

Abstract
Immune cell death caused by neutrophil extracellular traps (NETs), referred to as NETosis, can contribute to the pathogenesis of endotoxemia and organ damage. Although the mechanisms by which infection induces NETosis and how that leads to organ dysfunction remain largely unknown, NET formation is often found following citrullination of histone H3 (CitH3) by peptidylarginine deiminase (PAD). We hypothesized that lipopolysaccharide (LPS)-induced activation of PAD and subsequent CitH3-mediated NET formation increase endothelial permeability and pulmonary dysfunction and, therefore, that inhibition of PAD could mitigate damage and improve survival in lethal endotoxemia. Here, we showed that treatment with YW3-56, a PAD2/PAD4 inhibitor, significantly diminished PAD activation, blocked LPS-induced pulmonary vascular leakage, alleviated acute lung injury, and improved survival in a mouse model of lethal LPS-induced endotoxemia. We found CitH3 in the bloodstream 30 min after intraperitoneal injection of LPS (35 mg/kg) into mice. Additionally, CitH3 production was induced in cultured neutrophils exposed to LPS, and NETs derived from these LPS-treated neutrophils increased the permeability of endothelial cells. However, YW3-56 exposure reduced CitH3 production and NET formation by neutrophils following LPS exposure. Moreover, treatment with YW3-56 decreased the levels of circulating CitH3 and abolished neutrophil activation and NET formation in the lungs of mice with endotoxemia. These data suggest a novel mechanism by which PAD-NET-CitH3 can play a pivotal role in pulmonary vascular dysfunction and the pathogenesis of lethal endotoxemia.
Keywords: Neutrophil extracellular traps, Citrullinated histone H3, Peptidylarginine deiminase, Lipopolysaccharide, Microvascular permeability
1. Introduction
Neutrophils are a host’s first line of defense against microbes and play crucial roles in innate immunity. In response to microbial infection, neutrophils release networks of nuclear DNA and proteins, including histones and enzymes, which are known as neutrophil extracellular traps (NETs) (Brinkmann et al., 2004). The consequence of NET-regulated cell death is called NETosis (Kaplan and Radic, 2012), which generally plays a host-protective role in infection (Fuchs et al., 2007). So far, NETosis has been found in many pathological conditions, including appendicitis (Brinkmann et al., 2004), cystic fibrosis (Young et al., 2011), diabetes (Wong et al., 2015), small vessel vasculitis (Kessenbrock et al., 2009), systemic lupus erythematosus (Villanueva et al., 2011), and thrombosis (Fuchs et al., 2010). In addition, excessive NETs were observed in various organs of a murine sepsis model (Tanaka et al., 2014). However, despite growing evidence of their importance in disease pathology, the role of NETs in the pathogenesis of endotoxemia and sepsis is still not well understood.
It was recently revealed that the citrullination of histone H3 (CitH3) by peptidylarginine deiminase (PAD) serves as a convergence point for many diverse inflammatory signals that trigger NET formation (Neeli et al., 2008). We have reported that CitH3 was released into the circulation of mice three h after intraperitoneal injection of a lethal dose of lipopolysaccharide (LPS) (Li et al., 2011). Moreover, levels of CitH3 in the blood were formed to be significantly associated with the severity of the shock (Li et al., 2011), and the neutralization of circulating CitH3 with specific anti-CitH3 antibody significantly improved survival in a mouse cecal ligation and puncture (CLP) model (Li et al., 2014). These findings suggest that inhibition of PAD activity and subsequent CitH3 production can attenuate the consequences of endotoxemia or sepsis.
To date, five isozymes of PAD, numbered 1, 2, 3, 4 and 6, have been found (Wang and Wang, 2013), but only nuclear-localized PAD2 (Zhang et al., 2012) and PAD4 (Wang et al., 2009) can citrullinate histone H3 to induce NETosis. YW3-56 is a PAD2 and PAD4 inhibitor that was originally found as an anticancer compound with inhibitory activity against PAD2 (IC50: 0.5-1 μm) and PAD4 (IC50: 1-2 μm) (Wang et al., 2012b). However, it had not been determined whether inhibition of PAD2 and PAD4 by YW3-56 could decrease CitH3 production, inhibit NET formation, and attenuate LPS-induced shock.
We hypothesized that rapid and excessive production of NETs with CitH3 in response to an LPS insult would increase endothelial permeability, and that selective inhibition of the specific PAD isoforms PAD2 and PAD4 could improve survival in a mouse model of lethal endotoxemia. Consequently, we examined the effects of (i) the PAD2/ PAD4 inhibitor YW3-56 on survival in mice with lethal endotoxemia, (ii) LPS insult on PAD activation, NET formation, and vascular permeability in lung and endothelial cells, and (iii) YW3-56 treatment on the LPS-induced alterations. These results reveal a new and important role for the specific PAD inhibitor in the treatment of lethal endotoxemia.
2. Materials and Methods
2.1. Reagents
Human umbilical vein endothelial cells (HUVECs, Lonza catalog number CC-2519) and endothelial cell growth medium (EGM) BulletKit (Lonza catalog numbers CC-3121 & CC-4133) were purchased from Lonza (Walkersville, MD, USA). Human promyelocytic leukemia cells (HL-60) were obtained from American Type Culture Collection, and 4’, 6-diamidino-2-phenylindole dihydrochloride (DAPI), anti-fade reagent, and formalin were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) were obtained from Gibco-BRL (Grand Island, NY, USA). Anti-CitH3 (citrulline R2 + R8 + R17, ab5103), anti-PAD4 (ab96758), and anti-myeloperoxidase (MPO, ab9535) antibodies were supplied by Abcam (Cambridge, MA, USA). Donkey anti-mouse IgG Fab fragment, FITC-conjugated donkey anti-mouse IgG, tetramethyl rhodamine isothiocyanate (TRITC)-conjugated donkey anti-mouse IgG, and normal donkey serum were provided by Jackson Immuno Research Laboratories (West Grove, PA, USA). Citrate buffer was purchased from Electron Microscopy Sciences (Hatfield, PA, USA). LPS from Salmonella typhosa (Catalog #L6386, Lot# 063M4017A), dimethyl sulfoxide (DMSO), Evans blue (EB) dye, formamide, and anti-actin antibody (A2228) were purchased from Sigma-Aldrich (St. Louis, MO, USA), and 12 mm Transwells with 0.4 μm pore polyester membrane inserts were provided by Corning Life Sciences (Corning, NY, USA). The ECL Detection Kit was obtained from GE Healthcare (Buckinghamshire, UK), and nitrocellulose/filter paper was purchased from BIO-RAD (Hercules, CA, USA). YW3-56 (purity > 95%) was kindly provided by Dr. Yanming Wang from Pennsylvania State University (University Park, PA, USA) (Wang et al., 2012b).
2.2. Animals
The Animal Care and Use Committee at the University of Michigan approved all animal studies. C57BL/6J mice (6-8 weeks old), weighing 20–25 g, were purchased from Jackson Labs (Bar Harbor, ME). All animals were housed with a 12 h light/dark cycle and had access to food and water throughout the experiment. Mice were randomly divided into three groups (n = 8–10/group) for intraperitoneal (i.p.) injection with different reagents as follows: group 1, DMSO (1 μl/g mouse body weight); group 2, LPS (35 mg/kg) + DMSO; and group 3, LPS + YW3-56 (5 mg/kg) dissolved in DMSO, with the YW3-56 injected 30 min after LPS administration. Mouse survival was monitored for 10 days. In a parallel experiment, mice received the same treatments as described above, and their blood and lungs were collected at various time points after injection of LPS or DMSO. Samples of serum and lung tissues were stored at −80°C or fixed with formalin for future use.
2.3. Assay for pulmonary microvascular permeability
Pulmonary microvascular permeability was assessed via the EB assay as previously described (Wang et al., 2012a). In brief, 1% EB solution in normal saline (4 ml/kg) was injected into the tail veins of mice 12 h after LPS challenge with or without YW3-56 treatment. Thirty min later, the mice were killed. Lungs were perfused with phosphate-buffered saline (PBS), 25 ml per mouse, and then excised, washed, and frozen in liquid nitrogen. Frozen tissue was immediately homogenized in ice-cold PBS and then incubated in formamide (60°C, 16 h). After centrifugation (7,000 × g, 10 min), the supernatant was collected, and the optical density (OD) at A620 was measured. The EB extravasation was expressed as EB in lung tissues (μg/g).
2.4. Histological analysis of acute lung injury (ALI)
Lung tissues were harvested 12 h after treatment. The samples were fixed in formalin, dehydrated, and stored in 70% ethanol, prior to being embedded in paraffin and sliced into 5-¼m sections. Hematoxylin-eosin staining was performed according to standard procedures. A pathologist was assigned for grading ALI in a blinded fashion. The method for objective quantification of ALI has been previously described (Kim et al., 2012).
2.5. Immunohistochemical staining of PAD4
For immunohistochemistry staining, sections were deparaffinized and rehydrated in graded concentrations of ethanol. Antigen retrieval was achieved by treating sections with 10 mM citrate buffer and heating in a microwave oven for 15 min. Samples were blocked using peroxidase blocking reagent for 10 min followed by 5% BSA for 1 h at room temperature. Sections were incubated with anti-PAD4 antibody in a humidified chamber at 4°C overnight. After washing in 0.05% PBST (Tween-20), the samples were incubated with horseradish peroxidase-conjugated goat anti-rabbit antibody for 1 h at room temperature followed by incubation with diaminobenzidine in the dark. The slides were counterstained with hematoxylin and mounted. The percentage of positively stained cellular areas in each of eight randomly selected fields for each group was determined using ImageJ. The quantitative results for PAD4 expression were determined by the percentage of PAD4 positively stained cellular areas over the total area of cells in scanned fields.
2.6. Immunofluorescent staining of NETs
Lung sections were deparaffinized and rehydrated in graded concentrations of ethanol. Antigen retrieval was achieved by treating sections with 10 mM citrate buffer and heating in a microwave oven for 15 min. Samples were blocked with donkey anti-mouse IgG Fab fragment overnight at 4°C. After a thorough wash in 0.05% PBST (Tween-20), the sections were sequentially incubated with mouse anti-CitH3 monoclonal and rabbit anti-MPO polyclonal antibodies in a humidified chamber at 4°C overnight. After being washed again, the samples were incubated with FITC-conjugated donkey anti-rabbit and TRITC-conjugated donkey anti-mouse IgG for 1 h at room temperature. Following another wash, the sections were incubated with DAPI for 20 min and then mounted with anti-fade reagent. All the antibodies mentioned above were diluted in 10% normal donkey serum.
2.7. Cell culture and treatment
HL-60 cells were cultured in DMEM, supplemented with 20% FBS, and were differentiated into granulocytes by exposure to all-trans retinoic acid (ATRA; 1 μM) for 3 days. The HL-60 neutrophilic cells were treated with LPS (1 μg/ml) in the presence or absence of YW3-56 (5 μmol/l) for 6 h.
HUVEC were grown in EGM with supplements and treated with NET extracts (10 μg/ml) after they were growing in Transwell chambers.
2.8. NET collection
HL-60 neutrophilic cells were subjected to LPS (1 μg/ml) with or without YW3-56 (5 μmol/l) for 6 h. Cells in the sham group did not receive any treatment. NETs were formed and collected as previously described, with some modifications (Saffarzadeh et al., 2012). Briefly, HL-60 cells were grown in a flask (75 cm2) until they reached 80% confluence, and then 0.6 ml of cold PBS was added after the medium was removed. NETs (the smear on the wells) were detached by 15 min of vigorous shaking and subsequently collected into 15 ml tubes. After centrifugation at 20 × g for 5 min to remove cells and cell debris, the NETs suspended in the supernatant were collected. After the protein concentration was measured, the NETs were stored at −80°C.
2.9. Western blot analysis
Western blot analysis was performed to detect CitH3 in both the mouse serum and NETs. The serum samples were diluted at 1:1 using normal saline. Equal amounts of samples were electrophoresed using SDS-PAGE and electronically transferred onto nitrocellulose membranes. The membranes were probed with anti-CitH3 polyclonal antibody (1:1000), and anti-rabbit IgG antibody conjugated to horseradish peroxidase (1:3000) was used as a secondary antibody. β-actin was used as an internal control. The immunoblotting bands were visualized and quantified using Image J software.
2.10. Transwell permeability assay
For the in vitro permeability assay, HUVEC (5×105 cells /ml) were grown on a Transwell insert for 3 days to develop a confluent monolayer. On day 4, NETs (10 μg/ml) were added to the chambers for 16 h, and the chambers were then incubated in the presence of 10-kDa FITC-dextran (1 mg/ml, Life Technologies). The fluorescence levels of media (50 μl) in the lower chambers were determined using a GloMax-multi detection system (Promega, Madison, WI).
2.11. Statistical analysis
All values are expressed as the mean ± standard deviation (S.D.) and were analyzed using GraphPad Prism 6.0 statistical software (GraphPad Software, Inc., La Jolla, CA). Survival differences were compared by Kaplan-Meier curve with log-rank analysis. One-way analysis of variance (ANOVA) with Tukey’s post-test was used for multiple comparisons. P < 0.05 was considered statistically significant.
3. Results
3.1. Inhibition of PAD2/PAD4 with YW3-56 diminishes LPS-induced PAD activation in lungs and improves survival in a mouse model of lethal endotoxemia
We assessed the effect of YW3-56 on survival in a mouse model of lethal endotoxemia. Intraperitoneal injection of LPS (35 mg/kg) was universally lethal, with the majority of deaths occurring within 24 h (Fig. 1A). When compared to the LPS-only group, the YW3-56 treatment group had a significantly improved survival rate (survival rate of YW3-56 vs LPS: 50% vs 0%, P < 0.0001). In a parallel study, lungs were harvested from these groups at 3 h after LPS or DMSO injection, and immunohistochemistry (IHC) was performed to analyze PAD4 protein expression in the lungs. As shown in Fig. 1B, PAD4 expression was barely detectable in the lungs of DMSO-treated mice, whereas cells positive for PAD4 staining were observed widely in the alveolar space and pulmonary capillaries of LPS-treated mice. However, treatment with YW3-56 significantly decreased the LPS-induced alteration (P < 0.0001) (Fig. 1C). Our results indicate that YW3-56 treatment can improve survival and is associated with a decrease in the PADexpressing cells in the lungs, suggesting that the cells expressing high levels of PAD4 following LPS insult could be involved in the lethal endotoxemia.
Fig. 1. YW3-56 inhibited PAD4 expression and improved survival in a mouse model of lethal endotoxemia.
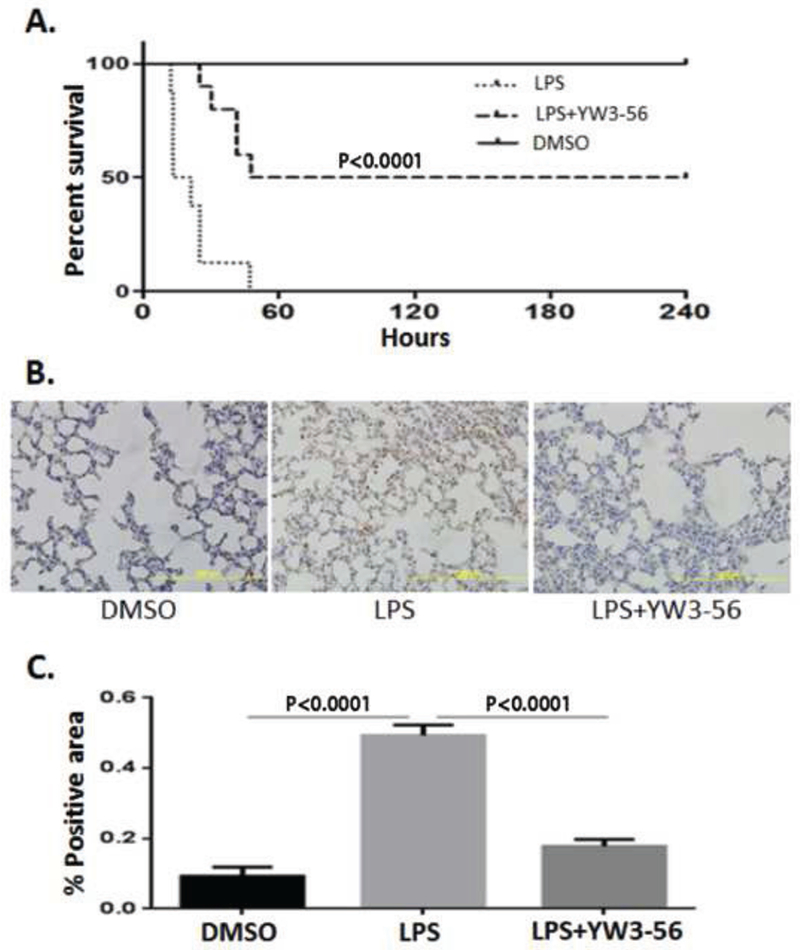
Mice were randomly divided into DMSO, LPS, and LPS+YW3-56 groups as described in Materials and Methods. The animals received i.p. injections with YW3-56 dissolved in DMSO (5 mg/kg, n = 10) or with DMSO (n = 8) 30 min after LPS administration (35 mg/kg). The dose and injection times of the reagents (LPS, YW-356) were maintained throughout the study, unless otherwise noted. (A) Kaplan-Meier curve illustrating survival rates over a 10-day period. All of the mice in the LPS group died within 48 h, whereas the YW3-56 treatment group had significantly improved survival compared to the LPS group (P < 0.0001). (B, C) Lungs were harvested 3 h after treatment and then sectioned for IHC staining of PAD4. Representative images (B) and quantification of PAD4 expression by ImageJ (C). Data are presented as mean ± S.D. (n = 8/group). PAD4, peptidylarginine deiminase 4; IHC, immunohistochemistry.
3.2. YW3-56 decreases LPS-induced NET formation in lungs
To further determine whether the NETs were formed after PAD activation in the lethal endotoxemia, we performed immunofluorescence staining on lung tissues of mice from different groups. Generally, NET formation can be detected by staining for MPO and other nuclear proteins (Cheng and Palaniyar, 2013). We harvested lung tissues 12 h after LPS or DMSO injections and examined colocalization of MPO with CitH3 for LPS-induced NET formation (Fig. 2). In our experiment, NET formation was seldom seen in the lungs of the DMSO-treated animals. The LPS insult, however, increased the staining for MPO and CitH3 with the widespread, web-like NETs. Treatment with YW3-56 eliminated these alterations. The result indicates that NETs are formed in lungs during the lethal endotoxemia and that inhibition of PAD2/PAD4 can abolish NET formation.
Fig. 2. YW3-56 significantly decreased NET formation in mouse lung.
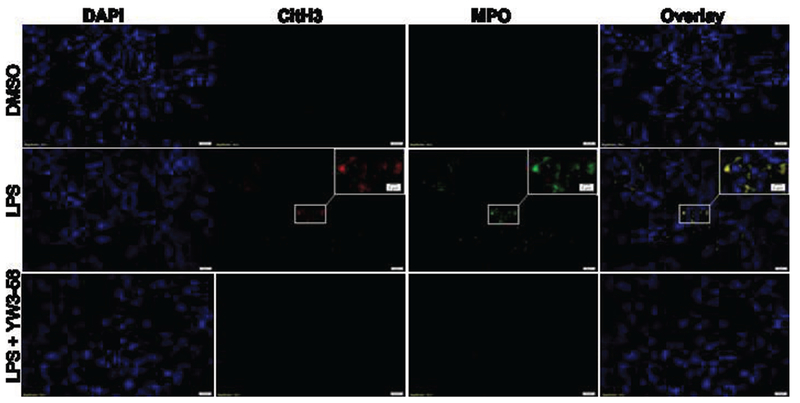
Mice were injected intraperitoneally with DMSO (vehicle control) or YW3-56 in DMSO 30 min after LPS administration. Lungs were harvested 12 h after treatment and then sectioned for immunofluorescence staining of nuclei (blue), CitH3 (red), and MPO (green). Representative immunofluorescence images of NET formation in the lung (n = 3/group) demonstrated colocalization of MPO and CitH3 (yellow) outside the nuclei. NETs, neutrophil extracellular traps; CitH3, citrullinated histone H3; IHC, immunohistochemistry; MPO, myeloperoxidase.
3.3. YW3-56 diminishes the LPS-induced increase of CitH3 and permeability of endothelial cells
We have previously demonstrated that levels of CitH3 are elevated in blood 3 h after intraperitoneal injection of LPS (35 mg/kg) (Li et al., 2011) and that the neutralization of circulating CitH3 can improve survival (Li et al., 2014). In the present study, we found that CitH3 was even detectable 30 min after the LPS insult, and treatment with YW3-56 dramatically decreased the CitH3 levels compared to the LPS-only group (Fig. 3A).
Fig. 3. YW3-56 significantly attenuated LPS-induced CitH3 production and decreased HUVEC permeability.
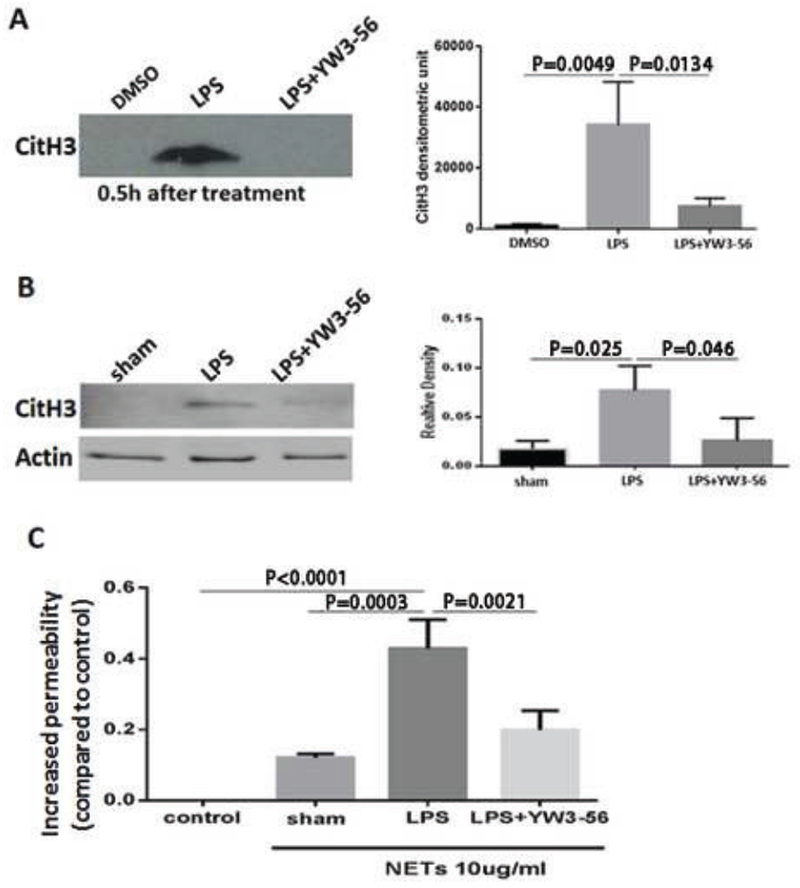
(A) Mice were injected intraperitoneally with DMSO only (sham), LPS+DMSO, or LPS+YW3-56, and then 30 min after treatment, sera were prepared for detection of CitH3 by Western blot. (B) HL-60 neutrophilic cells were treated with LPS (1 μg/ml) in the presence or absence of YW3-56 (5 μmol/1) for 6 h. The neutrophilic cells for the sham group did not receive LPS. NETs were collected and subjected to Western blotting for CitH3 analysis. Densitometric units of CitH3 bands were measured using Image Studio Lite (right panel). (C) HUVEC were grown in Transwell® plates until reaching confluence and then incubated with NETs collected as for Fig. 3B. HUVEC without any treatment were used as the control group. Cell permeability was assessed by measuring leakage of FITC-labeled dextran. Data are presented as mean ± S.D. (n = 3/group). NETs, neutrophil extracellular traps; CitH3, citrullinated histone H3.
To determine whether YW3-56 can inhibit CitH3 production and NET formation, HL-60 neutrophilic cells were treated with LPS (1 μg/ml) in the presence or absence of the PAD inhibitor (5 μmol/l) for 6 h. A small amount of NET was detected in the sham group in which neutrophilic cells did not receive any treatment (Fig. 3B, right panel). However, LPS significantly stimulated citrullination of histone H3 compared to levels in the sham group, whereas YW3-56 treatment attenuated the LPS-induced alteration (Fig. 3B, P < 0.05).
To find out whether they could affect endothelial cell function, we collected NETs from all three groups and analyzed the effect of the NETs on the permeability of HUVEC. We took equal amounts of NETs (10 μg/ml) from HL-60 neutrophilic cells in the aforementioned sham (no LPS and no treatment), LPS-only, and LPS + YW3-56 groups, and then treated the HUVEC in a Transwell plate for 16 h. In this experiment, HUVECs treated without NETs served as a control group. HUVECs treated with NETs from the sham-treated HL-60 cells were used as a sham group. The cell permeability was determined by measuring leakage of FITC-labeled dextran. The NETs derived from the LPS group increased the endothelial layer permeability by 42% ± 5%, whereas those derived from the sham group only enhanced permeability by 12% (Fig. 3C). Treatment with YW3-56 significantly decreased the permeability as compared to the LPS group (21 ± 3% vs 42 ± 5%). This result indicates that NETs can cause leakage of endothelial cells and that different NETs (spontaneously formed NETs, NETs from the LPS-only group, and NETs from the LPS+YW3-56 group) induce permeability to different extents. Moreover, treatment of HUVEC with YW3-56 protects endothelial cells from LPS-induced permeability, suggesting that a decrease of CitH3 levels could be one of the critical mechanisms.
3.4. YW3-56 alleviates LPS-induced pulmonary microvascular dysfunction
To further assess whether YW3-56 can prevent pulmonary microvascular dysfunction in the lungs during the lethal endotoxemia, we examined the levels of pulmonary tissue damage following exposure of mice to DMSO, LPS, or LPS +YW3-56. EB dye (1%, 4 ml/kg) was intravenously injected into the mice through the tail vein 12 h after they were exposed to the reagents, and lung tissues were harvested 30 min later. As shown in Fig. 4A, gross pigmentation increased in the LPS group as compared to the DMSO group, and treatment with YW3-56 significantly decreased the pigmentation. Examination of the left lung samples by light microscopy revealed numerous black spots in lungs from the LPS group (Fig. 4B), which were hardly detectable in lungs from the DMSO group. YW3-56 markedly attenuated the alteration cause by LPS (Fig 4B). To confirm these changes, the right lungs of mice from different groups were homogenized, and the OD of the supernatant was determined spectrophotometrically (Fig. 4C). The extravasation of EB into lung tissues was significantly increased in the LPS group compared to that in the DMSO group (46.2 ± 13 vs. 13.7 ± 0.5; P = 0.0045). For the LPS+YW3-56 group, the EB extravasation was significantly lower than those of the LPS group (17.7 ± 1.1 vs. 46.2 ± 13; P = 0.0085). These results indicate that YW3-56 alleviates LPS-induced pulmonary microvascular dysfunction.
Fig. 4. YW3-56 alleviated LPS-induced pulmonary microvascular dysfunction.
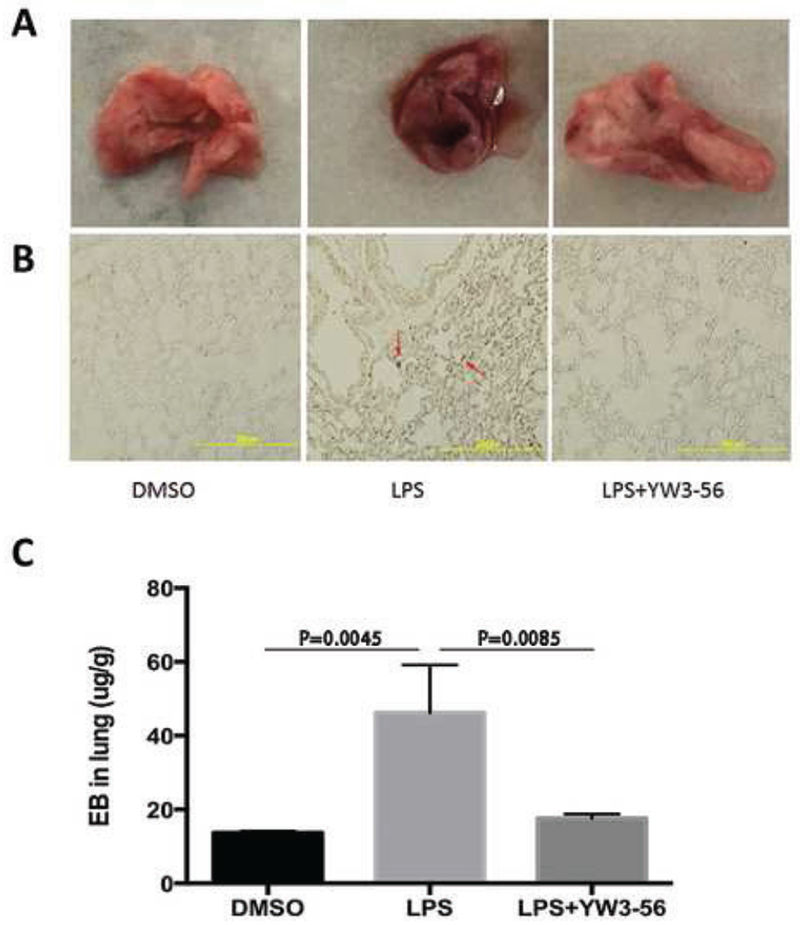
Mice were injected intraperitoneally with DMSO only (sham), LPS+DMSO, or LPS+YW3-56. EB dye (1%, 4 ml/kg) was intravenously injected 12 h after LPS, and the lungs were harvested 30 min after the dye injection. (A) Gross images of mouse lungs from DMSO, LPS, and LPS+YW3-56 groups. (B) Representative images of lung tissue sections from all three mouse groups, with EB leakage (red arrows) into alveolar spaces detected only with the LPS group. (C) EB extravasation was quantified by spectrophotometry, and expressed as described in Materials and Methods. Data are expressed as mean ± S.D. (n = 3/group). Evans blue dye albumin, EB.
3.5. YW3-56 alleviates LPS-induced ALI
To determine whether YW3-56 can protect animals against ALI during the lethal endotoxemia, lung tissues were harvested 12 h after treatment from all three groups. Pathological examination showed that the evidence of acute lung injury in the LPS group, with interstitial edema and infiltration of inflammatory cells (Fig. 5A). YW3-56 treatment, however, dramatically reversed the histological changes. Blinded ALI grading, performed by a pathologist, revealed an increased score for the LPS group compared to the DMSO group, and a decreased score for the YW3-56 treated group compared to the LPS group (Fig. 5B, P < 0.0001).
Fig. 5. YW3-56 alleviated LPS-induced ALI.
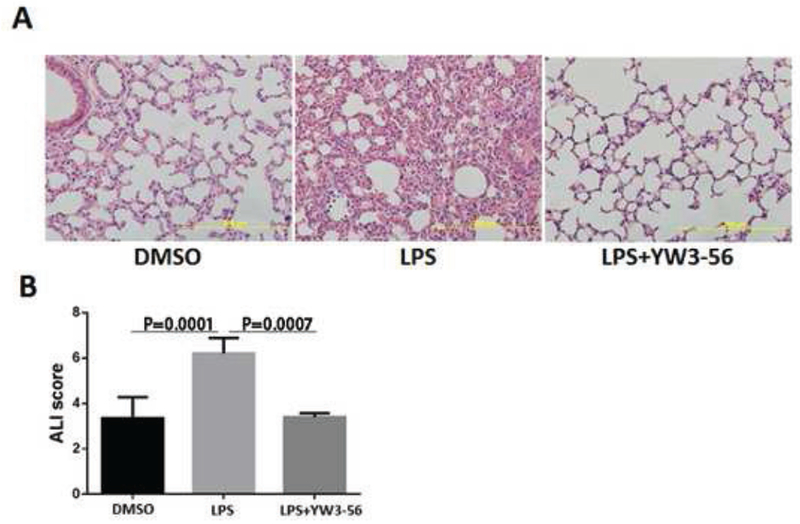
Mice were injected intraperitoneally with DMSO only, LPS+DMSO, or LPS+YW3-56. (A) Mouse lungs were harvested 12 h after treatment and then sectioned for H&E staining. (B) Histological ALI scores (n = 3/group). Data are expressed as mean ± S.D. ALI, acute lung injury.
4. Discussion
In the present study, we tested our hypothesis that treatment with a PAD2/PAD4 inhibitor can markedly improve the outcomes of mice subjected to lethal endotoxemia. We found that CitH3 can be detected in the circulation as early as 0.5 h after peritoneal injection of LPS (35 mg/kg). Inhibition of PAD2/PAD4 with YW3-56 can significantly decrease NET formation and CitH3 production in vivo and in vitro, maintain the integrity of endothelial cells and reduce pulmonary vascular dysfunction, alleviate ALI, and improve survival in a mouse model of lethal endotoxemia (Fig. 6).
Fig. 6. Schematic diagram showing the mechanism of action of YW3-56 on lethal LPS-induced shock.
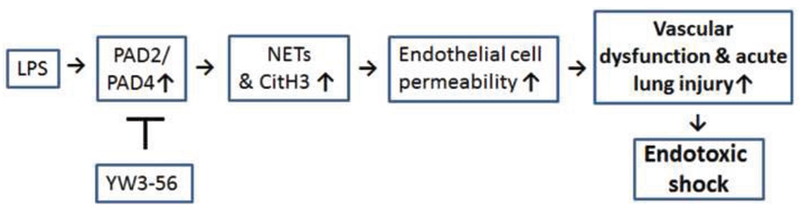
Activation of PAD by LPS can induce NET formation to release CitH3. CitH3 increases permeability of HUVEC, leading to ALI with pulmonary microvascular dysfunction. Inhibition of PAD2/PAD4 with YW3-56 can attenuate these LPS-induced alterations and improve survival.
To our knowledge, the present study is the first demonstration that inhibition of PAD2/PAD4 with YW3-56 can attenuate the effects of LPS. Recently, we have found that treatment of septic mice with the pan-PAD inhibitor Cl-amidine can also significantly improve survival (Li et al., 2014; Zhao et al., 2016). However, Cl-amidine also drives apoptosis of inflammatory cells in vitro and in vivo (Chumanevich et al., 2011), which could limit its therapeutic applicability in sepsis. Nonetheless, specific inhibition of nuclear PAD2/PAD4 could be a promising alternative therapeutic strategy.
We further investigated the effect of YW3-56 on LPS-induced NET formation, CitH3 production, and endothelial permeability in vitro and in vivo. NETs are web-like structures that consist of decondensed DNA, histones, enzymes, and antimicrobial peptides (Kaplan and Radic, 2012). NETs function as double-edged swords (Kaplan and Radic, 2012). Normally, NETs play an essential role in innate immunity, but they have also been associated with some harmful aspects (Doring et al., 2013; Grayson and Kaplan, 2016), including direct cytotoxic and thrombotic effects on vascular endothelium (Fuchs et al., 2010; Xu et al., 2009). In the current study, our data imply that citrullination of histone H3 may be involved in the dysfunction of endothelial cells, although there is still no direct evidence. First, we demonstrated that NETs can increase the permeability of HUVEC in vitro, in particular the NETs from neutrophilic cells pretreated with LPS. In these cells, LPS can induce NET formation with increased CitH3 levels. However, inhibition of PAD2/PAD4 with YW3-56 reversed the effects of LPS on NET formation and CitH3 production (Fig. 3). These findings were supported by our in vivo studies in which we showed that YW3-56 could attenuate LPS-induced PAD4 activation, CitH3 production, and NET formation in the lungs of mice. In addition, YW3-56 suppressed LPS-increased pulmonary permeability and ALI. Recently, we have reported that the treatment of septic mice with a PAD inhibitor or neutralization of blood CitH3 with a specific antibody can improve survival in a mouse model of lethal CLP (Li et al., 2014). As supported by our data in Fig. 2 and Fig. 3, increased expression of CitH3, a main component of LPS-induced NETs, increases the permeability of endothelium.
ALI/acute respiratory distress syndrome (ARDS) is one of the most common causes of morbidity and mortality in sepsis (Husak et al., 2010). ALI is characterized by the injury and dysfunction of the pulmonary microvasculature, which results in enhanced pulmonary microvascular sequestration, pulmonary infiltration of polymorphonuclear neutrophils (PMN), and the disruption of the normal alveolo-capillary permeability barrier, with a leak of albumin-rich fluid into the pulmonary interstitium and alveoli (edema). In a mouse CLP model, Gill et al reported that septic pulmonary microvascular dysfunction in vivo is due to pulmonary microvascular endothelial cell death, which is mediated through caspase-dependent apoptosis and iNOS/NADPH-oxidase dependent signaling (Gill et al., 2015). It has been widely accepted that LPS-induced ALI is neutrophil-dependent in a murine model of endotoxic shock (Rittirsch et al., 2008). In our present study, we showed that a lethal dose of LPS induces neutrophils to release NETs within 30 min (Fig. 3A) and causes ALI in 3 h (data not shown). It is conceivable that, in this neutrophil-dependent ALI, pulmonary microvascular dysfunction is primarily the result of the effects of NETs and CitH3 on microvascular endothelial cells. We observed abundant infiltration and accumulation of leukocytes, particularly neutrophils, in both the interstitial and alveolar spaces of the lungs (Fig. 5A). Treatment with YW3-56 markedly attenuated the LPS-induced ALI and led to significant reductions in PMN infiltration, NET formation, and CitH3 production. These findings suggest that pulmonary microvascular barrier dysfunction in a murine LPS model, as reflected by enhanced CitH3 production and albumin leakage in the lungs, is associated with the presence of both alveolar PMN and NETosis. Collectively, the activation of neutrophils, release of NETs, and increase of CitH3 levels may contribute to the pathogenesis of endotoxemia and progression of ALI in the lethal LPS mouse model. In the current study, we did not examine effects of YW3-56 on all organs. However, we have recently investigated effect of Cl-amidine, a derivative of YW3-56, on other organs such as bone marrow (BM) and liver. We found that Cl-amidine plays protective roles by significantly restoring innate immune cells in BM, increasing blood monocytes and blood/liver bacteria clearance, and attenuating pro-inflammatory cytokine production in a murine model of lethal sepsis (Zhao et al., 2016).
It has been reported that an anti-inflammatory agent acetylsalicylic acid can abrogate NETosis, which might be associated with inhibition of NF-κβ (Lapponi et al., 2013). PAD4 can induce protein citrullination and enhance translocation of citrullinated NF-kβ into nucleus, and then increases gene expression of pro-inflammatory cytokines IL-1β and TNFα (Sun et al., 2017). It is conceivable that YW3-56 can inhibit PAD activity and therefore suppress LPS-induced NETosis.
We have to acknowledge that our present study has certain limitations. The rodent endotoxemia and human sepsis are not synonymous. Recently, we have studied effect of YW3-56 on blood levels of CitH3 biomarker in a mouse endotoxemia model (Pan et al., 2017). To keep our research consistent, in the present study we took the mouse model of endotoxemia to determine whether YW3-56 can improve survival. Follow-up studies with a mouse model of CLP-induced sepsis will be needed, since CLP model is widely considered the gold standard in sepsis research (Lilley et al., 2015; Ruiz et al., 2016). In the present study, we did not test the time-effect of YW3-56. However, we have recently shown that intraperitoneal injection of PAD inhibitor YW3-56 (5 mg/kg, once) can effectively diminish circulating biomarker CitH3 for over 24 h compared to no inhibitor control (Pan et al., 2017). According to this finding we treated the endotoxic mice with YW3-56 only one time.
5. Conclusions
Our studies support the hypothesis that suppression of histone H3 citrullination and NETosis can prevent ALI and protect the host against lethal endotoxemia.
Supplementary Material
Acknowledgements
We thank Dr. Susan T. Howard for editing this manuscript.
Funding
This work was funded by grants from Mcubed U049657 and Kickstart N022142 to Dr. Yongqing Li, UMHS-PUHSC Joint Institute U050150 to Dr. Hasan B. Alam, and U.S. National Institutes of Health NIH 2-R01-GM-084127-06-A1 to Dr. Hasan B. Alam.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting galley proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Ethics approval and consent to participate
The Animal Care and Use Committee at the University of Michigan approved all animal studies.
Declarations of interest
None.
References
- Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A, 2004. Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535. [DOI] [PubMed] [Google Scholar]
- Cheng OZ, Palaniyar N, 2013. NET balancing: a problem in inflammatory lung diseases. Frontiers in immunology 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumanevich AA, Causey CP, Knuckley BA, Jones JE, Poudyal D, Chumanevich AP, Davis T, Matesic LE, Thompson PR, Hofseth LJ, 2011. Suppression of colitis in mice by CI-amidine: a novel peptidylarginine deiminase inhibitor. American journal of physiology. Gastrointestinal and liver physiology 300, G929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doring Y, Weber C, Soehnlein O, 2013. Footprints of neutrophil extracellular traps as predictors of cardiovascular risk. Arteriosclerosis, thrombosis, and vascular biology 33, 1735–1736. [DOI] [PubMed] [Google Scholar]
- Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A, 2007. Novel cell death program leads to neutrophil extracellular traps. The Journal of cell biology 176, 231–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr., Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD, 2010. Extracellular DNA traps promote thrombosis. Proceedings of the National Academy of Sciences of the United States of America 107, 15880–15885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SE, Rohan M, Mehta S, 2015. Role of pulmonary microvascular endothelial cell apoptosis in murine sepsis-induced lung injury in vivo. Respir Res 16, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson PC, Kaplan MJ, 2016. At the Bench: Neutrophil extracellular traps (NETs) highlight novel aspects of innate immune system involvement in autoimmune diseases. Journal of leukocyte biology 99, 253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husak L, Marcuzzi A, Herring J, Wen E, Yin L, Capan DD, Cernat G, 2010. National analysis of sepsis hospitalizations and factors contributing to sepsis in-hospital mortality in Canada. Healthc Q 13 Spec No, 35–41. [DOI] [PubMed] [Google Scholar]
- Kaplan MJ, Radic M, 2012. Neutrophil extracellular traps: double-edged swords of innate immunity. J Immunol 189, 2689–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, Grone HJ, Brinkmann V, Jenne DE, 2009. Netting neutrophils in autoimmune small-vessel vasculitis. Nature medicine 15, 623–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Li Y, Jin G, Chong W, Liu B, Lu J, Lee K, Demoya M, Velmahos GC, Alam HB, 2012. Effect of valproic acid on acute lung injury in a rodent model of intestinal ischemia reperfusion. Resuscitation 83, 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapponi MJ, Carestia A, Landoni VI, Rivadeneyra L, Etulain J, Negrotto S, Pozner RG, Schattner M, 2013. Regulation of neutrophil extracellular trap formation by anti-inflammatory drugs. J Pharmacol Exp Ther 345, 430–437. [DOI] [PubMed] [Google Scholar]
- Li Y, Liu B, Fukudome EY, Lu J, Chong W, Jin G, Liu Z, Velmahos GC, Demoya M, King DR, Alam HB, 2011. Identification of citrullinated histone H3 as a potential serum protein biomarker in a lethal model of lipopolysaccharide-induced shock. Surgery 150, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu Z, Liu B, Zhao T, Chong W, Wang Y, Alam HB, 2014. Citrullinated histone H3: A novel target for the treatment of sepsis. Surgery 156, 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley E, Armstrong R, Clark N, Gray P, Penny H, Mason K, López-Salesansky N, Stark A-K, Jackson S, Thiemermann C, Nandi M, 2015. Refinement of animal models of sepsis and septic shock. Shock 43, 304–316. [DOI] [PubMed] [Google Scholar]
- Neeli I, Khan SN, Radic M, 2008. Histone deimination as a response to inflammatory stimuli in neutrophils. Journal of immunology 180, 1895–1902. [DOI] [PubMed] [Google Scholar]
- Pan B, Alam HB, Chong W, Mobley J, Liu B, Deng Q, Liang Y, Wang Y, Chen E, Wang T, Tewari M, Li Y, 2017. CitH3: a reliable blood biomarker for diagnosis and treatment of endotoxic shock. Sci Rep 7, 8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittirsch D, Flierl MA, Day DE, Nadeau BA, McGuire SR, Hoesel LM, Ipaktchi K, Zetoune FS, Sarma JV, Leng L, Huber-Lang MS, Neff TA, Bucala R, Ward PA, 2008. Acute lung injury induced by lipopolysaccharide is independent of complement activation. Journal of immunology 180, 7664–7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz S, Vardon-Bounes F, Merlet-Dupuy V, Conil JM, Buleon M, Fourcade O, Tack I, Minville V, 2016. Sepsis modeling in mice: ligation length is a major severity factor in cecal ligation and puncture. Intensive Care Med Exp 4, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, Lohmeyer J, Preissner KT, 2012. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PloS one 7, e32366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Dwivedi N, Bechtel TJ, Paulsen JL, Muth A, Bawadekar M, Li G, Thompson PR, Shelef MA, Schiffer CA, Weerapana E, Ho IC, 2017. Citrullination of NF-kappaB p65 promotes its nuclear localization and TLR-induced expression of IL-1beta and TNFalpha. Sci Immunol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Koike Y, Shimura T, Okigami M, Ide S, Toiyama Y, Okugawa Y, Inoue Y, Araki T, Uchida K, Mohri Y, Mizoguchi A, Kusunoki M, 2014. In vivo characterization of neutrophil extracellular traps in various organs of a murine sepsis model. PloS one 9, e111888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, Rubin CJ, Zhao W, Olsen SH, Klinker M, Shealy D, Denny MF, Plumas J, Chaperot L, Kretzler M, Bruce AT, Kaplan MJ, 2011. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. Journal of immunology 187, 538–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Taneja R, Razavi HM, Law C, Gillis C, Mehta S, 2012a. Specific role of neutrophil inducible nitric oxide synthase in murine sepsis-induced lung injury in vivo. Shock 37, 539–547. [DOI] [PubMed] [Google Scholar]
- Wang S, Wang Y, 2013. Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim Biophys Acta 1829, 1126–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, Allis CD, Coonrod SA, 2009. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 184, 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li P, Wang S, Hu J, Chen XA, Wu J, Fisher M, Oshaben K, Zhao N, Gu Y, Wang D, Chen G, Wang Y, 2012b. Anticancer peptidylarginine deiminase (PAD) inhibitors regulate the autophagy flux and the mammalian target of rapamycin complex 1 activity. The Journal of biological chemistry 287, 25941–25953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, Kahn CR, Wagner DD, 2015. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nature medicine 21, 815–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, Esmon CT, 2009. Extracellular histones are major mediators of death in sepsis. Nature medicine 15, 1318–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RL, Malcolm KC, Kret JE, Caceres SM, Poch KR, Nichols DP, Taylor-Cousar JL, Saavedra MT, Randell SH, Vasil ML, Burns JL, Moskowitz SM, Nick JA, 2011. Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: evidence of acquired resistance within the CF airway, independent of CFTR. PloS one 6, e23637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bolt M, Guertin MJ, Chen W, Zhang S, Cherrington BD, Slade DJ, Dreyton CJ, Subramanian V, Bicker KL, Thompson PR, Mancini MA, Lis JT, Coonrod SA, 2012. Peptidylarginine deiminase 2-catalyzed histone H3 arginine 26 citrullination facilitates estrogen receptor alpha target gene activation. Proc Natl Acad Sci U S A 109, 13331–13336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Pan B, Alam HB, Liu B, Bronson RT, Deng Q, Wu E, Li Y, 2016. Protective effect of CI-amidine against CLP-induced lethal septic shock in mice. Sci Rep 6, 36696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.