth factor receptor‐1 stimulates phosphorylation of the beta2‐adrenergic receptor in vivo on sites distinct from those phosphorylated in response to insulin. J Biol Chem 1996; 271:29347–52. [DOI] [PubMed] [Google Scholar]
- 5. Karoor V, Baltensperger K, Paul H, Czech MP, Malbon CC. Phosphorylation of tyrosyl residues 350/354 of the beta‐adrenergic receptor is obligatory for counterregulatory effects of insulin. J Biol Chem 1995; 270:25305–8. [DOI] [PubMed] [Google Scholar]
- 6. Doronin S, Shumay E, Wang HY, Malbon CC. Akt mediates sequestration of the beta(2)‐adrenergic receptor in response to insulin. J Biol Chem 2002; 277:15124–31. [DOI] [PubMed] [Google Scholar]
- 7. Cruse G, Yang W, Duffy SM et al Counterregulation of beta(2)‐adrenoceptor function in human mast cells by stem cell factor. J Allergy Clin Immunol 2010; 125:257–63, e1–5. [DOI] [PubMed] [Google Scholar]
- 8. Lewis RJ, Chachi L, Newby C, Amrani Y, Bradding P. Bidirectional counterregulation of human lung mast cell and airway smooth muscle beta2 adrenoceptors. J Immunol 2016; 196:55–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chachi L, Shikotra A, Duffy SM et al Functional KCa3.1 channels regulate steroid insensitivity in bronchial smooth muscle cells. J Immunol 2013; 191:2624–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cruse G, Singh SR, Duffy SM et al. Functional KCa3.1 K+ channels are required for human fibrocyte migration. J Allergy Clin Immunol 2011; 128:1303–9 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cooper PR, Panettieri RA Jr. Steroids completely reverse albuterol‐induced beta(2)‐adrenergic receptor tolerance in human small airways. J Allergy Clin Immunol 2008; 122:734–40. [DOI] [PubMed] [Google Scholar]
- 12. Bosse Y, Rola‐Pleszczynski M. FGF2 in asthmatic airway‐smooth‐muscle‐cell hyperplasia. Trends Mol Med 2008; 14:3–11. [DOI] [PubMed] [Google Scholar]
- 13. Freund‐Michel V, Frossard N. Overexpression of functional TrkA receptors after internalisation in human airway smooth muscle cells. Biochim Biophys Acta 2008; 1783:1964–71. [DOI] [PubMed] [Google Scholar]
- 14. Kazi AS, Lotfi S, Goncharova EA et al Vascular endothelial growth factor‐induced secretion of fibronectin is ERK dependent. Am J Physiol Lung Cell Mol Physiol 2004; 286:L539–45. [DOI] [PubMed] [Google Scholar]
- 15. Schuliga M, Javeed A, Harris T et al Transforming growth factor‐beta‐induced differentiation of airway smooth muscle cells is inhibited by fibroblast growth factor‐2. Am J Respir Cell Mol Biol 2013; 48:346–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nogami M, Romberger DJ, Rennard SI, Toews ML. TGF‐beta 1 modulates beta‐adrenergic receptor number and function in cultured human tracheal smooth muscle cells. Am J Physiol 1994; 266:L187–91. [DOI] [PubMed] [Google Scholar]
- 17. Kim JH, Jain D, Tliba O et al TGF‐beta potentiates airway smooth muscle responsiveness to bradykinin. Am J Physiol Lung Cell Mol Physiol 2005; 289:L511–20. [DOI] [PubMed] [Google Scholar]
- 18. Baltensperger K, Karoor V, Paul H, Ruoho A, Czech MP, Malbon CC. The beta‐adrenergic receptor is a substrate for the insulin receptor tyrosine kinase. J Biol Chem 1996; 271:1061–4. [DOI] [PubMed] [Google Scholar]
- 19. Sanderson MJ. Exploring lung physiology in health and disease with lung slices. Pulm Pharmacol Ther 2011; 24:452–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Casaburi R, Adame D, Hong CK. Comparison of albuterol to isoproterenol as a bronchodilator for use in pulmonary function testing. Chest 1991; 100:1597–600. [DOI] [PubMed] [Google Scholar]
- 21. Amrani Y, Bradding P. Beta2‐adrenoceptor function in asthma. Adv Immunol 2017; 136:1–28. [DOI] [PubMed] [Google Scholar]
- 22. Chachi L, Gavrila A, Tliba O, Amrani Y. Abnormal corticosteroid signalling in airway smooth muscle: mechanisms and perspectives for the treatment of severe asthma. Clin Exp Allergy 2015; 45:1637–46. [DOI] [PubMed] [Google Scholar]
- 23. Chanez P, Wenzel SE, Anderson GP et al Severe asthma in adults: what are the important questions? J Allergy Clin Immunol 2007; 119:1337–48. [DOI] [PubMed] [Google Scholar]
- 24. Chang PJ, Bhavsar PK, Michaeloudes C, Khorasani N, Chung KF. Corticosteroid insensitivity of chemokine expression in airway smooth muscle of patients with severe asthma. J Allergy Clin Immunol 2012; 130:877–85.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]