

Abstract
Background
Age of onset is linked to variations in clinical phenotypes and natural history in Crohn’s disease (CD). We aim to define etiologically more homogenous subgroups in CD based on ages of onset.
Methods
We examined the distribution of CD polygenetic risk score (PRS) across ages of diagnosis in a Caucasian cohort of 2344 independent CD patients. We identified subgroups with a distinct distribution of PRS and compared those groups in genetics, demographic characteristics, clinical subphenotypes, and serological markers. The results were replicated in an independent cohort of 13,065 CD patients from the International Inflammatory Bowel Diseases Genetic Consortium (IIBDGC).
Results
We identified a late-onset (LO) subgroup in CD (age at diagnosis ≥ 55 years) with significantly lower PRS compared with the intermediate group (age at diagnosis between 5 and 55 years) in both cohorts. Smoking cessation, a risk factor for ulcerative colitis (UC) and protective factor for CD, had a higher rate in this LO subgroup in comparison with the intermediate group. We also compared the LO group with the intermediate group, and, consistent with previous reports, the LO group more often had colonic CD, had less penetrating disease behavior, and had less need for surgery. Serological analysis showed that LO CD patients were more antineutrophil cytoplasmic antibody positive and less antisaccharomyces cerevisiae antibody positive compared with the intermediate group. Variance component analysis indicated that overall genetic contribution to LO CD was lower relative to the middle group, and genetic heterogeneity testing indicated that LO CD was different from the middle group in underlying genetic architecture.
Conclusions
Late-onset CD is subgroup distinct in genetic and behavioral risk factors with UC-like characteristics.
Keywords: Crohn’s disease, genetics, smoking, late-onset
INTRODUCTION
Crohn’s disease (CD), a debilitating gastrointestinal disorder affecting more than a million people in United States,1, 2 is a complex disorder with a wide spectrum of observed phenotypic heterogeneity.3 Understanding this heterogeneity is important for more accurate prognosis assessment, better treatment strategies, and ultimately the development of tailored and targeted therapeutics.
Previous investigations indicate that clinical phenotypes and natural history of CD might differ according to age at diagnosis. Several studies have reported that early-onset CD patients have more severe disease,4, 5 more upper gastrointestinal issues,6, 7 and a greater need for aggressive treatment.5, 8, 9 In a number of studies, it has been reported that late-onset CD might differ in terms of disease location and natural history,6, 10–12 although the findings have been inconsistent.
In the widely applied Montreal13, 14 and Paris classifications,15 CD patients are classified into subgroups based on age at diagnosis. However, those classifications are largely based on the heterogeneity in clinical features, and little is known about the potentially different mechanisms contributing to onset of symptoms and age at diagnosis. Instead of focusing on the heterogeneity in clinical features, one could identify patient subsets from an etiological point of view, which might provide insight to the underlying mechanisms for this complex human diseases and lead to novel intervention strategies.
In this study, we examined the distribution of genetic factors based on 172 CD loci (including loci associated with overall IBD) identified in 2 recent large-scale association studies16, 17 across a European ancestry cohort of CD patients with different ages of onset. We calculated the polygenetic risk score (PRS),18 which reflects the overall genetic burden in CD, and examined its distribution across age groups. We identified a late-onset (LO) subgroup (defined as age at diagnosis ≥55 years) characterized by significantly lower PRS, which we replicated in the large International IBD Genetic Consortium (IIBDGC) cohort.16, 17 We further identified specific clinical and demographic features, including a significant role of smoking cessation in the LO group. Moreover, we examined differences in serological markers in the index cohort to explore the difference in innate/adaptive immunity in the identified subgroup.
METHODS
Subjects
The details of subject recruitment in the Cedars-Sinai cohort have been described previously.19, 20 Briefly, CD patients were recruited at Cedars-Sinai Medical Center (CSMC) from 1985 to 2015. The diagnosis of each patient was based on standard endoscopic, histologic, and radiographic features, as previously described.19, 20 Blood samples were collected at the time of enrollment. The study protocol and data collection, including DNA preparation/genotyping and antibody measurement, were approved by the CSMC Institutional Review Board. Written informed consent was obtained from all study participants.
Subject recruitment in the IIBDGC cohort is documented elsewhere.16–18 Briefly, 17,302 CD patients and controls were recruited from 15 countries in Europe, North America, and Oceania. Diagnosis of IBD was based on accepted radiological, endoscopic, and histopathological evaluation. All included cases fulfilled clinical criteria for IBD and gave written consent.
Clinical and Serologic Phenotyping
Clinical data, including patients’ current age, sex, age at diagnosis, current disease location and behavior (according to the Montreal Classification), surgical history, and smoking status at diagnosis, were collected as previously described.18 In the Cedars cohort, IBD-associated serologies, including antinuclear cytoplasmic antibody (ANCA), antisaccharomyces cerevisiae antibodies (ASCA IgG and IgA), antiflagellin (anti-CBir1), anti-outer membrane protein C (anti-OmpC), and antipseudomonas fluorescens–related protein (anti-I2), were measured by enzyme-linked immunosorbent assay, as previously described.21, 22 All assays were performed blinded to patient clinical characteristics. Based on the measured serological markers, we further calculated the quartile sum score (QSS) of Anti-CBir1, Anti-I2, Anti-OmpC, IgA-ASCA, and IgG-ASCA.21
Genotyping and Genotype Quality Control
Genotyping of the Cedars cohort was performed at CSMC using Illumina ImmunoChip (IChip) array. Individual and genotype missingness, allele frequencies, and deviations from Hardy-Weinberg equilibrium were calculated using the PLINK software package.23 Individual-level quality control (QC) thresholds include genotyping call rate >95%, inbreeding coefficient <0.05, and lack of cryptic relatedness (Pi-hat > 0.25 in PLINK). Ethnicity outliers identified using Admixture software24 were also removed. Single nucleotide polymorphisms (SNPs) with a call rate <0.95, with minor allele frequency (MAF) <0.01, and that strongly deviated from Hardy-Weinberg equilibrium (P < 1 × 10–7) were also removed. After QC, there were 2344 CD cases and 118,611 SNPs available for analysis in this cohort.
Genotyping and QC in the IIBDGC cohort have been described elsewhere.16–18 In brief, the IIBDGC IChip samples were genotyped in 36 batches, and genotype calling was performed separately for each batch. Similar QC was performed, which removed SNPs with a call rate lower than 98% across all genotyping batches or 90% in 1 of the genotyping batches, but not in 1000 Genomes Project Phase I, failing Hardy-Weinberg equilibrium (false discovery rate [FDR] < 1 × 10-5 across all samples or within each genotyping batch), or monomorphic SNPs. Individuals were assigned to different populations based on principal components, and those not in the Caucasian cluster or with a low call rate (<98%), outlying heterozygosity rate (FDR < 0.01), or cryptic relatedness (identity by decent > 0.4) were removed. After QC, 152,232 SNPs and 13,065 CD cases were included in current analysis.
PRS Calculation
CD PRS were calculated as a weighted sum of the number of risk alleles carried by each individual (0, 1, or 2) at known CD loci (n = 172, including 126 loci also associated with overall IBD), with weights proportional to the effect estimates from the previously published large-scale association studies.16–18 The PRS were then normalized separately in the Cedars and IIBDGC cohorts to have mean of 0 and standard deviation of 1. We also calculated a UC PRS, in which known UC loci (n = 157, including 126 loci associated with IBD) were included in the score calculation. As there is strong overlap in loci used to construct UC and CD PRS, we further calculated a UC-only PRS, in which variants only associated with UC but not with CD or IBD in previous reported large-scale association studies (n = 31) were included. Details of the loci included in score calculation can be found in Supplementary Table 1.
Statistical Analysis
Ages at diagnosis were first grouped by 5-year increments to identify distinct subgroups, and analysis of variance (ANOVA) was used to examine the difference of PRS across different groups. Structural changes in PRS as ages of diagnosis increased were evaluated using the strucchange package in R25 based on the method proposed by Zeileis, Shah, and Patnaik.26 We thereby divided CD patients into subgroups based on the identified changing point.
Thereafter, logistic regression was performed to compare the difference between the identified subgroups in demographic, clinical, and serological characteristics. The Cedars cohort was used as a discovery cohort, and replication was performed in the IIBDGC cohort when applicable. Serological analysis was performed only in the Cedars cohort as there were no serological data available in IIBDGC. To account for the correlation of the clinical and serological factors, identified variables in univariate analyses were put in a joint model to identify independently associated factors, and Akaike information criterion (AIC)–based stepwise model selection was used to identify variables in the final model.
Associations of Ichip SNPs and the LO CD were performed using the software PLINK23 separately in the Cedars and IIBDGC cohorts, and a meta-analysis was performed to combine results from both cohorts. Mixed-model based association27 was utilized to evaluate pathway level difference between the LO and intermediate groups, with pathways defined based on the KEGG pathway database.28
We also calculated the genetic variance contribution from all SNPs genotyped on ImmunoChip for the LO and middle groups separately in the Cedars and IIBDGC cohorts using GCTA software.29 The difference in underlying genetic architecture between the LO and middle groups was further examined using the pseudo-likelihood ratio (PLR) approach proposed by Liley et al.30 The same approach was also utilized to examine the genetic similarity between LO CD and UC.
In all analyses, principal components from population stratification analysis31 were included as covariates. In the smoking analyses, current age was also included to control for potential confounding effects from patients’ age. To confirm that the association of smoking behavior with the LO group was not due to confounding of the age-cohort effect, we further performed a matched analysis in which patients from the LO group were matched based on 5-year groups in current age. Conditional logistic regression analysis was performed to examine the association of smoking behaviors with the LO CD in the matched analysis. All statistical analyses were performed using R 3.2.0.25
RESULTS
Clinical characteristics for the 2 cohorts are shown in Table 1. In both cohorts, the majority of the CD patients were diagnosed between age 5 and 55 years; 106 (4.52%) and 845 CD patients were diagnosed after age 55 years in the Cedars and IIBDGC cohorts, respectively.
TABLE 1:
Clinical Characteristics of CD Patients in the Current Study
| Cedars-Sinai Cohort | IIBDGC Cohort | ||||||
|---|---|---|---|---|---|---|---|
| Total No. | No. (Yes) | % | Total No. | No. (Yes) | % | ||
| Sex | Female | 2344 | 1088 | 46.42 | 13,065 | 7334 | 56.13 |
| Age at diagnosis, y | <5 | 2344 | 36 | 1.54 | 13,065 | 37 | 0.3 |
| 5–55 | 2344 | 2202 | 93.94 | 13,065 | 12,183 | 93.2 | |
| ≥55 | 2344 | 106 | 4.52 | 13,065 | 845 | 6.5 | |
| Disease location | L1 | 2094 | 488 | 23.30 | 12,773 | 4020 | 31.47 |
| L2 | 2094 | 422 | 20.15 | 12,773 | 2824 | 22.25 | |
| L3 | 2094 | 1184 | 56.54 | 12,773 | 5517 | 43.58 | |
| L4 | 1950 | 339 | 17.38 | 10,070 | 1066 | 10.59 | |
| Perianal | 2019 | 632 | 31.30 | 10,664 | 2960 | 27.76 | |
| Disease behavior | B1 | 2095 | 1002 | 47.83 | 12,235 | 5722 | 46.77 |
| B2 | 2095 | 574 | 27.40 | 12,235 | 3023 | 24.71 | |
| B3 | 2095 | 519 | 24.77 | 12,235 | 3490 | 28.52 | |
| Surgery | 2210 | 1129 | 51.09 | 12,691 | 6579 | 51.84 | |
B1, nonstricturing, nonpenetrating disease; B2, stricutring disease; B3, penetrating disease; L1, ileal-only affection; L2, colon-only affection; L3, ileocolonic affection; L4, upper GI affection; Perianal, perianal disease location; Surgery, surgery for Crohn’s disease.
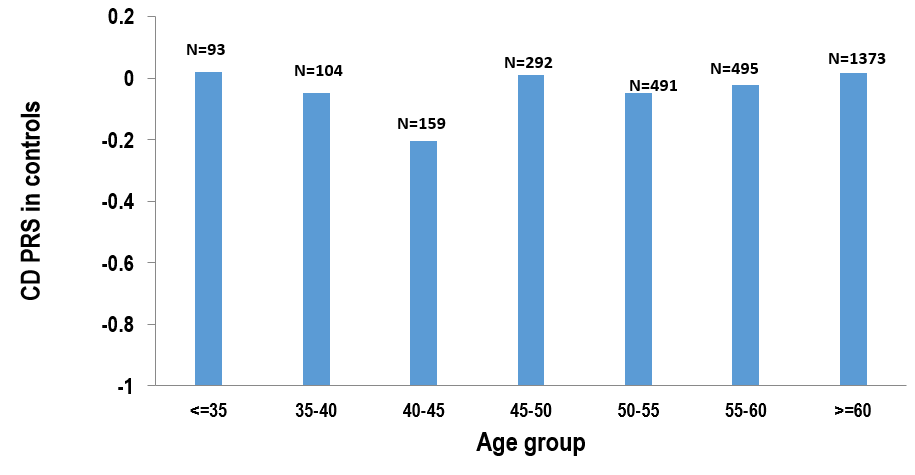
We first examined the distribution of PRS with different ages at diagnosis (grouped by 5 years) in the Cedars cohort (Fig. 1) and observed statistically significant differences (P = 1.17 × 10-6) between groups. Structural change tests indicated that there is statistically significant change in the slope of PRS as age at diagnosis increases (F-value, 29.59; P = 1.23E-5). This analysis also demonstrated that there is 1 changing point in the slope of PRS in the age group ≥55 years. We thereby regrouped patients with age at diagnosis ≥55 years as the LO group. To exclude potential confounding from the very early-onset (VEO) CD patients, who have monogenic forms of IBD, we defined the patients with age at diagnosis <5 years as the VEO group and patients with age at diagnosis of 5–55 years as the intermediate group.
FIGURE 1.

Distribution of CD PRS according to age at diagnosis of disease.
Thereafter we examined the mean PRS and observed a significantly lower PRS in the LO group compared with the intermediate group (0.15 vs 0.55, P = 2.99 × 10-5) (Table 2). This difference was replicated in the IIBDGC cohort (0.25 vs 0.48, P = 1.35 × 10-11) (Table 2). No significant difference was observed when comparing VEO CD patients PRS with the intermediate group, in either of the cohorts. Moreover, the distribution of CD PRS in patients was not observed in non-IBD patients (grouped based on current age) (Supplementary Fig. 1).
TABLE 2:
CD Genetic Burden in the VEO, LO, and Intermediate Groups
| Groups of Age at Diagnosis | No. | PRS, Mean±SD | Beta (95% CI)a | P | |
|---|---|---|---|---|---|
| Cedars cohort | VEO | 36 | 0.37 ± 0.81 | –0.14 (–0.45 to 0.16) | 0.36 |
| Intermediate | 2212 | 0.55 ± 0.96 | – | – | |
| LO | 106 | 0.15 ± 0.92 | –0.40 (–0.57 to –0.21) | 2.99E-05 | |
| IIBDGC | VEO | 37 | 0.53 ± 0.85 | 0.15 (–0.2 to 0.39) | 0.51 |
| Intermediate | 12,183 | 0.48 ± 0.92 | – | – | |
| LO | 845 | 0.25 ± 0.96 | –0.21 (–0.28 to –0.15) | 1.35E-11 |
VEO, age at diagnosis <5 years; Intermediate group, 5 years ≤ age at diagnosis < 55 years; LO, age at diagnosis ≥55 years.
aCompared with the intermediate group.
We also compared the allele frequencies of known CD SNPs16, 17 between the LO and intermediate groups (details in Supplementary Table 2). The NOD2 frameshift mutation (rs5743293) is the only known CD-associated SNP that was associated, after correction for multiple testing, with the LO CD group when compared with the intermediate group (OR, 0.58; P = 3.98 × 10-6 in the meta-analysis), with the direction of association in the inverse direction of the reported association with CD. Interestingly, the log of odds ratios (ORs) for the known CD variants in the LO vs intermediate analysis are negatively correlated (r = –0.67; P = 3.54 × 10-25) with the association observed in the intermediate group vs non-IBD controls (Fig. 2). This negative correlation remained when SNPs with strong effects (including variants in NOD2, ATG16L1, and IL23R) were excluded (r = –0.43; P = 9.90 × 10-7).
FIGURE 2.

Negative correlation between the log(ORs) of the LO CD group vs the intermediate group and the middle group vs non-IBD controls. Intermediate group: 5 years ≤ age at diagnosis < 55 years; LO: age at diagnosis ≥55 years.
We further examined sex and smoking behavior in the LO and intermediate groups (Table 3). There was no statistically significant difference between the LO and intermediate groups in sex in the Cedars cohort (OR, 0.87; P = 0.49) or the IIBDGC cohort (OR, 0.97; P = 0.76). In the Cedars cohort, there were significantly fewer current smokers (OR, 0.03; P = 4.60 × 10-4), and there were more ex-smokers (OR, 4.38; P = 3.30 × 10-6) in the LO group, even after controlling for current age. A similar phenomenon was observed in the IIBDGC cohort (OR, 0.72 and 1.65; P = 5.26 × 10-3 and 2.17 × 10-5, respectively). There was no statistically significant difference for ever-smoker in either cohort. In the matched analysis in which patients from the LO group were matched based on 5-year age groups in current age, a similar trend was observed (for ex-smoking, hazard ratio [HR], 4.22 and 1.34; P = 7.39 × 10-5 and 5.91 × 10-3; in the Cedars and IIBDGC cohorts, respectively; for current-smoking, HR = 0.08 and 0.75; P = 0.02 and 0.01; in the Cedars and IIBDGC cohorts, respectively).
TABLE 3:
Smoking Behavior and Sex in the LO Group Compared With the Intermediate Group
| Phenotype | In LO | In Intermediate Group | OR (95% CI) | P | |||||
|---|---|---|---|---|---|---|---|---|---|
| Yes | No | % | Yes | No | % | ||||
| Cedars cohort | Sex (F) | 46 | 60 | 43.40 | 1028 | 1174 | 46.68 | 0.87 (0.59 to 1.30) | 0.492 |
| Current smoker | 1 | 60 | 1.64 | 188 | 1327 | 12.41 | 0.03 (0.00 to 0.21) | 4.60E-04 | |
| Ex-smoker | 32 | 29 | 52.46 | 104 | 1411 | 6.86 | 4.38 (2.35 to 8.17) | 3.30E-06 | |
| Ever smoker | 33 | 28 | 54.10 | 292 | 1223 | 19.27 | 1.01 (0.56 to 1.79) | 0.986 | |
| IIBDGC | Sex | 492 | 353 | 58.22 | 6881 | 5302 | 56.48 | 0.97 (0.81 to 1.16) | 0.760 |
| Current smoker | 166 | 509 | 24.59 | 2824 | 6299 | 30.95 | 0.72 (0.57 to 0.90) | 5.26E-03 | |
| Ex-smoker | 232 | 443 | 34.37 | 1261 | 7862 | 13.82 | 1.63 (1.30 to 2.04) | 2.17E-05 | |
| Ever smoker | 398 | 277 | 58.96 | 4085 | 5038 | 44.78 | 1.14 (0.92 to 1.41) | 0.225 | |
Intermediate group, 5 years ≤ age at diagnosis < 55 years; LO, age at diagnosis ≥55 years.
Distribution of disease location in the LO and intermediate groups is shown in Table 4. There was no statistically significant difference in L1 between the LO and intermediate groups in the Cedars cohort, although the difference was marginally significant in the IIBDGC cohort (OR, 1.21; P = 0.013). The proportion of L2 was higher in the LO group compared with the intermediate group in the Cedars cohort (OR, 2.00; P = 2.58 × 10-3) and also in the IIBDGC cohort (OR, 2.48; P = 7.74 × 10-32). In contrast, the proportion of L3 was lower for LO in both the Cedars cohort (OR, 0.57; P = 7.90 × 10-3) and the IIBDGC cohort (OR, 0.32; P = 7.93 × 10-36). There was no statistically significant difference for L4 and perianal location in the Cedars cohort, but the results were significant in IIBDGC (OR, 0.31; P = 2.22 × 10-8; and OR, 0.38; P = 6.11 × 10-18; respectively, for upper gastrointestinal [GI] and perianal location).
TABLE 4:
Disease Location and Behavior in the LO Group Compared With the Intermediate Group
| Phenotype | In LO | In Intermediate Group | OR | P | |||||
|---|---|---|---|---|---|---|---|---|---|
| Yes | No | % | Yes | No | % | ||||
| Cedars cohort | L1 | 23 | 70 | 24.73 | 462 | 1509 | 23.44 | 1.06 (0.66 to 1.73) | 0.802 |
| L2 | 30 | 63 | 32.26 | 380 | 1591 | 19.28 | 2.00 (1.27 to 3.14) | 2.58E-03 | |
| L3 | 40 | 53 | 43.01 | 1129 | 842 | 57.28 | 0.57 (0.37 to 0.86) | 7.90E-03 | |
| L4 | 9 | 70 | 11.39 | 323 | 1518 | 17.54 | 0.62 (0.30 to 1.25) | 0.177 | |
| Perianal | 24 | 60 | 28.57 | 596 | 1309 | 31.29 | 0.87 (0.54 to 1.42) | 0.586 | |
| B2vsB1 | 30 | 57 | 34.48 | 541 | 926 | 36.88 | 0.90 (0.57 to 1.41) | 0.636 | |
| B3vsB1 | 8 | 57 | 12.31 | 503 | 926 | 35.20 | 0.26 (0.12 to 0.54) | 3.48E-04 | |
| B2B3vsB1 | 38 | 57 | 40.00 | 1044 | 926 | 52.99 | 0.59 (0.38 to 0.89) | 0.013 | |
| Surgery | 38 | 62 | 38.00 | 1078 | 999 | 51.90 | 0.56 (0.37 to 0.85) | 6.30E-03 | |
| IIBDGC | L1 | 277 | 509 | 35.24 | 3533 | 7733 | 31.36 | 1.21 (1.04 to 1.41) | 0.013 |
| L2 | 315 | 466 | 40.33 | 2349 | 8877 | 20.92 | 2.48 (2.13 to 2.88) | 7.74E-31 | |
| L3 | 163 | 618 | 20.87 | 5094 | 6109 | 45.47 | 0.32 (0.27 to 0.39) | 7.93E-36 | |
| L4 | 25 | 607 | 3.96 | 1010 | 8247 | 10.91 | 0.31 (0.21 to 0.47) | 2.22E-08 | |
| Perianal | 98 | 611 | 13.82 | 2789 | 6650 | 29.55 | 0.38 (0.31 to 0.48) | 6.11E-18 | |
| B2vsB1 | 190 | 441 | 30.11 | 2655 | 4978 | 34.78 | 0.84 (0.70 to 0.99) | 0.047 | |
| B3vsB1 | 107 | 441 | 19.53 | 3166 | 4978 | 38.88 | 0.40 (0.32 to 0.49) | 1.30E-16 | |
| B2B3vsB1 | 297 | 441 | 40.24 | 5821 | 4978 | 53.90 | 0.59 (0.51 to 0.70) | 4.90E-11 | |
| Surgery | 270 | 511 | 34.57 | 6025 | 5327 | 53.07 | 0.47 (0.41 to 0.55) | 7.65E-22 | |
B1, nonstricturing, nonpenetrating disease; B2, stricutring disease; B3, penetrating disease; Intermediate group, 5 years ≤ age at diagnosis < 55 years; L1, ileal-only affection; L2, colon-only affection; L3, ileocolonic affection; L4, upper GI affection; LO, age at diagnosis ≥55 years; Perianal, perianal disease location; Surgery, surgery for Crohn’s disease.
In Table 4, we also compared disease behavior (measured based on Montreal classification13, 14) and need for surgery between the LO and intermediate groups. In both cohorts, the difference in disease behavior was observed in B3 (penetrating disease) vs B1 (nonstricturing nonpenetrating disease), with an OR of 0.26 (P = 3.84 × 10-4) in the Cedars cohort and 0.40 (P = 1.30 × 10-16) in the IIBDGC cohort. The difference in B2 (stricturing disease) vs B1 was not significant in the Cedars cohort but showed borderline significance in IIBDGC (OR, 0.84; P = 0.047). In both cohorts, patients had less need for surgery in the LO group (OR, 0.56; P = 6.30 × 10-3; in the Cedars cohort; OR, 0.47; P = 7.65 × 10-22; in IIBDGC). Similar results were observed if the length of follow-up was included as a covariate in both cohorts (details not shown).
We further examined serological markers in the LO and intermediate groups in the Cedars cohort (Table 5). In the LO group, a higher proportion of ANCA+ (OR, 1.57; P = 0.034) and a lower proportion of both anti-CBir1+ (OR, 0.61; P = 0.020) and both IgA-ASCA+ (OR, 0.21; P = 2.27 × 10-6) and IgG-ASCA+ (OR, 0.29; P = 1.43 × 10-5) were observed. We also calculated the QSS of serological markers using Anti-CBir1, Anti-I2, Anti-OmpC, IgA-ASCA, and IgG-ASCA, and lower QSS was observed in the LO group (11.67 ± 2.96 in the LO group vs 12.68 ± 3.85 in the intermediate group; P = 0.027).
TABLE 5:
Serological Characteristics in the LO Group Compared With the Intermediate Group
| Markers | In LO | In Intermediate Group | OR | P | ||||
|---|---|---|---|---|---|---|---|---|
| + | - | % | + | - | % | |||
| ANCA | 36 | 67 | 34.95 | 516 | 1501 | 25.58 | 1.57 (1.03 to 2.38) | 0.034 |
| CBir1 | 36 | 65 | 35.64 | 966 | 1067 | 47.52 | 0.61 (0.40 to 0.93) | 0.020 |
| I2 | 36 | 39 | 48.00 | 657 | 982 | 40.09 | 1.38 (0.87 to 2.20) | 0.174 |
| OmpC | 30 | 73 | 29.13 | 531 | 1488 | 26.30 | 1.14 (0.74 to 1.77) | 0.547 |
| IgA-ASCA | 10 | 93 | 9.71 | 676 | 1316 | 33.94 | 0.21 (0.11 to 0.40) | 3.27E-06 |
| IgG-ASCA | 15 | 88 | 14.56 | 737 | 1255 | 37.00 | 0.29 (0.17 to 0.50) | 1.43E-05 |
ANCA, antinuclear cytoplasmic antibody; CBir, antiflagellin antibody; I2, antipseudomonas fluorescens-related protein; IgA and IgG-ASCA, antisaccharomyces cerevisiae antibodies IgA and IgG; Intermediate group, 5 years ≤ age at diagnosis < 55 years; LO, age at diagnosis ≥55 years; OmpC, anti-outer membrane protein C.
We subsequently examined the difference between the LO and intermediate groups with demographic, serological (only in the Cedars cohort), and clinical characteristics (B3, L2, L3, and surgery) jointly in a multivariate model. Interestingly, in the Cedars cohort, differences in 3 factors (ex-smoking, ASCA positivity, and CD PRS) were observed in the joint model (P = 5.64 × 10-7, 3.87 × 10-4, and 0.046, respectively), whereas the difference in B3 and L2 was not significant at all. To mimic the situation in the IIBDGC cohort, in which serological markers were not available, we re-examined the joint model after excluding the serological markers, and marginal differences in 2 more variables, B3 and L2 (P = 0.036 and 0.099, respectively), were observed in addition to ex-smoking and CD PRS in the joint model. A similar pattern was observed in the IIBDGC joint model, with differences in ex-smoking, B3, L2, and CD PRS observed (P = 2.0 × 10-3, 3.14 × 10-9, 5.04 × 10-15, and 2.10 × 10-7, respectively).
With the UC-like features of the LO CD group, we calculated a UC GRS in which all known SNPs associated with UC (with SNPs associated with both CD and UC included) were included and weighted based on association with UC. A similar pattern was observed with lower UC PRS in the LO CD group (Supplementary Fig. 2). We further constructed a UC-only PRS, in which only SNPs that are associated with UC but not CD or IBD were included (based on SNPs reported to be associated only with UC in Jostin et al.16 and Liu et al.17), and there was no statistically significant difference between the LO and intermediate groups in UC-only PRS in the Cedars cohort (P = 0.29) or in the IIBDGC cohort (P = 0.75).
After excluding the known loci, no statistically significant signal survived the genome-wide significance threshold (data not shown) for single SNPs in iChip comparing the LO group with the intermediate group. The pathway-level association suggests that the NOD-like receptor signaling pathway (P = 1.07 × 10-4) (Supplementary Table 3) might contribute to the difference between the LO and intermediate groups.
We also examined the correlation in the association signals of the LO group vs the intermediate group, and those in UC vs CD. A surprisingly strong correlation in log(ORs) of the 2 independent associations was observed in SNPs with P values <1.0E-3 in UC vs CD (r = 0.57; P = 1.68E-13) (Fig. 3; details in Supplementary Table 4) after pruning for SNPs in linkage disequilibrium (LD) with each other. Similar patters were observed when using different P value cutoffs from 0.05 to 5.0E-8 in UC vs CD (details not shown).
FIGURE 3.

Positive correlation between the log(ORs) of the LO CD group vs the intermediate group and the UC group vs the CD group. Intermediate group: 5 years ≤ age at diagnosis < 55 years; LO: age at diagnosis ≥55 years.
We used the software GCTA to estimate the genetic contribution from all SNPs in ImmunoChip to the LO CD and middle groups. After control for principal component analysis, the genetic contributions to the LO and middle groups were estimated to be 6.2% (P = 0.15) and 10.24% (P = 2.02E-90) for the 109,525 variants that passed the QC procedures in the Cedars cohort and 11.5% (P = 2.15E-17) and 17.83% (P < 4.94E-324) for the 156,499 variants that passed the QC procedures in the IIBDGC cohort.
We further tested the hypothesis that the overall underlying genetic architecture is different between the LO and middle groups using the PLR approach.30 We observed a PLR of 44.57 in the Cedars cohort for the heterogeneity between the LO CD and middle groups, and after 2000 permutations, we observed a P value of 0.17. In IIBDGC, the PLR and P value after permutation were 771.56 and 5.05E-6, respectively.
We also compared the underlying genetic architecture of LO and UC using the PLR approach. In the Cedars cohort, the PLR was 32.52 with a P value of 0.19. In IIBDGC cohort, the PLR and P value were 3374.34 and 2.40E-6, respectively, for the genetic heterogeneity between LO CD and UC.
DISCUSSION
In the current study, we examined the CD PRS in CD patients with different ages at diagnosis and identified that LO patients with age at diagnosis ≥55 years have lower PRS. LO CD patients differ in disease location, disease behavior, and need for surgery, consistent with previous reports. Moreover, we demonstrated UC-like smoking behaviors (more ex-smokers, fewer current smokers) and UC-like serological patterns (high ANCA, low ASCA) in the LO group. Our analyses also illustrated that in an LO group vs intermediate group comparison, there is a parallel pattern in single-SNP level signals to UC vs CD. Our findings clearly demonstrate that the late-onset CD group is a distinct subgroup with UC-like features that could impact future clinical practice.
For more than 30 years, clinicians have observed that late-onset CD patients might have different characteristics than their younger peers. There have been a number of studies comparing LO with other CD patients,6, 10–12 with conflicting results, probably due to small sample sizes and inconsistent and often arbitrary definitions of “late onset.” In the current study, we started from an etiological point of view by examining the genetic burden in patients with different ages at diagnosis, and further expanded the comparison to include clinical and immunological features. The main findings were replicated in an independent, large cohort, indicating that we’ve identified a distinct subgroup of CD. The way we identified this subgroup can be viewed as a model mechanism for patient stratification in other complex diseases.
The observed lower PRS in the LO group, in both cohorts, indicates differences in underlying pathogenic mechanisms in this subgroup. The cause of CD remains unclear, but it’s generally agreed that the interplay of genetic factors, innate/adaptive immunity, microbiome, and environmental triggers are contributing factors.32–34 The much lower PRS in the LO group indicates that known genetic variants, which have been demonstrated to be critical in disease development in most CD cases, might play much less important roles in the subgroup of LO CD. This may imply distinct underlying mechanisms in LO patients. Those results are further supported by the heterogeneity test that indicates strong difference in genetic structure underlying the LO and middle groups, with significant results in the PLR test in the IIBDGC cohort, although the test for difference in genetic structure in the Cedars cohort was not significant, probably due to a relatively small sample size.
It is important to determine whether the difference of PRS in LO is due to a subset of the known associated SNPs. When examining the differences of allele frequencies of single known SNPs between the LO and intermediate groups, the only variant that was statistically significant was the NOD2 frameshift mutation, with a much lower frequency in the LO group. Moreover, we observed a strong negative correlation between log(ORs) in the LO vs intermediate groups and the intermediate group vs non-IBD controls, even after excluding variants like NOD2. Thus, for the majority of known variants, the LO group has lower allele frequencies for risk variants and higher frequencies for protective variants, although our sample size may not to be large enough to capture, with high statistical confidence, any association with other SNPs beyond NOD2. This suggests that the low PRS in the LO group is not due to a single or a few known SNPs, but an overall weaker genetic burden, which is further supported by the lower overall genetic contribution from all SNPs on the ImmunoChip in the LO group.
Cigarette smoking is one of the few widely accepted environmental risk factors for CD35, 36 and is associated with more complicated diseases,32, 37–43 need for surgery,32, 37–39, 42, 43 and relapse after treatments.32, 39–41, 44 Conversely, smoking cessation decreases the risk of developing CD but is associated with increased risk of UC.35, 36 Unexpectedly, we observed a lower prevalence of current smokers and more ex-smokers in the LO group of CD patients, which is a UC-like feature. As age was included as a covariate in smoking behavior analysis, the observed effects of smoking behaviors cannot be explained by the age-cohort effect. This is also supported by the similar results observed when examining age-matched analysis. Smoking cessation can lead to profound changes in the composition of the gut microbiome,45 and the gut microbiome is well known to play important roles in the development of CD.46, 47 Still, it is intriguing to observe opposite effects of smoking behaviors in CD at different ages of onset, indicating, further, a complex role for smoking as an environmental factor in IBD. Further investigation is warranted to demonstrate the role of smoking in the pathogenesis of this subgroup and, moreover, whether it shares similar etiology as UC. Furthermore, it would be worthwhile to explore the potential treatment options related to this unique and modifiable environmental factor.
Distinction of LO CD is also reflected by our analysis examining the serological markers in the LO group, with higher ANCA and lower anti-CBir1 and ASCA observed. Higher ANCA has long been associated with UC44, 48 and with UC-like features in CD, including L2 disease location.49 The observed higher ANCA in LO CD indicates a common intestinal mucosal inflammatory process between LO CD and UC. Higher ANCA has also been linked to nonresponse to anti–tumor necrosis factor (anti-TNF) treatments in both CD and UC,50, 51 and given the higher adverse event rate that has been reported in the elderly on anti-TNF therapies, these data, collectively, suggest that a dedicated trial of the risks and benefits of anti-TNF therapy in late-onset Crohn’s disease is warranted. Anti-CBir1 and ASCA reflect innate/adaptive immunity to selected microbiome agents,52, 53 and both are associated with a more complicated CD phenotype. Lower anti-CBir1 and much lower ASCA clearly indicate an alternative route of shifted genetic/environmental factors leading to abnormal immune response in the gut microbiome, which in turn influences disease onset, leading to different clinical phenotypes, and defines a separate disease subgroup. However, the observed serological characteristics of LO CD, which were identified in the Cedars cohort, cannot be replicated in the IIBDGC cohort as there are no serology measures in the IIBDGC phenotype data set. Independent replications on the serological measures in LO CD are warranted to validate the serological findings in the current study.
In the current study, we have demonstrated that there is more isolated colonic (L2) and less ileocolonic disease in the LO group. This is consistent with previous observations in elderly-onset CD patients.6, 11, 12, 54, 55 We also observed that LO CD patients tend to have less need for surgery and less penetrating behavior, consistent with previous reports that isolated colonic CD tends to be less severe.6, 10–12, 55 It is also critical to develop specific intervention strategies for this subgroup. For example, the LO CD cohort may be a subgroup of CD patients who respond to mesalazine or even to anti-MADCAM therapy, both of which seem to be more effective therapies for UC than CD.56, 57
With more colon-only affection, UC-like smoking behaviors (more ex-smokers, fewer current smokers), and UC-like serological patterns (high ANCA and low ASCA), the LO group is likely a distinct subgroup of CD with UC-like characteristics in both clinical presentation and disease etiology. This is also supported by the strong correlation of single-SNP signals in LO vs intermediate and CD vs UC comparisons. These findings further illustrate the heterogeneous nature of CD and the need for more personalized treatment strategies in clinical practice.
Interestingly, even with the UC-like clinical and environmental characteristics, heterogeneity analysis using a PLR approach30 indicates that the overall underlying genetic structure is different between LO CD and UC. The seemingly contradicting results are not very surprising as the LO CD patients were diagnosed as CD but not UC based on standard clinical and endoscopic criteria, indicating submucosal or transmural inflammations in those patients, which might be related to different genetic mutations. One possibility is that UC and LO CD might have related or overlapping but still distinct pathogenesis mechanisms; more research is needed to solve this complex puzzle.
Of note, conditioning on etiological factors (smoking behavior and CD PRS) and serological markers reflecting host response to the microbiome, the difference in clinical presentations (disease location and behavior) is no longer significant in the Cedars cohort. This observation seems to indicate that the observed clinical characteristics in the LO group are mainly due to the difference in underlying pathogenesis, which again suggests that LO CD may be a unique IBD subgroup. This observation cannot be replicated in the IIBDGC cohort, though, as serological markers are not measured in this cohort, and additional cohorts are needed to replicate this particular observation.
We did not know the exact age of disease onset for each CD patient in this current study. Instead, we used age at diagnosis as a proxy for age of onset. The diagnosis of CD often presents a challenge for clinicians, probably due to similar presenting symptoms as functional digestive pathologies such as irritable bowel disease.58, 59 This delay in diagnosis has been reported to be 18–24 months on average58, 60 and in certain cases can be multiple years. This could potentially bias our results, in particular in the IIBDGC cohort, in which centers from different areas of world are involved. However, this delay of diagnosis would only make patients with age of onset before age 55 years classified as late-onset in our analysis, which would likely bias our results toward the null hypothesis. In this sense, our findings may be more conservative.
Note that in the previously established Montreal and Paris classification of CD,13–15 the A3 group was defined as CD patients with age at diagnosis >40 years. The identified LO subgroup, with its distinct clinical and etiological characteristics, indicates that adding an additional subgroup for a later age of onset is warranted. It is possible that defining disease subgroups based on age of onset or age at diagnosis, which is probably a proxy to the difference in underlying disease-causing mechanisms, cannot be exact by nature. Better characterization of the underlying disease-causing mechanisms might help to identify more homogenous subgroups in CD.
It is worth noting that in the current study, the Cedars cohort mainly consists of CD patients from the southwestern United States, whereas the IIBDGC cohort (after excluding overlapping samples) consists of patients from different locations in North America, Europe, and Australia, with the majority of the samples from Europe.16 Between the 2 cohorts, the source populations will likely have distinct demographic characteristics with different disease diagnosis/treatment guidelines applied in clinical practice. This might explain the observation that the effect sizes vary between the 2 cohorts in the comparison between the LO and intermediate groups. This is particularly true for smoking behavior–based analysis as previous epidemiology studies have indicated a lower smoking prevalence rate in the United States in comparison with Europe.61, 62 Moreover, the smoking prevalence rate in California, where most subjects in the Cedars cohort are from, was reported to be the lowest in the United States.63 This partially explain the dramatic difference in the proportion of current smokers and ever smokers between the Cedars and IIBDGC cohorts. Despite these differences, we still observed a highly consistent pattern in the 2 cohorts, strongly suggesting that the distinct characteristics we observed in the LO group are genuine.
In summary, we identified late-onset CD patients as a distinct subgroup with different genetic, clinical, environmental, and serological characteristics. The features in this subgroup are more UC-like, and further investigations on the underlying mechanism(s) and specific treatment strategies are warranted.
SUPPLEMENTARY DATA
Supplementary data are available at Inflammatory Bowel Diseases online.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Conflicts of interest: None.
Supported by: U01DK062413 (D.P.B.M.), P01DK046763 (S.R.T., D.P.B.M.), R01HS021747 (D.P.B.M.), U01AI067068 (D.P.B.M.), and R21DK108140 (D.L.). The International IBD Genetic Consortium Data Coordinating Center is supported by National Institute of Diabetes and Digestive and Kidney Diseases grant DK062429.
Author contributions: D.L. proposed and performed the analysis. D.P.B.M., S.R.T., and M.D. supervised the whole project. T.H. performed the genotyping. T.H., A.P., and D.L. performed the QC of the genotype data. S.Y. and D.P.B.M. collected the clinical phenotypes. C.L. measured the serological markers. D.L. and H.H. calculated the genetic risk score. Primary drafting of the manuscript: D.L., S.R.T., D.P.B.M. All authors read, had the opportunity to comment on, and approved the final draft.
REFERENCES
- 1. Kappelman MD, Moore KR, Allen JK et al. Recent trends in the prevalence of Crohn’s disease and ulcerative colitis in a commercially insured US population. Dig Dis Sci. 2013;58:519–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dahlhamer JM, Zammitti EP, Ward BW et al. Prevalence of inflammatory bowel disease among adults aged ≥18 years - United States, 2015. MMWR Morb Mortal Wkly Rep. 2016;65:1166–1169. [DOI] [PubMed] [Google Scholar]
- 3. Di Sabatino A, Rovedatti L, Vidali F et al. Recent advances in understanding Crohn’s disease. Intern Emerg Med. 2013;8:101–113. [DOI] [PubMed] [Google Scholar]
- 4. Gower-Rousseau C, Vasseur F, Fumery M et al. Epidemiology of inflammatory bowel diseases: new insights from a French population-based registry (EPIMAD). Dig Liver Dis. 2013;45:89–94. [DOI] [PubMed] [Google Scholar]
- 5. Gasparetto M, Guariso G, Pozza LV et al. Clinical course and outcomes of diagnosing inflammatory bowel disease in children 10 years and under: retrospective cohort study from two tertiary centres in the United Kingdom and in Italy. BMC Gastroenterol. 2016;16:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Duricova D, Burisch J, Jess T et al. ; ECCO-EpiCom Age-related differences in presentation and course of inflammatory bowel disease: an update on the population-based literature. J Crohns Colitis. 2014;8:1351–1361. [DOI] [PubMed] [Google Scholar]
- 7. Crocco S, Martelossi S, Giurici N et al. Upper gastrointestinal involvement in paediatric onset Crohn’s disease: prevalence and clinical implications. J Crohns Colitis. 2012;6:51–55. [DOI] [PubMed] [Google Scholar]
- 8. Turner D, Levine A, Escher JC et al. ; European Crohn’s and Colitis Organization; European Society for Paediatric Gastroenterology, Hepatology, and Nutrition Management of pediatric ulcerative colitis: joint ECCO and ESPGHAN evidence-based consensus guidelines. J Pediatr Gastroenterol Nutr. 2012;55:340–361. [DOI] [PubMed] [Google Scholar]
- 9. Dignass A, Van Assche G, Lindsay JO et al. ; European Crohn’s and Colitis Organisation (ECCO) The second European evidence-based Consensus on the diagnosis and management of Crohn’s disease: current management. J Crohns Colitis. 2010;4:28–62. [DOI] [PubMed] [Google Scholar]
- 10. Wagtmans MJ, Verspaget HW, Lamers CB et al. Crohn’s disease in the elderly: a comparison with young adults. J Clin Gastroenterol. 1998;27:129–133. [DOI] [PubMed] [Google Scholar]
- 11. Softley A, Myren J, Clamp SE et al. Inflammatory bowel disease in the elderly patient. Scand J Gastroenterol Suppl. 1988;144:27–30. [PubMed] [Google Scholar]
- 12. Hou JK, Feagins LA, Waljee AK. Characteristics and behavior of elderly-onset inflammatory bowel disease: a multi-center US study. Inflamm Bowel Dis. 2016;22:2200–2205. [DOI] [PubMed] [Google Scholar]
- 13. Satsangi J, Silverberg MS, Vermeire S et al. The Montreal classification of inflammatory bowel disease: controversies, consensus, and implications. Gut. 2006;55:749–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Silverberg MS, Satsangi J, Ahmad T et al. Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a Working Party of the 2005 Montreal World Congress of Gastroenterology. Can J Gastroenterol. 2005;19:5A–36A. [DOI] [PubMed] [Google Scholar]
- 15. Levine A, Griffiths A, Markowitz J et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: the Paris classification. Inflamm Bowel Dis. 2011;17:1314–1321. [DOI] [PubMed] [Google Scholar]
- 16. Jostins L, Ripke S, Weersma RK et al. ; International IBD Genetics Consortium (IIBDGC) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liu JZ, van Sommeren S, Huang H et al. ; International Multiple Sclerosis Genetics Consortium; International IBD Genetics Consortium Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cleynen I, Boucher G, Jostins L et al. ; International Inflammatory Bowel Disease Genetics Consortium Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: a genetic association study. Lancet. 2016;387:156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Franke A, McGovern DP, Barrett JC et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McGovern DP, Jones MR, Taylor KD et al. ; International IBD Genetics Consortium Fucosyltransferase 2 (FUT2) non-secretor status is associated with Crohn’s disease. Hum Mol Genet. 2010;19:3468–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dubinsky MC, Kugathasan S, Mei L et al. ; Western Regional Pediatric IBD Research Alliance; Pediatric IBD Collaborative Research Group; Wisconsin Pediatric IBD Alliance Increased immune reactivity predicts aggressive complicating Crohn’s disease in children. Clin Gastroenterol Hepatol. 2008;6:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Devlin SM, Yang H, Ippoliti A et al. NOD2 variants and antibody response to microbial antigens in Crohn’s disease patients and their unaffected relatives. Gastroenterology. 2007;132:576–586. [DOI] [PubMed] [Google Scholar]
- 23. Purcell S, Neale B, Todd-Brown K et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. R Core Team(2013). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. http://www.R-project.org/ [Google Scholar]
- 26. Zeileis A, Shah A, Patnaik I. Testing, monitoring, and dating structural changes in exchange rate regimes. Comput Stat Data An. 2010;54:1696–1706. [Google Scholar]
- 27. Lee S, Emond MJ, Bamshad MJ et al. ; NHLBI GO Exome Sequencing Project—ESP Lung Project Team Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. 2012;91:224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kanehisa M, Goto S, Furumichi M et al. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang J, Lee SH, Goddard ME et al. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liley J, Todd JA, Wallace C. A method for identifying genetic heterogeneity within phenotypically defined disease subgroups. Nat Genet. 2017;49:310–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Price AL, Patterson NJ, Plenge RM et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. [DOI] [PubMed] [Google Scholar]
- 32. Vavricka SR, Rogler G. Recent advances in the etiology and treatment of Crohn’s disease. Minerva Gastroenterol Dietol. 2010;56:203–211. [PubMed] [Google Scholar]
- 33. Lapaquette P, Brest P, Hofman P, Darfeuille-Michaud A. Etiology of Crohn’s disease: many roads lead to autophagy. J Mol Med (Berl). 2012;90:987–996. [DOI] [PubMed] [Google Scholar]
- 34. Loddo I, Romano C. Inflammatory bowel disease: genetics, epigenetics, and pathogenesis. Front Immunol. 2015;6:551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ananthakrishnan AN. Epidemiology and risk factors for IBD. Nat Rev Gastroenterol Hepatol. 2015;12:205–217. [DOI] [PubMed] [Google Scholar]
- 36. Rosenfeld G, Bressler B. The truth about cigarette smoking and the risk of inflammatory bowel disease. Am J Gastroenterol. 2012;107:1407–1408. [DOI] [PubMed] [Google Scholar]
- 37. van der Heide F, Dijkstra A, Weersma RK et al. Effects of active and passive smoking on disease course of Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis. 2009;15:1199–1207. [DOI] [PubMed] [Google Scholar]
- 38. Seksik P, Nion-Larmurier I, Sokol H et al. Effects of light smoking consumption on the clinical course of Crohn’s disease. Inflamm Bowel Dis. 2009;15:734–741. [DOI] [PubMed] [Google Scholar]
- 39. van der Heide F, Wassenaar M, van der Linde K et al. Effects of active and passive smoking on Crohn’s disease and ulcerative colitis in a cohort from a regional hospital. Eur J Gastroenterol Hepatol. 2011;23:255–261. [DOI] [PubMed] [Google Scholar]
- 40. Joyce MR, Hannaway CD, Strong SA et al. Impact of smoking on disease phenotype and postoperative outcomes for Crohn’s disease patients undergoing surgery. Langenbecks Arch Surg. 2013;398:39–45. [DOI] [PubMed] [Google Scholar]
- 41. Gustavsson A, Magnuson A, Blomberg B et al. Smoking is a risk factor for recurrence of intestinal stricture after endoscopic dilation in Crohn’s disease. Aliment Pharmacol Ther. 2013;37:430–437. [DOI] [PubMed] [Google Scholar]
- 42. Lunney PC, Kariyawasam VC, Wang RR et al. Smoking prevalence and its influence on disease course and surgery in Crohn’s disease and ulcerative colitis. Aliment Pharmacol Ther. 2015;42:61–70. [DOI] [PubMed] [Google Scholar]
- 43. To N, Gracie DJ, Ford AC. Systematic review with meta-analysis: the adverse effects of tobacco smoking on the natural history of Crohn’s disease. Aliment Pharmacol Ther. 2016;43:549–561. [DOI] [PubMed] [Google Scholar]
- 44. Forcione DG, Rosen MJ, Kisiel JB et al. Anti-saccharomyces cerevisiae antibody (ASCA) positivity is associated with increased risk for early surgery in Crohn’s disease. Gut. 2004;53:1117–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Biedermann L, Zeitz J, Mwinyi J et al. Smoking cessation induces profound changes in the composition of the intestinal microbiota in humans. PLoS One. 2013;8:e59260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gevers D, Kugathasan S, Denson LA et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hofer U. Microbiome: bacterial imbalance in Crohn’s disease. Nat Rev Microbiol. 2014;12:312. [DOI] [PubMed] [Google Scholar]
- 48. Kuna AT. Serological markers of inflammatory bowel disease. Biochem Med (Zagreb). 2013;23:28–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vasiliauskas EA, Plevy SE, Landers CJ et al. Perinuclear antineutrophil cytoplasmic antibodies in patients with Crohn’s disease define a clinical subgroup. Gastroenterology. 1996;110:1810–1819. [DOI] [PubMed] [Google Scholar]
- 50. Taylor KD, Plevy SE, Yang H et al. ANCA pattern and LTA haplotype relationship to clinical responses to anti-TNF antibody treatment in Crohn’s disease. Gastroenterology. 2001;120:1347–1355. [DOI] [PubMed] [Google Scholar]
- 51. Lew D, Yoon SM, Yan X et al. Genetic associations with adverse events from anti-tumor necrosis factor therapy in inflammatory bowel disease patients. World J Gastroenterol. 2017;23:7265–7273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sandborn WJ. Serologic markers in inflammatory bowel disease: state of the art. Rev Gastroenterol Disord. 2004;4:167–174. [PubMed] [Google Scholar]
- 53. Beniwal P, Harrell L. The status of diagnostic markers for inflammatory bowel disease. Curr Gastroenterol Rep. 2010;12:479–484. [DOI] [PubMed] [Google Scholar]
- 54. Shapiro PA, Peppercorn MA, Antoniolo DA et al. Crohn’s disease in the elderly. Am J Gastroenterol. 1981;76:132–137. [PubMed] [Google Scholar]
- 55. Rhodes J, Rose J. Crohn’s disease in the elderly. Br Med J (Clin Res Ed). 1985;291:1149–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vermeire S, Sandborn WJ, Danese S et al. Anti-MAdCAM antibody (PF-00547659) for ulcerative colitis (TURANDOT): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:135–144. [DOI] [PubMed] [Google Scholar]
- 57. Pillai N, Dusheiko M, Burnand B et al. A systematic review of cost-effectiveness studies comparing conventional, biological and surgical interventions for inflammatory bowel disease. PLoS One. 2017;12:e0185500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Vavricka SR, Spigaglia SM, Rogler G et al. ; Swiss IBD Cohort Study Group Systematic evaluation of risk factors for diagnostic delay in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:496–505. [DOI] [PubMed] [Google Scholar]
- 59. Maconi G, Orlandini L, Asthana AK et al. The impact of symptoms, irritable bowel syndrome pattern and diagnostic investigations on the diagnostic delay of Crohn’s disease: a prospective study. Dig Liver Dis. 2015;47:646–651. [DOI] [PubMed] [Google Scholar]
- 60. Zaharie R, Tantau A, Zaharie F et al. ; IBDPROSPECT Study Group Diagnostic delay in Romanian patients with inflammatory bowel disease: risk factors and impact on the disease course and need for surgery. J Crohns Colitis. 2016;10:306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Phillips E, Wang TW, Husten CG et al. Tobacco product use among adults - United States, 2015. MMWR Morb Mortal Wkly Rep. 2017;66:1209–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gallus S, Lugo A, La Vecchia C et al. Pricing Policies And Control of Tobacco in Europe (PPACTE) project: cross-national comparison of smoking prevalence in 18 European countries. Eur J Cancer Prev. 2014;23:177–185. [DOI] [PubMed] [Google Scholar]
- 63. Odani S, Armour BS, Graffunder CM et al. State-specific prevalence of tobacco product use among adults - United States, 2014-2015. MMWR Morb Mortal Wkly Rep. 2018;67:97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.