

Abstract
Recent genome-wide DNA methylation analyses of insect genomes accentuate an intriguing contrast compared with those in mammals. In mammals, most CpGs are heavily methylated, with the exceptions of clusters of hypomethylated sites referred to as CpG islands. In contrast, DNA methylation in insects is localized to a small number of CpG sites. Here, we refer to clusters of methylated CpGs as “methylation islands (MIs),” and investigate their characteristics in seven hymenopteran insects with high-quality bisulfite sequencing data. Methylation islands were primarily located within gene bodies. They were significantly overrepresented in exon–intron boundaries, indicating their potential roles in splicing. Methylated CpGs within MIs exhibited stronger evolutionary conservation compared with those outside of MIs. Additionally, genes harboring MIs exhibited higher and more stable levels of gene expression compared with those that do not harbor MIs. The effects of MIs on evolutionary conservation and gene expression are independent and stronger than the effect of DNA methylation alone. These results indicate that MIs may be useful to gain additional insights into understanding the role of DNA methylation in gene expression and evolutionary conservation in invertebrate genomes.
Keywords: DNA methylation, whole-genome bisulfite sequencing, gene body methylation, methylation islands, gene expression
Introduction
Methylation of cytosine residues is a widespread epigenetic modification in eukaryotes. In animal genomes, the primary targets of DNA methylation are cytosines in the context of CpG dinucleotides. DNA methylation of CpGs (often referred to as CpG methylation) has been extensively investigated in mammalian model systems to reveal its critical roles in key regulatory processes such as genomic imprinting, disease, and development (Razin and Cedar 1994; Tremblay et al. 1995; Robertson and Wolffe 2000; Saze et al. 2003; Beck 2018). In the recent decade, the phylogenetic scope of CpG methylation studies has dramatically expanded, thanks to the advances of sequencing methods to profile whole genome methylomes. The influx of whole genome methylation data from previously little explored taxa, in turn, has further advanced our understanding of the genomic as well as phylogenetic distribution of DNA methylation.
For example, even though DNA methylation has been traditionally viewed as a repressive marker, it has now become clear that the functional consequences of DNA methylation depend on the genomic target of DNA methylation. Specifically, DNA methylation near transcription start sites is associated with transcriptional repression of downstream genes (Jones 2012; Schübeler 2015). Methylation of repetitive elements curbs the activity of these sequences, thereby protecting the genome from the harmful effects of transposition of these elements (Yoder et al. 1997; Schübeler 2015). DNA methylation of gene bodies, on the other hand, is generally associated with active transcription of genes, even though the exact cause and effect relationship of this association is not yet resolved (Jjingo et al. 2012; Jones 2012).
Recent whole genome profiling of diverse species has further revealed that DNA methylation is phylogenetically more widespread than previously envisioned (Feng et al. 2010; Zemach et al. 2010). The lack of DNA methylation in some prominent model organisms (i.e., fruit flies and Caenorhabditis elegans) represents lineage-specific loss of DNA methylation (Glastad et al. 2011; Yi 2012; Rosic et al. 2018). With the new wealth of methylome data from closely related species, some lineages, such as hymenopteran insects (bees, wasps, and ants) and nematodes, are emerging as useful model systems to understand the evolution and function of DNA methylation (Lyko et al. 2010; Wang et al. 2013; Greer et al. 2015; Rosic et al. 2018).
Notably, the pattern of genomic DNA methylation is highly variable among different animals. In vertebrates (especially in mammals), the majority of genomic CpG dinucleotides are heavily methylated. Exceptions to this are found in clusters of hypomethylated CpGs, referred to as “CpG islands” (Bird et al. 1985; Bird 1992). DNA methylation of CpG islands is associated with regulation of gene expression (Schübeler 2015; Mendizabal et al. 2016). Consequently, CpG islands have been widely used as units of investigation for DNA methylation studies. Furthermore, commercially available DNA methylation chips tend to target CpGs found in CpG islands.
In contrast, genomic CpG dinucleotides in invertebrates are typically devoid of DNA methylation (Suzuki and Bird 2008; Feng et al. 2010; Zemach and Zilberman 2010). For example, in hymenopteran genomes, DNA methylation is limited to a subset of CpG dinucleotides, often found within genes (Wang et al. 2013; Beeler et al. 2014; Bewick et al. 2017). Figure 1 is a representative example of the differences between the methylomes of humans and honey bees. An interesting observation is that in honey bees and other hymenopteran species, methylated CpGs tend to localize in clusters, as seen in figure 1 (Huh et al. 2014; Wang et al. 2016). Therefore, whereas the heavily methylated human methylome is punctuated by hypomethylated CpG clusters (CpG islands), the lowly methylated honey bee methylome is punctuated by clusters of hypermethylated CpGs (fig. 1).
Fig. 1.

—Contrasting methylation landscapes found in honey bees and humans. Methylated CpGs are sparse but clustered in honey bees and other hymenopteran insects. These “Methylation Islands” are ∼250 bp in length and stand out in the otherwise lowly methylated insect genomes. In contrast, the human methylation landscape is heavily methylated throughout with breaks of hypomethylated CpG islands that are typically ∼1 kb in length.
As studies using CpG islands as units of epigenetic variation have been highly successful in illuminating the functional implications of epigenetic variation, here we investigated the distribution and functional implications of clusters of methylated CpGs in insect genomes. We refer to them as “methylation islands (MIs).” In this study, we analyzed seven methylomes of hymenopteran insects, which offer well-annotated genomes and high quality methylomes, to define MIs and characterize their genomic distribution, and investigated potential functional consequences using RNA-seq data. Our analyses show that clusters of hypomethylated CpGs, namely MIs, have profound associations with gene sequence conservation and gene expression.
Materials and Methods
Bisulfite-Sequencing and RNA-Seq Data Analysis
Raw bisulfite-sequencing data of selected species were obtained from the SRA, and accession numbers can be found in supplementary file, Supplementary Material online. Reads were subjected to quality as well as adapter trimming using Trim-galore and subsequently aligned and deduplicated to their respective reference genomes using Bismark v0.14.4 (Krueger and Andrews 2011; Martin 2011). Additionally, reads were aligned to the unmethylated lambda phage genome (NCBI reference NC_001416.1) to estimate the bisulfite conversion efficiency for each sample. Alignment summaries and conversion rates can be found in supplementary file S1, Supplementary Material online.
RNA-Seq data of Apismellifera, Nasoniavitripennis, and Trichogrammapretiosum were downloaded from the SRA, and accession numbers can be found in supplementary file S1, Supplementary Material online. Transcriptome sequencing reads were preprocessed using FastQC to assess average quality scores (Andrews 2010). We removed potential adaptor sequences using Trimmomatic (Bolger et al. 2014). Tophat2 was used to align transcriptome reads to a reference genome and FeatureCount was used to quantify transcripts (Kim et al. 2013; Liao et al. 2014). We removed lowly expressed genes that had less than five read counts.
Identification of mCGs and MIs
Methylated CpGs (mCGs) were first identified using the Bis-Class algorithm, which takes into account correlation of methylation levels for adjacent CpGs (Huh et al. 2014). Our custom script for identifying methylation islands scans for clustered mCGs using the following steps (supplementary fig. S2, Supplementary Material online):
- Each scaffold is scanned (5′ -> 3′) in 200-bp windows. Each window is evaluated for its mCG fraction, which is defined as:
If the mCG fraction of the window is below the threshold of 0.02, the algorithm moves onto the next downstream mCG and uses it as the starting position for the new 200-bp window and evaluates its mCG fraction (supplementary fig. S2A, Supplementary Material online). This process is repeated until a window’s mCG fraction is greater than or equal to the threshold.
A 200-bp window satisfying the mCG fraction threshold is extended by 50 bp and re-evaluated for its mCG fraction. This extension continues until the mCG fraction of the extended window falls below the threshold, after which the last mCG in the previously evaluated window is chosen as the ending position of the methylation island (supplementary fig. S2B, Supplementary Material online). Therefore, the starting and ending positions of methylation islands are always mCGs.
Once a methylation island has been identified, the algorithm begins evaluating 200-bp windows again starting at the next downstream mCG. Steps 2 and 3 are repeated until the last mCG on the scaffold is reached (supplementary fig. S2C, Supplementary Material online).
Computing the Conservation Score
Orthologous gene sets were generated using ProteinOrtho (ver. 5.1.6) with the default setting (Lechner et al. 2011). The orthologous gene sets including protein sequences from all species were further processed to calculate conservation scores. Clustal-Omega (ver. 1.2.4) was used for sequence alignment (Sievers et al. 2014). The conservation score of each amino acid position was quantified based on the Jensen–Shannon (JS) divergence, which is a robust method for identifying protein sequence conservation (Lin 1991; Capra and Singh 2007). Conservation scores were analyzed with a linear mixed model using amino acid location (outside-MI or inside-MI) and the existence of corresponding DNA methylation sites (unmethylated CpGs or methylated CpGs) as the main factors and the interaction and random factors of gene and species information. To ensure adequate representation, we only analyzed genes that had at least five amino acids with mCpGs and non-mCpGs and five amino acids inside and outside of MIs.
Results
Identification of Methylation Islands in Invertebrate Genomes
We selected seven Hymenopteran insects (A.mellifera, Camponotus floridanus, Harpegnathos saltator, N.vitripennis, Polistes canadensis, Solenopsis invicta, and T.pretiosum) with well-annotated genome information and available deep-coverage whole-genome bisulfite sequencing (BS-seq) data for this study (table 1). The average fractional methylation of methylated CpGs (mCGs) varies among species ranging from 0.44 to 0.74 (table 1). The global average fractional methylation of all CpG sites was ranged from 0.008 to 0.025 in all seven species (table 1). Previously it was shown that methylated CpGs in some species tend to occur in clusters (Wang et al. 2013; Huh et al. 2014). Indeed, mCGs in our data set exhibited clustering (supplementary fig. S1, Supplementary Material online). Specifically, the distances between two adjacent mCGs were significantly shorter than the distance between two randomly selected CGs from the genome, for all species considered (supplementary table S3, Supplementary Material online).
Table 1.
Genome Composition of the Species Used in This Study Along with Methylation Statistics
| Species | Genome Size (Mb) | # Protein-Coding Genes | # of mCGs (% of all CGs) | Avg. Fractional Methylation of mCGs |
|---|---|---|---|---|
| Apis mellifera | 234.07 | 15,314 | 78,846 (0.78%) | 0.584 |
| Camponotus floridanus | 232.68 | 11,042 | 85,746 (0.84%) | 0.635 |
| Harpegnathos saltator | 294.46 | 11,838 | 112,212 (0.53%) | 0.662 |
| Nasonia vitripennis | 295.78 | 13,354 | 114,261 (0.85%) | 0.737 |
| Polistes canadensis | 211.21 | 9,876 | 15,744 (0.24%) | 0.386 |
| Solenopsis invicta | 396.02 | 14,451 | 157,829 (0.98%) | 0.526 |
| Trichogramma pretiosum | 196.22 | 13,200 | 60,298 (0.60%) | 0.345 |
To capture the clusters of mCGs, henceforth referred to as methylation islands (MIs), we utilized a sliding window approach to identify regions of high mCG density and employ them as units of measurement to explore DNA methylation (detailed description of MI definition and search can be found in supplementary file S1, Supplementary Material online). Specifically, our algorithm identified MIs as regions harboring >2% of mCGs (∼3-fold enrichment compared with the genome average, table 1) in windows longer than 200 bp (Materials and Methods).
Characteristics of MIs
Our approach identified thousands of MIs in the seven species. As expected from the pattern of clustering, a large portion of mCGs was located within the MIs, even though the total lengths of MIs were only a minute fraction of the genome size (table 2). The average length and number of the identified MIs were positively correlated with the total number of mCGs (Pearson correlation coefficients = 0.97) rather than the size of the genome (tables 1 and 2). For example, P. canadensis had the lowest number of MIs (n = 1,342) even though its genome size is 20 Mb larger than that of T. pretiosum (the number of MIs in T. pretiosum is 4,889). Similarly, the average MI length was the longest in S. invicta, the species with the most mCGs, and the shortest in P. canadensis, the species with the least mCGs.
Table 2.
Summary Statistics of MIs Detected in Each Species
| Apis mellifera | Camponotus floridanus | Harpegnathos saltator | Nasonia vitripennis | Polistes canadensis | Solenopsis invicta | Trichogramma pretiosum | |
|---|---|---|---|---|---|---|---|
| # of predicted MIs | 5,126 | 6,327 | 8,375 | 9,644 | 1,342 | 10,574 | 4,889 |
| # of mCGs in MIs (% of total mCGs) | 29,254 (37.1%) |
47,804 (55.8%) |
78,490 (69.9%) |
85,007 (74.4%) |
8,293 (52.7%) |
112,819 (71.5%) |
30,141 (50%) |
| Total MI length (bp) (% of genome) |
1,043,247 (0.45%) |
1,803,969 (0.77%) |
2,969,693 (1.01%) |
3,355,006 (1.13%) |
210,235 (0.099%) |
4,291,930 (1.08%) |
1,136,846 (0.58%) |
| Avg. MI length (bp) | 213.15 | 286.12 | 355.59 | 348.88 | 157.66 | 406.89 | 233.53 |
| Avg. mCG density per MI (# of mCGs/MI length) | 0.03 | 0.02 | 0.03 | 0.02 | 0.07 | 0.03 | 0.03 |
| # of MIs overlapping with genesa (% of all MIs) | 4,958 (96.7%) |
6,082 (96.1%) |
7,845 (93.7%) |
9,079 (94.1%) |
1,020 (76%) |
9,843 (93.1%) |
4,603 (94.2%) |
| # of MIs overlapping exclusively with genesa (% of all MIs) | 4,788 (93.4%) |
5,961 (94.2%) |
7,606 (90.8%) |
8,873 (92.0%) |
1,010 (75.3%) |
9,477 (89.6%) |
4,469 (91.4%) |
| # of MIs overlapping with exons/exclusively with exons (% of all MIs) | 4,830/3,117 (94.2%/60.8%) |
5,763/2,634 (91.1%/41.6%) |
7,319/2,704 (87.4%/32.3%) |
8,184/3,381 (84.9%/35.1%) |
741/524 (55.2%/39.0%) |
8,839/3,412 (83.6%/32.3%) |
4,433/2,926 (90.7%/59.8%) |
| # of MIs overlapping with introns/exclusively with introns (% of all MIs) | 1,794/178 (35.0%/3.5%) |
3,404/382 (53.8%/6.0%) |
5,011/592 (59.8%/7.1%) |
5,739/1,206 (59.5%/12.5%) |
478/273 (35.6%/20.3%) |
6,300/1,160 (59.6%/11.0%) |
1,881/242 (38.5%/4.9%) |
| # of MIs overlapping with exon–intron boundaries/only one exon–intron boundary (% of all MIs) | 1,637/705 (31.9%/13.8%) |
3,051/1,312 (48.2%/20.7%) |
4,461/1,690 (53.3%/20.2%) |
4,672/1,635 (48.4%/17.0%) |
205/92 (15.3%/6.9%) |
5,252/2,123 (49.7%/20.1%) |
1,649/611 (33.7%/12.5%) |
| # of MIs overlapping with promoters (% of all MIs) | 172 (3.4%) |
117 (1.8%) |
146 (1.7%) |
199 (2.1%) |
30 (2.2%) |
308 (2.9%) |
213 (4.4%) |
Defined as the region spanning the transcript start site to the transcription termination site.
In the honey bee, clusters of DNA methylation predominantly overlap with gene bodies (96.7%, defined as the region between the transcription start and transcription termination sites), particularly exons (94.2%; table 2). Of all the MIs found in the honey bee, 60.8% were exclusively residing (100% overlap) within exons (supplementary fig. S4, Supplementary Material online). MIs are also found in introns, though less frequently. In fact, only 3.5% of honey bee MIs were found exclusively in introns. Interestingly, 31.9% of the honey bee MIs extended across an exon–intron boundary. Previous studies have speculated on the role of DNA methylation at exon–intron boundaries in signaling splice junctions and/or playing a role in alternative splicing (Lyko et al. 2010; Herb et al. 2012; Li-Byarlay et al. 2013; Galbraith et al. 2015). Consequently, we asked whether MIs were preferentially located at exon–intron boundaries. To answer this question, we randomly permuted MIs onto concatenated genes, and examined how often MIs were found in exon–intron boundaries (detailed methodology can be found in supplementary file, Supplementary Material online). We found significant (empirically determined P value <0.001) enrichment of MIs at exon–intron boundaries for all seven species (supplementary fig. S5, Supplementary Material online).
It was previously shown that mCGs in some hymenopteran insects, including honey bees (A. mellifera), tended to occur near the 5′ end of a gene (Lyko et al. 2010; Hunt et al. 2013a; Wang et al. 2013; Galbraith et al. 2015; Lindsey et al. in press). Interestingly, MIs from A. mellifera as well as T. pretiosum were found slightly biased to the 3′ end of a gene (fig. 2B). On the other hand, MIs in C. floridanus, H. saltator, P. canadensis, and N. vitripennis were 5′ biased (fig. 2B).
Fig. 2.

—Characterization of MIs across genic regions in seven hymenopteran species. (A) Box plots displaying the coverage of MIs across different types of genic regions (gene body, introns, and exons). (B) Violin plots comparing the relative position of MIs with regards to the TSS for genes with MIs.
MIs Are Enriched in Evolutionarily Conserved Genes and the Amino Acids inside of MIs Are More Conserved than the Amino Acids outside of MIs
Previous studies often classified genes as methylated or unmethylated, typically based on the mean fractional methylation (Lyko et al. 2010; Sarda et al. 2012; Wang et al. 2013; Glastad et al. 2016). These studies have shown that methylated genes are more evolutionarily conserved than unmethylated genes (Lyko et al. 2010; Wang et al. 2013; Galbraith et al. 2015; Glastad et al. 2016). Here, we further examined evolutionary conservation of genes based on the presence or absence of MIs. We first processed protein sequences of all species to identify orthologous gene sets (Materials and Methods). This yielded a total of 12,249 gene sets out of which 5,403 (44%) were single copy orthologs found in all seven species and thus termed as the Complete Orthologous (CO) gene set. Of the remaining gene sets, 6,429 (52%) were found in two or more species and classified as the Incomplete Orthologous (IO) gene set. Genes unique to each species (n = 21,523) were termed as the Unique Gene (UG) set (fig. 3A).
Fig. 3.

—MIs are enriched in evolutionarily conserved genes. (A) Bar plots illustrating the number of genes in each gene type (all genes [AG], complete orthologous genes [CO], incomplete orthologous genes [IO], and unique genes in each species [UG]). (B) Bar plots indicating the proportion of genes having different types of methylation features.
We then examined the frequencies of 1) genes with MI, 2) genes without MI but with at least one mCG, and 3) genes without both MI and mCG in each type of gene set. The frequency of genes with MIs is higher in the CO compared with those in the IO and UG while the frequency of genes without MI but with mCGs is similar between CO and IO (fig. 3B). We tested if genes with MIs were overrepresented in CO compared with IO using Fisher’s exact test, which yielded an average odds ratio of 2.84. In comparison, the same test using the number of genes without MI but with mCGs yields an average odds ratio of 1.31 (supplementary table S1, Supplementary Material online). These two odds ratios are statistically significantly different, indicating that clusters of mCGs (which by definition are MIs), more so than individual mCGs, tend to be highly enriched in conserved gene sets (table 3 and supplementary table S1, Supplementary Material online).
Table 3.
Statistical Significance of Differences between Odds Ratios (OR) of Genes with and without MI Using Z Approximation
| Species | OR of Genes w/MIa | OR of Genes w/o MI but w/mCGb | Difference of Log. OR (δ) | SE(δ) | P Value |
|---|---|---|---|---|---|
| Apis mellifera | 2.31 | 1.65 | 0.34 | 0.07 | 3.8E-07 |
| Camponotus floridanus | 2.79 | 1.33 | 0.74 | 0.07 | 2.2E-16 |
| Harpegnathos saltator | 3.23 | 0.93 | 1.25 | 0.08 | 2.2E-16 |
| Nasonia vitripennis | 3.29 | 1.14 | 1.06 | 0.09 | 2.2E-16 |
| Polistes Canadensis | 1.44 | 1.23 | 0.16 | 0.10 | 5.7E-02 |
| Solenopsis invicta | 3.84 | 1.19 | 1.17 | 0.07 | 2.2E-16 |
| Trichogramma pretiosum | 2.97 | 1.74 | 0.53 | 0.08 | 1.2E-11 |
Note.—Odds ratios were calculated and summarized in supplementary table S1, Supplementary Material online.
Odds ratio of the number of genes with MIs and the number of the remaining genes between CO and IO types, respectively, were tested using Fisher’s exact test.
Odds ratio of the number of genes without MIs but with mCGs and the number of the remaining genes between CO and IO types, respectively, were tested using Fisher’s exact test.
We further examined whether the existence of DNA methylation and/or MI have effects on the conservation of specific amino acids. To do so, we mapped genomic coordinates of mCGs occurring in the coding regions of the corresponding positions in the protein sequence and quantified the conservation scores using the Jensen–Shannon (JS) divergence of protein sequence conservation (Lin 1991; Capra and Singh 2007). A linear mixed model was fitted to predict the conservation scores of amino acids based on the existence of mCG sites in the corresponding DNA coding sequence and the location of amino acids positioned either inside or outside of MIs (Materials and Methods). We found that amino acids harboring mCGs had higher conservation scores compared with amino acids without mCGs (fig. 4). Specifically, amino acids positioned inside MIs showed higher conservation scores compared with amino acids positioned outside of MIs (P value <2.2×10−16). Strikingly, amino acids that did not have mCG sites but were located inside MIs had similar or even higher conservation scores than amino acids in MIs with mCG sites (fig. 4; see also supplementary fig. S6, Supplementary Material online). Even though the exact relationship between conservation scores and the location in regard to MIs was variable in different hymenopteran species (supplementary fig. S6, Supplementary Material online), sites within MIs consistently exhibited higher conservation than those outside of MIs. These results demonstrate that protein sequence conservation has a stronger association with methylation islands than individually methylated CG sites.
Fig. 4.
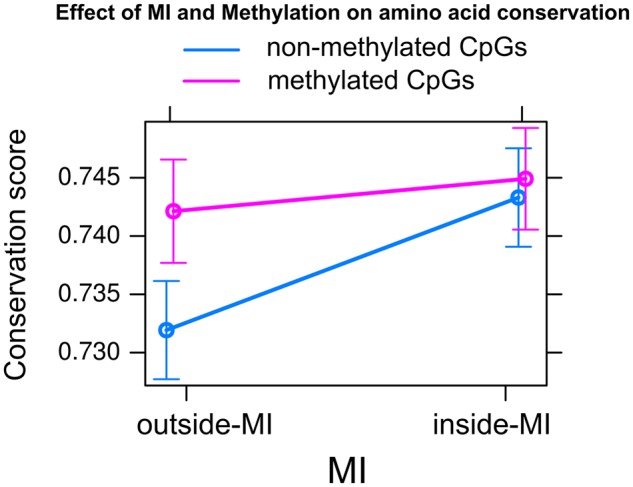
—Effect of MI and DNA methylation on amino acid conservation. Linear mixed models were fitted to estimate the conservation score of amino acid sites using amino acid location (outside-MI or inside-MI) and the existence of corresponding DNA methylation sites (nonmethylated CpGs or methylated CpGs) as the main factors with the interaction and random factors being gene and species information, respectively. The conservation score of each amino acid position is quantified based on the Jensen–Shannon (JS) divergence.
Gene Expression Is Influenced by the Presence of MIs
Previous studies demonstrated that gene body methylation is prevalent in evolutionarily conserved genes and correlates with constitutive and high gene expression (Elango et al. 2009; Lyko et al. 2010; Wang et al. 2013; Galbraith et al. 2015; Glastad et al. 2016). Here, we investigated expression patterns with respect to the presence of MIs. We compared normalized gene expression levels between MI- and non-MI-genes for three of the species that we had RNA-seq data for (fig. 5), and observed that expression levels of MI-genes were higher than that of non-MI genes in all cases. In addition, highly conserved genes (i.e., CO) tend to have higher expression levels than lowly conserved genes across all species (i.e., IO and UG), indicating that gene expression increases according to the degree of conservation. These results align with other studies that have shown gene body methylation to be associated with sequence conservation and robust expression (Sarda et al. 2012; Huh et al. 2013; Hunt et al. 2013b). Interestingly, while expression levels of non-MI genes clearly decreased as conservation level decreased, expression levels of MI genes remained consistent regardless of gene sequence conservation (fig. 5).
Fig. 5.

—Gene expression levels between MI- and non-MI-genes in each gene type. The y-axis is log2-transformed gene expression level (normalized by gene length). The x-axis represents gene types of all genes (AG), complete orthologous genes (CO), incomplete orthologous genes (IO), and unique genes in each species (UG).
We sought to characterize expression change in relation to gain or loss of MIs in conserved genes. Since gene expression varies extensively among species and conditions, a direct comparison between species is difficult. To overcome this limitation, we tested how a change in the MI state of CO genes associated with the overall correlation of gene expression between species. We first assigned each gene pair a binary classification for its MI state. A gene pair is considered to have the “same MI state” if it lacks an MI in both species or it possesses at least one MI in both. Conversely, a gene is labeled as having a “different MI state” if only one species in the pair has an MI. There are greater number of “same MI state” genes than “different MI state” genes in the orthologous gene pairs, consistent with the previous observation that conserved genes tend to share similar MI states (table 4). We then conducted pairwise comparisons of gene expression for each species pair. We observed a significant difference in Spearman’s rank correlation coefficients for all three pairwise comparisons between genes with “same MI state” and “different MI state.” Specifically, “same MI state” genes exhibited stronger correlations, indicating that the presence of MIs in conserved genes is associated with stable and constitutive transcriptional activity in insects (table 4).
Table 4.
Statistical Significance between Pairwise Correlation Coefficients of “Same State MI” and “Different State MI” Genes
| Same State MIs |
Different State MIs |
||||
|---|---|---|---|---|---|
| Spearman’s ρ | Number of Genes | Spearman’s ρ | Number of Genes | P value | |
| Apis mellifera–Nasonia vitripennis | 0.607 | 3,590 | 0.557 | 1,768 | 9.30E-03 |
| Apis mellifera–Trichogramma pretiosum | 0.374 | 3,587 | 0.301 | 1,779 | 4.50E-03 |
| Nasonia vitripennis–Trichogramma pretiosum | 0.468 | 3,927 | 0.351 | 1,431 | 2.20E-16 |
Note.—The correlation coefficients were estimated between two species’ gene expression level using Spearman’s rho correlations.
To further test whether the existence of MIs affected gene expression levels, we compared relative expression levels of exons located in MIs (MI-exon) and that of exons located outside of MIs (non-MI-exon) within the same gene. The median expression level of MI-exons was generally higher than that of non-MI-exons (fig. 6). This was particularly evident for the CO and IO gene groups where the locally weighted smoothing line was >0 for all three species, suggesting expression bias inclined toward MI-exons. Overall, we consistently observed expression bias toward higher expression of MI-exons regardless of gene type and species, indicating a robust relationship between MI-exons and increased gene expression.
Fig. 6.

—Comparison of average expression levels between exons located in MIs (MI-exon) and exons located outside of MIs (non-MI-exon) within the same gene. For complete orthologous genes (CO), incomplete orthologous genes (IO), and unique genes in each species (UG), we calculated the expression fold change (log2 transformed) between MI-exons and non-MI-exons within the same gene, where each dot represents one gene. A locally weighted smoothing line is included to map the general direction bias of relative expression change; when the line is >0 it indicates higher expression in MI-exons compared with non-MI-exons and vice versa for when the line dips <0. We repeated this analysis for three species, (A) Apis mellifera, (I) Nasonia vitripennis, and (C) Trichogramma pretiousum.
DNMT3 Knockdown Further Highlights Significance of MIs on Alternative Splicing
We performed additional analyses to further explore roles of MI using data from a previous knockdown experiment of DNMT3, the enzyme responsible for de novo methylation, in A. mellifera (Li-Byarlay et al. 2013). There was a modest drop in both mCGs and MIs in the knockdown sample (table 5), which is consistent with the role of DNMT3 in DNA methylation. In total, 89.8% of mCGs remained consistent between control and knockdown bees, which was similarly reflected in the MI measurements where 83.2% of MIs remained the same (table 5). In the 205 genes that lost MIs in the knockdown sample, we found no significant expression differences between control and knockdown genes (supplementary fig. S7, Supplementary Material online). We further examined genes that were similarly methylated in both conditions but lost MIs in the knockdown bee. Gene ontology analysis revealed functions related to nucleotide binding (P value = 0.017) and methyltransferase activity (P value = 0.032), though none of the terms were statistically significant following adjustment for the false discovery rate (supplementary table S4, Supplementary Material online).
Table 5.
Summary Statistics of DNA Methylation and MIs in Control and dnmt3 Gene Knockdown Honey Bees
| Control | dnmt3 Gene Knockdown | |
|---|---|---|
| # total mCG sites | 78,846 | 75,897 |
| # genes with mCG sites | 6,308 | 6,277 |
| Avg. # of mCGs per gene | 12.3 | 11.9 |
| # MIs (MI genes) | 5,126 (3,280) | 4,946 (3,207) |
| # MIs only present in group (MI genes) | 501 (222) | 372 (147) |
| # MIs at exon–intron boundary only present in group | 116 | 38 |
Interestingly, among the 501 MIs lost in the knockdown bees, 116 (23.1%) were on exon–intron boundaries. The observation that MIs in exon–intron boundaries tend to be excluded from those that were lost in the knockdown (P < 0.05, Fisher’s exact test) is consistent with the importance of DNA methylation, and MIs, residing at splice sites (Li-Byarlay et al. 2013). In the 372 MIs that were gained in the knockdown, those residing in exon–intron boundaries were significantly underrepresented (P value <0.0001, Fisher’s exact test), which might indicate that regulation of splicing is overall impeded in knockdown bees (Li-Byarlay et al. 2013).
Discussion
One of the most remarkable classical findings of DNA methylation studies in mammals is that unmethylated CpGs tend to occur in clusters, or “CpG islands (CGIs)” (Bird et al. 1985; Bird 1992; Suzuki and Bird 2008). They have been used as central “markers” to study epigenetic variation for several decades (Suzuki and Bird 2008; Illingworth and Bird 2009; Yi 2017). As methylome data from nonmodel species including many invertebrates begin to accumulate, the intriguing contrast between methylomes of mammals and invertebrates (fig. 1) becomes clearer, motivating us to ask several questions: given that methylated CpGs are marked exceptions to the generally unmethylated invertebrate genomes, do methylated cytosines occur in clusters in these species, and if so, do they carry specific functional consequences?
As the first step to answer these questions, we used relatively well-characterized genomes and methylomes of seven hymenopteran insects in this study. Previous analyses of hymenopteran methylomes often defined methylated and unmethylated genes based on whether they harbor or lack methylated cytosines, or used the average methylation level of a gene as a summary statistic for comparisons (Bonasio et al. 2012; Smith et al. 2012; Wang et al. 2013). Even though these prior studies were successful in illuminating novel aspects of invertebrate DNA methylation, because the proportion of methylated sites in each gene is typically small, taking averages could dilute the true signal of DNA methylation (Lyko et al. 2010; Bonasio et al. 2012; Smith et al. 2012; Wang et al. 2013). In addition, some studies indicated that methylated CpGs in these species occur in clusters (Wang et al. 2013; Huh et al. 2014), which we show is true in the seven species analyzed here.
We used a sliding window approach that is conceptually similar to the algorithms used to identify CpG islands in mammalian genomes. We reasoned that if MIs represented functional units, they might occur in similar numbers across closely related species as in the case of mammals (Illingworth et al. 2010). This idea led to identifying MIs as regions exhibiting >3-fold enrichment of methylated CpGs compared with the rest of the genome. Interestingly, mammalian CGI algorithms typically use 3-fold enrichment of unmethylated CpGs as one of the criteria (Gardiner-Garden and Frommer 1987; Takai and Jones 2002). This similarity is another interesting parallel between the methylomes of mammals and hymenopteran insects. Nevertheless, it is known that the criteria to define CGIs require some adjustments when used in nonhuman species, to account for species-specific nucleotide compositions (Matsuo et al. 1993; Aerts et al. 2004). Similarly, the criteria we used here likely will require finer adjustments according to specific genomes that will be targets of the study.
An important discovery regarding CGIs is that genes harboring CGIs in their promoters are more highly and more stably expressed compared with genes that lack CGIs in the promoters (Antequera 2003; Saxonov et al. 2006; Elango and Yi 2008) Moreover, this trend was consistently observed in diverse vertebrates (Elango and Yi 2008). We show that MIs in hymenopteran insects also have deep implications for gene expression. First, MIs are significantly overrepresented in exon–intron boundaries, which is consistent with their presumed role in regulation of intron splicing or alternative splicing (Flores et al. 2012; Herb et al. 2012; Li-Byarlay et al. 2013; Maunakea et al. 2013; Galbraith et al. 2015). This discovery has potential uses in aiding annotation of previously unannotated genes, particularly exonic regions. Interestingly, when DNMT3 was knocked down (Li-Byarlay et al. 2013), MIs occurring on exon–intron boundaries tended to be maintained in a higher frequency than expected. Second, genes harboring MIs exhibit higher and more stable levels of gene expression compared with those without MIs, a pattern that was also evident at the exon level and may be indicative of their inclusion in alternative transcripts. We also investigated whether the gain or loss of MIs could be connected to these expression traits, thus getting us closer to understanding the cause-and-effect relationship between MIs and expression. Regrettably, currently available data sets are limited to moderately diverged species trios (A. mellifera, N. vitripennis, and T. pretiosum), prohibiting accurate identification of orthology of individual MIs. Nevertheless, we could clearly demonstrate that expression levels across species were more strongly correlated when MIs were maintained in coding sequences. It is notable that the effect of MIs on expression is in the same direction as the effect of CGIs in case of mammals (promoting higher and more stable gene expression). These findings suggest that characterizing DNA methylation in insects beyond singular CpGs or broader regions could offer additional insights for understanding the functional role of DNA methylation.
Supplementary Material
Acknowledgments
This study was supported by a grant from the National Science Foundation (MCB-1615664) and funds from Georgia Institute of Technology to S.V.Y.
Literature Cited
- Aerts S, Thijs G, Dabrowski M, Moreau Y, De Moor B.. 2004. Comprehensive analysis of the base composition around the transcription start site in metazoa. BMC Genomics 5:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data.
- Antequera F. 2003. Structure, function and evolution of CpG island promoters. Cell Mol Life Sci. 60(8):1647–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck S. 2018. Implications of CpG islands on chromosomal architectures and modes of global gene regulation. Nucleic Acids Res. 46(9):4382–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler SM, et al. 2014. Whole-genome DNA methylation profile of the jewel wasp (Nasonia vitripennis). G3 (Bethesda) 4:383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bewick AJ, Vogel KJ, Moore AJ, Schmitz RJ.. 2017. Evolution of DNA methylation across insects. Mol Biol Evol. 34(3):654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. 1992. The essentials of DNA methylation. Cell 70(1):5–8. [DOI] [PubMed] [Google Scholar]
- Bird A, Taggart M, Frommer M, Miller OJ, Macleod D.. 1985. A fraction of the mouse genome that is derived from islands of nonmethylated, Cpg-rich DNA. Cell 40(1):91–99. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonasio R, et al. 2012. Genome-wide and caste-specific DNA methylomes of the ants Camponotus floridanus and Harpegnathos saltator. Curr Biol. 22(19):1755–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra JA, Singh M.. 2007. Predicting functionally important residues from sequence conservation. Bioinformatics 23(15):1875–1882. [DOI] [PubMed] [Google Scholar]
- Elango N, Hunt BG, Goodisman MA, Yi SV.. 2009. DNA methylation is widespread and associated with differential gene expression in castes of the honeybee, Apis mellifera. Proc Natl Acad Sci U S A. 106(27):11206–11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elango N, Yi SV.. 2008. DNA methylation and structural and functional bimodality of vertebrate promoters. Mol Biol Evol. 25(8):1602–1608. [DOI] [PubMed] [Google Scholar]
- Feng S, et al. 2010. Conservation and divergence of methylation patterning in plants and animals. Proc Natl Acad Sci U S A. 107(19):8689–8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores K, et al. 2012. Genome-wide association between DNA methylation and alternative splicing in an invertebrate. BMC Genomics 13(1):480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith DA, Yang X, Niño EL, Yi S, Grozinger C.. 2015. Parallel epigenomic and transcriptomic responses to viral infection in honey bees (Apis mellifera). PLoS Pathog. 11(3):e1004713.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M.. 1987. CpG islands in vertebrate genomes. J Mol Biol. 196(2):261–282. [DOI] [PubMed] [Google Scholar]
- Glastad KM, Gokhale K, Liebig J, Goodisman MA.. 2016. The caste- and sex-specific DNA methylome of the termite Zootermopsis nevadensis. Sci Rep. 6(1):37110.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glastad KM, Hunt BG, Yi SV, Goodisman MA.. 2011. DNA methylation in insects: on the brink of the epigenomic era. Insect Mol Biol. 20(5):553–565. [DOI] [PubMed] [Google Scholar]
- Greer EL, et al. 2015. DNA Methylation on N6-Adenine in C. elegans. Cell 161(4):868–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herb BR, et al. 2012. Reversible switching between epigenetic states in honeybee behavioral subcastes. Nat Neurosci. 15(10):1371–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh I, Yang X, Park T, Yi SV.. 2014. Bis-class: a new classification tool of methylation status using bayes classifier and local methylation information. BMC Genomics 15(1):608.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh I, Zeng J, Park T, Yi SV.. 2013. DNA methylation and transcriptional noise. Epigenet Chromatin 6(1):9.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt BG, Glastad KM, Yi SV, Goodisman MA.. 2013a. The function of intragenic DNA methylation: insights from insect epigenomes. Integr Comp Biol. 53(2):319–328. [DOI] [PubMed] [Google Scholar]
- Hunt BG, Glastad KM, Yi SV, Goodisman MA.. 2013b. Patterning and regulatory associations of DNA methylation are mirrored by histone modifications in insects. Genome Biol Evol. 5(3):591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth RS, Bird AP.. 2009. CpG islands – ‘a rough guide’. FEBS Lett. 583(11):1713–1720. [DOI] [PubMed] [Google Scholar]
- Illingworth RS, et al. 2010. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS Genet. 6(9):e1001134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK.. 2012. On the presence and role of human gene-body DNA methylation. Oncotarget 3(4):462–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA. 2012. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 13(7):484–492. [DOI] [PubMed] [Google Scholar]
- Kim D, et al. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14(4):R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR.. 2011. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27(11):1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner M, et al. 2011. Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics 12(1):124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W.. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30(7):923–930. [DOI] [PubMed] [Google Scholar]
- Li-Byarlay H, et al. 2013. RNA interference knockdown of DNA methyl-transferase 3 affects gene alternative splicing in the honey bee. Proc Natl Acad Sci U S A. 110:12750–12755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH. 1991. Divergence measures based on the Shannon entropy. IEEE Trans Inform Theory 37(1):145–151. [Google Scholar]
- Lindsey ARI, et al. Comparative genomics of the miniature wasp and pest control agent Trichogramma pretiosum. BMC biology 16.1(2018):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyko F, et al. 2010. The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol. 8(11):e1000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17(1):10–12. [Google Scholar]
- Matsuo K, Clay O, Takahashi T, Silke J, Schaffner W.. 1993. Evidence for erosion of mouse CpG islands during mammalian evolution. Somat Cell Mol Genet. 19(6):543–555. [DOI] [PubMed] [Google Scholar]
- Maunakea AK, Chepelev I, Cui K, Zhao K.. 2013. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 23(11):1256–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendizabal I, et al. 2016. Comparative methylome analyses identify epigenetic regulatory loci of human brain evolution. Mol Biol Evol. 33(11):2947–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razin A, Cedar H.. 1994. DNA methylation and genomic imprinting. Cell 77(4):473–476. [DOI] [PubMed] [Google Scholar]
- Robertson KD, Wolffe AP.. 2000. DNA methylation in health and disease. Nat Rev Genet. 1(1):11–19. [DOI] [PubMed] [Google Scholar]
- Rosic S, et al. 2018. Evolutionary analysis indicates that DNA alkylation damage is a byproduct of cytosine DNA methyltransferase activity. Nat Genet. 50(3):452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarda S, Zeng J, Hunt BG, Yi SV.. 2012. The evolution of invertebrate gene body methylation. Mol Biol Evol. 29(8):1907–1916. [DOI] [PubMed] [Google Scholar]
- Saxonov S, Berg P, Brutlag DL.. 2006. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A. 103(5):1412–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saze H, Scheid OM, Paszkowski J.. 2003. Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat Genet. 34(1):65–69. [DOI] [PubMed] [Google Scholar]
- Schübeler D. 2015. Function and information content of DNA methylation. Nature 517(7534):321–326. [DOI] [PubMed] [Google Scholar]
- Sievers F, et al. 2014. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 7(1):539.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CR, et al. 2012. Patterns of DNA methylation in development, division of labor and hybridization in an ant with genetic caste determination. PLoS One 7(8):e42433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, Bird A.. 2008. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 9(6):465–476. [DOI] [PubMed] [Google Scholar]
- Takai D, Jones PA.. 2002. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 99(6):3740–3745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay KD, Saam JR, Ingram RS, Tilghman SM, Bartolomei MS.. 1995. A paternal-specific methylation imprint marks the alleles of the mouse H19. Gene Nat Genet. 9(4):407–413. [DOI] [PubMed] [Google Scholar]
- Wang X, et al. 2013. Function and evolution of DNA methylation in Nasonia vitripennis. PLoS Genet. 9(10):e1003872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Werren JH, Clark AG.. 2016. Allele-specific transcriptome and methylome analysis reveals stable inheritance and Cis-regulation of DNA methylation in Nasonia. PLoS Biol. 14(7):e1002500.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi S. 2012. Birds do it, bees do it, worms and ciliates do it too: dNA methylation from unexpected corners of the tree of life. Genome Biol. 13(10):174.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi SV. 2017. Insights into epigenome evolution from animal and plant methylomes. Genome Biol Evol. 9(11):3189–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder JA, Walsh CP, Bestor TH.. 1997. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 13(8):335–340. [DOI] [PubMed] [Google Scholar]
- Zemach A, McDaniel IE, Silva P, Zilberman D.. 2010. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328(5980):916–919. [DOI] [PubMed] [Google Scholar]
- Zemach A, Zilberman D.. 2010. Evolution of eukaryotic DNA methylation and the pursuit of safer sex. Curr Biol. 20(17):R780–R785. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.