

Summary
Signal transducer and activator of transcription 1 (STAT‐1) gain‐of‐function (GOF) mutations cause chronic mucocutaneous candidiasis (CMC), a disease associated with Candida albicans and Staphylococcus aureus infection. Patients suffer from dysegulated immune responses due to aberrant cell programming and function. We investigated the effect of inhibitory molecules targeting histone deacetylases (HDACi) on the immune responses of peripheral blood mononuclear cells (PBMCs) of healthy controls and patients with CMC towards microbes relevant for CMC. PBMCs cells were pretreated with HDACi and challenged with C. albicans or S. aureus. Innate and adaptive cytokines were measured in cell culture supernatants by enzyme‐linked immunosorbent assay (ELISA). We assessed the effect of HDAC inhibitors on T helper type 1 (Th1) and Th17 cells and measured STAT‐1 and STAT‐3 phosphorylation using flow cytometry. Panobinostat, a pan‐HDAC inhibitor, strongly inhibits innate and adaptive cytokines upon challenge with C. albicans or S. aureus. Specific inhibitors (entinostat or RGFP966) also had a tendency to lower production of most innate cytokines in CMC patient cells. Entinostat and RGFP966 increased the production of interleukin (IL)‐22 specifically after S. aureus challenge in patient cells. In healthy and control cells, entinostat and RGFP966 treatment down‐regulated STAT‐1 phosphorylation while pSTAT‐3 levels remained stable. HDACi modulate cytokine production in response to C. albicans and S. aureus. Pan‐inhibitors lower overall cytokine production, whereas specific inhibitors confer a selective effect. Entinostat and RGFP966 are promising therapeutic candidates to treat STAT‐1 GOF due to their capacity to restore IL‐22 production and decrease STAT‐1 phosphorylation; however, their inhibition of innate cytokines poses a possible risk to secondary infections.
Keywords: chronic mucocutaneous candidiasis, cytokines, histone acetylation
Introduction
Chronic mucocutaneous candidiasis (CMC) is characterized by recurrent or persisting fungal infections of nails, skin or mucosal areas. Patients suffering from CMC can be divided into two categories: CMC disease (CMCD) caused predominantly by gain‐of‐function mutation (GOF) in signal transducer and activator of transcription 1 (STAT‐1) and syndromic CMC (sCMC), such as the hyper‐immunoglobulin (Ig)E syndrome caused by defects in STAT‐3 [1]. All patients suffering from CMC, whether CMCD or sCMC, share certain immunological abnormalities associated with decreased function or loss of T helper type 17 (Th17) responses and increased susceptibility to bacterial infections, due mainly to Staphylococcus aureus. Taken together, these manifestations indicate a common link in disturbed programming of cell fate and regulation of gene expression.
Gene expression is controlled primarily by the accessibility of chromatin to transcription machinery and trans‐acting factors. The dynamic constituent assembly of DNA and various nucleoproteins, including histones, spatially regulates transcriptional competency by structural adaptation and genome compartmentalization [2]. Covalent epigenetic modifications facilitate this functional modulation and chromatin reorganization by various mechanisms. One of the best‐characterized mechanisms governing chromatin architecture is via the acetylation of lysine residues on histone proteins, which is associated ubiquitously with transcriptional competency and is regulated by the opposing activities of histone acetyltransferases (HATs) and histone deacetylases (HDACs). Conversely, lysine residues of non‐histone proteins such as transcription factors can also be acetylated, modification of which can affect activity, stability and protein–protein interactions [3]. The balance between acetylation and deacetylation is fundamental to many chromatin‐dependent and ‐independent processes and is increasingly explored as a pharmacological target for the treatment of numerous diseases.
Considerable effort has been directed to defining the exact role of HDACs in immune regulation. Innate as well as adaptive immunity are impacted strongly by HDACs. For example, HDAC expression is induced in murine macrophages upon lipopolysaccharide (LPS) stimulation and to selectively down‐regulate proinflammatory gene expression of Ccl2, Ccl7 and End1 [4]. HDACs are also critical for adaptive immunity, humoral as well as cellular responses: deletion of both Hdac1 and Hdac2 prevents normal B cell maturation [5] and HDAC7 is crucial for negative selection during T cell development [6]. As HDACs control fundamental cellular processes, small inhibitory molecules interfering with HDAC function have been investigated extensively during past years to assess their therapeutic potential, especially in cancer therapy [7]. Also, HDAC inhibitors (HDACi) have been explored as additive HIV treatment [8] and were proved to induce expression of anti‐microbial peptides effectively at mucosal surfaces, helping to clear bacterial infections [9].
HDAC inhibition has been proposed as a promising therapeutic approach to treat CMC [10]. We have shown recently that inhibition of HDACs (specifically HDAC1 and HDAC2) in STAT‐1 GOF mutation cells can re‐establish normal STAT‐3‐dependent gene expression [11]. However, the precise interplay between STAT acetylation and phosphorylation varies greatly, depending on the position of the modified lysine residue in the protein and cell type [3]. Here we investigate systematically the modulatory effect of several HDAC inhibitors on the pro‐ and anti‐inflammatory innate cytokine response of peripheral mononuclear blood cells (PBMCs) induced by Candida albicans and Staphylococcus aureus, two of the most relevant pathogens in CMC. Specifically, we investigated panobinostat [a pan‐inhibitor of HDACs including class I (HDAC1, 2, 3, 8), class IIa (HDAC4, 5, 7, 9), class IIb (HDAC6, 10), class IV (HDAC11) ], entinostat (a partial class I inhibitor, HDAC1 and HDAC3) and RGFP966 (a selective HDAC3 inhibitor). Our data suggest that proinflammatory as well as anti‐inflammatory innate immune responses are modulated by some, but not all, HDAC inhibitors, and these observations must be taken into account when HDAC inhibition is explored in patients with CMC.
Methods and materials
Peripheral blood mononuclear cell isolation
Human PBMCs were isolated from healthy volunteers (Sanquin Blood Bank, Nijmegen, the Netherlands) by density‐gradient centrifugation over Ficoll‐Paque (GE Healthcare, Chicago, IL, USA), as described previously [12]. PBMCs were washed with phosphate‐buffered saline (PBS) and maintained in RPMI‐1640 Dutch‐modified culture medium (RPMI medium; Invitrogen, Carlsbad, CA, USA) supplemented with 10 μg/ml gentamicin (Centraform), 10 mmol/l glutamax (Invitrogen) and 10 mmol/l pyruvate (Invitrogen). For all experiments, cells were stimulated per well in 96‐well round‐bottomed plates. In order to measure adaptive cytokines [interferon (IFN)‐γ, interleukin (IL)‐17 and IL‐22], PBMCs were incubated with C. albicans or S. aureus for 6 days. At day 6, supernatants were removed. C. albicans and S. aureus stimuli were prepared and added to the cells either with or without HDAC inhibitors. On day 7 (24 h later), supernatants were collected and stored at –20°C for future analysis.
Inhibitors and stimuli
All HDAC inhibitors were dissolved in dimethylsulphoxide (DMSO), according to the manufacturer’s instructions (Selleckchem, Houston, TX, USA). Concentrations used in experiments range from 1000 to 1 nM. The manufacturer provided half maximal inhibitory concentration (IC50) values based on cell‐free assays: 5 nM for panobinostat, 0·51 μM (HDAC1)–1·7 μM (HDAC3) for entinostat and 0·08 μM for RGFP966.
For panobinostat, various pharmacokinetic studies have been performed in patients. Most of these studies are summarized in a review [13] indicating very rapid metabolism of the drug (up to 99%). In several studies, panobinostat concentration in patient plasma were measured after administration. An exposure of Cmax between 20 and 70 ng/ml was reported. Considering the molecular weight of panobinostat (349·43 g/mol), a plasma concentration of 50 ng/ml equals [0·05 g/(349·43 g/mol) ] = 143·09 μM. As panobinostat is rapidly metabolized by the liver in vivo, we decided to use lower concentrations for our experiments (100–1000 nM).
Entinostat levels in patients plasma were also assessed after treatment [14]. The authors found plasma concentrations of approximately 1 ng/ml, depending on the dosage of entinostat treatment. Entinostat has a molecular weight of 376·41 g/mol, therefore plasma levels are of 1 ng/ml equal to approximately 2000 nM. As entinostat’s IC50 is approximately ×1.5 higher for HDAC3 than HDAC1 in cell free‐assays (information provided by the manufacturer), we aimed to lower the concentration in our experimental setting by a factor of 1·5 to estimate reasonable working concentrations.
To our knowledge, no patient studies have been published on the pharmacokinetics of RGFP966. Cell‐free assays indicated an IC50 of approximately 80 nM. As that value is similar to the lower concentration used for other inhibitors, we decided to also use 100 and 1000 nM for RGFP966 in our experiments.
C. albicans UC820 conidia were grown in Sabouraud broth (Sigma, St Louis, MO, USA), S. aureus in brain heart infusion broth (BHI) (Sigma) at 37°C, 5% CO2 overnight. Morphology was checked by light microscopy the following day, cells were washed ×3 in sterile PBS, resuspended in sterile PBS and concentration was adjusted to 1 × 108 cells/ml (counting chamber). Micro‐organisms were heat‐killed for 60 min at 95°C; cell viability was checked by plating the suspension on Sabouraud or BHI agar at 37°C, 5% CO2 overnight. Micro‐organisms were stored at –20°C until further use.
Enzyme‐linked immunosorbent assay (ELISA)
Cytokine production was measured in supernatants by enzyme‐linked immunosorbent assay (ELISA), according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN, USA, IL‐1β, IL‐1Ra, IL‐17, IL‐22 and tumour necrosis factor (TNF)‐α; Sanquin, Amsterdam, the Netherlands, IL‐6, IL‐10 and IFN‐γ). IL‐6 and IL‐10 in patients’ samples were measured using ELISA kits supplied by R&D Systems.
Flow cytometry
Flow cytometry was used to determine cell viability during treatment with HDAC inhibitors. Briefly, cells were collected following stimulation for 24 h and washed in PBS containing 1% bovine serum albumin (BSA). Annexin V‐fluorescein isothiocyanate (FITC) (ITK‐Diagnostics, Uithoorn, the Netherlands) was added to RPMI‐1640 (gibco, Carlsbad, CA, USA) supplemented with 5 mM CaCl2. Samples were incubated for 15 min in the dark on ice. One μl of propidium iodine (Life Technologies, Paisley, UK) per sample was added and incubated for another 5 min on ice.
Phosphorylated STAT‐1 and STAT‐3 were measured in freshly isolated PBMCs after 15 min stimulation with IFN‐γ (50 ng/ml) or IL‐6 (20 ng/ml). Cells were fixed and permeabilized using BD Fix&Perm solution (BD Biosciences, San Jose, CA, USA) according to instructions provided by the manufacturer and stored in 100% methanol at –20°C for at least 12 h. Before staining cells with pSTAT‐1‐phycoerythrin (PE) or phosphorylated (p)STAT‐3‐PE (1 : 10, eBioscience, San Diego, CA, USA) in combination with CD45‐knockout (KO) (1 : 10, Beckman Coulter, Brea, CA, USA) and CD4‐allophycocyanin (APC) (1 : 5 ITK), samples were washed twice in PBS containing 1% BSA.
For intracellular detection of IFN‐γ and IL‐17, freshly isolated PBMCs were treated with HDACi (1 h) prior to general activation by phytohaemagglutinin (PHA) (10 μg/ml, 4 h). After stimulation, all samples were fixed and permeabilized using BD Fix&Perm solution, as recommended by the manufacturer. Cells were stained with IFN‐γ‐PE (1 : 80, ImmunoSource, Schilde, Belgium) and IL‐17‐FITC (1:250, ImmunoSource) to detect intracellular cytokines. The gating strategy is shown in the Supporting information (Fig. S1). Samples were measured on the FC500 (Beckman Coulter) using CXP software.
Statistics
Statistics were performed with Prism Graph Pad version 5.0 software, using a Wilcoxon signed‐rank test or two‐way analysis of variance (anova) where applicable. All n numbers provided refer to the number of independent donors used. A P‐value < 0·05 (*) was considered statistically significant, (**)P < 0·01, (***)P < 0·001. Data represent mean ± s.e.m. or median, as indicated.[
Results
HDAC inhibitors down‐regulate C. albicans‐induced TNF‐α
Healthy immune cells produce proinflammatory cytokines when they encounter C. albicans. In order to investigate the impact of HDAC inhibitors on cytokine production, PBMCs isolated from buffy coats were stimulated for 24 h in the presence of different HDAC inhibitors (1000–1 nM) and heat‐killed C. albicans. As a broad HDAC inhibitor, panobinostat interferes strongly with cytokine production normally produced in response to C. albicans, especially TNF‐α (Fig. 1). In contrast, IL‐1β (Fig. 1 ) and IL‐6 (Fig. 1 ) release is strongly impaired only at high concentrations of panobinostat. Entinostat, which selectively inhibits class I HDACs, attenuated TNF‐α secretion induced by C. albicans at all concentrations tested (Fig. 1). No effect could be detected on IL‐1β production, whereas IL‐6 showed a trend similar to that of TNF‐α; however, these changes did not reach statistical significance (Fig. 1). RGFP966, a specific HDAC3 inhibitor, did not significantly alter any of the cytokines induced by C. albicans. Production of TNF‐α was slightly decreased at 1000 and 100 nM (Fig. 1), but no changes could be observed for production of IL‐1β or IL‐6.
Figure 1.
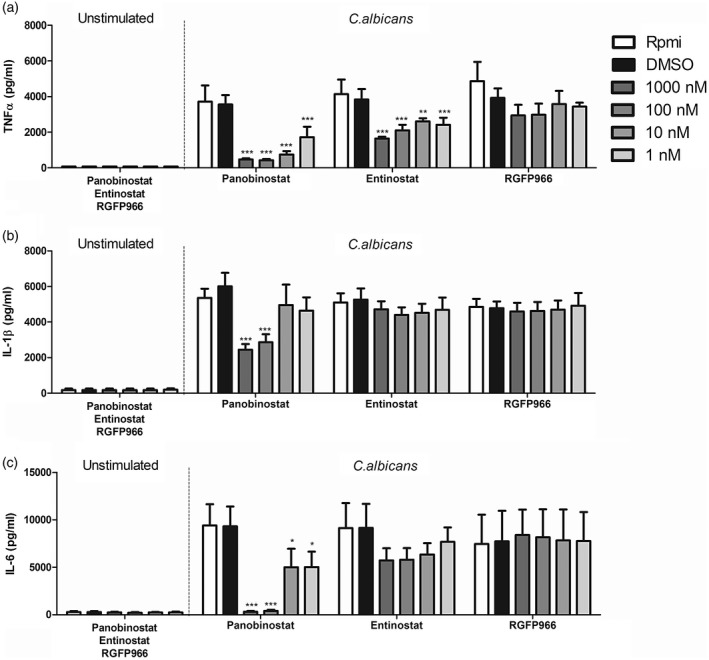
Histone deacetylase (HDAC) inhibitors modulate proinflammatory immune response to Candida albicans. (a–c) Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats provided by Sanquin Blood Bank, Nijmegen, the Netherlands. Cells were preincubated for 1 h with three different HDAC inhibitors and then stimulated for 24 h in the presence or absence of heat‐killed C. albicans conidia [1 × 106 colony‐forming units (CFU)/ml]. Supernatants were collected and stored at –20ᵒC until tumour necrosis factor (TNF)‐α (a), interleukin (IL)‐1β (b) and IL‐6 (c) induction of panobinostat, etinostat and RGFP966 were measured by enzyme‐linked immunosorbent assay (ELISA) [mean ± standard error of the mean (s.e.m.), n = 5, *P < 0·05, **P < 0·01, two‐way analysis of variance (anova) with Bonferroni post‐test].
Anti‐inflammatory cytokines are strongly down‐regulated in the presence of particular HDACs
To assess the full effect of HDAC inhibitors on early innate immune responses, we also examined differences in the secretion of potent anti‐inflammatory cytokines IL‐1Ra (Fig. 2) and IL‐10 (Fig. 2). Panobinostat inhibited IL‐1Ra as well as IL‐10 release at concentrations of 1000 and 100 nM. Lower concentrations effectively decreased IL‐1Ra release, but did not affect IL‐10 levels. Entinostat diminished production of IL‐1Ra at 1000 nM, however, did not alter anti‐inflammatory cytokine production significantly at lower concentrations. Candida‐induced IL‐10 tended to be slightly lower in the presence of 1000 nM entinostat. Incubation with RGFP966 did not significantly change IL‐1Ra or IL‐10 production, but showed a trend towards increase of IL‐10.
Figure 2.
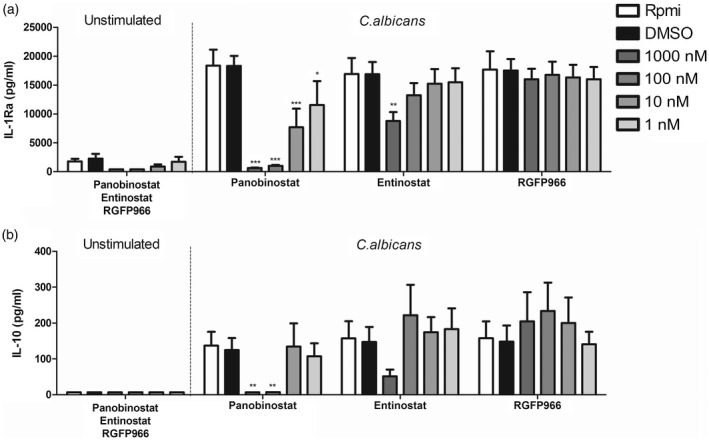
Panobinostat disrupts anti‐inflammatory response to Candida albicans. (a,b) Buffy coat peripheral blood mononuclear cells (PBMCs) were isolated, cells were pretreated with panobinostat, etinostat and RGFP966 (1 h) and then incubated for 24 h with heat‐killed C. albicans conidia [1 × 106 colony‐forming units (CFU)/ml]. Supernatants were collected and stored at –20ᵒC until interleukin (IL)‐1Ra (a) and IL‐10 (b) induction was measured by enzyme‐linked immunosorbent assay (ELISA) [mean ± standard error of the mean (s.e.m.), n = 5 *P < 0·05, **P < 0·01, two‐way analysis of variance (anova) with Bonferroni post‐test].
Panobinostat compromises cell viability
To exclude cell death as the underlying cause of differential cytokine release, PBMC viability was assessed using concentrations of 1000 and 100 nM annexin V/PI staining using flow cytometry. No differences in any conditions tested could be observed for entinostat and RGFP966, whereas an increase of 10% (24 h stimulation) (Fig. 3) up to 25% cell death (6 days stimulus + 24 h HDACi) (Supporting information, Fig. S2) was observed in panobinostat‐treated samples. Cytotoxicity assays measuring lactate dehydrogenase (LDH) in cell culture supernatants indicated no significant differences between HDAC inhibitor‐treated and ‐untreated cells (Fig. 3). Cell death analysis of cells derived from CMC patients revealed similar results (Supporting information, Fig. S2)
Figure 3.
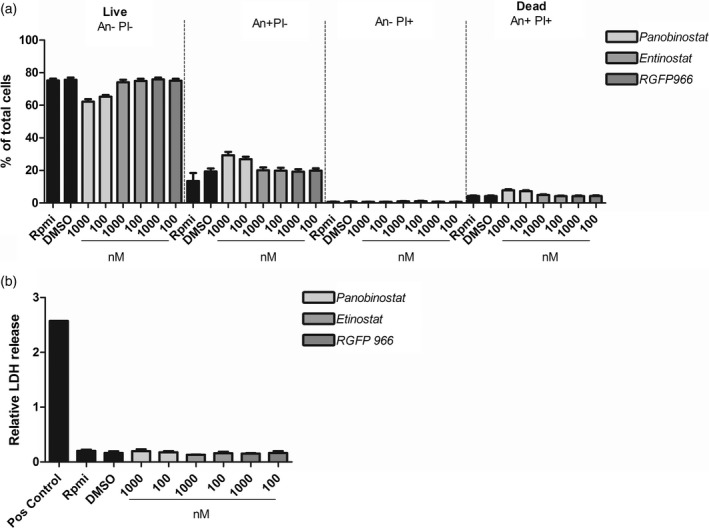
Peripheral blood mononuclear cell (PBMC) viability. (a) Viability of PBMCs in experimental settings was assessed using flow cytometry. Here, PBMCs stimulated for 24 h were collected and stained with annexin V‐fluorescein isothiocyanate (FITC) and propidium iodide (PI). (b) Supernatants from the same experiment were used to assess cytotoxicity/membrane damage based on lactate dehydrogenase (LDH) release [mean ± standard error of the mean (s.e.m.), n = 4, *P < 0·05, **P < 0·01, Wilcoxon’s signed‐rank test].
S. aureus and C. albicans‐induced cytokine profiles are modulated similarly in the presence of HDACi
In order to assess whether the effect of HDAC inhibitors on innate immune responses are specific to fungal pathogens, the same HDAC inhibitors were tested for a CMC‐relevant bacterial pathogen, S. aureus. Similar to C. albicans, TNF‐α levels produced after 24 h of S. aureus challenge were reduced in the presence of panobinostat, most prominently at high concentrations (1000–100 nM) (Fig. 4). Production of IL‐1β also tended to be lower at 1000–100 nM panobinostat; however, this effect was not statistically significant (Fig. 4). Production of IL‐6 was strongly decreased when high concentrations of panobinostat were used, but this effect was lost at lower concentrations (Fig. 4). The pattern of inhibition at high concentrations was also seen for production of IL‐1Ra and IL‐10 (Fig. 4). Entinostat reduced cytokines levels similarly to that observed when C. albicans was used as a stimulus. TNF‐α secretion was strongly impaired; however only at 1000–100 nM (Fig. 4).The other innate cytokines measured followed the same trend; however, the changes did not reach statistical significance (Fig. 4). No clear effect was observed on any cytokine tested in the presence of RGPF966 (Fig. 4). At a concentration of 1000 nM, RGFP966 resulted in a trend towards reduced TNF‐α and increased IL‐10 secretion after S. aureus challenge; however, the overall effect was not statistically significant.
Figure 4.
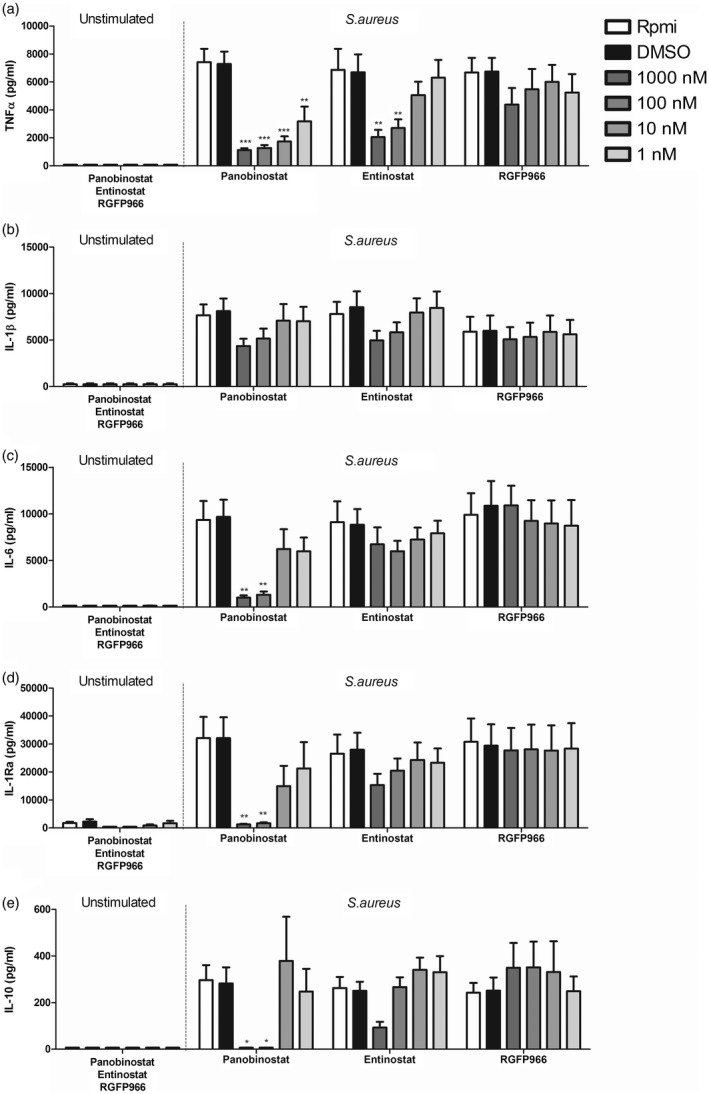
Histone deacetylase (HDAC) inhibitors also affect early immune responses to Staphylococcus aureus. (a–e) As in previous experiments, peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats. Cells were preincubated with three different HDAC inhibitors (1 h) and subsequently incubated for 24 h either in the presence or absence of heat‐killed S. aureus [1 × 107 colony‐forming units (CFU)/ml]. Supernatants were collected and stored at –20ᵒC until tumour necrosis factor (TNF)‐α (a), interleukin (IL)‐1β (b), IL‐6 (c), IL‐1Ra (d) and IL‐10 (e) levels were measured [mean ± standard error of the mean (s.e.m.), n = 5, *P < 0·05, **P < 0·01, two‐way analysis of variance (anova) with Bonferroni post‐test].
Entinostat modulates adaptive immunity in healthy control cells
Loss of functional Th17 cells is an immunological hallmark of CMC. Absence of IL‐17 has been reported to be one of the major factors causing insufficient anti‐fungal defence [15]. When investigating adaptive cytokines produced in response to C. albicans and S. aureus in the presence of entinostat, we observed a decrease of C. albicans induced IFN‐γ (Fig. 5) and IL‐17 (Fig. 5) production, while there was a slight increase of IL‐22 at 1000 nM entinostat (Fig. 5). Compared to C. albicans, S. aureus elicited a much weaker IFN‐γ response, which remained similar in the presence of entinostat (Fig. 5). IL‐17 was not induced by S. aureus alone, and also not in the presence of entinostat (Fig. 5). IL‐22 secretion showed a very subtle increase when 1000 nM entinostat was present (Fig. 5).
Figure 5.
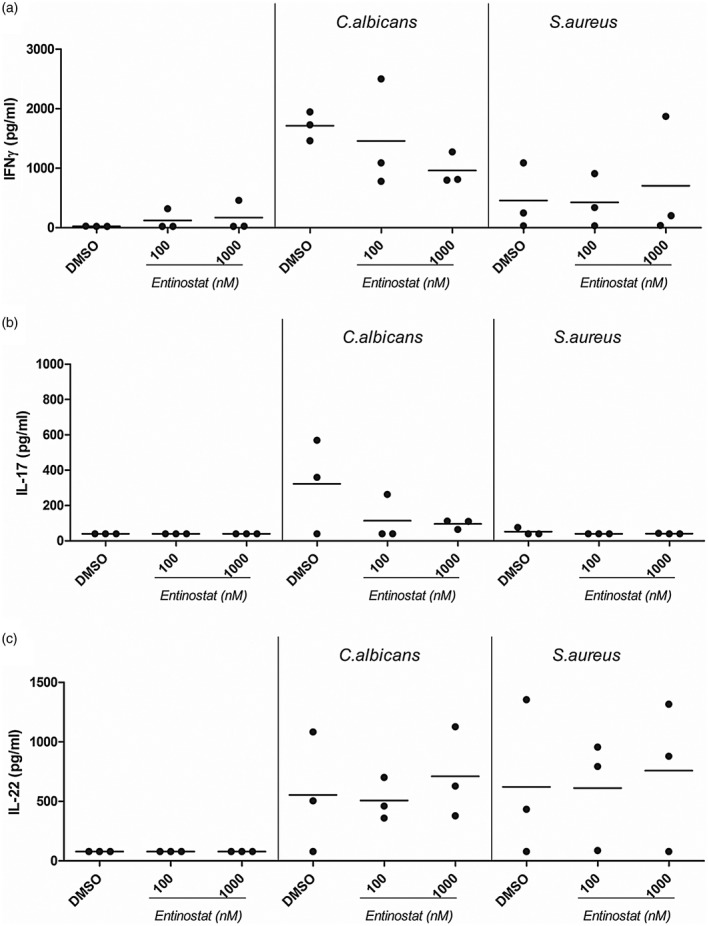
Entinostat differentially modulates adaptive cytokine immune responses to Staphylococcus aureus in healthy control cells. (a–c) Peripheral blood mononuclear cells were freshly isolated from healthy controls. Cells were incubated for 6 days in the presence or absence of Candida albicans [1 × 106 colony‐forming units (CFU)/ml] or heat‐killed S. aureus (1 × 107 CFU/ml). At day 6, supernatants were removed and replaced with freshly prepared stimuli (C. albicans or S. aureus) in combination with or without entinostat. Cells were maintained in these conditions for an additional 24 h before supernatants were collected and stored at –20ᵒC until (a) interferon (IFN)‐γ, (b) interleukin (IL)‐17 or (c) IL‐22 production was measured by enzyme‐linked immunosorbent assay (ELISA) (median, n = 3, no statistical analysis was performed).
Pretreatment of PBMCs with RGFP966 did not change IFN‐γ (Fig. 6). For IL‐17 (Fig. 6), only one of three donors showed a slight increase of IL‐17 in the presence of 1000 nM RGFP966 after C. albicans stimulation. S. aureus stimulated cells did not produce IL‐17, regardless of the presence or absence of RGFP966 (Fig. 6). C. albicans‐induced IL‐22 secretion was slightly increased in two of three donors when cell were pretreated with RGFP966; however, this effect was only seen for one donor after S. aureus challenge (Fig. 6). All adaptive cytokines in PBMCs treated with panobinostat (Supporting information, Fig. S3) were strongly decreased.
Figure 6.
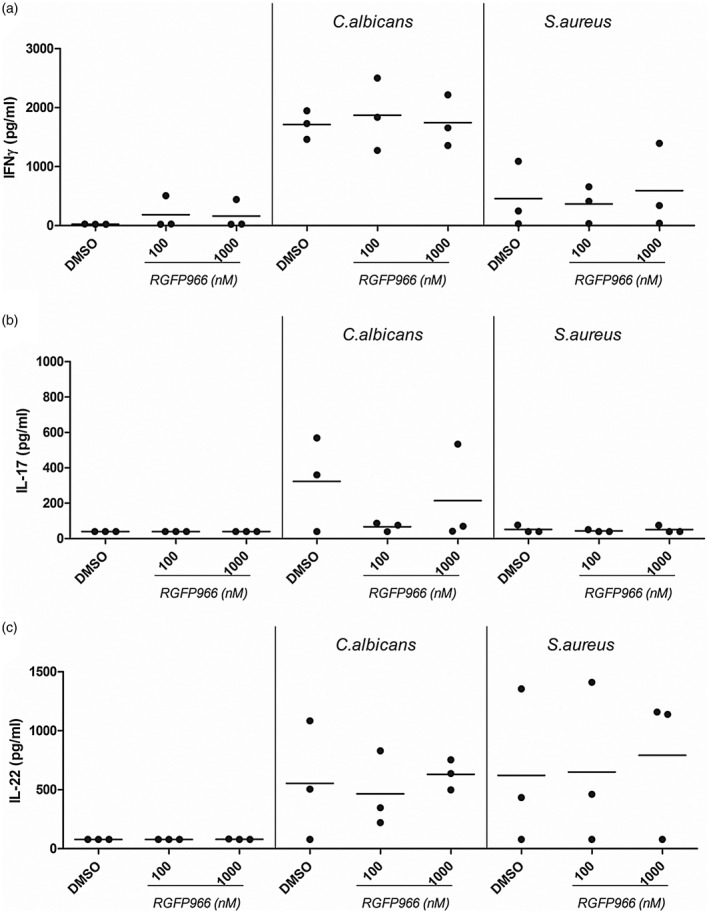
RGFP966 does not modulate adaptive cytokine immune responses to Candida albicans or Staphylococcus aureus in healthy control cells. (a–c) Peripheral blood mononuclear cells (PBMCs) were freshly isolated from healthy controls. Cells were incubated for 6 days in the presence or absence of C. albicans [1 × 106 colony‐forming units (CFU)/ml] or heat‐killed S. aureus (1 × 107 CFU/ml). At day 6, supernatants were removed and replaced with freshly prepared stimuli (C. albicans or S. aureus) in combination with or without RGFP966. Cells were maintained in these conditions for an additional 24 h before supernatants were collected and stored at –20ᵒC until (a) interferon (IFN)‐γ, (b) interleukin (IL)‐17 or (c) IL‐22 production was measured by enzyme‐linked immunosorbent assay (ELISA) (median, n = 3, no statistical analysis was performed).
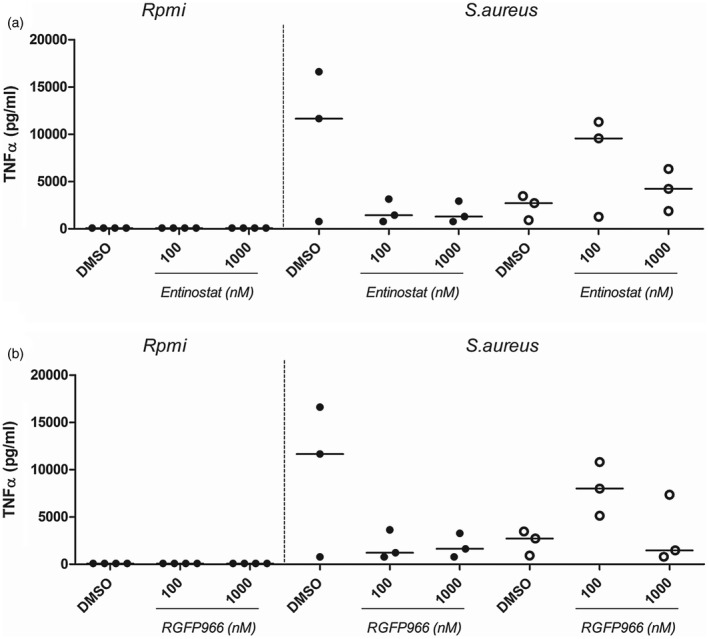
Entinostat and RGFP966 increase TNF‐a responses in STAT‐1 GOF cells
In order to predict possible outcomes for patient treatment with HDAC inhibitors, we isolated PBMCs from STAT‐1 GOF patients and measured cytokine responses. Because of the strong inhibitory effect on cytokine production and increased apoptosis observed for panobinostat, we decided to focus upon entinostat and RGFP966 instead. Additionally, C. albicans‐ and S. aureus‐stimulated cells responded very similarly to HDACi treatment, therefore we chose to only use S. aureus stimulation to assess how HDAC inhibitors modulate innate cytokine profiles in STAT‐1 GOF cells. Both entinostat (Fig. 7) and RGFP966 (Fig. 7) augmented TNF‐α responses towards S. aureus in STAT‐1 GOF cells. This effect is different compared to healthy control cells, where both HDAC inhibitors lowered TNF‐α responses. Both entinostat and RGFP966 lowered IL‐1β and IL‐6 responses dose‐dependently in healthy controls and patients (Supporting information, Fig. S4). Interestingly, IL‐10 production was increased dose‐dependently by RGFP966 in patients in response to S. aureus, whereas IL‐1Ra was unaffected. Innate cytokines were also measured in response to panobinostat; however, almost all were inhibited dramatically in healthy volunteers as well as in patients (Supporting information, Fig. S5).
Figure 7.
The tumour necrosis factor (TNF)‐α response of signal transducer and activator of transcription 1 (STAT‐1) gain‐of‐function (GOF) cells towards Staphylococcus aureus is increased in the presence of entinostat and RGFP966. Peripheral blood mononuclear cells were freshly isolated from healthy controls (n = 3 black) or STAT‐1 GOF patients (n = 3 clear). Cells were pretreated with or without entinostat (1 h) and subsequently incubated for 24 h in the presence or absence of heat‐killed S. aureus [1 × 106 colony‐forming units (CFU)/ml]. Supernatants were collected and stored at –20ᵒC until TNF‐α was measured by enzyme‐linked immunosorbent assay (ELISA) (no statistical analysis was performed).
Entinostat and RGFP966 increase IL‐22 in response to S. aureus in STAT‐1 GOF cells
In order to assess the effect of entinostat and RGFP966 on adaptive immune responses, we measured IFN‐γ, IL‐17 and IL‐22 production. In response to C. albicans stimulation, the presence of entinostat had almost no effect (Fig. 8). However, in response to S. aureus, entinostat increased IFN‐γ (100 and 1000 nM) as well as IL‐22 production (100 nM) (Fig. 8,c), while IL‐17 concentrations remained undetectable (Fig. 8). Cells treated with RGFP966 showed the same trend with increased IFN‐γ and IL‐22 at 100 nM (Fig. 8). Panobinostat decreased all cytokines measured (Supporting information, Fig. S3). In addition to secreted protein levels, we also measured intracellular IFN‐γ and IL‐17. PBMCs were stimulated with a non‐specific stimulus (PHA) known to generally activate T cells and thus induce cytokine production. Entinostat and RGFP966 influenced intracellular cytokine accumulation only mildly in PHA activated cells. While IFN‐γ was slightly decreased, increasing numbers of IL‐17 positive cells were detected (Supporting information, Fig. S7).
Figure 8.
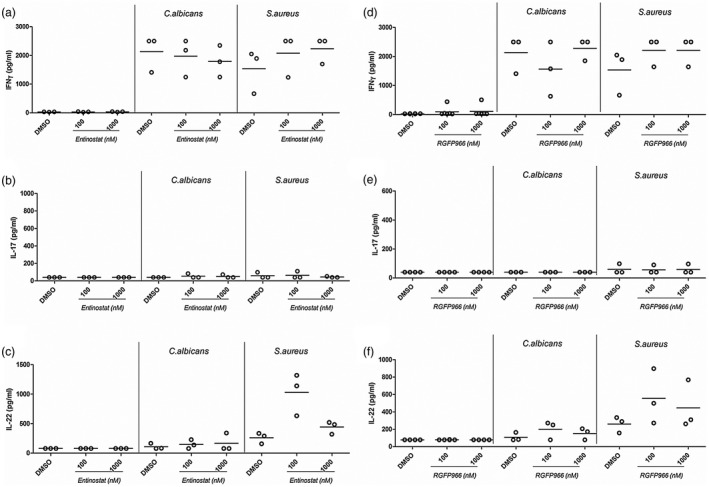
Interleukin (IL)‐22 secretion upon long‐term Staphylococcus aureus stimulation is increased in the presence of 100 nM entinostat. (a–c) Peripheral blood mononuclear cells were freshly isolated from signal transducer and activator of transcription 1 (STAT‐1) gain‐of‐function (GOF) patients. Cells were incubated for 6 days in the presence or absence of Candida albicans [1 × 106 colony‐forming units (CFU)/ml] or heat‐killed S. aureus (1 × 107 CFU/ml). At day 6, supernatants were removed and replaced with freshly prepared stimuli (C. albicans or S. aureus) in combination with or without entinostat. Cells were maintained in these conditions for an additional 24 h before supernatants were collected and stored at –20ᵒC until (a) interferon (IFN)‐γ, (b) IL‐17 or (c) IL‐22 production was measured by enzyme‐linked immunosorbent assay (ELISA) (median, n = 3, no statistical analysis was performed).
Entinostat and RGFP966 lower intracellular pSTAT‐1 levels while maintaining normal pSTAT‐3
While STAT‐1 is crucial for Th1 development, STAT‐3 is involved in Th17 differentiation by mediating IL‐6 and IL‐23 signalling. Therefore, we aimed to assess the affect of HDAC inhibitors on the activation of these molecules to gain more insight into the mode of action of entinostat and RGFP966. Data show a decreased IFN‐γ‐induced STAT‐1 phosphorylation in the presence of both entinostat (Fig. 9) and RGFP966 (Fig. 9), while activated phosphorylated STAT‐3 levels remained stable with or without treatment (Fig. 9). Panobinostat showed the same trend (Supporting information, Fig. S6).
Figure 9.
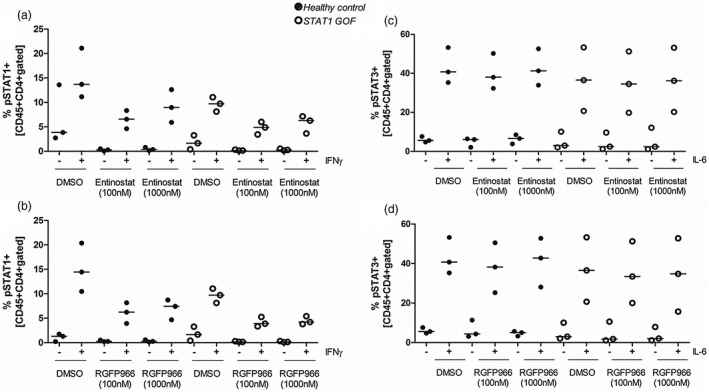
Entinostat lowers phosphorylated signal transducer and activator of transcription 1 (STAT‐1) but not phosphorylated STAT‐3 levels within CD4+ cells. (a,b) Intracellular levels of phosphorylated STAT‐1 (pSTAT‐1) and (c,d) phosphorylated STAT‐3 (pSTAT‐3) were measured in freshly isolated peripheral blood mononuclear cells of healthy controls and STAT‐1 gain‐of‐function (GOF) patients. Peripheral blood mononuclear (PBMCs) were pretreated for one hour with entinostat, RGFP966 or vehicle (DMSO) control. Subsequently, cells were stimulated with interferon (IFN)‐γ (50 ng/ml) or interleukin (IL)‐6 (20 ng/ml) for 15 min to induce STAT‐1 or STAT‐3 phosphorylation, respectively. All samples were stained for CD45 and CD4 in combination with either anti‐pSTAT‐1 or anti‐pSTAT‐3 and measured by flow cytometry (median, n = 3 for patients and controls, no statistical analysis was performed). (a) Entinostat reduces IFN‐γ‐induced pSTAT‐1 in healthy and STAT‐1 GOF cells. (b) No effect could be observed of STAT‐3 phosphorylation (median, n = 3 for patients and controls, no statistical analysis was performed).
Discussion
Infectious complications in patients suffering from CMC due to STAT‐1 GOF mutation are caused mainly by C. albicans and S. aureus [16]. HDACi treatment has been suggested as possible treatment for STAT‐1 GOF CMC patients; however, studies directly investigating potential benefits or complications associated with HDACi treatment in CMC patients in relation to infection are rare. Here we assess how different HDAC inhibitors modulate the innate and adaptive immune responses of healthy and STAT‐1 GOF PBMCs in response to the two micro‐organisms associated most frequently with CMC. We demonstrate that HDACi influence general pro‐ and anti‐inflammatory cytokine production of PBMCs in response to C. albicans and S. aureus. Effects were dependent upon the specific target of the HDACi investigated. Panobinostat, a non‐specific panHDACi, decreased production of all cytokines measured in healthy controls as well as patient cells: TNF‐α, IL‐1β, IL‐1Ra, IL‐6, IL‐10, IFN‐γ, IL‐17 and IL‐22. Notably, the annexin V staining showed that a 10–25% increase in cell death was dependent upon the experimental setting. In comparison, cells in the presence of entinostat or RGFP966 did not show differences in apoptosis compared to untreated cells in any of the conditions tested. Entinostat and RGFP699 decreased TNF‐α production in healthy cells, while other cytokine responses remained largely unaffected. Most interestingly, entinostat and RGFP966 appeared to have a different effect on STAT‐1 GOF cells, where we demonstrated increased TNF‐α production in response to S. aureus stimulation. When assessing the effect of HDACi on crucial anti‐fungal responses, Th1 and Th17, both entinostat and RGFP966 were able to restore defective IL‐22 production in cells from patients with STAT‐1 GOF. Moreover, intracellular staining of phosphorylated transcription factors STAT‐1 and STAT‐3 showed that these HDAC inhibitors down‐regulated pSTAT‐1 levels while maintaining normal pSTAT‐3 levels, which might play a role in the increased IL‐22 production.
The effect of HDACi on immune cell function has been reviewed previously in detail by Kroesen et al. [17]. In line with our observations, panHDAC inhibitors were described to generally suppress cytokine production by macrophages, dendritic cells (DCs) and/or PBMCs upon Toll‐like receptors (TLR)‐3 and ‐4 (panobinostat) [18] or TLR‐2, ‐3 and ‐9 (givinostat) [19] engagement. We confirmed these results by showing that panobinostat reduces proinflammatory cytokine production (TNF‐α, IL‐6 and IL‐1β) after C. albicans challenge. Panobinostat also decreased the anti‐inflammatory cytokines IL‐1Ra and IL‐10, further supporting general cytokine inhibition. Dysregulated immune profiles have long been described in CMC patients, specifically in response to C. albicans [20]. While panobinostat is able to down‐regulate IFN‐γ responses which could, theoretically, promote Th17 cell development, the somewhat drastic inhibitory effect and overall dampening of all cytokine responses is likely to cause secondary problems with a broad array of additional infections. Additionally, increased cell death percentages raise concerns about adverse effects due to cytotxicity and thus advocate against panobinostat as treatment of choice in CMC patients.
The HDACi class I inhibitor entinostat has been reported to reduce inflammation and bone loss in an experimental murine arthritis model [21]. Additionally, entinostat proved to be effective in mitigating the phenotypes of bacterial infection models with salmonella and Escherichia coli [22] and demonstrated beneficial effects in a rabbit in‐vivo model of cholera infection [23]. Often studied at single loci, indirect enrichment of histone acetylation and concomitant transcriptional activation is the paradigmatic mechanism of action. However, the biology of lysine acetylation is much more complex. Genomewide analyses also reveal broad deacetylation conferred by HDAC inhibitors in vascular endothelial cells, mediated by the loss of EP300/CREBBP binding [24]. Such findings, as well as the transcription‐modulating effects of lysine acetylation on transcription factors, highlight the fact that certain acetylation events regulating gene expression are in fact chromatin‐independent. In support of this hypothesis, we demonstrate that only STAT‐1, but not STAT‐3, phosphorylation is modified by entinostat. It is tempting to speculate that this difference in regulation might confer beneficial effects to CMC patients when treated with entinostat: lowering STAT‐1 phosphorylation could attenuate IFN‐γ responses and therefore support STAT‐3‐driven Th17 development. In line with these data we observed increased IL‐22 production by S. aureus in the presence of entinostat in patients with STAT‐1 GOF, which supports the hypothesis that increased STAT‐1 phosphorylation can suppress IL‐22 responses. We did not observe any increase in IL‐17 production in STAT‐1 GOF cell in the presence of entinostat; however, results for intracellular cytokines show a trend towards increased IL‐17, while IFN‐γ is down‐regulated. These data support the notion that entinostat may partially restore defective Th17 responses in STAT‐1 GOF cells.
A recent report elucidates anti‐microbial protein up‐regulation via STAT‐3 activation as one of the mechanisms by which entinostat aids to combat bacterial infections [25]. Moreover, specific blocking of HDAC1 with siRNA in cells of patients with STAT‐1 GOF restored defective STAT3 signalling associated with deficient Th17 responses. Therefore, specific HDAC class I inhibition poses a possible treatment option for CMC due to STAT‐1 GOF. However, while increasing TNF‐α in STAT‐1 GOF cells, which is a relevant protective cytokine in the host defence against S. aureus and C. albicans, entinostat in fact down‐regulated IL‐6 and IL‐1β production in STAT‐1 GOF cells. Therefore, although entinostat seems to have beneficial effects on IL‐22 production and decreases STAT‐1 phosphorylation, HDAC class I inhibition might also weaken overall protective innate immune responses.
RGFP966 is a specific HDAC3 inhibitor linked to down‐regulation of phagocytic function of neutrophils in an Acinetobacter actinomycetemcomitans in‐vitro assay, whereas cytokine production by neutrophils was largely unaffected, except for lower levels of TNF‐α [26]. Earlier this year another study assessed the effect of RGFP966 on primary microglia and found that immune pathways linked to the TLR signalling and STAT‐3 and STAT‐5 were impacted strongly by RGFP966 [27]. However, the importance for HDAC3 in cytokine production of myeloid and lymphoid cells in the context of Candida or S. aureus is yet to be established. In healthy control cells, we observed minor effects of HDAC3 inhibition on the cytokine production of PBMCs after C. albicans or S. aureus, which basically followed the same trend observed for entinostat. Strikingly, in STAT‐1 GOF cells, RGFP966 heightened TNF‐α and IL‐22 production specifically in response to S. aureus stimulation. Our results suggest that targeting HDAC3 might thus have protective effects on S. aureus host defence by increasing both innate and adaptive immune responses of PBMCs, and in this regard would be a candidate to explore in CMC. However, we have reported previously that HDAC3 inhibition did not restore STAT‐3‐dependent IL‐22 production in contrast to HDAC1 and HDAC2 inhibition [11]. These differences might be due to the stimulus used. In current study we used the pathogen, and previously we used the cytokine IL‐23 to induce IL‐22.
Mechanistically, we show that IFN‐γ‐induced pSTAT‐1 levels are markedly decreased in the presence of either entinostat or RGFP966, while pSTAT‐3 remained stable. As increased pSTAT‐1 is a hallmark in the pathophysiology of STAT‐1 GOF, the dampening effect of the HDAC inhibitors tested in this study provides an additional rationale for a possible CMC treatment. Moreover, we show that pSTAT‐3 induction is not inhibited by entinostat and RGFP966, theoretically allowing for Th17 development under treatment conditions.
Taken together, our study summarizes the effects of different HDAC inhibitors on healthy and STAT‐1 GOF PBMCs in response to C. albicans and S. aureus, the two most common micro‐organisms associated with CMC. We show that panHDAC inhibitors have a major impact upon innate and adaptive immune response, whereas class I inhibitors (entinostat) or RGFP966 (HDAC3 inhibitor) show differential effects. Specific HDACi class I inhibition has been already been proposed to be beneficial due to its ability to restore STAT‐3 defects. We support these data by demonstrating that both entinostat and RGFP966 inhibit cytokine production only very selectively, while restoring some immunological defects in CMC. Therefore, entinostat and RGFP966 appear to be the best candidates to further explore in patients with CMC due to STAT‐1 GOF.
However, even if inhibitory effects on most cytokines are minimal, possible consequences need to be taken into account. Complications due to secondary infection have to be anticipated and need to be monitored closely when such a strategy is explored. In addition, tissue‐specific roles of individual HDACs must be considered in context of therapeutic interventions [2].
Disclosures
None.
Author contributions
B. R. performed experiments and wrote the manuscript; X. W. designed the experiment; S. T. K. edited the manuscript; L. A. B. J. and M. G. N. provided financial support and intellectual input; F. v.d. V. eesigned the study, edited the manuscript and provided financial support.
Supporting information
Fig. S1. Freshly isolated peripheral blood mononuclear cells were stained with CD45‐KO, CD4‐APC pSTAT1‐PE or pSTAT3‐PE. The debris was removed from the cell population based on SSc and FSc. From the cell population, CD45+ cells were defined as leukocytes and within this population CD4+ cells were identified. Finally, percentages of pSTAT1 or pSTAT3 positive cells were assessed as shown above.
Fig. S2. The viability of peripheral blood mononuclear cells was assessed after 6 days of stimulation followed by 24 hours of co‐incubation of fresh stimuli (Rpmi, C. albicans or S. aureus) and HDAC inhibitors. (a‐c) Healthy control cells showed increased apoptosis in the presence of Panobinostat. Entinostat as well as RGFP966 do not induce apoptosis when compared to vehicle (DMSO) controls. (d‐f) STAT1 GOF cells behave similar to healthy control cells. Panobinostat induced cell death, whereas Entinostat and RGFP966 are comparable to DMSO control. In general, patient cells showe slightly higehr percentages of apoptotic cells compared to healthy control cells.
Fig. S3. Peripheral blood mononuclear cells were freshly isolated from healthy controls (black) or STAT1 GOF patients (clear). Cells were incubated for 6 days in the presence or absence of C. albicans (1*106 CFU/ml) or heat killed S. aureus (1*107 CFU/ml). At day 6, supernatants were removed and replaced with freshly prepared stimuli (C. albicans or S. aureus) in combination with or without Panobinostat. Cells were kept in these conditions for an additional 24 hours before supernatants were collected and stored at ‐20ᵒC until (a and b) IFN‐γ, (c and d) IL‐17 or (e and f) IL‐22 induction was measured by ELISA (median, n = 3 for patients and controls, no statistical analysis was performed).
Fig. S4. Peripheral blood mononuclear cells were freshly isolated from healthy controls (black) or STAT1 GOF patients (clear). Cells were pre‐incubated for one hour with Entinostat (a‐d) or RGFP966 (e‐h) and subsequently stimulated for an additional 24 hours in the presence or absence of heat killed S. aureus (1*107 CFU/ml). Supernatants were collected and stored at ‐20ᵒC until IL‐1β, IL‐6, IL‐1Ra and IL‐10 production were measured by ELISA (negative controls for controls and patients were pooled. Median, n = 3 for patients and controls, no statistical analysis was performed).
Fig. S5. Peripheral blood mononuclear cells were freshly isolated from healthy controls (black) or STAT1 GOF patients (clear). Cells were pre‐incubated for one hour with Panobinostat and the stimulated with or without heat killed S. aureus (1*107 CFU/ml) for 24 hours. Supernatants were collected and stored at ‐20ᵒC until TNF‐α (a), IL‐6 (b), IL‐1β (c), IL‐1Ra (d) and IL‐10 (e) production were measured by ELISA (median, n = 3 for patients and controls, no statistical analysis was performed).
Fig. S6. Intracellular levels of (a) phosphorylated STAT1 (pSTAT1) upon IFN‐γ (50 ng/ml) and (b) phosphorylated STAT3 (pSTAT3) upon IL‐6 (20 ng/ml) were measured in freshly isolated peripheral blood mononuclear cells of healthy controls (black) and STAT1 GOF patients (clear). Peripheral blood mononuclear cells were pre‐treated for one hour with Panobinostat or DMSO control. All samples were stained for CD45 and CD4 in combination with either anti‐pSTAT1 or anti‐pSTAT3 and measured by flow cytometry (median, n = 3 for patients and controls, no statistical analysis was performed (median, n = 3 for patients and controls, no statistical analysis was performed).
Fig. S7. (a‐c) Intracellular levels of IFN‐γ, (d‐f) IL‐17 or (g‐i) IL‐17/ IFN‐γ were measured in freshly isolated peripheral blood mononuclear cells of healthy controls and STAT1 GOF patients. Cells were pre‐incubated for one hour with HDAC inhibitors and subsequently all samples were activated with phytohaemagglutinin (PHA) (10 μg/ml) for 4 hours. Cells were collected immediately and stained for intracellular IL‐17 and IFN‐γ. All samples were measured on the same day by flow cytometry (median, n = 3 for patients and controls, no statistical analysis was performed).
Acknowledgements
This project was supported by ZonMW under the name EURO‐CMC frame of E‐Rare‐2, the ERA‐Net for Research on Rare Diseases. L. A. B. J. and M. G. N. received funding from the European Union Horizon 2020 research and innovation program under grant agreement no. 667837, and the IN‐CONTROL grant from the Heart Foundation Netherlands. M. G. N. is supported by an ERC Consolidator grant (no. 310372) and a Spinoza grant of the Netherlands Organization for Scientific Research. X. W. was supported by the National Natural Science Foundation of China (81501727).
References
- 1. Okada S, Puel A, Casanova JL, Kobayashi M. Chronic mucocutaneous candidiasis disease associated with inborn errors of IL‐17 immunity. Clin Transl Immunology 2016; 5:e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Keating ST, El‐Osta A. Epigenetics and metabolism. Circ Res 2015; 116:715–36. [DOI] [PubMed] [Google Scholar]
- 3. Zhuang S. Regulation of STAT signaling by acetylation. Cell Signal 2013; 25:1924–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aung HT, Schroder K, Himes SR et al LPS regulates proinflammatory gene expression in macrophages by altering histone deacetylase expression. FASEB J 2006; 20:1315–27. [DOI] [PubMed] [Google Scholar]
- 5. Yamaguchi T, Cubizolles F, Zhang Y et al Histone deacetylases 1 and 2 act in concert to promote the G1‐to‐S progression. Genes Dev 2010; 24:455–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dequiedt F, Van Lint J, Lecomte E et al Phosphorylation of histone deacetylase 7 by protein kinase D mediates T cell receptor‐induced Nur77 expression and apoptosis. J Exp Med 2005; 201:793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li Z, Zhu WG. Targeting histone deacetylases for cancer therapy: from molecular mechanisms to clinical implications. Int J Biol Sci 2014; 10:757–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abdel‐Hameed EA, Ji H, Shata MT. HIV‐induced epigenetic alterations in host cells. Adv Exp Med Biol 2016; 879:27–38. [DOI] [PubMed] [Google Scholar]
- 9. Yedery RD, Jerse AE. Augmentation of cationic antimicrobial peptide production with histone deacetylase inhibitors as a novel epigenetic therapy for bacterial infections. Antibiotics (Basel) 2015; 4:44–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van de Veerdonk FL, Netea MG. Treatment options for chronic mucocutaneous candidiasis. J Infect 2016; 72 (Suppl):S56–60. [DOI] [PubMed] [Google Scholar]
- 11. Zheng J, van de Veerdonk FL, Crossland KL et al Gain‐of‐function STAT1 mutations impair STAT3 activity in patients with chronic mucocutaneous candidiasis (CMC). Eur J Immunol 2015; 45:2834–46. [DOI] [PubMed] [Google Scholar]
- 12. Becker KL, Rosler B, Wang X et al Th2 and Th9 responses in patients with chronic mucocutaneous candidiasis and hyper‐IgE syndrome. Clin Exp Allergy 2016; 46:1564–74. [DOI] [PubMed] [Google Scholar]
- 13. Srinivas NR. Clinical pharmacokinetics of panobinostat, a novel histone deacetylase (HDAC) inhibitor: review and perspectives. Xenobiotica 2017; 47:354–68. [DOI] [PubMed] [Google Scholar]
- 14. Gore L, Rothenberg ML, O'Bryant CL et al A phase I and pharmacokinetic study of the oral histone deacetylase inhibitor, MS‐275, in patients with refractory solid tumors and lymphomas. Clin Cancer Res 2008; 14:4517–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Netea MG, Joosten LA, van der Meer JW, Kullberg BJ, van de Veerdonk FL. Immune defence against Candida fungal infections. Nat Rev Immunol 2015; 15:630–42. [DOI] [PubMed] [Google Scholar]
- 16. Toubiana J, Okada S, Hiller J et al Heterozygous STAT1 gain‐of‐function mutations underlie an unexpectedly broad clinical phenotype. Blood 2016; 127:3154–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kroesen M, Gielen P, Brok IC, Armandari I, Hoogerbrugge PM, Adema GJ. HDAC inhibitors and immunotherapy; a double edged sword? Oncotarget 2014; 5:6558–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Song W, Tai YT, Tian Z et al HDAC inhibition by LBH589 affects the phenotype and function of human myeloid dendritic cells. Leukemia 2011; 25:161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reddy P, Sun Y, Toubai T et al Histone deacetylase inhibition modulates indoleamine 2,3‐dioxygenase‐dependent DC functions and regulates experimental graft‐versus‐host disease in mice. J Clin Invest 2008; 118:2562–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lilic D, Gravenor I, Robson N et al Deregulated production of protective cytokines in response to Candida albicans infection in patients with chronic mucocutaneous candidiasis. Infect Immun 2003; 71:5690–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cantley MD, Fairlie DP, Bartold PM, Marino V, Gupta PK, Haynes DR. Inhibiting histone deacetylase 1 suppresses both inflammation and bone loss in arthritis. Rheumatology (Oxf) 2015; 54:1713–23. [DOI] [PubMed] [Google Scholar]
- 22. Ariffin JK, das Gupta K, Kapetanovic R et al Histone deacetylase inhibitors promote mitochondrial reactive oxygen species production and bacterial clearance by human macrophages. Antimicrob Agents Chemother 2015; 60:1521–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sarker P, Banik A, Stromberg R, Gudmundsson GH, Raqib R, Agerberth B. Treatment with entinostat heals experimental cholera by affecting physical and chemical barrier functions of intestinal epithelia. Antimicrob Agents Chemother 2017; 61:e02570–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rafehi H, Balcerczyk A, Lunke S et al Vascular histone deacetylation by pharmacological HDAC inhibition. Genome Res 2014; 24:1271–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miraglia E, Nylen F, Johansson K et al Entinostat up‐regulates the CAMP gene encoding LL‐37 via activation of STAT3 and HIF‐1alpha transcription factors. Sci Rep 2016; 6:33274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Correa RO, Vieira A, Sernaglia EM et al Bacterial short‐chain fatty acid metabolites modulate the inflammatory response against infectious bacteria. Cell Microbiol 2017; 19:1–4. [DOI] [PubMed] [Google Scholar]
- 27. Xia M, Zhao Q, Zhang H et al Proteomic analysis of HDAC3 selective inhibitor in the regulation of inflammatory response of primary microglia. Neural Plast 2017; 2017:6237351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Freshly isolated peripheral blood mononuclear cells were stained with CD45‐KO, CD4‐APC pSTAT1‐PE or pSTAT3‐PE. The debris was removed from the cell population based on SSc and FSc. From the cell population, CD45+ cells were defined as leukocytes and within this population CD4+ cells were identified. Finally, percentages of pSTAT1 or pSTAT3 positive cells were assessed as shown above.
Fig. S2. The viability of peripheral blood mononuclear cells was assessed after 6 days of stimulation followed by 24 hours of co‐incubation of fresh stimuli (Rpmi, C. albicans or S. aureus) and HDAC inhibitors. (a‐c) Healthy control cells showed increased apoptosis in the presence of Panobinostat. Entinostat as well as RGFP966 do not induce apoptosis when compared to vehicle (DMSO) controls. (d‐f) STAT1 GOF cells behave similar to healthy control cells. Panobinostat induced cell death, whereas Entinostat and RGFP966 are comparable to DMSO control. In general, patient cells showe slightly higehr percentages of apoptotic cells compared to healthy control cells.
Fig. S3. Peripheral blood mononuclear cells were freshly isolated from healthy controls (black) or STAT1 GOF patients (clear). Cells were incubated for 6 days in the presence or absence of C. albicans (1*106 CFU/ml) or heat killed S. aureus (1*107 CFU/ml). At day 6, supernatants were removed and replaced with freshly prepared stimuli (C. albicans or S. aureus) in combination with or without Panobinostat. Cells were kept in these conditions for an additional 24 hours before supernatants were collected and stored at ‐20ᵒC until (a and b) IFN‐γ, (c and d) IL‐17 or (e and f) IL‐22 induction was measured by ELISA (median, n = 3 for patients and controls, no statistical analysis was performed).
Fig. S4. Peripheral blood mononuclear cells were freshly isolated from healthy controls (black) or STAT1 GOF patients (clear). Cells were pre‐incubated for one hour with Entinostat (a‐d) or RGFP966 (e‐h) and subsequently stimulated for an additional 24 hours in the presence or absence of heat killed S. aureus (1*107 CFU/ml). Supernatants were collected and stored at ‐20ᵒC until IL‐1β, IL‐6, IL‐1Ra and IL‐10 production were measured by ELISA (negative controls for controls and patients were pooled. Median, n = 3 for patients and controls, no statistical analysis was performed).
Fig. S5. Peripheral blood mononuclear cells were freshly isolated from healthy controls (black) or STAT1 GOF patients (clear). Cells were pre‐incubated for one hour with Panobinostat and the stimulated with or without heat killed S. aureus (1*107 CFU/ml) for 24 hours. Supernatants were collected and stored at ‐20ᵒC until TNF‐α (a), IL‐6 (b), IL‐1β (c), IL‐1Ra (d) and IL‐10 (e) production were measured by ELISA (median, n = 3 for patients and controls, no statistical analysis was performed).
Fig. S6. Intracellular levels of (a) phosphorylated STAT1 (pSTAT1) upon IFN‐γ (50 ng/ml) and (b) phosphorylated STAT3 (pSTAT3) upon IL‐6 (20 ng/ml) were measured in freshly isolated peripheral blood mononuclear cells of healthy controls (black) and STAT1 GOF patients (clear). Peripheral blood mononuclear cells were pre‐treated for one hour with Panobinostat or DMSO control. All samples were stained for CD45 and CD4 in combination with either anti‐pSTAT1 or anti‐pSTAT3 and measured by flow cytometry (median, n = 3 for patients and controls, no statistical analysis was performed (median, n = 3 for patients and controls, no statistical analysis was performed).
Fig. S7. (a‐c) Intracellular levels of IFN‐γ, (d‐f) IL‐17 or (g‐i) IL‐17/ IFN‐γ were measured in freshly isolated peripheral blood mononuclear cells of healthy controls and STAT1 GOF patients. Cells were pre‐incubated for one hour with HDAC inhibitors and subsequently all samples were activated with phytohaemagglutinin (PHA) (10 μg/ml) for 4 hours. Cells were collected immediately and stained for intracellular IL‐17 and IFN‐γ. All samples were measured on the same day by flow cytometry (median, n = 3 for patients and controls, no statistical analysis was performed).