

Summary
Dipeptidyl peptidase 4 (DPP4, CD26) is a serine protease that is expressed constitutively by many haematopoietic and non‐haematopoietic tissues. It exists as a membrane‐associated protein, as well as in an active, soluble form (herein called sDPP4), present at high concentrations in bodily fluids. Despite the proposed use of sDPP4 as a biomarker for multiple diseases, its cellular sources are not well defined. Here, we report that individuals with congenital lymphocyte immunodeficiency had markedly lower serum concentrations of sDPP4, which were restored upon successful treatment and restoration of lymphocyte haematopoiesis. Using irradiated lymphopenic mice and wild‐type to Dpp4–/– reciprocal bone marrow chimeric animals, we found that haematopoietic cells were a major source of circulating sDPP4. Furthermore, activation of human and mouse T lymphocytes resulted in increased sDPP4, providing a mechanistic link between immune system activation and sDPP4 concentration. Finally, we observed that acute viral infection induced a transient increase in sDPP4, which correlated with the expansion of antigen‐specific CD8+ T cell responses. Our study demonstrates that sDPP4 concentrations are determined by the frequency and activation state of lymphocyte populations. Insights from these studies will support the use of sDPP4 concentration as a biomarker for inflammatory and infectious diseases.
Keywords: chemokines, immunodeficiency diseases, T cells
Introduction
Dipeptidyl peptidase 4 (DPP4, CD26, EC 3.4.14.5) is a multi‐functional protein involved in the regulation of both physiological and pathological responses. DPP4 has many reported functions, including protease activity, direct interaction with adenosine deaminase (ADA), cell surface co‐receptor for viral entry and regulation of intracellular signal transduction in T cells 1, 2, 3. Cell‐associated DPP4 has a molecular weight of 110 kDa and is active as a homodimer with an extracellular enzymatic protease domain and a short cytoplasmic region 4. Both soluble and membrane‐bound DPP4 cleave dipeptides from the N‐terminus of proteins that contain a proline or alanine in the penultimate position. DPP4 substrates include chemokines, neuropeptides and incretin hormones 5, 6, 7, 8, 9. The functional consequences of DPP4‐mediated post‐translational modification may vary among substrates, but notably DPP4 cleavage eliminates the biological activity of the incretin hormone glucagon‐like protein 1 (GLP‐1). This observation led to the development of DPP4 inhibitors that were established successfully as therapeutic agents in type 2 diabetes patients, as they prevent DPP4‐mediated inactivation of GLP‐1, and thus enhance cellular insulin sensitivity by prolonging the half‐life of GLP‐1 10.
Human DPP4 forms a molecular complex with ADA, which is a cell surface enzyme, catalyzing the irreversible deamination of adenosine 11. ADA–DPP4 interactions act as co‐stimulatory signals during T cell receptor signalling, resulting in enhanced secretion of interferon gamma (IFNγ), tumor necrosis factor alpha (TNFα) and interleukin 6 (IL‐6) 2, 12. Recent studies have shown that DPP4 can also regulate T cell migration negatively through cleavage of proinflammatory chemokines, such as C‐X‐C motif chemokine 10 (CXCL10) 13, 14, 15. Our previous work demonstrated that inhibition of DPP4 results in the protection of the active, full‐length form of CXCL10 in healthy individuals and hepatitis C patients 16. This mechanism of CXCL10 protection is relevant in the context of preclinical cancer models, as it increases chemokine‐mediated T cell migration into the tumour parenchyma, improving tumour immunity and immunotherapy 15, 17.
DPP4 is expressed in immune cells such as T lymphocytes, and is also found in non‐ haematopoietic tissues such as the kidney epithelium, prostate, lung, liver and small intestine 18, 19, 20. Moreover, a soluble active form of DPP4 (sDPP4) is also present in many biological fluids such as plasma, urine, bile and semen 21. Notably, variations in the concentration and activity of sDPP4 have been associated with many diseases, and these parameters are considered informative biomarkers for solid and haematological cancers, autoimmune disorders and chronic infectious diseases such as hepatitis C virus (HCV), where high plasma levels of sDPP4 are associated positively with liver fibrosis 22, 23, 24, 25, 26. Highlighting its importance in clinical development programmes, sDPP4 is under evaluation as a predictive marker in anti‐IL‐13 therapeutic trials for the treatment of asthma 27. Strikingly, despite its apparent utility as a biomarker, the mechanisms mediating DPP4 release from cells are poorly defined, and may involve shedding and/or secretion 28. Importantly, the cellular origin of sDPP4 is also not well defined.
Herein, we report that lymphocytes are a major source of sDPP4, and that T cell activation accounts for the dynamic changes observed in its plasma concentration. This discovery followed from our hypothesis that patients with ADA deficiency would have perturbed sDPP4 expression, but observed instead that lower levels were found in all types of severe combined immunodeficiency (SCID) patients. In both humans and experimental mouse models, sDPP4 concentration and activity correlated with the number of circulating lymphocytes. Reciprocal bone marrow chimera experiments, using wild‐type (WT) and Dpp4–/– mice, demonstrated that bone marrow haematopoietic cells were the major source of plasma sDPP4. Finally, mouse and human T cell stimulation, in the context of in‐vivo infection or in‐vitro T cell receptor (TCR) cross‐linking, respectively, induced release of sDPP4. Together, these findings support that sDPP4 may serve as a surrogate for the number and activation state of lymphocytes in health and disease.
Methods
Human plasma collection and ethical consideration
Human bioresources (serum and plasma samples and associated data such as age and health status) were collected from healthy volunteers of the Investigation Clinique et Accès aux Resources Biologiques (ICAReB) platform (Centre de Recherche Translationnelle, Institut Pasteur, Paris, France) as part of the Diagmicoll protocol, which has been approved by the French Ethical Committee (CPP) Ile‐de‐France. Peripheral blood from SCID patients diagnosed by genetic analysis was obtained after informed consent on the occasion of other blood testing in the context of a research protocol at Ospedale Pediatrico Bambino Gesù, Roma, Italy. The research protocol was approved by the Institutional Ethical Committee of Ospedale Pediatrico Bambino Gesù and informed consent forms were signed by all subjects' parents or their legal guardian.
ADA–SCID patients and treatment
ADA–SCID patients refer to San Raffaele Hospital or patients' parents signed informed consent on anonymized data collection for research studies conducted at San Raffaele Hospital (Tiget02). A portion of the analyses, shown herein, was performed as a baseline evaluation for patients who subsequently underwent further treatment with pegademase bovine (PEG)–ADA, bone marrow transplantation (BMT) or haematopoietic stem cell gene therapy (HSC‐GT). PEG–ADA‐treated and ADA–SCID patients received 10–80 U/kg/week Adagen (PEG; Enzon Pharmaceuticals, Piscataway, NJ, USA). BMT ADA–SCID patients received bone marrow transplantation as described previously 29. Patients with ADA–SCID undergoing HSC‐GT were enrolled in Phases I/II clinical protocols approved by the San Raffaele Scientific Institute's Ethical Committee and Italian National Regulatory Authorities. Gene therapy treatment was performed as described previously 30. Data from patients undergoing HSC‐GT were collected from 2008 to 2011. Since April 2012, GlaxoSmithKline has been a sponsor of the ADA–SCID long‐term follow‐up trial no. 115611 (HSC‐GT) conducted at Tiget.
sDPP4 ELISA
DPP4 quantification was performed with the human or mouse DPPIV/CD26 DuoSet ELISA kit (R&D Systems). Human plasma samples were diluted 1/1000 or 1/2000; mouse plasma sample were diluted 1/100 and 1/500 by serial dilution. Culture supernatants were diluted twofold. All samples were tested in duplicate, following the manufacturer's instructions. Plates were read with a Labsystems Multiskan MS (Thermo Fisher Scientific, Waltham, MA, USA) device set at 450 nm. Eight‐point standard curves, using twofold serial dilutions with a high standard of 4000 pg/ml, were performed in duplicate.
DPP4 activity in plasma samples
DPP4 activity was measured with the DPPIV‐Glo protease Assay (Promega, Madison, WI, USA). This assay provides a luminogenic DPP substrate, Gly‐Pro‐aminoluciferin. After cleavage of the proximal two amino acids from the substrate by sDPP4, the aminoluciferin engages luciferase. Briefly, in a white plate (Greiner Bio‐One, Monroe, NC, USA), 50 μl of kit reagent containing the luciferase substrate was added to 50 μl of sample diluted in 10 mM Tris‐HCl pH 8, 0·1% prionex (Calbiochem, San Diego, CA, USA). Maximal signal is reached within 30 min and is stable for 3 h. Recombinant human DPP4 (Sigma D4943; Sigma Aldrich, St Louis, MO, USA) was used as a reference. A seven‐point standard curve, using twofold serial dilutions with a high standard of 125 ng/ml, was performed in duplicate. DPP activity was expressed in units/ml based on supplier information after subtraction of the background signal [phosphate‐buffered saline (PBS) or medium only]. Supplier unit definition was: one unit will produce 1·0 micromole of P‐nitroaniline from Gly‐Pro‐P‐nitroanilide/min at pH 7·6 at 37°C. Samples were tested in duplicate with the same range of dilutions as reported above for ELISA. Plates were read in a Tristar LB941 device (Berthold Technologies, Bad Wildbad, Germany).
Animal experiments
All experimental protocols were approved by the Comité d'Éthique pour l'Expérimentation Animale (Institut Pasteur, Paris, France), protocol number: B 75 15‐06. Experimental animals had access to food and water ad libitum. Mice were 7–12 weeks old and were allowed to acclimate for 1 week prior to any manipulation. Mice were anaesthetized with 100 mg/kg ketamine and 5 mg/kg xylazine. WT C57BL/6 CD45.1, Rag2–/–, Rag2–/–γc–/– and Dpp4–/– (CD45.2) mice on a C57BL/6 strain background were bred in the animal facility of Institut Pasteur. Wild‐type C57BL/6 CD45.2 mice were purchased from Charles River Laboratories (Wilmington, MA, USA). All mice were specifically pathogen‐free; 100 μl of blood was collected by submandibular bleed into tubes containing 10 μl of 100 mM ethylenediamine tetraacetic acid (EDTA) at indicated time‐points.
Chimeric mouse experiments
Mice were exposed to a single lethal dose of 9 gray gamma irradiation. To generate chimeric animals, 3 × 106 bone marrow cells were injected intravenously in a volume of 100 μl into recipient mice 5–6 h after irradiation. Mice were bled by submandibular bleed at the indicated time‐points. Fifty μl of blood was collected into tubes containing 10 μl of 100 mM EDTA for evaluation of haematopoietic cell reconstitution and 50 μl of blood was collected with heparin‐coated capillaries for evaluation of plasma‐associated sDPP4 concentration and activity. Mice differing in their congenic CD45 allele (CD45.1 versus CD45.2) were used to track host and recipient cells.
Influenza infection
Mice were injected intraperitoneally (i.p.) with 3 × 105 haemagglutinating unit (HAU) of influenza A/PR/8/34 (Charles River Laboratories) resuspended in 100 μl of sterile PBS. As a control, mice were injected i.p. with 100 μl of PBS. Seven days after infection 50 μl of blood was collected from each mouse and circulating influenza virus‐specific CD8+ T lymphocytes were quantified by flow cytometry using tetramers specific to nucleoprotein (NP) and polymerase protein (PA) antigens (see below for details).
Flow cytometry of whole blood and absolute cell counts
Fluorochrome‐conjugated anti‐mouse CD45.1 (clone 20), CD45.2 (clone 104), CD3 (clone145‐2C11), CD8 (clone 53‐ 6.7) and CD4 (clone RM4‐5) were from eBiosciences (San Diego, CA, USA) and BD Biosciences (San Jose, CA, USA). Tetrameric complexes of H‐2Db/influenza PA224–233 and H‐2Db/influenza NP366–374 were made in house. Monomers were prepared using a modified version of that described and tetramerization was performed prior to use with R‐phycoerythrin (PE) or allophycocyanin (APC)‐conjugated streptavidin (Invitrogen, Carlsbad, CA, USA) 31. Reagents were combined to assess the total PA and NP‐specific T cells present in mice. For determination of absolute cell numbers of immune cells by cytometry, 10 μl of AccuCheck beads (Invitrogen) were added to 10 µl of EDTA‐treated whole blood incubated into 200 µl of Fix/Lyse buffer (eBiosciences) prior to acquisition. To identify cell populations, blood was incubated with 1·6% NH4Cl to lyse red blood cells. Cells were then incubated with fluorescence activated cell sorter (FACS) buffer (PBS, 2% fetal calf serum, 0·01% azide), FcBlock (BD Biosciences) and subsequently stained with specific antibodies. Samples were acquired on a BD LSRFortessa using diva software (Becton Dickinson) and data were analysed by FlowJo software (Tree Star Inc., Ashland, OR, USA).
Peripheral blood mononuclear cell (PBMC) stimulation
Human PBMCs were purified from fresh whole blood [Etablissement Français du Sang (EFS), Paris] by Ficoll–Paque Plus (GE Healthcare, Little Chalfont, UK) density gradient separation. Two million PBMCs were stimulated in a 48‐well plate in 500 μl of AIMV medium (Invitrogen). Anti‐CD3 antibody (clone HIT3a) (BD Biosciences) was used at a final concentration of 0·5 μg/ml. Superantigen toxic shock syndrome toxin 1 (TSST1), kindly given by the laboratory of Andres Alcover (Institut Pasteur), were used at a final concentration of 0·2 μg/ml. Brefeldin A (BFA; Sigma Aldrich) was used at a final concentration of 5 μg/ml.
Statistical analyses
Statistical analyses were performed using GraphPad Prism version 6.0 software. The Mann‐Whitney U‐test was used to determine whether a difference existed between two groups of individuals. The Kruskal–Wallis test, with Dunn's multiple comparison post‐test, was used to compare samples in experiments with three or more groups. A P‐value < 0·05 was considered to be statistically significant. Spearman's correlation (r s) was determined for analysis in association studies.
Results
SCID patients have reduced levels of sDPP4
It was demonstrated previously that the ADA enzyme (ecto‐ADA) binds to DPP4 in human cells 2, 32. These observations led us to hypothesize that a genetic defect in ADA might impact DPP4 protein turnover at the surface of DPP4‐expressing cells, such as T lymphocytes. To test this prediction, we measured the concentration and activity of sDPP4 in serum samples from ADA‐deficient patients. The ADA‐deficient cohort was composed of untreated (ADA UT) and treated patients (ADA TR) who were stratified for the analysis. The serum of healthy, aged‐matched subjects (healthy) and patients with severe combined immunodeficiency (SCID) caused by genetic defects other than ADA deficiency were analysed as comparator groups. While sDPP4 concentration and enzymatic activity were reduced by 32 and 29%, respectively, in untreated ADA patients compared to healthy controls, the differences were not statistically significant (Fig. 1a,b). Notably, in treated ADA patients, sDPP4 concentrations were restored to levels similar to those observed in healthy donors. We were surprised, however, to observe that sDPP4 concentration and DPP4 activity in non‐ADA SCID patients were also significantly lower than in healthy donors, with an absolute reduction of 58 and 47%, respectively. These results suggested that ADA expression did not account for reduced sDPP4 levels in immune‐deficient patients. Instead, these data indicated that the presence versus relative absence of lymphocytes might determine circulating sDPP4 concentration.
Figure 1.
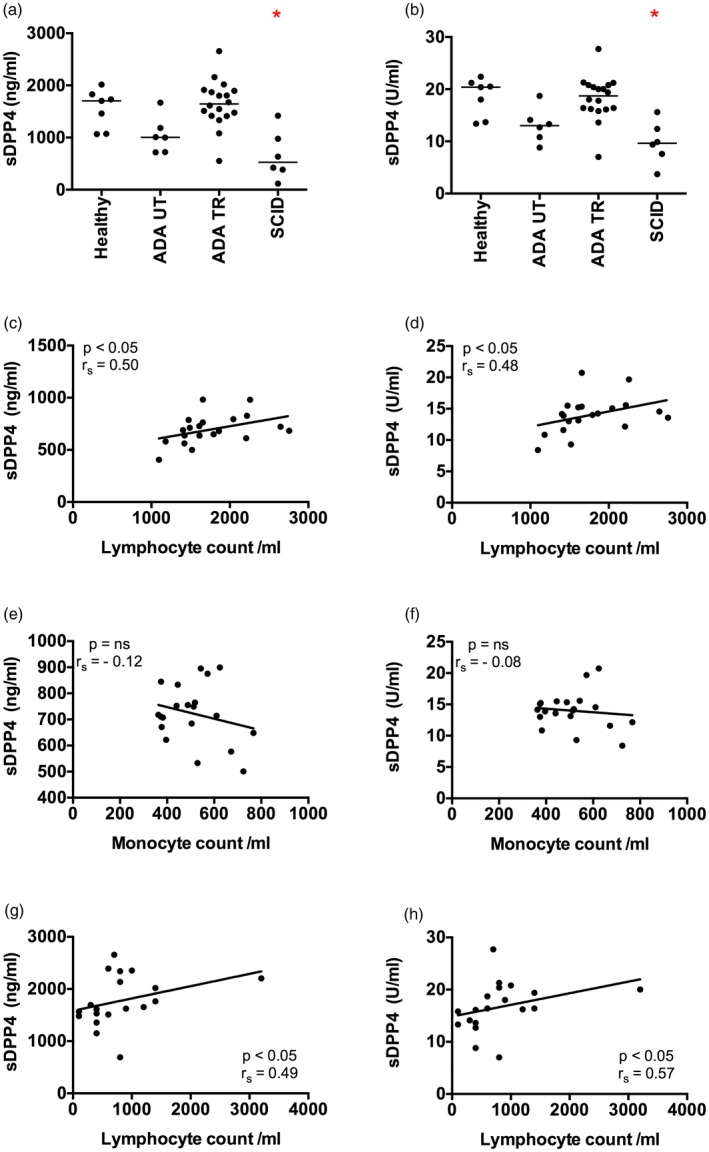
Patients with severe combined immunodeficiency have low levels of serum soluble dipeptidyl peptidase 4 (sDPP4). (a,b) The concentration (a) and activity level (b) of sDPP4 was measured in the serum of healthy children (healthy), untreated adenosine deaminase (ADA)‐deficient patients (ADA UT), treated ADA‐deficient patients (ADA TR) and severe combined immunodeficiency (SCID) patients. (c–f) Graphs show circulating lymphocyte or monocyte numbers versus sDPP4 concentration (c,e) or activity (d,f) in the serum of adult healthy donors. (g,h) Graphs show circulating lymphocyte number versus sDPP4 concentration (g) or activity (h) in the serum of treated ADA‐deficient patients. Each dot represents the mean value of one healthy donor or patient measured three times. Horizontal lines represent the median of the group. (a,b) Differences between cohorts were tested using the Kruskal–Wallis test followed by Dunn's multiple comparison test. *P < 0·05. (c–h) Spearman's correlation (r s) and P‐value for each analysis are shown.
To test this hypothesis, we compared the number of circulating immune cell subsets to sDPP4 levels and enzymatic activity. Within the adult healthy donor group, we observed a positive correlation between the number of circulating lymphocytes and sDPP4 (Fig. 1c,d), but no correlation was observed with circulating monocytes (Fig. 1e,f) or other immune cell subsets (data not shown). A similar result was obtained when analysing samples from ADA patients, as the level of immune reconstitution was correlated positively with sDPP4 concentration and activity (Fig. 1g,h). Of note, sDPP4 concentration and activity was lower in healthy adults compared to healthy children (Fig. 1). Together, these data suggested that plasma‐associated sDPP4 might originate from lymphocytes.
Immunodeficient mice have reduced sDPP4 concentration and activity
To extend our observations from human samples we employed experimental mouse models, with the notable caveats that DPP4 does not bind mouse ADA, and that the pattern of DPP4 expression on haematopoetic cells is somewhat different between mouse and human 19. To assess the impact of lymphocyte deficiency on sDPP4 concentration and corresponding plasma enzymatic activity, we compared WT mice to Rag2–/–/γc–/– mice, which lack B, T and natural killer (NK) cells 33. Supporting our findings in human SCID patients, we found a statistically significant difference between WT and Rag2–/–/γc–/– mice, with the latter showing marked reduction in both sDPP4 concentration and activity (Fig. 2a,b).
Figure 2.
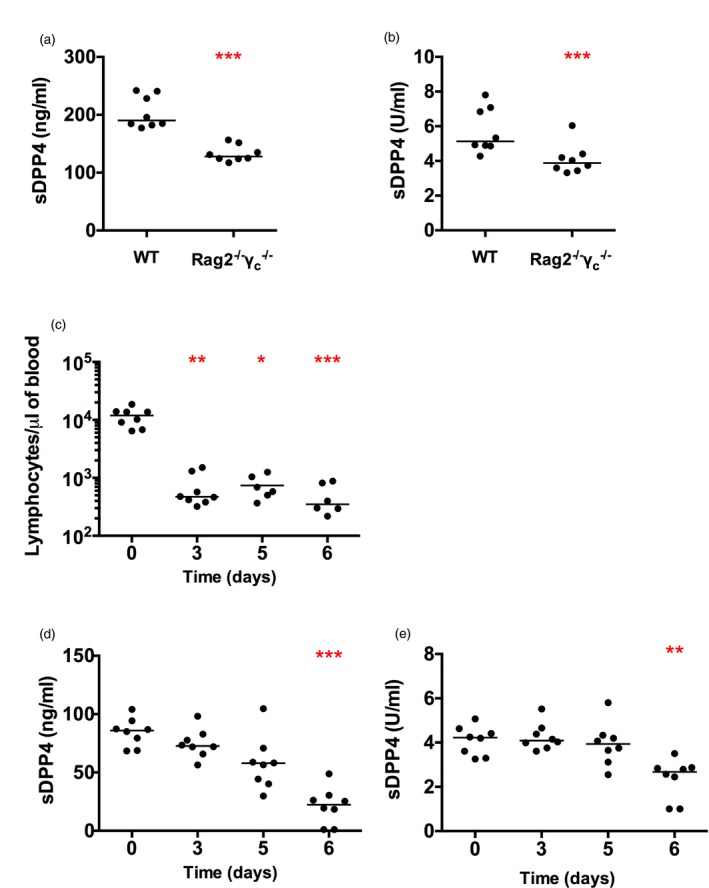
Soluble dipeptidyl peptidase 4 (sDPP4) level and activity are decreased in immunodeficient mice and after lymphocyte depletion by irradiation. Graphs show (a) sDPP4 concentration and (b) sDPP4 activity in wild‐type (WT) and Rag –/– γc –/– mice. (c–e) Mice were exposed to a lethal dose of irradiation. Graphs depict (c) the number of circulating lymphocytes, (d) the sDPP4 concentration and (e) sDPP4 activity in irradiated mice over time. Each dot represents one mouse. The experiment was performed twice. Horizontal lines represent the median. (a,b) Differences between groups were tested for statistical significance using the Mann–Whitney test (c–e). Differences among groups were tested for statistical significance using the Kruskal–Wallis test followed by Dunn's multiple comparison test, comparing all time‐points to day 0. *P < 0·05; **P < 0·01; ***P < 0·001.
Next, we induced immunodeficiency by whole‐body irradiation of WT mice to determine the impact of bone marrow depletion on the half‐life of plasma sDPP4. Lymphocyte numbers were measured by flow cytometry and the concentration and activity of sDPP4 were analysed in parallel. Three days after irradiation the number of circulating lymphocytes dropped sharply, from a mean frequency of 11 536 cells/μl [standard deviation (s.d.) = 4 146] to a mean frequency of 681 cells/µl (s.d. = 459) (Fig. 2c). Concurrently, the concentration of sDPP4 declined progressively, with a pre‐irradiation mean concentration of 84 ng/ml (s.d. = 12), which decreased to 21 ng/ml (s.d. = 16) 6 days post‐irradiation (Fig. 2d). Dipeptidyl peptidase activity showed a similar decrease, from a mean activity of 4·1 U/ml (s.d. = 0·6) before irradiation to a post‐irradiation activity of 2·1 U/ml (s.d. = 1·3) (Fig. 2e). As the irradiation dose used was lethal, we could not extend our sDPP4 evaluation beyond 6 days. However, based on these findings, approximately 75% of circulating sDPP4 originates from bone marrow‐derived cells. Moreover, these data suggest that sDPP4 is produced constitutively, with a plasma half‐life of ~3 days. Importantly, we also observed that mouse housing conditions impacted baseline concentrations of sDPP4. In the course of our studies, our colony was relocated to a new, cleaner breeding facility and we observed up to twofold differences in sDPP4 concentration levels in naive WT mice (compare Fig. 2a with 2d). In all experiments, matched controls from the same mouse facility were used.
Bone marrow‐derived cells are a significant source of plasma sDPP4
While our human and mouse studies and others supported that lymphocyte numbers correlate with sDPP4 concentration, they did not formally establish a direct source of the enzyme. To investigate the contribution of haematopoietic cells to sDPP4 concentration, we established reciprocal bone marrow chimeras using congenic WT and Dpp4–/– mice. We generated four groups of chimeric mice: (1) mice lacking DPP4 on haematopoietic cells (Dpp4–/– → WT), (2) mice lacking DPP4 on non‐haematopoietic cells (WT → Dpp4–/–), (3) positive control mice expressing DPP4 on both haematopoietic and non‐haematopoietic cells (WT → WT) and (4) negative control mice, deficient for DPP4 in both compartments (Dpp4–/– → Dpp4–/–) (Fig. 3a). To confirm chimerism, we assessed peripheral blood from these animals, tracking congenic markers with specific antibodies. Mice were bled once per week to follow immune cell reconstitution and sDPP4 concentrations in the plasma. Six weeks after bone marrow transfer, > 90% of the haematopoietic cells were donor‐derived (Fig. 3b).
Figure 3.
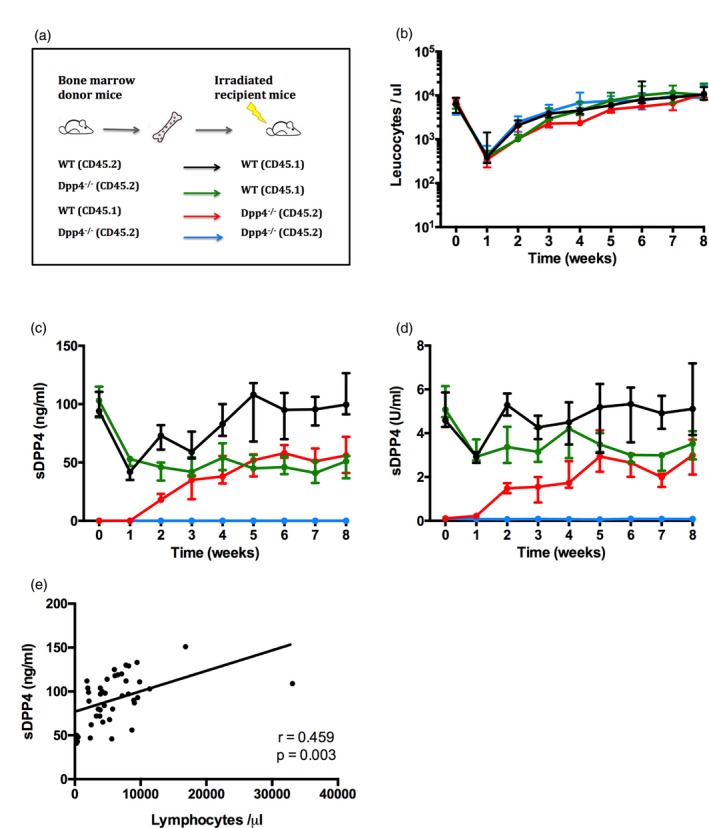
Bone marrow‐derived cells are a major source of soluble dipeptidyl peptidase 4 (sDPP4) in plasma. (a) Schematic representation of the experimental model. (b–e) Mice were lethally irradiated and reconstituted with bone marrow according to the experimental design as illustrated in (a). (b) Graph shows the number of donor leucocytes in circulation in recipient mice. (c) The graphs depict sDPP4 concentration and (d) sDPP4 activity in the plasma of recipient mice. Lines and dots show the mean for each group, with standard deviations. The experiment was performed twice, with three to five mice per group. One representative experiment is shown. (e) Graph shows circulating lymphocyte number versus sDPP4 concentration in [wild‐type (WT) → WT] group mice during the experimental follow‐up.
In WT → WT control mice, sDPP4 concentration and activity decreased initially to approximately 50% of pre‐irradiation levels, and then returned to baseline levels by week 5, consistent with the timing of immune reconstitution. As expected, sDPP4 was not detected at any time in the Dpp4–/– → Dpp4–/– group (Fig. 3c,d). In the Dpp4–/– → WT cohort, sDPP4 decreased rapidly then remained stable at approximately 50% of WT levels (Fig. 3c,d). In WT → Dpp4–/– mice, sDPP4 was initially absent from the plasma, but reached measurable quantities 1 week after bone marrow transplant, concurrent with the appearance of circulating donor cells (Fig. 3b–d). The concentration and activity levels plateaued at approximately 50% of WT levels 5 weeks post‐transplant. Notably, while haematopoietic cells expressed significant levels of DPP4 and served as a robust source of sDPP4, the concentration in WT → Dpp4–/– never reached that of WT → WT controls, suggesting that other cellular sources may also contribute to circulating sDPP4 levels. Notably, as observed in patient plasma, there was a positive correlation (r s = 0·46) between sDPP4 concentration and the number of circulating lymphocytes in reconstituted mice (Fig. 3e).
T cell stimulation results in increased release of sDPP4
Prior studies have suggested a functional relationship between DPP4 and TCR activation (reviewed in Gorrel et al. 28). However, no mechanistic link has been made between T cell activation and the release of lymphocyte‐derived sDPP4. We therefore tested whether T cell activation induced sDPP4 release. Using PBMCs derived from healthy donors, we activated lymphocytes in vitro using CD3 cross‐linking or stimulation with superantigen. Next, we measured sDPP4 concentration and activity in culture supernatants after 20 or 44 h incubation (Fig. 4a,b). Both α‐CD3 antibody treatment and Staphylococcus aureus TSST1 superantigen stimulation led to induction of sDPP4 (83 and 63% for concentration, 91 and 77% for activity, respectively). Treatment with brefeldin A (BFA) inhibited the release of sDPP4, suggesting that secretory activity is required for the mechanism underlying lymphocyte‐derived sDPP4 release.
Figure 4.
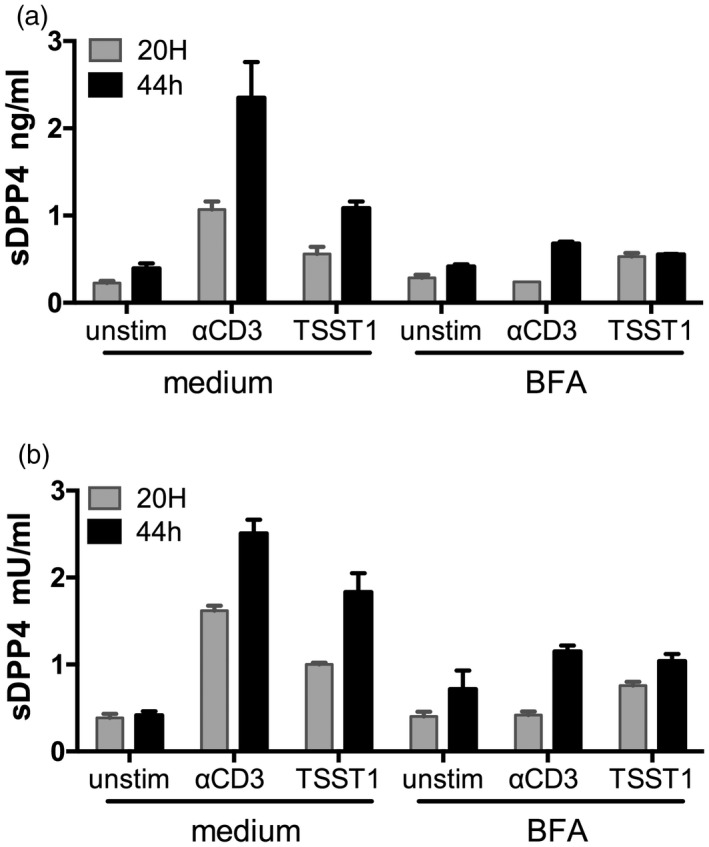
Peripheral blood mononuclear cell (PBMC) stimulation by anti‐CD3 and superantigen toxic shock syndrome toxin 1 (TSST1) induces release of soluble dipeptidyl peptidase 4 (sDPP4). PBMCs were isolated from healthy donors and stimulated as indicated and treated or not with brefeldin A (BFA). (a) sDPP4 concentration and (b) activity were measured in supernatants after 20 or 44 h of culture. Bars represent errors of experimental duplicates. A representative experiment of seven independent experiments with different healthy donors is shown.
Finally, we employed in‐vivo influenza infection to test whether antigen‐specific T cell expansion influences sDPP4 concentration. C57Bl/6 mice were injected intraperitoneally with influenza A/PR/8/34 and we sampled blood on days 5, 7 and 9 post‐infection, measuring sDPP4 concentration and activity. By day 7 post‐infection, we observed a statistically significant increase in the concentration and activity of sDPP4 in influenza‐infected mice compared to non‐infected animals (Fig. 5a,b). Moreover, influenza‐specific CD8+ T cells, enumerated using tetramer analysis (Fig. 5c–g), showed a clear correlation with sDPP4 (Fig. 5h). These results establish a functional relationship between T cell activation and the concentration of sDPP4, providing a rationale for why sDPP4 serves as a prognostic or predictive biomarker in various immune‐mediated disease settings.
Figure 5.
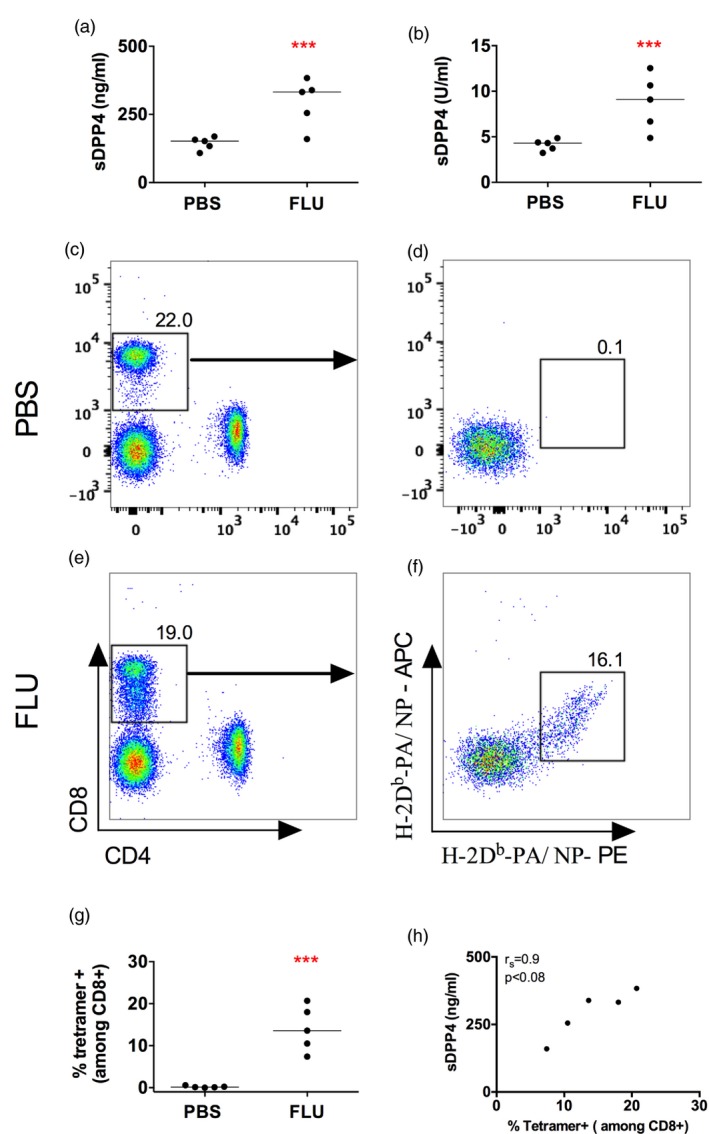
Induced soluble dipeptidyl peptidase 4 (sDPP4) by influenza virus is correlated with the intensity of tetramer CD8+ T cell‐specific responses. Mice were infected with influenza virus PR8 (influenza). (a,b) Graphs show (a) concentration and (b) activity of sDPP4 in blood of mice 7 days after infection. (c–f) Flow cytometry dot‐plots show combined tetramer (H‐2Db‐PA)‐ and (H‐2Db‐NP)‐specific cells in a representative (c,d) phosphate‐buffered saline (PBS)‐treated and (e,f) influenza‐infected mouse. (g) Graph shows the percentage of tetramer‐positive T cells in a group of five PBS‐treated and five influenza‐infected mice. (h) Graph shows the percentage of circulating tetramer‐positive cells versus sDPP4 levels. A representative experiment of three performed is shown. Spearman's correlation (r s) and P‐value are shown; ***P < 0·001.
Discussion
Dipeptidyl peptidase 4 expression is distributed widely among different cell types throughout multiple tissues within the body, participating in multiple biological functions. At least two major forms of the enzyme exist: a membrane‐bound homodimeric form located on the surface of cells and a soluble form that is found in multiple bodily fluids. Plasma or serum sDPP4 measurements have been used widely as a biomarker. For example, Cordero et al. have reviewed studies pertaining to the origin and altered concentration of sDPP4 in cancer patients 34. To exploit further the potential of DPP4 as a biomarker, insight into the mechanisms leading to variable sDPP4 concentration is needed. Thus, it is important to establish the tissue or cellular source of sDPP4, as well as the circumstances that provoke altered levels during disease 26.
We report a comprehensive analysis of sDPP4 measurements in a human cohort with primary immune deficiency, before and after treatment, and additional analyses in preclinical experimental models demonstrating that bone marrow‐derived cells contribute substantially to circulating sDPP4. Furthermore, we describe a clear correlation between sDPP4 and circulating lymphocyte numbers, establishing a relationship between the secretion of DPP4 and the activation status of lymphocytes. Similar observations in the context of disease, such as rheumatoid arthritis and type 2 diabetes, have been reported 35, 36.
In humans, but not in rodents, DPP4 can bind ADA on the surface of T cells and can mediate co‐stimulation during T cell activation 37, 38. We hypothesized that patients with an ADA deficiency would have perturbed DPP4 expression and activity. As predicted, we observed lower levels of sDPP4 in these patients, who were restored to normal levels after treatment. However, low levels of sDPP4 could also be detected in non‐ADA SCID patients. These data suggested that the number of lymphocytes in circulation, and not ADA expression per se, determined plasma sDPP4 levels. Indeed, this interpretation was supported by experiments in irradiated mice in which DPP4 concentration and activity decreased substantially and in chimeric mice, demonstrating that WT bone marrow transplantation into Dpp4–/– mice contributed to an appreciable increase in levels of sDPP4. Using a similar approach in rats, others have demonstrated that sDPP4 is derived in part from bone marrow cells 39. Importantly, however, reconstitution of a Dpp4 –/– mouse with WT bone marrow did not fully reached sDPP4 concentration of WT mice, suggesting that other cell sources also contribute to the soluble form of this enzyme. It is important to note that DPP4 expression in the haematopoietic compartment differs between human and mouse. Indeed, in humans, DPP4 is expressed mainly by T cells, whereas in mice, dendritic cells, B cells, and NK cells also express DPP4 40. Additional studies are needed to determine the inflammatory conditions that lead to increased expression and/or activity and the precise mechanisms of its release.
Both the kidney and liver express high amounts of DPP4, and it has been suggested that these tissues are the source of sDPP4. In the kidney, DPP4 is expressed mainly in the cortex, and not in the medulla, on the cell membranes of glomerular podocytes, but not on the endothelial or mesangial cells 20. DPP4 is also present on the brush borders of the proximal tubule cells and along the entire proximal tubule 20. Cleaved sDPP4 may, therefore, accumulate in the urine from this location, accounting for its high expression in this bodily fluid 41. Consistent with this interpretation, Wang and colleagues showed recently that blood sDPP4 does not originate from the kidney in rats, demonstrated using reciprocal kidney transplantation between DPP4‐deficient and WT rats 39. The hepatobiliary system may also be a source of sDPP4 in the blood circulation. Fukui and colleagues quantified DPP4 in rat liver cells by immunogold localization. In hepatocytes, DPP4 localized mainly to the bile canalicular surface and lysosomal limiting membranes 42. DPP4 was also observed on the plasma membrane and lysosomal membrane of endothelial and Kupffer cell 42. Supporting the idea that the liver might be a source of sDPP4, the activity level of this enzyme is elevated in serum from patients with liver diseases, such as hepatitis and cirrhosis, and correlates with liver‐associated enzyme concentrations 14, 34. Additionally, aberrant DPP4 expression in human hepatocellular carcinoma and liver cirrhosis has been reported 43, 44. Interestingly, rat bile duct ligation experiments showed that hepatocyte‐associated DPP4 is transported in transcytotic vesicles in the bile and serum‐associated DPP4 activity is elevated in patients with primary biliary cirrhosis 45, 46. We may speculate, therefore, that in normal physiological conditions, DPP4 would be secreted in the bile and during liver perturbation, DPP4 levels might increase in the blood. However, our experiments support an alternative interpretation, where increased sDPP4 may derive from activated T cells that are the mediators of liver inflammation and liver cell damage.
DPP4 was characterized originally as a T cell differentiation antigen and the expression of this enzyme increases following activation (e.g. upon stimulation with antigen, α‐CD3 and IL‐2, or with mitogens such as phytohaemagglutinin (PHA) 47, 48. Indeed, both the percentage of cells expressing DPP4 and the number of molecules per cell are increased following T cell activation 28, 49. Our results demonstrate that stimulation of human lymphocytes with α‐CD3 or superantigen induces DPP4 release in vitro and that this release is via a putative secretory pathway. Furthermore, in‐vivo T cell activation resulted in increased circulating sDPP4, demonstrated in influenza virus infection. Therefore, activation of lymphocytes is associated directly with higher levels of sDPP4 measured in blood.
The mechanism behind release of sDPP4 from the cell surface is still under investigation. In our experiments, incubation with brefeldin A abolished the release of sDPP4. Brefeldin A causes structural and functional alterations of the trans‐Golgi network, suggesting that secretion may facilitate sDPP4 release 50. Alternatively, brefeldin A may be inhibiting the secretion of a protease necessary for DPP4 shedding. Röhrborn and colleagues described sDPP4 release from adipocytes and smooth muscle cells via shedding by matrix metalloproteinase (MMP)9 and MMP1, MMP2 and MMP14, respectively 51. However, in their studies, DPP4 secretion from adipocytes and smooth cells was not impacted by brefeldin A treatment. Nargis et al. recently reported an alternative mechanism whereby kallikrein‐related peptidase 5 (KLK5) was shown to mediate enzymatic cleavage of DPP4 from the surface of IL‐17‐producing CD4+ T helper type 17 (Th17) cells 52. Thus, the mechanism of DPP4 secretion may be tissue‐dependent and additional biochemical analysis of activated lymphocytes may provide additional insights.
Tanaka and colleagues reported that recombinant sDPP4 enhances proliferative responses of peripheral blood lymphocytes stimulated with soluble antigen 53, 54. More recently, and using various engineered isoforms of DPP4, Gorrel and colleagues demonstrated that sDPP4 enhances human lymphocyte proliferation in vitro, independently of both its enzyme activity and adenosine deaminase binding properties 55. Subsequently, it was demonstrated that sDPP4 induces T cell proliferation through CD86 up‐regulation on antigen‐presenting cells 56. As DPP4 plays many biological roles, including the truncation of chemokines that directly impact T cell functions, its influence on lymphocyte activation requires further investigation 57. Indeed, we have demonstrated previously that DPP4 inhibition enhances lymphocyte trafficking to solid tumours, resulting in increased immunity and delayed tumour growth 15. Therefore, increased sDPP4 expression may have immunological consequences, as the release of sDPP4 after TCR stimulation would alter the proliferation and migration of lymphocytes.
There is some discrepancy in the literature regarding the usage of sDPP4 as a biomarker in disease 34. This may be due in part to the contribution of proteins other than DPP4 with DPP4‐like activity 58, 59, 60. While a potential caveat, DPP4 contributes to more than 90% of the overall dipeptidyl peptidase activity in the serum and plasma of healthy people 15, 57, 61. One important consideration is the use of assays that are optimized for measuring the concentration or activity of DPP4. Throughout our study, we measured protein concentration with an ELISA assay specific for DPP4 and quantified sDPP4 activity using a defined DPP4‐selective substrate, demonstrating that activity is well correlated with soluble protein concentrations. To confirm sDPP4 as a biomarker in human disease, it will be necessary to harmonize methods used to measure sDPP4 among various clinical situations. Additional complexity in analysing sDPP4 may arise, as in rheumatoid arthritis and systemic lupus erythematosus, where autoantibodies to DPP4 are present and may interfere with the detection of sDPP4 62, 63.
In sum, our study identified lymphocytes as a major cellular source of sDPP4 and reinforces the critical role of T cell activation in the fluctuation in plasma sDPP4 concentration. Together, a standardized approach for DPP4 measurements and the integration of lymphocyte status for normalization of the biomarker data may help to clarify the role of sDPP4 as a biomarker, and advance it for use in the clinical monitoring of immune‐related disorders.
Disclosures
M. L. A and R. B. d. S. are employees of Genentech. The remaining authors declare no competing financial interests.
Author contributions
A. C. and R. B. d. S. performed the experiments and analysed the data; M. L. A. supervised the experiments; A. S., A. A., M. T. A, S. S. R, ICAReB and M. A. I. provided patient samples and/or critically reviewed the data and manuscript; A. C. and M. L. A. designed the study. A. C., M. A. I. and M. L. A. wrote the manuscript.
Acknowledgements
The authors thank the numerous patients and healthy volunteers who kindly donated peripheral blood for these experiments. We thank Immacolata Brigida for data management and H. Saklani for providing tetramers. We thank James Di Santo and Darragh Duffy for critical reading of the manuscript. We would like to acknowledge Marie‐Noelle Unheheuer for her role in leadership of the Clinical Investigation and Access to BioResources platform (ICAReB). Funding was provided by the Institut Pasteur (Pasteur‐Roux post‐doctoral fellowship to R. B. S.), the Ligue Contre le Cancer (M. L. A.), the Fondation ARC pour la recherche sur le cancer (M. L. A.), the Italian Ministero della Salute (GR‐2011‐02346985 to A. V. S.), the European Union Seventh Framework Programme Marie Curie Action (PCIG11‐GA‐2012‐3221170 to M. A. I.) and the French government's Invest in the Future Program, managed by the Agence Nationale de la Recherche (LabEx Immuno‐Onco to A. C., R. B. d. S., M. A. I. and M. L. A.).
References
- 1. McCaughan GW, Wickson JE, Creswick PF, Gorrell MD. Identification of the bile canalicular cell‐surface molecule GP110 as the ectopeptidase dipeptidyl peptidase‐IV – an analysis by tissue distribution, purification and N‐terminal amino‐acid‐sequence. Hepatology 1990; 11:534–44. [DOI] [PubMed] [Google Scholar]
- 2. Kameoka J, Tanaka T, Nojima Y, Schlossman SF, Morimoto C. Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science 1993; 261:466–9. [DOI] [PubMed] [Google Scholar]
- 3. Raj VS, Mou H, Smits SL et al Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus – EMC. Nature 2013; 495:251–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chien CH, Huang LH, Chou CY et al One site mutation disrupts dimer formation in human DPP‐IV proteins. J Biol Chem 2004; 279:52338–45. [DOI] [PubMed] [Google Scholar]
- 5. Durinx C, Lambeir AM, Bosmans E et al Molecular characterization of dipeptidyl peptidase activity in serum: soluble CD26/dipeptidyl peptidase IV is responsible for the release of X‐Pro dipeptides. Eur J Biochem 2000; 267:5608–13. [DOI] [PubMed] [Google Scholar]
- 6. Mentlein R, Schiemann F, Ludwig A, Brandt E. Modification of the biological activity of chemokines by dipeptidyl peptidase IV – a side effect in the use of inhibitors? Adv Exp Med Biol 2003; 524:37–47. [DOI] [PubMed] [Google Scholar]
- 7. Lambeir AM, Durinx C, Proost P et al Kinetic study of the processing by dipeptidyl‐peptidase IV/CD26 of neuropeptides involved in pancreatic insulin secretion. FEBS Lett 2001; 507:327–30. [DOI] [PubMed] [Google Scholar]
- 8. Zhu L, Tamvakopoulos C, Xie D et al The role of dipeptidyl peptidase IV in the cleavage of glucagon family peptides – in vivo metabolism of pituitary adenylate cyclase‐activating polypeptide‐(1–38). J Biol Chem 2003; 278:22418–23. [DOI] [PubMed] [Google Scholar]
- 9. Wagner L, Kaestner F, Wolf R et al Identifying neuropeptide Y (NPY) as the main stress‐related substrate of dipeptidyl peptidase 4 (DPP4) in blood circulation. Neuropeptides 2016; 57:21–34. [DOI] [PubMed] [Google Scholar]
- 10. Mulvihill EE, Drucker DJ. Pharmacology, physiology, and mechanisms of action of dipeptidyl peptidase‐4 inhibitors. Endocr Rev 2014; 35:992–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abbott CA, McCaughan GW, Levy MT, Church WB, Gorrell MD. Binding to human dipeptidyl peptidase IV by adenosine deaminase and antibodies that inhibit ligand binding involves overlapping, discontinuous sites on a predicted beta propeller domain. Eur J Biochem 1999; 266:798–810. [DOI] [PubMed] [Google Scholar]
- 12. De Meester I, Vanham G, Kestens L et al Binding of adenosine deaminase to the lymphocyte surface via CD26. Eur J Immunol 1994; 24:566–70. [DOI] [PubMed] [Google Scholar]
- 13. Proost P, Struyf S, Loos T et al Coexpression and interaction of CXCL10 and CD26 in mesenchymal cells by synergising inflammatory cytokines: CXCL8 and CXCL10 are discriminative markers for autoimmune arthropathies. Arthritis Res Ther 2006; 8:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Casrouge A, Decalf J, Ahloulay M et al Evidence for an antagonist form of the chemokine CXCL10 in patients chronically infected with HCV. J Clin Invest 2011; 121:308–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barreira Da Silva R, Laird ME, Yatim N et al Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nat Immunol 2015; 16:850–8. [DOI] [PubMed] [Google Scholar]
- 16. Decalf J, Tarbell KV, Casrouge A et al Inhibition of DPP4 activity in humans establishes its in vivo role in CXCL10 post‐translational modification: prospective placebo‐controlled clinical studies. EMBO Mol Med 2016; 8:679–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mortier A, Gouwy M, VanDamme J, Proost P, Struyf S. CD26/dipeptidylpeptidase IV—chemokine interactions: double‐edged regulation of inflammation and tumor biology. J Leukoc Biol 2016; 99:955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heike M, Möbius U, Knuth A, Meuer S, Meyer zum Büschenfelde KH. Tissue distribution of the T cell activation antigen Ta1. Serological, immunohistochemical and biochemical investigations. Clin Exp Immunol 1988; 74:431–4. [PMC free article] [PubMed] [Google Scholar]
- 19. Dinjens WN, ten Kate J, Wijnen JT et al Distribution of adenosine deaminase‐complexing protein in murine tissues. J Biol Chem 1989; 264:19215–20. [PubMed] [Google Scholar]
- 20. Mentzel S, Dijkman HB, VanSon JP, Koene RA, Assmann KJ. Organ distribution of aminopeptidase A and dipeptidyl peptidase IV in normal mice. J Histochem Cytochem 2017; 44:445–61. [DOI] [PubMed] [Google Scholar]
- 21. Mentlein R. Dipeptidyl‐peptidase IV CD26) – role in the inactivation of regulatory peptides. Regul Pept 1999; 85:9–24. [DOI] [PubMed] [Google Scholar]
- 22. Yu DMT, Yao T‐W, Chowdhury S et al The dipeptidyl peptidase IV family in cancer and cell biology. FEBS J 2010; 277:1126–44. [DOI] [PubMed] [Google Scholar]
- 23. Klemann C, Wagner L, Stephan M, von Hörsten S. Cut to the chase: a review of CD26/dipeptidyl peptidase‐4's (DPP4) entanglement in the immune system. Clin Exp Immunol 2016; 185:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ragab D, Laird M, Duffy D et al CXCL10 antagonism and plasma sDPPIV correlate with increasing liver disease in chronic HCV genotype 4 infected patients. Cytokine 2013; 63:105–12. [DOI] [PubMed] [Google Scholar]
- 25. Tejera‐Alhambra M, Casrouge A, de Andrés C et al Low DPP4 expression and activity in multiple sclerosis. Clin Immunol 2014; 150:170–83. [DOI] [PubMed] [Google Scholar]
- 26. Yazbeck R, Jaenisch SE, Abbott CA. Potential disease biomarkers: dipeptidyl peptidase 4 and fibroblast activation protein. Protoplasma 2017; 23:1–12. [DOI] [PubMed] [Google Scholar]
- 27. Nieto‐Fontarigo JJ, González‐Barcala FJ, San José E et al CD26 and asthma: a comprehensive review. Clin Rev Allergy Immunol 2016; 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gorrell MD, Gysbers V, McCaughan GW. CD26: a multifunctional integral membrane and secreted protein of activated lymphocytes. Scand J Immunol 2001; 54:249–64. [DOI] [PubMed] [Google Scholar]
- 29. Cancrini C, Ferrua F, Scarselli A et al Role of reduced intensity conditioning in T‐cell and B‐cell immune reconstitution after HLA‐identical bone marrow transplantation in ADA‐SCID. Haematologica 2010; 95:1778–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aiuti A, Cattaneo F, Galimberti S et al Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med 2009; 360:447–58. [DOI] [PubMed] [Google Scholar]
- 31. Altman JD, Moss PA, Goulder PJ et al Phenotypic analysis of antigen‐specific T lymphocytes. Science 1996; 274:94–6. [PubMed] [Google Scholar]
- 32. Dong RP, Kameoka J, Hegen M et al Characterization of adenosine deaminase binding to human CD26 on T cells and its biologic role in immune response. J Immunol 1996; 156:1349–55. [PubMed] [Google Scholar]
- 33. Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol 2007; 7:118–30. [DOI] [PubMed] [Google Scholar]
- 34. Cordero OJ, Salgado FJ, Nogueira M. On the origin of serum CD26 and its altered concentration in cancer patients. Cancer Immunol Immunother 2009; 58:1723–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cordero OJ, Varela‐Calviño R, López‐González T et al CD26 expression on T helper populations and sCD26 serum levels in patients with rheumatoid arthritis. PLOS One 2015; 10:e0131992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee SA, Kim YR, Yang EJ et al CD26/DPP4 levels in peripheral blood and T cells in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab 2013; 98:2553–61. [DOI] [PubMed] [Google Scholar]
- 37. Iwaki‐Egawa S, Watanabe Y, Fujimoto Y. CD26/dipeptidyl peptidase IV does not work as an adenosine deaminase‐binding protein in rat cells. Cell Immunol 1997; 178:180–6. [DOI] [PubMed] [Google Scholar]
- 38. Dong RP, Tachibana K, Hegen M et al Determination of adenosine deaminase binding domain on CD26 and its immunoregulatory effect on T cell activation. J Immunol 1997; 159:6070–6. [PubMed] [Google Scholar]
- 39. Wang Z, Grigo C, Steinbeck J et al Soluble DPP4 originates in part from bone marrow cells and not from the kidney. Peptides 2014; 57:109–17. [DOI] [PubMed] [Google Scholar]
- 40. Heng TSP, Painter MW. The Immunological Genome Project Consortium. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 2008; 9:1091–4. [DOI] [PubMed] [Google Scholar]
- 41. Mitic B, Lazarevic G, Vlahovic P, Rajic M, Stefanovic V. Diagnostic value of the aminopeptidase N, N‐acetyl‐beta‐D‐glucosaminidase and dipeptidylpeptidase IV in evaluating tubular dysfunction in patients with glomerulopathies. Ren Fail 2008; 30:896–903. [DOI] [PubMed] [Google Scholar]
- 42. Fukui Y, Yamamoto A, Kyoden T, Kato K, Tashiro Y. Quantitative immunogold localization of dipeptidyl peptidase‐IV (DPP‐IV) in rat‐liver cells. Cell Struct Funct 1990; 15:117–25. [DOI] [PubMed] [Google Scholar]
- 43. Stecca BA, Nardo B, Chieco P et al Aberrant dipeptidyl peptidase IV (DPP IV/CD26) expression in human hepatocellular carcinoma. J Hepatol 1997; 27:337–45. [DOI] [PubMed] [Google Scholar]
- 44. Hascelik G, Asan E, Hamaloglu E, Tatar G IV. Dipeptidyl peptidase IV (DDP IV) in Nash patients. Ann Hepatol 2007; 6:242–50. [PubMed] [Google Scholar]
- 45. Barr VA, Hubbard AL. Newly synthesized hepatocyte plasma membrane proteins are transported in transcytotic vesicles in the bile duct‐ligated rat. Gastroenterology 1993; 105:554–71. [DOI] [PubMed] [Google Scholar]
- 46. Lakatos PL, Firneisz G, Borcsiczky D et al Elevated serum dipeptidyl peptidase IV (CD26, EC 3.4.14.5) activity in experimental liver cirrhosis. Eur J Clin Invest 2000; 30:793–7. [DOI] [PubMed] [Google Scholar]
- 47. Morimoto C, Schlossman SF. The structure and function of CD26 in the T‐cell immune response. Immunol Rev 1998; 161:55–70. [DOI] [PubMed] [Google Scholar]
- 48. Ohnuma K, Dang NH, Morimoto C. Revisiting an old acquaintance: CD26 and its molecular mechanisms in T cell function. Trends Immunol 2008; 29:295–301. [DOI] [PubMed] [Google Scholar]
- 49. Salgado FJ, Pérez Díaz A, Villanueva NM et al CD26: a negative selection marker for human Treg cells. Cytometry Part A 2012; 81:843–55. [DOI] [PubMed] [Google Scholar]
- 50. Wagner M, Rajasekaran AK, Hanzel DK, Mayor S, Rodriguez‐Boulan E. Brefeldin A causes structural and functional alterations of the trans‐Golgi network of MDCK cells. J Cell Sci 1994; 107:933–43. [DOI] [PubMed] [Google Scholar]
- 51. Röhrborn D, Eckel J, Sell H. Shedding of dipeptidyl peptidase 4 is mediated by metalloproteases and up‐regulated by hypoxia in human adipocytes and smooth muscle cells. FEBS Lett 2014; 588:3870–7. [DOI] [PubMed] [Google Scholar]
- 52. Nargis T, Kumar K, Ghosh AR et al KLK5 induces shedding of DPP4 from circulatory Th17 cells in type 2 diabetes. Mol Metab 2017; 6:1529–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tanaka T, Kameoka J, Yaron A, Schlossman SF, Morimoto C. The costimulatory activity of the CD26 antigen requires dipeptidyl peptidase‐IV enzymatic‐activity. Proc Natl Acad Sci USA 1993; 90:4586–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tanaka T, Duke‐Cohan JS, Kameoka J et al Enhancement of antigen‐induced T‐cell proliferation by soluble CD26/dipeptidyl peptidase IV. Proc Natl Acad Sci USA 1994; 91:3082–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yu DMT, Slaitini L, Gysbers V et al Soluble CD26 / dipeptidyl peptidase IV enhances human lymphocyte proliferation in vitro independent of dipeptidyl peptidase enzyme activity and adenosine deaminase binding. Scand J Immunol 2011; 73:102–11. [DOI] [PubMed] [Google Scholar]
- 56. Ohnuma K, Munakata Y, Ishii T, et al. Soluble CD26/dipeptidyl peptidase IV induces T cell proliferation through CD86 up‐regulation on APCs. J Immunol 2001; 167:6745–55. [DOI] [PubMed] [Google Scholar]
- 57. Durinx C, Lambeir AM, Bosmans E et al Molecular characterization of dipeptidyl peptidase activity in serum soluble CD26/dipeptidyl peptidase IV is responsible for the release of X‐Pro dipeptides. Eur J Biochem 2000; 267:5608–13. [DOI] [PubMed] [Google Scholar]
- 58. Abbott CA, Yu D, Woollatt E et al Cloning, expression and chromosomal localization of a novel human dipeptidyl peptidase (DPP) IV homolog, DPP8. Eur J Biochem 2000; 267:6140–50. [DOI] [PubMed] [Google Scholar]
- 59. Maes M‐B, Dubois V, Brandt I et al Dipeptidyl peptidase 8/9‐like activity in human leukocytes. J Leukoc Biol 2007; 81:1252–7. [DOI] [PubMed] [Google Scholar]
- 60. Wagner L, Klemann C, Stephan M, vonHörsten S. Unravelling the immunological roles of dipeptidyl peptidase 4 (DPP4) activity and/or structure homolog (DASH) proteins. Clin Exp Immunol 2016; 184:265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Friedrich D, Hoffmann T, Bär J et al Does human attractin have DP4 activity? Biol Chem 2007; 388:155–62. [DOI] [PubMed] [Google Scholar]
- 62. Cuchacovich M, Gatica H, Pizzo SV, Gozalez‐Gronow M. Characterization of human serum dipeptidyl peptidase IV (CD26) and analysis of its autoantibodies in patients with rheumatoid arthritis and other autoimmune diseases. Clin Exp Rheumatol 2001; 19:673–80. [PubMed] [Google Scholar]
- 63. Cordero OJ, Varela‐Calviño R, López‐González T et al Anti‐CD26 autoantibodies are involved in rheumatoid arthritis and show potential clinical interest. Clin Biochem 2017; 50:903–10. [DOI] [PubMed] [Google Scholar]