

Summary
The objective of the present study was to evaluate the role of CDKN1A in rheumatoid arthritis (RA). Related gene expression data screened from Gene Expression Omnibus (GEO) were processed with network analysis. Protein–protein interaction was analysed through string database. Quantitative reverse transcription–polymerase chain reaction (qRT–PCR) was used to measure mRNA and microRNA expression. Cell proliferation and cell cycle were tested by MTT assay and flow cytometry, respectively. Transwell migration and invasion assay was used to test cell migration and invasion. CDKN1A screened by bioinformatics methods showed differential expression in RA cells compared with healthy controls (HC), and was at an important position in the protein–protein interaction network of RA. Compared with the HC group, CDKN1A was down‐regulated in human RA synovium tissues and human fibroblast‐like synoviocytes (HFLS). Contrary to CDKN1A silencing, CDKN1A over‐expression significantly inhibited the proliferation and invasion of HFLS‐RA, arrested HFLS‐RA in G0/G1 phase and down‐regulated the expressions of tumour necrosis factor (TNF)‐α and interleukin (IL)‐6, while it up‐regulated the expression of IL‐10. CDKN1A over‐expression could also suppress phosphorylated signal transducers and activators of transcription 1 (pSTAT‐1) expression. MiR‐146a, highly expressed in RA tissues, could regulate CDKN1A negatively. Anti‐146a suppressed cell proliferation and invasion, and at the same time enhanced IL‐10 expression but inhibited IL‐6, TNF‐α and pSTAT‐1 expression. The results indicated that CDKN1A over‐expression, which could be enhanced by miR‐146a suppression, inhibited the proliferation of invasion in HFLS‐RA. This was probably a result of suppressed pSTAT‐1, IL‐6 and TNF‐α expression and enhanced IL‐10 expression.
Keywords: CDKN1A, human fibroblast‐like synoviocytes, MiR‐146a, rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease characterized by synovial hyperplasia, inflammatory infiltration and cartilage degradation. Tumour necrosis factor (TNF)‐α pathway activation caused interleukin (IL)‐6 and other proinflammatory cytokine production, usually found in RA, driving both synovial inflammation and joint destruction 1. There are two cell types in normal synovium: macrophage‐like synoviocytes (MLS) and human fibroblast‐like synoviocytes (HFLS) 2. In RA these FLS become hyperplastic, characterized by enhanced migratory potential and invasiveness, leading ultimately to cartilage and bone destruction 3. To reveal the mechanism underlying RA progression more clearly, this study focused on the miR/mRNA axis and the activated proinflammatory pathway.
HFLS, as an essential effector cell in RA, proliferates and produces large amounts of IL‐6, IL‐8 and cyclooxygenase (COX)‐2 4. Enhanced signal transducer and activator of transcription (STAT)‐1 phosphorylation was also found in RA HFLS 5. Tumour suppressor p53 presented as wild‐type (WT) in non‐RA patients, while the mutant type was presented in synovial tissues and synoviocytes of RA patients 6. Increasing evidence indicates that active HFLS‐RA exhibits malignant characteristics 7. For example, HFLS‐RA display abnormal proliferation, low apoptotic ability, pannus and cartilage invasion 1, 8. This study investigates the mechanisms accounting for malignant characteristics.
Cyclin‐dependent kinase inhibitor 1A (CDKN1A), as a main target of tumour suppressor gene p53, can encode p21 protein, which is a cyclin‐dependent kinase inhibitor, and is able to arrest cells in G1/S phase by blocking cyclin E‐CDK2 9. CDKN1A over‐expression, which occurs in a p53‐independent manner, inhibits cell proliferation in breast cancer 10. CDKN1A inhibits the proliferation, migration and invasion in hepatocarcinoma cells and thyroid cancer cells under the negative regulation of miR‐93 and miR‐4295, respectively 11, 12. The inflammatory process is accelerated in p21‐deficient mice that develop spontaneous epithelial tumours 13. Activation of the p53‐p21 signalling axis prevents inflammation induced by proinflammatory cytokines 14. Enhancement of p21 limits the activation response of macrophages and suppresses the development and persistence of RA 15. However, the underlying mechanism of CDKN1A in HFLS‐RA has not been explored thoroughly.
In this study, the inhibitory role of CDKN1A was suspected in RA synovium and HFLS and validated with in‐vitro experiments. We then hypothesized the mechanism as activation of the inflammatory pathway, which included tumour necrosis factor (TNF)‐α, IL‐6 and IL‐10 cytokines. The upstream regulator of CDKN1A, miR‐146a, was revealed. These findings may provide a new regulatory mechanism in RA as the CDKN1A/miR‐146a axis and suppressed inflammatory cytokines.
Materials and methods
Gene chip data extraction and network‐based meta‐analysis
Gene expression data were downloaded from Gene Expression Omnibus (GEO) with ‘rheumatoid arthritis’ (RA) as keywords. Four data sets that contained gene expression data of both RA patients and healthy controls (HC) were selected. GSE55457 and GSE55235 were submitted by the same author, but were different in their GSM numbers and gene expression data.
The downloaded gene expression data were classified into HC and RA groups, and analysed comparatively by network‐based meta‐analysis to screen out differentially expressed genes (DEGs). The probe ID in the microarray data was converted to the corresponding gene name through the corresponding platform using the conversion tool. After matching, the data were normalized by log2 with limma with variance [interquartile range (IQR)] set at 15%. It could then be viewed through the block diagram to ensure that the samples had the same distribution. The standard thresholds for screening DEGs were set as antibodies > 1 and adjusted P‐value < 0·05.
Collection of RA clinical samples
From 2015 to 2017, synovial tissues from RA patients undergoing total knee joint replacement surgery were selected from the Second Hospital of Jilin University. Healthy synovial biopsies were collected from traumatic knee patients and used as normal controls. The experimental protocol was approved in advance by the Ethics Committees of the Second Hospital of Jilin University. All patients gave their written informed consent.
Cell culture
293T HFLS and HFLS‐RA were bought from BeNa Culture Collection (Beijing, China) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad, CA, USA) for 4 h at 37°C in 5% CO2. After centrifugation, the cells were incubated in DMEM medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C in 5% CO2, 21% O2 and 75% N2.
Quantitative reverse transcription–polymerase chain reaction (qRT–PCR)
The total RNA from synovial tissues in the RA patient and HC groups were extracted by Trizol (Invitrogen). The obtained RNA was reverse‐transcribed to cDNA using the RevertAid First‐Strand cDNA Synthesis Kit (Thermo, Shanghai, China), and Power SYBR Green PCR Master Mix (Thermo, Shanghai, China) was used for determination. The PCR programme comprised predenaturation at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s, annealing at 60°C for 30 s and extension at 72°C for 30 s. Subsequently, analysis of the dissolution curve of PCR products was performed with glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) expression as the standard for mRNA and U6 expression as the standard for miRNA; relative mRNA and miRNA expression was calculated by the 2–ΔΔCt method. The primers supplied are shown in Table 1.
Table 1.
Primers for quantitative reverse transcription–polymerase chain reaction (qRT–PCR)
| Gene | Forward primer (5′→3′) | Reverse primer (5′→3′) |
|---|---|---|
| CDKN1A | TGCCGAAGTCAGTTCCTTGT | GCATGGGTTCTGACGGACAT |
| STAT‐1 | TACAAACCTCAAGCCAGCCT | GCAGTAACACGGGGATCTCA |
| IL‐6 | TCAATATTAGAGTCTCAACCCCCA | GAGAAGGCAACTGGACCGAA |
| IL‐10 | TGTTCTTTGGGGAGCCAACA | GGGCTCCCTGGTTTCTCTTC |
| TNF‐α | AGAACTCACTGGGGCCTACA | AGGAAGGCCTAAGGTCCACT |
| GAPDH | AGCCACATCGCTCAGACAC | GCCCAATACGACCAAATCC |
| MiR‐146a | TGGTACTGATGTGATGGACT | TCATATCACACAGCACCGAT |
| U6 | CTCGCTTCGGCAGCACA | AACGCTTCACGAATTTGCGT |
CKDN1A = cyclin‐dependent kinase inhibitor 1A; STAT‐1 = signal transducer and activator of transcription 1; IL = interleukin; TNF = tumour necrosis factor; GAPDH = glyceraldehyde‐3‐phosphate dehydrogenase; MIR‐146a = MiR‐146a.
Western blot
Total proteins were obtained with RAPI (Beyotime, Shanghai, China), 100 μg of which were separated using sodium dodecyl sulphate‐polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were incubated in Tris‐buffered saline with Tween 20 (TBST) containing 5% non‐fat milk for 1 h, and then incubated with anti‐CDKN1A (ab102013, 1/1000; Abcam, Cambridge, MA, USA), anti‐STAT‐1 (ab31369, 1/500; Abcam), anti‐phosphorylated STAT‐1 (pSTAT)‐1 (ab30645, 1/500; Abcam) and anti‐GAPDH (ab8245, 1/1000; Abcam) at 4°C overnight. The membrane was washed with TBST three times and incubated with secondary antibodies at room temperature for 1·5 h. After washing three times with TBST, the membrane was subjected to colour reaction by ECL Plus from Life Technology, and GAPDH was detected as the internal control.
Vector construction and cell transfection
According to the human CDKN1A gene sequence in GenBank (Accession number NM_001291549.1), CDKN1A‐specific PCR primers were: forward primer 5′‐ GAATTCATGTGGGGAGTATTCAGGA‐3′ and reverse primer 5′‐ CTCGAGTTAGGGCTTCCTCTTGGAG ‐3′. The cDNA was then subcloned into the pcDNA3.1(+) (Thermo Fisher Scientific, Waltham, MA, USA) vector with the restriction enzymes EcoR1 and XhoI to establish plasmid over‐expression. Cell transfection with plasmid over‐expression and empty vector was carried out separately in HFLS using lipofectamine 2000 (Invitrogen, Shanghai, China). Small interfering (si) RNA of CDKN1A (siR‐CDKN1A) and si‐NC were purchased from GenScript Co., Ltd (Nanjing, China). The siR‐CDKN1A sequence was 5′‐GUCCUGUGUUCCAACUAUAGU‐3′.
MTT assay
The effect of CDKN1A and miR‐146a on cell proliferation was detected using the 3‐(4,5‐dimethylthiazol‐2‐Yl)‐2,5‐diphenyltetrazolium bromide (MTT) kit (Ruitai Biology Co., Ltd, Beijing, China). HFLS‐RA was inoculated for 24, 48 and 72 h in three identical 96‐well plates at a density of 1 × 103 cells/well; 10 μl MTT reagent was then added to each well at each time‐point, and cultured at 37°C in 5% CO2 for 2 h. The supernatant was then removed, and 100 μl dimethylsulphoxide (DMSO) was added to each well. Optical density (OD) was detected at 570 nm using an enzyme‐linked immune‐monitor.
Transwell assay
The HFLS‐RA migration was determined with Transwell chambers (8‐μm pores) (Corning, New York, NY, USA). HFLS‐RA (1 × 105 cells per chamber) was plated in the upper chambers in duplicate filters. DMEM containing 10% FBS was added to the lower chamber as a chemoattractant. Cells were incubated for 24 h under normoxic conditions. Non‐migrating cells were removed from the upper surface and filters were stained with crystal violet. Migrated cells were counted in five representative microscopic fields.
In order to assess the invasion ability of HFLS‐RA, HFLS‐RA (5 × 104 cells per chamber) were seeded onto Transwell chambers precoated with matrigel. The lower chambers were filled with DMEM medium containing 10% FBS. Cells were allowed to invade for 24 h under normoxic conditions. Cells on the top membrane surface were removed. Cells which had penetrated to the bottom were stained with crystal violet and counted as described above.
Flow cytometry
To synchronize the cells in the conversion period between G0/G1 and G2/M phases, HFLS‐RA were collected after transfection for 48 h and centrifugation. The cells were harvested and washed once with PBS, fixed in ice‐cold 70% ethanol and stored at 4°C. Prior to analysis, the cells were washed once again with PBS, suspended in 1 ml of cold propidium iodide (PI; Sigma, St Louis, MO, USA) solution containing 100 µg/ml RNase A, 50 µg/ml PI, 0·1% (w/v) sodium citrate and 0·1% (v/v) NP‐40, and incubated further on ice for 30 min in the dark. Flow cytometric analyses were carried out using a flow cytometer (FACScalibur; Becton‐Dickinson, San Jose, CA, USA).
Enzyme‐linked immunosorbent assay (ELISA)
HFLS‐RA culture supernatants were collected after transfection and diluted for the following experiment. Concentrations of the relative cytokines of TNF‐α, IL‐6 and IL‐10 were evaluated with commercial ELISA kits (R&D Systems, Lille, France). A standard curve method was performed for each plate and used to calculate the absolute concentrations of the cytokines indicated.
Dual‐luciferase reporter gene assay
Luciferase reporter vector recombinant plasmids inserted with CDKN1A 3′‐untranslated region (3′‐UTR) WT and mutant type (MUT) were constructed, named as MiR‐146a mimics, or control, were used for co‐transfection with CDKN1A‐WT and CDKN1A‐MUT into 293T (BeNa Culture Collection, Beijing, China) using lipofectamine 2000 (Invitrogen). The Luciferase Dual Assay Kit (Thermo Fisher Scientific) was used for dual‐luciferase reporter gene assay 48 h after cells were transfected.
Statistical analysis
GraphPad Prism 6 (GraphPad, La Jolla, CA, USA) software was used for statistical analysis. The data were expressed as average ± standard deviation (AV ± s.d.). Student’s t‐test was used to detect differences between experimental groups. P < 0·05 was considered to be statistically significant.
Results
Screening of DEGs
Gene expression data in the RA and HC groups were analysed and DEGs with adjusted P < 0·01 and antibodies (logFC) > 1 were selected as shown in Supporting information, Table S1 (other DEGs are shown in Supporting information, Gene tables [Link], [Link], [Link], [Link]). In GSE55235, there were 1119 DEGs in the RA group compared with the HC group. Using comparative analysis, 12 common DEGs, including CDKN1A, were found among the four data sets, which might be associated with RA disease (Fig. 1a).
Figure 1.
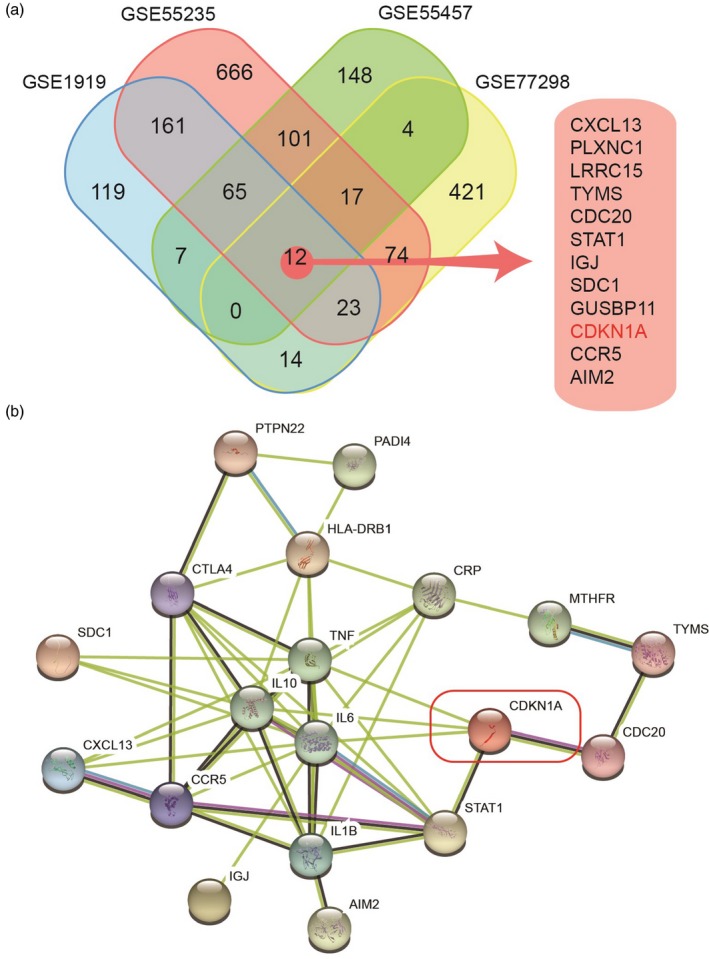
Screening of differentially expressed genes (DEGs) and core genes in rheumatoid arthritis (RA). (a) Analysis of differentially expressed genes in four data sets. Four sets of gene expression data containing the samples in the RA group and the healthy control (HC) group were downloaded from the Gene Expression Omnibus (GEO) database for analysis. Twelve common DEGs, including CDKN1A, were found among the four data sets, which might be associated with RA disease. (b) Analysis of gene–gene interaction was performed between the DEGs and hot genes of RA through the string website. Cluster analysis was carried out using k‐means clustering, and it was found that CDKN1A, signal transducers and activators of transcription 1 (STAT‐1) and C‐C chemokine receptor type 5 (CCR5) were at the core position in the cluster network of RA hot genes.
Core genes in RA
DisGeNET is a free public platform for searching human disease‐related genes and variants. RA‐related hot genes were retrieved from the DisGeNET database with ‘rheumatoid arthritis’ as the keywords. The top 10 hot genes of RA are shown in Table 2.
Table 2.
The top 10 hot genes of rheumatoid arthritis (RA) from the DisGeNET database
| Gene | Gene name | Score | PMIDs |
|---|---|---|---|
| PTPN22 | Protein tyrosine phosphatase, non‐receptor type 22 | 0·567 | 151 |
| TNF | Tumour necrosis factor | 0·506 | 570 |
| HLA‐DRB1 | Major histocompatibility complex, class II, DR beta 1 | 0·44 | 614 |
| IL‐1β | Interleukin‐1 beta | 0·323 | 155 |
| CRP | C‐reactive protein | 0·315 | 75 |
| CTLA‐4 | Cytotoxic T lymphocyte‐associated protein 4 | 0·299 | 61 |
| PADI4 | Peptidyl arginine deiminase 4 | 0·298 | 90 |
| IL‐6 | Interleukin‐6 | 0·297 | 198 |
| MTHFR | Methylenetetrahydrofolate reductase | 0·287 | 53 |
| IL‐10 | Interleukin‐10 | 0·286 | 95 |
Total number of PubMed identifiers (PMIDs) supporting the association.
Analysis of gene–gene interaction was performed between the DEGs and hot genes of RA through the string website. Cluster analysis was carried out using k‐means clustering, and it was found that CDKN1A, STAT‐1 and C‐C chemokine receptor type 5 (CCR5) were at the core position in the cluster network of RA hot genes (Fig. 1b). There are many studies on the role of STAT‐1 and CCR5 in RA, while few studies have been conducted concerning the cell growth‐related gene CDKN1A in RA. However, CDKN1A was associated closely with RA hot genes, which indicated that CDKN1A may be a potential influencing factor in RA.
CDKN1A expression was decreased in RA tissues and HFLS
CDKN1A expression was detected in clinical samples. Consistent with the results of bioinformatics analysis, qRT–PCR detection validated that CDKN1A mRNA was decreased significantly in the RA group compared with the HC group (Fig. 2a). The levels of CDKN1A mRNA and protein in HFLS were also detected in our experiments. Our results showed that the level of CDKN1A was inhibited in the HFLS‐RA group (Fig. 2b). Western blot assay illustrated that, compared with the HFLS group, the CDKN1A protein level was also inhibited in the HFLS‐RA group (Fig. 2c).
Figure 2.
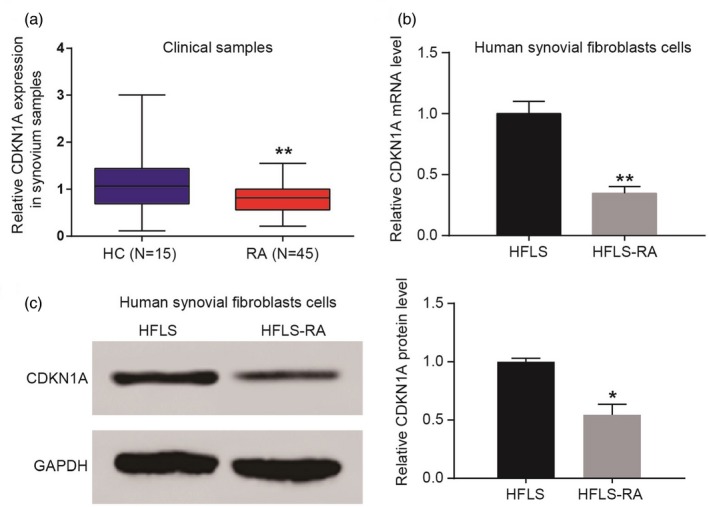
Cyclin‐dependent kinase inhibitor 1A (CDKN1A) expression in rheumatoid arthritis (RA) tissues and human fibroblast‐like synoviocytes (HFLS). (a) CDKN1A mRNA was decreased significantly in the RA group compared with the healthy control (HC) group. (b) The level of CDKN1A was inhibited in the HFLS‐RA group. (c) Compared with the HFLS group, the CDKN1A protein level was also inhibited in the HFLS‐RA group. *P < 0·05; **P < 0·01.
CDKN1A inhibited the proliferation, migration and invasion of HFLS‐RA and hindered the cell cycle of HFLS‐RA
The effect of CDKN1A on the proliferation of HFLS‐RA was investigated in this study. In a scramble group, both CDKN1A+ and CDKN1A– were added. The HFLS‐RA group was named the ‘NC group’. MTT assay found that the growth rate in the CDKN1A+ group was significantly lower than that in the scramble and NC groups. When compared with the scramble and NC groups, the growth rate in the CDKN1A– group was increased significantly (P < 0·05), and the difference in cell proliferation was greater with the prolongation of culture time (Fig. 3a). Flow cytometry illustrated that the majority of cells in the CDKN1A+ group were arrested in the G1/S phase compared with the scramble and NC groups, which suggested that cell division was hindered by CDKN1A over‐expression (Fig. 3b). The results of the Transwell assay showed that compared with the scramble and NC groups, migration and invasion of HFLS‐RA were increased significantly by CDKN1A silencing and clearly inhibited by CDKN1A over‐expression (Fig. 3c).
Figure 3.
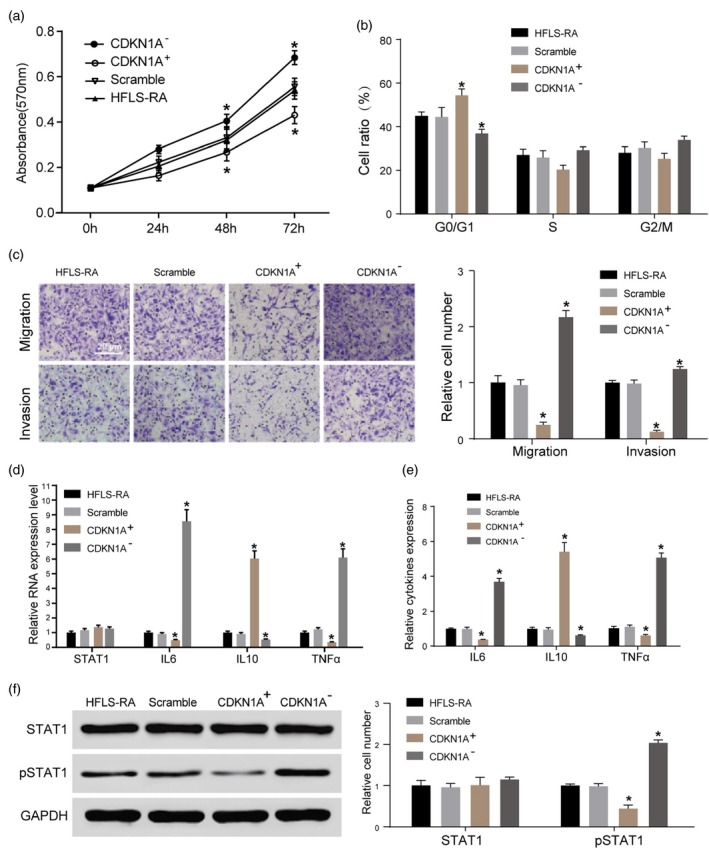
The role of cyclin‐dependent kinase inhibitor 1A (CDKN1A) in proliferation, migration and invasion of human fibroblast‐like synoviocytes rheumatoid arthritis (HFLS‐RA). (a) Compared with the scramble and NC groups, the growth rate in the CDKN1A– group was increased significantly (*P < 0·05), and the difference in cell proliferation was greater with the prolongation of culture time. (b) The majority of cells in the CDKN1A+ group were arrested in the G1/S phase compared with the scramble and NC groups. (c) Compared with the scramble and NC groups, migration and invasion of HFLS‐RA were increased significantly by CDKN1A silencing and inhibited clearly by CDKN1A over‐expression. (d) The expression of tumour necrosis factor (TNF)‐α and interleukin (IL)‐6 was lower and the expression of IL‐10 was higher to different degrees after over‐expressing CDKN1A in HFLS‐RA (*P < 0·05; **P < 0·01). There was no obvious change in the level of signal transducers and activators of transcription 1 (STAT‐1). (e) Compared with the negative siRNA scramble cell line, the expression of TNF‐α and IL‐6 was higher and the level of IL‐10 was lower after CDKN1A silencing. (f) Over‐expression of CDKN1A reduced the protein expression of phosphorylated STAT‐1 (pSTAT‐1) significantly, while CDKN1A silencing increased it. *P < 0·05.
CDKN1A inhibited the expression of TNF‐α and IL‐6 and promoted the expression of IL‐10 in HFLS‐RA
We detected mRNA expression of TNF‐α, IL‐6, IL‐10 and STAT‐1 in the four groups. It was found that the expression of TNF‐α and IL‐6 was attenuated and IL‐10 expression was enhanced to different degrees after over‐expressing CDKN1A in HFLS‐RA (P < 0·05, P < 0·01). There was no obvious change in the level of STAT‐1 (Fig. 3d). When compared with the negative siRNA scramble cell line, CDKN1A silencing promoted the expression of TNF‐α and IL‐6 and attenuated the level of IL‐10 (Fig. 3e). Western blot results illustrated that over‐expression of CDKN1A inhibited the protein expression of pSTAT‐1 significantly, while CDKN1A silencing promoted pSTAT‐1 expression (Fig. 3f).
MiR‐146a targeted CDKN1A directly and correlated negatively with CDKN1A
To explore the role of miR‐146a in RA, we detected the expression of miR‐146a in clinical samples. Relative miR‐146a expression in synovium samples was higher in the RA group compared with the HC group (Fig. 4a). Relative miR‐146a expression and relative CDKN1A expression showed a negative correlation in synovium samples (R 2 = 0·8105, *P < 0·05) (Fig. 4b). The level of miR‐146a was also found to be higher in HFLS‐RA than that in HFLS (Fig. 4c). Dual‐luciferase reporter gene assay results indicated that luciferase activity only decreased in the CDKN1‐WT + miR‐146a group, which corroborated that miR‐146a regulated CDKN1A directly (Fig. 4d). After transfection with anti‐146a the level of miR‐146a was suppressed in HFLS‐RA cells (Fig. 4e), while the expression of CDKN1A mRNA was promoted (Fig. 4f). The expression of CDKN1A protein was up‐regulated in the anti‐146a group, while anti‐146a + CDKN1A– could rescue the effect of anti‐146a (Fig. 4g).
Figure 4.
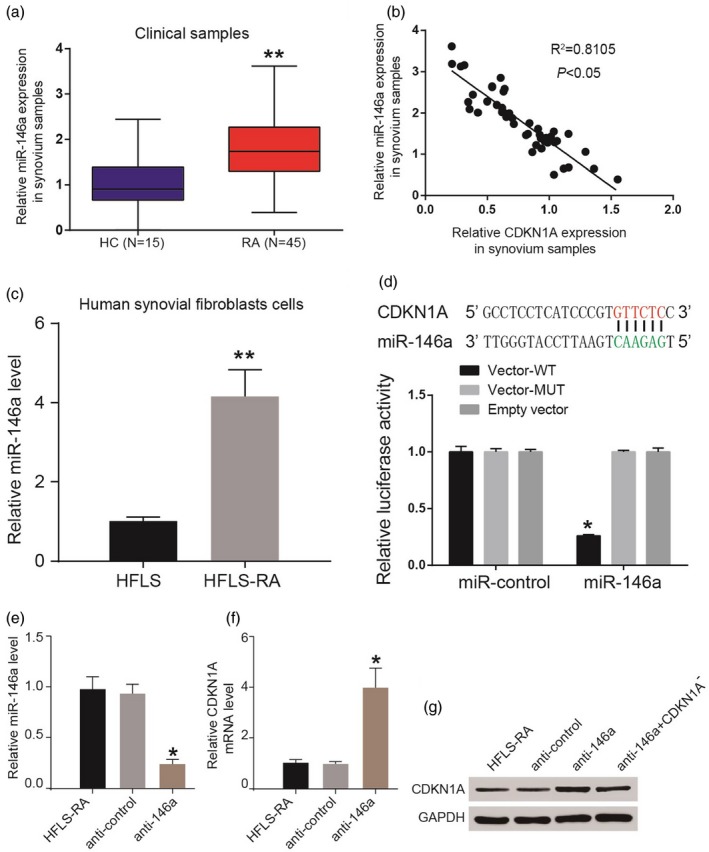
MiR‐146a directly targeted cyclin‐dependent kinase inhibitor 1A (CDKN1A). (a) Relative miR‐146a expression in synovium samples was higher in the rheumatoid arthritis (RA) group compared with the healthy control (HC) group. (b) Relative miR‐146a expression and relative CDKN1A expression showed a negative correlation in synovium samples. (c) The level of miR‐146a was also found higher in human fibroblast‐like synoviocytes (HFLS)‐RA than that in HFLS. (d) Luciferase activity decreased only in the CDKN1‐wild‐type (WT)+ miR‐146a group. (e) After transfected with anti‐146a, the level of miR‐146a was suppressed in HFLS‐RA. (f) The expression of CDKN1A mRNA was promoted. (g) The expression of CDKN1A protein was up‐regulated in the anti‐146a group while anti‐146a+ CDKN1A– reversed it. *P < 0·05; **P < 0·01.
Inhibition of miR‐146a reversed the effects of CDKN1A silencing on HFLS‐RA
We investigated the effects of miR‐146a on HFLS‐RA. Anti‐146a + CDKN1– was named the ‘scramble group’ for convenience and HFLS‐RA was named the ‘NC group’. The growth rate in the anti‐146a group showed clear down‐regulation compared with the scramble and NC groups, while it was reversed in the anti‐146a + CDKN1A– group. The difference in cell proliferation was greater with the prolongation of culture time (Fig. 5a). Compared with the scramble and NC groups, cells in the anti‐146a group were arrested mainly in the G1/S phase while anti‐146a + CDKN1A– reversed it (Fig. 5b). Results of the Transwell assay showed that compared with the scramble and NC groups, migration and invasion of HFLS‐RA were inhibited significantly in the anti‐146a group and clearly reversed in the anti‐146a + CDKN1A– group (Fig. 5c). We further detected the mRNA expression of TNF‐α, IL‐6, IL‐10 and STAT‐1 in HFLS‐RA. It was shown that expression of TNF‐α and IL‐6 was suppressed in the anti‐146a group, while expression of IL‐10 was promoted. These influences were reversed significantly in the anti‐146a + CDKN1A– group (Fig. 5d,e). The protein levels of STAT‐1 and pSTAT‐1 were detected in our study. We found a significant inhibition of pSTAT‐1 protein in the anti‐146a group and there was no significant difference in STAT‐1 protein. Anti‐146a + CDKN1A– reversed the inhibition of pSTAT‐1 protein in HFLS‐RA (Fig. 5f).
Figure 5.
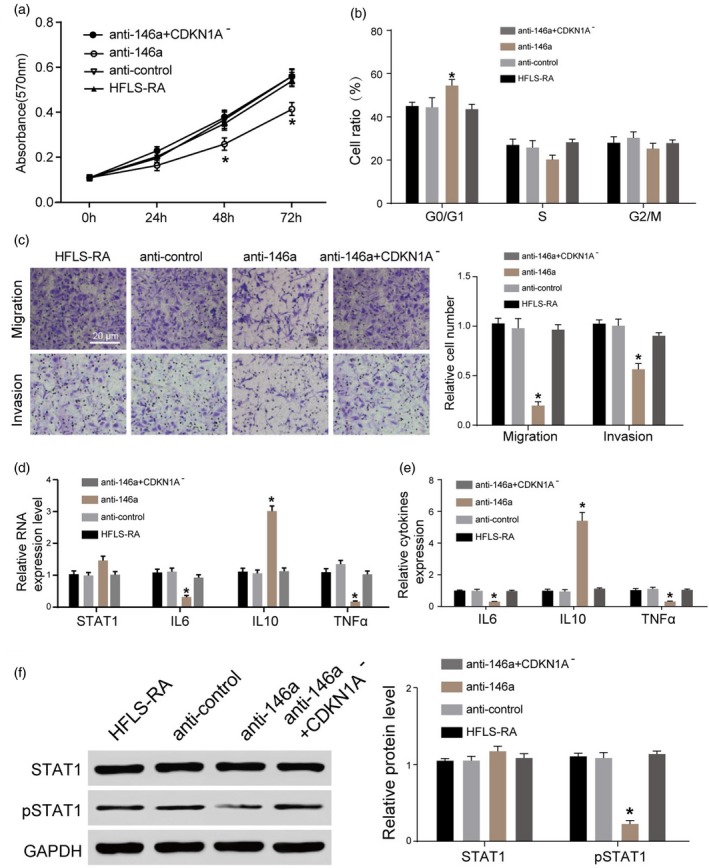
Inhibition of miR‐146a reversed the effects of cyclin‐dependent kinase inhibitor 1A (CDKN1A) silencing on human fibroblast‐like synoviocytes rheumatoid arthritis (HFLS‐RA). (a) The growth rate in the anti‐146a group showed an obvious down‐regulation compared with the scramble and NC groups, while it was reversed in the anti‐146a+ CDKN1A– group. (b) The cells in the anti‐146a group were arrested mainly in the G1/S phase, while anti‐146a+ CDKN1A– reversed it. (c) Migration and invasion of HFLS‐RA was inhibited significantly in the anti‐146a group and clearly reversed in anti‐146a+ CDKN1A–. (d,e) The expression of tumour necrosis factor (TNF)‐α and interleukin (IL)‐6 was suppressed in the anti‐146a group, while the expression of IL‐10 was promoted. These influences were reversed significantly in the anti‐146a+ CDKN1A– group. (f) The expression level of phosphorylated signal transducer and activator of transcription 1 (pSTAT‐1) protein was lower in the anti‐146a group and there was no significant difference in STAT‐1 protein. Anti‐146a+ CDKN1A– reversed the inhibition of pSTAT‐1 protein in HFLS‐RA. *P < 0·05.
Discussion
In this study, GEO data and the string database indicated CDKN1A as an important DEG gene in RA tissues. CDKN1A was expressed sparsely in RA tissues and HFLS cells; high expression suppressed cell proliferation, migration and invasion via inflammatory cytokines suppression. MiR‐146a was an upstream regulator of CDKN1A and its suppression, which resulted in CDKN1A enhancement, finally suppressing HFLS cell motility.
CDKN1A was important in the regulation of various tumours. Previous study has verified the obvious change of CDKN1A in RA patients. Spurlock et al. indicated that RA patients expressed reduced baseline levels of CDKN1A, CDKN1B and CHEK2 mRNAs 16. Furthermore, various studies report that CDKN1A participates in the proliferation and migration of tumour cells under the direct or indirect regulation of microRNA or long non‐coding RNA (lncRNA). Suppressed p21 transcription inhibits fibroblast‐like synoviocytes from RA patients 17. Elevating inflammatory cytokine expression enhances HFLS proliferation 18. This study confirmed that CDKN1A inhibited the proliferation of HFLS‐RA, arresting the cell cycle in the G1/S phase, and its over‐expression could inhibit the invasion and migration of HFLS‐RA. Our results are consist with previous reports. CDKN1A was also regulated negatively by miR‐146a, a novel discovery in this study.
HFLS are a primary component of the hyperplastic synovial pannus. In the occurrence of RA, cytokine TNF‐α and interleukin IL‐1 activate HFLS. Once activated, HFLS secrete IL‐6, chemokines mediate the adjacent cells to cause inflammatory infiltration and endothelial cells secrete MMPs to stimulate cartilage degradation 19, 20, 21, 22. Studies have found that IL‐6 could promote HFLS to produce a variety of cytokines to participate in the development of inflammation 23, stimulate the proliferation and activation of T lymphocytes, activate osteoclasts and regulate the formation of acute‐phase proteins 24, 25. In this study, TNF‐α and IL‐6 were down‐regulated to different degrees after over‐expressing CDKN1A, while IL‐10 was up‐regulated. Based on previous studies and the results of this study, it was concluded that high concentrations of TNF‐α in the extracellular matrix activated HFLS‐RA to induce the secretion of IL‐6, which mediated cell inflammation.
There are limitations to this study. For example, in‐vivo experiments could be performed to validate the discovered mechanism. Exact cells, such as B, T or macrophages which produced CDKN1A, could be ascertained. Furthermore, tissue samples and blood samples could all be examined for differentially expressed genes for better‐targeted therapies.
In conclusion, our study demonstrates that CDKN1A regulates negatively the proliferation and invasion of HFLS‐RA and the expression of TNF‐α and IL‐6 in HFLS‐RA and regulates positively the expression of IL‐10. We also indicate that miR‐146a regulates CDKN1A directly and has a negative correlation with CDKN1A, which means that miR‐146a plays a positive role in HFLS‐RA.
Disclosure
There is no financial or commercial conflicts of interests.
Supporting information
Table S1. Gene expression data in the rheumatoid arthritis (RA) and healthy control (HC) groups were analysed and differentially expressed genes (DEGs) with adjusted P < 0·01 and antibodies (logFC) > 1.
Gene table S1. 1igGene_RA_vs_HC_GSE1019.csv
Gene table S2. 2gGene_RA_vs_HC_GSE55235.csv
Gene table S3. 3SigGene_RA_vs_HC_GSE55457.csv
Gene table S4. igGene_RA_vs_HC_GSE77298.csv
Acknowledgements
We would like to give our sincere appreciation to the reviewers for their helpful comments on this paper.
References
- 1. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet 2010; 376:1094–108. [DOI] [PubMed] [Google Scholar]
- 2. Svarts N. Etiology and pathogenesis of rheumatoid arthritis. Ter Arkh 1975; 47:19–24. [PubMed] [Google Scholar]
- 3. Firestein GS. Invasive fibroblast‐like synoviocytes in rheumatoid arthritis. Passive responders or transformed aggressors? Arthritis Rheum 1996; 39:1781–90. [DOI] [PubMed] [Google Scholar]
- 4. Meinecke I, Rutkauskaite E, Gay S, Pap T. The role of synovial fibroblasts in mediating joint destruction in rheumatoid arthritis. Curr Pharm Des 2005; 11:563–8. [DOI] [PubMed] [Google Scholar]
- 5. Angiolilli C, Kabala PA, Grabiec AM et al Histone deacetylase 3 regulates the inflammatory gene expression programme of rheumatoid arthritis fibroblast‐like synoviocytes. Ann Rheum Dis 2017; 76:277–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc Natl Acad Sci USA 1997; 94:10895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Roivainen A, Jalava J, Pirila L, Yli‐Jama T, Tiusanen H, Toivanen P. H‐ras oncogene point mutations in arthritic synovium. Arthritis Rheum 1997; 40:1636–43. [DOI] [PubMed] [Google Scholar]
- 8. Chen DP, Wong CK, Tam LS, Li EK, Lam CW. Activation of human fibroblast‐like synoviocytes by uric acid crystals in rheumatoid arthritis. Cell Mol Immunol 2011; 8:469–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bartek J, Lukas J. Pathways governing G1/S transition and their response to DNA damage. FEBS Lett 2001; 490:117–22. [DOI] [PubMed] [Google Scholar]
- 10. Bianco S, Jangal M, Garneau D, Gevry N. LRH‐1 controls proliferation in breast tumor cells by regulating CDKN1A gene expression. Oncogene 2015; 34:4509–18. [DOI] [PubMed] [Google Scholar]
- 11. Shao M, Geng Y, Lu P et al miR‐4295 promotes cell proliferation and invasion in anaplastic thyroid carcinoma via CDKN1A. Biochem Biophys Res Commun 2015; 464:1309–13. [DOI] [PubMed] [Google Scholar]
- 12. Ohta K, Hoshino H, Wang J et al MicroRNA‐93 activates c‐Met/PI3K/Akt pathway activity in hepatocellular carcinoma by directly inhibiting PTEN and CDKN1A. Oncotarget 2015; 6:3211–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saiz‐Ladera C, Lara MF, Garin M et al p21 suppresses inflammation and tumorigenesis on pRB‐deficient stratified epithelia. Oncogene 2014; 33:4599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gamboni F, Anderson C, Mitra S et al Hypertonic saline primes activation of the p53–p21 signaling axis in human small airway epithelial cells that prevents inflammation induced by pro‐inflammatory cytokines. J Proteome Res 2016; 15:3813–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mavers M, Cuda CM, Misharin AV et al Cyclin‐dependent kinase inhibitor p21, via its C‐terminal domain, is essential for resolution of murine inflammatory arthritis. Arthritis Rheum 2012; 64:141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Spurlock CF, 3rd, Tossberg JT, Fuchs HA, Olsen NJ, Aune TM. Methotrexate increases expression of cell cycle checkpoint genes via JNK activation. Arthritis Rheum 2012; 64:1780–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fan L, Zong M, Gong R et al PADI4 epigenetically suppresses p21 transcription and inhibits cell apoptosis in fibroblast‐like synoviocytes from rheumatoid arthritis patients. Int J Biol Sci 2017; 13:358–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ni S, Miao K, Zhou X et al The involvement of follistatin‐like protein 1 in osteoarthritis by elevating NF‐kappaB‐mediated inflammatory cytokines and enhancing fibroblast like synoviocyte proliferation. Arthritis Res Ther 2015; 17:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Berckmans RJ, Nieuwland R, Kraan MC et al Synovial microparticles from arthritic patients modulate chemokine and cytokine release by synoviocytes. Arthritis Res Ther 2005; 7:R536–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mor A, Abramson SB, Pillinger MH. The fibroblast‐like synovial cell in rheumatoid arthritis: a key player in inflammation and joint destruction. Clin Immunol 2005; 115:118–28. [DOI] [PubMed] [Google Scholar]
- 21. Miyazawa K, Mori A, Miyata H, Akahane M, Ajisawa Y, Okudaira H. Regulation of interleukin‐1beta‐induced interleukin‐6 gene expression in human fibroblast‐like synoviocytes by p38 mitogen‐activated protein kinase. J Biol Chem 1998; 273:24832–8. [DOI] [PubMed] [Google Scholar]
- 22. Miyazawa K, Mori A, Yamamoto K, Okudaira H. Constitutive transcription of the human interleukin‐6 gene by rheumatoid synoviocytes: spontaneous activation of NF‐kappaB and CBF1. Am J Pathol 1998; 152:793–803. [PMC free article] [PubMed] [Google Scholar]
- 23. Emery P, Keystone E, Tony HP et al IL‐6 receptor inhibition with tocilizumab improves treatment outcomes in patients with rheumatoid arthritis refractory to anti‐tumour necrosis factor biologicals: results from a 24‐week multicentre randomised placebo‐controlled trial. Ann Rheum Dis 2008; 67:1516–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Takeda K, Kaisho T, Yoshida N, Takeda J, Kishimoto T, Akira S. Stat3 activation is responsible for IL‐6‐dependent T cell proliferation through preventing apoptosis: generation and characterization of T cell‐specific Stat3‐deficient mice. J Immunol 1998; 161:4652–60. [PubMed] [Google Scholar]
- 25. Scheid C, Young R, McDermott R, Fitzsimmons L, Scarffe JH, Stern PL. Immune function of patients receiving recombinant human interleukin‐6 (IL‐6) in a phase I clinical study: induction of C‐reactive protein and IgE and inhibition of natural killer and lymphokine‐activated killer cell activity. Cancer Immunol, Immunother: CII 1994; 38:119–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Gene expression data in the rheumatoid arthritis (RA) and healthy control (HC) groups were analysed and differentially expressed genes (DEGs) with adjusted P < 0·01 and antibodies (logFC) > 1.
Gene table S1. 1igGene_RA_vs_HC_GSE1019.csv
Gene table S2. 2gGene_RA_vs_HC_GSE55235.csv
Gene table S3. 3SigGene_RA_vs_HC_GSE55457.csv
Gene table S4. igGene_RA_vs_HC_GSE77298.csv