

Abstract
Most vaccines depend on co-administration of antigens and adjuvants that activate antigen-presenting cells (APCs). Nanoparticles (NPs) have emerged as an attractive vehicle for synchronized delivery of antigens and adjuvants to APCs and can be targeted to specific cell types such as dendritic cells (DCs), which are potent APCs. However which subset of human DCs should be targeted for optimal activation of T cell immunity remains unknown. Here we describe a poly-lactic-co-glycolic acid (PLGA)-based NP platform, wherein avidin-decorated NPs can be targeted to multiple human DC subsets via biotinylated antibodies. Both BDCA3+ and monocyte-derived DC-SIGN+ NP-loaded DCs were equally effective at generating antigen-specific human T cells in culture, including against complex peptide mixtures from viral and tumor antigens across multiple MHC molecules. Antibody-mediated targeting of NPs to distinct DC subsets led to enhanced T cell immunity. However combination targeting to both DC-SIGN and BDCA3+ DCs led to significantly greater activation of T cells compared to targeting either DC subset alone. Enhanced T cell activation following combination targeting depended on DC-mediated cytokine release and was IL-15 dependent. These data demonstrate that simultaneous targeting of multiple DC-subsets may improve NP vaccines by engaging DC-crosstalk and provides a novel approach to improve vaccines against pathogens and tumors.
Introduction:
Dendritic cells (DCs) play a central role in regulating innate and adaptive immunity and hence there is great interest in targeting these cells to improve the effectiveness of vaccines both against pathogens as well as cancer. The existence of different DC subsets with distinct functions as well as the ability of DCs to undergo phenotypic and functional changes in response to external stimuli allows them to regulate diverse types of immune responses (1, 2). Most of the adjuvants in current vaccines are thought to act in part via activating DCs. Due in part to their potency, several investigators have tried to target antigens to DCs in vivo to boost immunity and improve vaccines (3, 4). One approach involves protein antigens coupled to DC-targeting antibodies (such as DEC-205), which is currently in clinical trials (5). Another strategy involves coupling DC-targeting strategy to other antigen delivery vehicles (6).
Synchronized delivery of antigen and adjuvants to APCs is thought to be critical for vaccine design. In addition to chemical cross-linking, several vehicles such as PLGA polymeric nanoparticles, liposomes, nanocrystals, virus-like particles and 3D-scaffolds have been explored as vehicles for delivering antigens and adjuvants to APCs (7–10). Polymeric NPs fabricated from FDA approved polymers such as PLGA are an attractive platform for vaccines due to their established safety in human studies, lack of off-target effects and ease of production (6, 8, 11, 12). Several studies have explored targeting of NPs to human or murine DCs or DC-subsets via antibodies against receptors expressed on DCs / subsets such as DC-SIGN, DEC-205, CLEC9A, DCIR, BDCA-2 and CD32 (13–18) or more generally against pathogen-associated molecular patterns (19). The rationale for targeting different DC subsets derives in part from differences in their functional properties. For example, in mice, CD8α+ subset of DCs is specialized at cross-presentation of exogenous antigens to generate cytolytic T cells (20, 21). BDCA3+ myeloid dendritic cells (MDCs) were identified as human counterparts of CD8α+ DCs and potentially attractive targets for DC-targeting vaccines (22). However recent studies suggest that several subsets of lymph node resident human DCs may be equally efficient at cross presentation of soluble antigen (23). Cross-presentation of antibody targeted antigen by human BDCA3+ MDCs was instead shown to depend on the nature of endocytic compartment targeted (24). Therefore, at least some aspects of the biology of murine DC subsets may not translate readily to human DCs and the nature of optimal DC subsets for NP-mediated targeting in humans remains to be determined (25). Here we have utilized a novel PLGA-NP platform wherein the particles are decorated with avidin (26, 27) and loaded with clinically relevant viral and tumor antigens, allowing facile exploration of antibody-mediated targeting of different DC subsets via NPs. These data demonstrate for the first time, the potential advantages of NP vaccines for multivalent antigen delivery and simultaneous targeting of several DC subsets.
Methods:
Generation of peptide loaded nanoparticles:
PLGA (Poly-lactic-co-glycolic acid) nanoparticles containing avidin on the surface were prepared (see Figure 1a-1c for characterization of NPs and supplemental table 1 for NP composition), using methods as described earlier (27, 28). The NPs prepared included blank NP (no peptide), coumarin-labeled blank NP (NP-coumarin), NP-FMP (incorporating HLA A2.1 Flu matrix peptide sequence GILGFVFTL), NP-CEF (incorporating CEF pool peptide, pool of 32 peptides from EBV, CMV and influenza virus, Anaspec) and NP-SOX2 (22 15mer SOX2 peptides, Supplemental Table 2). The amount of each peptide in the nanoparticles was as follows: NPFMP (9μg/mg of NP); NP-CEF (0.56μg/mg of NP) and NP-SOX2 (4.1μg/mg of NP) (Supplemental Table 1). In some experiments TLR and/or antibody-coated nanoparticles were prepared by adding biotinylated lipopolysaccharide (LPS, InvivoGen), Poly (I:C) (InvivoGen), BDCA3 antibody (Miltenyi Biotec) or DC-SIGN antibody (Miltenyi Biotec) at the concentration of 5ug of antibody per mg of nanoparticles. The vials were gently rotated for 15 minutes. They were then centrifuged at 1200 rpm for 5 minutes to remove the supernatant and washed twice to remove any soluble ligand prior to using in experiments.
Figure 1. Structure and characterization of nanoparticles.
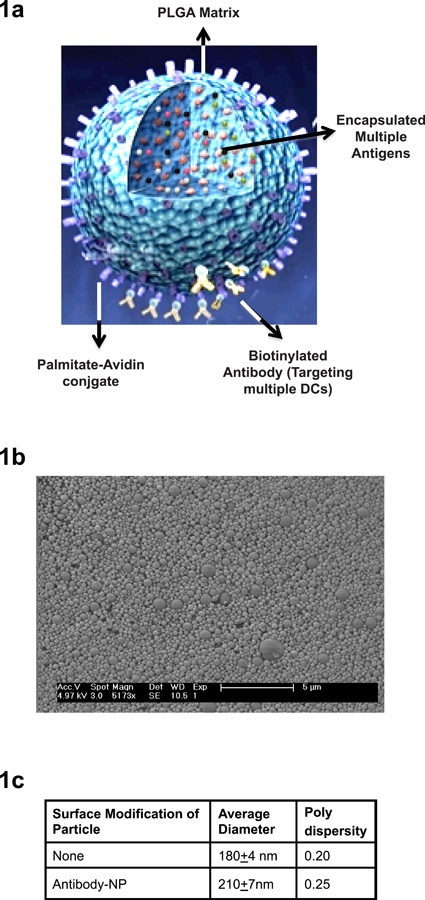
1a) Structure of the nanoparticles. NPs consist of PLGA matrix encapsulating antigens of interest. The surface of the NPs is decorated with palmitate avidin conjugate which allows easy conjugation of targeting antibodies coupled to biotin.
1b) Characterization of NPs using scanning electron microscope.
1c) NPs were sized using Dynamic Light Scattering (Malvern Zetasize).
Nanoparticle uptake experiments:
NPs loaded with coumarin as a marker were added to PBMCs for 30 minutes at varying concentrations (5.5, 55 and 110μg/ml) at either 37°C or 4°C. Flow cytometry was performed and MFI-coumarin analyzed to evaluate the uptake of nanoparticles by monocytes, myeloid DCs, B cells, NK cells and T cells using anti-human CD14, BDCA3, CD19, CD56 and CD3 antibodies respectively.
Generation of dendritic cells:
Monocyte-derived DCs were generated from peripheral blood mononuclear cells as described (29). Briefly, CD14+ monocytes were isolated from PBMCs by immunomagnetic bead selection using CD14 beads following the manufacturer’s protocol (Miltenyi Biotec). CD14+ cells were suspended in 1% healthy donor plasma in RPMI (Cellgro), supplemented with IL-4 (25μg/ml; R&D Systems) and GM-CSF (20ng/ml; Leukine Sargamostim, Genzyme) on days 0, 2 and 4 of culture. Immature monocyte-derived dendritic cells (Mo-DCs) were harvested on Days 5–6 and used for experiments described below. The CD14 negative fraction of PBMCs was cultured in the presence of 5% Pooled Human Serum (PHS, Labquip Ltd.) in RPMI. For some experiments BDCA3+ myeloid dendritic cells were isolated from the PBMCs using BDCA3 MACS beads (Miltenyi Biotec).
Antigen specific T cell stimulation:
Day 6 immature Mo-DCs or freshly isolated BDCA3+ MDCs were loaded with either blank nanoparticles or nanoparticles encapsulated with Flu matrix peptide; NP-FMP (1 hour), CEF pool peptide; NP-CEF (1 hour) or SOX2 pool peptide; NP-SOX2 (4 hours) at 37°C. After overnight culture in 1% plasma, nanoparticle loaded DCs were used to stimulate autologous T cells at a DC:T cell ratio of 1:30 in the presence of IL-2 (10u/ml at days 4 and 7; Chiron). After 10–12 days in culture, flow cytometry was performed to detect the presence of antigen specific T cells using A2.1 FMP tetramer (Beckman Coulter) as well as intracellular cytokine secretion assay for IFN-γ using the peptides used for initial T cell stimulation in the presence of anti-CD28 and anti-CD49D (1mcg/ml).
For experiments with NP-SOX2, T cells were re-stimulated with nanoparticle loaded DCs on days 7 and 14 in the presence of IL-2 (10u/ml) as well as IL-7 and IL-15 (both at 5u/ml, R&D). For some experiments DCs were matured overnight with LPS (50ng/ml, Sigma) or Poly (I:C) (25μg/ml, Sigma) or cytokine cocktail [IL-6 (0.01μg/ml, R&D), IL-1β (0.01μg/ml, R&D), TNF-α (0.01μg/ml, R&D) & PGE2 (1μg/ml, Sigma)] after loading with nanoparticles.
For targeting experiments, BDCA3 or DC-SIGN antibody coated nanoparticles were added to PBMCs at 4°C for 1 hour, washed and cultured for 10–14 days in the presence of IL-2 (10u/ml on days 4 and 7). In some experiments, these were compared with a combinatorial targeting approach in which both BDCA3 and DC-SIGN coated nanoparticles were added at half concentrations. In additional experiments, neutralizing antibodies against cytokines IL-15, IFN-lambda, IL-6 or isotype control mouse IgG1 (all 10μg/ml, R&D) were added to the combinatorial targeting condition. Flow cytometry was performed for the detection of antigen specific T cells as described above. The flow cytometry data was acquired using Cellquest (BD) software on FACSCalibur. The data was then analyzed using Flow-jo software (Tree Star Inc.).
Cytokines produced by BDCA3+ MDCs:
BDCA3+ MDCs isolated from healthy donor buffy coats or DCSIGN positive monocyte derived DCs were loaded with NP-FMP at 37°C and supernatants were collected after 24 hours. Cytokines were quantified by using VeriPlex Human Cytokine ELISA (PBL Interferon source) and data was analyzed by Q-View 2.160 software (Quansys Biosciences).
Statistical analysis:
Two-tailed paired or ratio paired t test were used to investigate significance among the results. P value <0.05 was considered as significant.
Results:
Uptake and presentation of antigen loaded NPs by human DCs / subsets:
We first characterized the relative uptake of NPs by different cellular components of human PBMCs. Coumarin-labeled NPs cultured with PBMCs were preferentially taken up by APCs including BDCA3+ myeloid DCs as well as CD14+ monocytes as compared to B cells, NK cells and T cells in a concentration dependent fashion (Figure 2a, and b). Uptake of NPs by DCs was an active process and inhibited by incubation at 4°C (Figure 2c). Next, we compared the capacity of monocyte-derived DCs (Mo-DCs) and BDCA3+ MDCs to stimulate influenza-matrix peptide (FMP)-specific T cells following uptake of NP-FMP. Both Mo-DCs as well as BDCA3+ MDCs were equally efficient at inducing proliferation of FMP-tetramer+ T cells with the ability to secrete interferon-γ in response to peptide stimulation (Figure 2d-2e).
Figure 2. Nanoparticles are taken up by antigen presenting cells preferentially and peptide loaded NPs lead to induction of antigen specific T cell response.
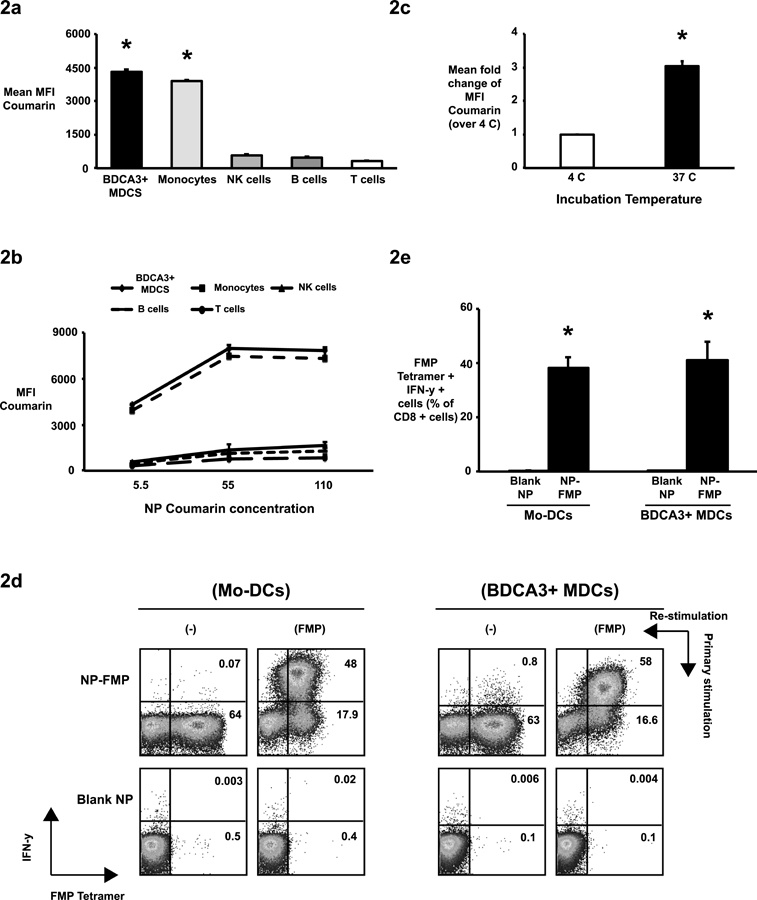
(2a-c). Coumarin-labeled nanoparticles (NP-coumarin) were used to study nanoparticle uptake by mononuclear cells using flow cytometry. Mean fluorescence intensity (MFI) of coumarin was used to compare uptake by different cells.
(2a). Peripheral blood mononuclear cells were incubated with NP-coumarin (5.5μg/ml) for 30 minutes. NP uptake by BDCA3+ myeloid dendritic cells (BDCA3+ MDCs), CD14+ monocytes CD19+ B cells, CD3-CD56+ NK cells and CD3+ T cells was studied using flow cytometry to examine MFI of coumarin as an indicator of NP uptake. The bar graph shows mean coumarin MFI ± standard error of the mean from experiments with 3 healthy donors. *p<0.05 BDCA3+ DCs and Monocytes compared with B cells, NK cells and T cells.
(2b). NP uptake by BDCA3+ DCs, monocytes, B cells, NK cells and T cells at different NP concentrations (n=3).
(2c). NP uptake by BDCA3+ MDCs at different incubation temperatures is shown (n=3).
(2d-e). Nanoparticles encapsulating flu matrix peptide (NP-FMP) stimulate a specific CD8 T cell functional response.
Mo-DCs or BDCA3+ MDCs from HLA A2.1+ healthy donors were loaded with NP-FMP at flu matrix peptide (FMP) concentration of 5μg/ml and then co-cultured with autologous T cells. DCs loaded with blank nanoparticles (Blank NP) were used as negative controls. After 10–12 days, the cells were re-stimulated with soluble FMP (5μg/ml) in the presence of anti-CD28 and anti-CD49D and analyzed for production of interferon-gamma (IFN-γ) by flow cytometry. Data shown in the figure are gated on CD3+ CD8+ T cells.
(2d). Representative figure from healthy donor showing expansion of FMP tetramer positive IFN-γ producing T cells by Mo-DCs loaded with either blank-NP or NP-FMP (left panel) and representative figure from healthy donor showing expansion of FMP tetramer positive IFN-γ producing T cells by BDCA3+ MDCs loaded with either blank-NP or NP-FMP.
(2e). The graphs shows mean percentage ± standard error of the mean of IFN-γ producing FMP tetramer positive CD8 cells from healthy donors stimulated with NP-FMP loaded Mo-DC (n=8, *p= 1.8 × 10−5, Blank NP vs NP-FMP) and BDCA3+ MDCs (n=4; *p= 8.7 × 10−3, Blank NP vs NP-FMP).
Vaccines with a single peptide epitope are by definition restricted to a single HLA type. In order to examine if the current platform could be extended to complex peptide mixtures to induce reactivity against multiple epitopes, we developed PLGA nanoparticles containing a pool of 32 peptides from Cytomegalovirus (CMV), Epstein-Barr virus (EBV) and Influenza (Flu) viruses (NP-CEF) recognized by different HLA haplotypes. Both Mo-DCs as well as BDCA3+ MDCs were able to stimulate antigen-specific IFN-γ secreting T cells in response to NP-CEF (Figure 3a-c). Importantly, the elicited immune response included reactivities against multiple peptides within the mix (Figure 3d).
Figure 3. Complex peptide nanoparticles encapsulating CEF pool peptide (NP-CEF) stimulate a specific and multivalent CD8 T cell functional response.

Mo-DCs or BDCA3+ MDCs were loaded with NP-CEF at individual peptide concentration of 0.5μg/ml and then co-cultured with CD14 negative fraction of PBMCs. DCs loaded with blank nanoparticles (Blank NP) were used as negative controls. After 10–12 days, the cells were re-stimulated with soluble CEF pool peptide (5ug/ml) and analyzed for production of interferon-gamma (IFN-γ) by flow cytometry. FACS plots shown in the figure are gated on CD3+ cells.
(3a). Representative figure from healthy donor showing induction of antigen-specific IFN-γ producing CD8 lymphocytes in response to stimulation with either Blank NP or NP-CEF loaded autologous Mo-DCs.
(3b). Representative figure from healthy donor showing induction of antigen-specific IFN-γ producing CD8 lymphocytes in response to stimulation with either Blank NP or NP-CEF loaded autologous BDCA3+ MDCs.
(3c). The graph shows mean percentage (± SEM) of CD8 cells producing IFN-γ when stimulated by Mo-DCs (n=8) or BDCA3+ MDCs (n=3) loaded with either blank NP or NP-CEF. *p= 6 × 10−4 (Blank NP vs NP-CEF for Mo-DCs) and p= 4.9 × 10−2 (Blank NP vs NP-CEF for BDCA3+ MDCs)
(3d). The multivalent nature of response was confirmed by re-stimulating NP-CEF stimulated cells with individual peptide components (Pep1 - Pep18; described in Supplemental Table 3) of CEF pool peptide. Representative examples with 2 different donors (Sample 1 and Sample 2) are shown. Blank NP and NP-CEF re-stimulated with CEF pool peptide were used as negative and positive controls respectively.
Next we analyzed if this platform could be utilized to generate T cells against a tumor-associated antigen. To this end, we loaded NPs with an overlapping peptide library derived from SOX2 (NP-SOX2). SOX2 has emerged as an important oncogene in several cancers including lung cancer (30). In recent studies, we and others have implicated this antigen in protective immunity in the context of monoclonal gammopathy and in patients with lung cancer treated with anti-programmed death (PD1) receptor antibody (31, 32). We stimulated T cells using NP-SOX2 loaded autologous DCs. Since the peptides are 15 amino acid long they require active processing for antigen-presentation. DCs loaded with NP-SOX2 were able to stimulate both SOX2-specific CD4 and CD8+ T cells in culture (Figure 4). Taken together these data demonstrate that both BDCA3+ and monocyte-derived DC-SIGN+ NP-loaded DCs are equally effective at generating antigen-specific human T cells in culture, including against complex peptide mixtures from viral and tumor antigens across multiple MHC molecules.
Figure 4. DCs targeted with nanoparticles containing complex mixture of long peptides from tumor antigen SOX2 leads to stimulation of antigen specific CD4 as well as CD8 T cells.
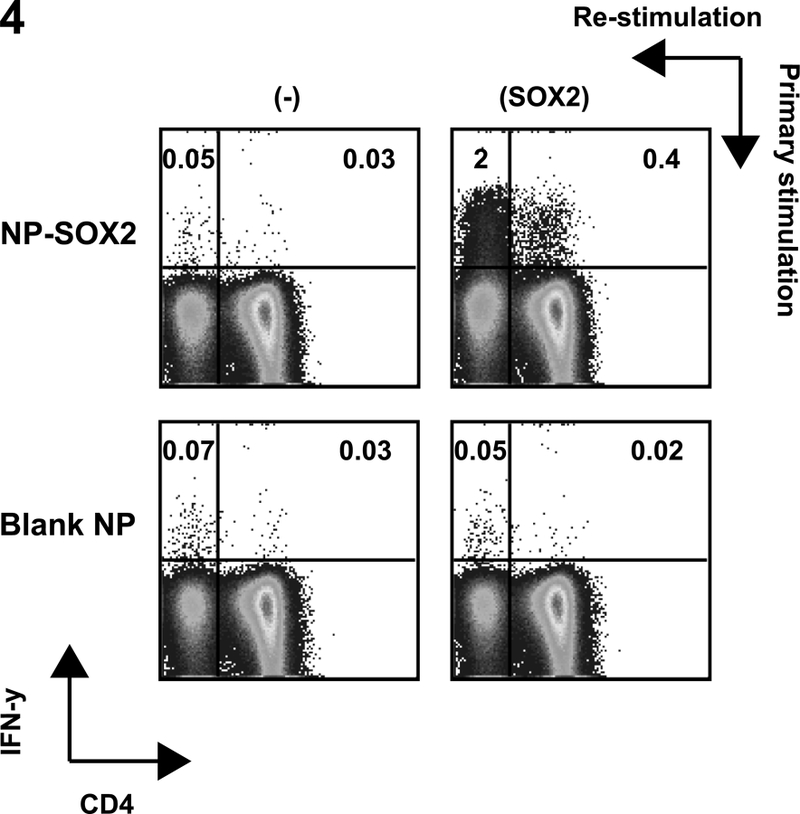
Day 5 immature Mo-DCs were cultured with SOX2 peptide containing NP (NP-SOX2, individual peptide concentration of 5μg/ml) matured (using inflammatory cytokine cocktail), and used to stimulate autologous T cells in the presence of IL-2 (10u/ml) and IL-7 and IL-15 (both at 10ng/ml). The T cells were re stimulated with NP-SOX2 loaded DCs on days 7 and 14. On day 21 of culture T cells were analyzed for their ability to secrete IFN-γ in response to stimulation with SOX2 peptides incorporated in the nanoparticles. Blank NP loaded DCs were used to stimulate T cells as negative controls. Figure is representative of experiments with three different healthy donors.
Targeting NPs to DC subsets:
As noted earlier, the NPs are decorated with avidin on their surface, allowing facile coupling to biotinylated antibodies for targeting. This coupling on the surface of nanoparticles was confirmed by staining with a rat antibody against the constant region of the biotinylated antibody (Supplemental Figure 1). Specificity of targeting to the BDCA3+ and DCSIGN+ APCs was examined using flow cytometry to detect uptake of coumarin labeled antibody decorated NPs. Enhanced uptake of NPs was seen in the targeted APCs compared to non-targeted APCs (Supplemental Figure 1b). Exposure of BDCA3+ DCs and DC-SIGN+ DCs to NPs leads to phenotypic maturation with up regulation of CD83, CD80 and CD86. Addition of Poly:IC coated NPs to BDCA3+ DCs did not lead to further increase in expression of CD80, CD83 or CD86. Addition of LPS coated particles to DC-SIGN DCs however, led to further increase in CD86 (supplemental figure 1c) when compared to non-coated NPs. For targeting experiments, peptide loaded NPs were used at 100 fold lower concentrations to avoid non-specific uptake of NPs by APCs. In contrast to prior studies wherein targeting of human DCs in culture was tested in the context of purified DCs, we examined the ability of antibody targeted NPs to target DC subsets in the context of bulk mononuclear cells, wherein DC subsets, particularly BDCA3+ MDCs, comprise only a minor fraction. In spite of this, targeting of NPs to either DC-SIGN+ subset or BDCA3+ MDCs led to enhanced activation of FMP specific T cells (Figure 5a-5b). Both BDCA3 and DC-SIGN targeted NPs led to similar increases in antigen specific T cells over non-targeted NPs (Figure 5c).
Figure 5. Combinatorial targeting of nanoparticles to BDCA3+ and DC SIGN+ dendritic cell subsets potentiates immune response.
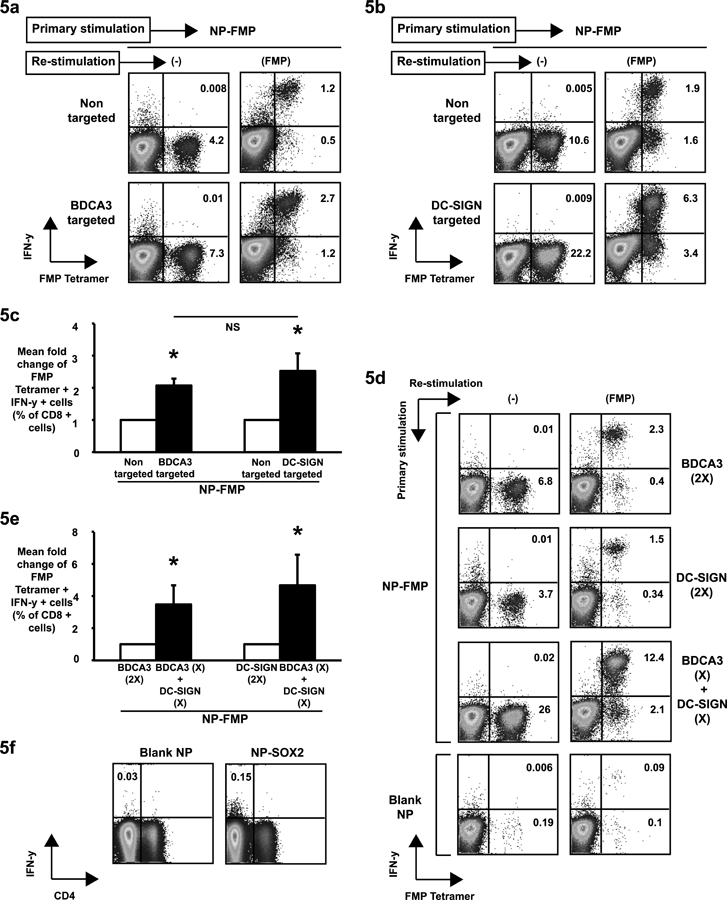
NP-FMP were conjugated with anti-BDCA3 or anti-DC-SIGN antibodies through avidin-biotin interactions. They were then added to peripheral blood mononuclear cells to evaluate the effect of targeting. Non-targeted NP-FMP were used as negative controls.
(5a). PBMCs (n=3) were pulsed with BDCA3-targeted or non-targeted NP-FMP (peptide concentration of 0.05μg/ml). After 12–14 days of culture, they were re-stimulated with soluble flu matrix peptide to evaluate IFN-γ production by FMP tetramer positive CD8 T cells using flow cytometry. Figure shows a representative dot plot.
(5b). PBMCs (n=4) were pulsed with DC SIGN targeted or non-targeted NP-FMP (peptide concentration of 0.05μg/ml). After 12–14 days of culture, they were re-stimulated with soluble flu matrix peptide to evaluate IFN-γ production by FMP tetramer positive CD8 T cells using flow cytometry. Figure shows a representative dot plot.
(5c). Figure shows induction of FluMP specific T cells using FluMP loaded NPs or FluMP loaded NPs decorated with either anti- BDCA3+(n=3) or anti-DCSIGN antibodies (n=4). Bar graph shows mean fold change (compared to non-targeted NPs) of FluMP tetramer positive interferon gamma secreting CD8 T cells generated under different stimulation conditions. * p<0.05
(5d). BDCA3-targeted NP-FMP (2X = peptide concentration of 0.05μg/ml), DC-SIGN-targeted NP-FMP (2X = peptide concentration of 0.05μg/ml) or a combination of the two (X+X = peptide concentrations of 0.025μg/ml each) were added to PBMCs to study the effect of combinatorial targeting. BDCA3-tageted & DC-SIGN-targeted NP-FMP were conjugated with biotin-LPS and biotin-Poly (I:C) respectively. Blank nanoparticles (Blank NP) were used as negative controls. After 10–14 days, the cells were re-stimulated with soluble flu matrix peptide (FMP, 5ug/ml) in the presence of anti-CD28 and anti-CD49d and analyzed for production of IFN-γ by FMP tetramer positive T cells using flow cytometry. Figure shows dot plot from a representative experiment. Cells are gated on CD3+ CD8+ T cells.
(5e). Figure shows data from 7 different healthy donors. Bars show mean fold increase in FMP tetramer positive IFN-γ secreting CD8+ T cells in cultures stimulated by combinatorial DC targeting with BDCA3 and DC-SIGN conjugated NP-FMP compared to those cultures stimulated with only BDCA3 targeted NP-FMP or DC-SIGN targeted NP-FMP. * p= 0.03 for PBMCs stimulated with either BDCA3 targeted NP-FMP or DC-SIGN targeted NP- FMP vs those stimulated with BDCA3 targeted NP-FMP and DC SIGN targeted NP-FMP.
(5f). Blank NP or SOX2 peptide loaded DCs (5mcg/ml individual peptide) were coated with anti-BDCA3 or anti-DC-SIGN antibody and a combination of BDCA3+ and DCSIGN+ coated NPs (as in figure 5d) were used to stimulate PBMCs from cancer patients (n=4) in the presence of IL2, IL7 and IL15. 15 days later the cells were re-stimulated with soluble SOX2 peptide mix (5ug/ml) in the presence of anti-CD28 and anti-CD49D and analyzed for production of IFN-γ using flow cytometry. Figure shows dot plot from a representative experiment where SOX2 reactive T cells could be stimulated. Cells are gated on CD3+ T cells.
Combinatorial targeting of DCs by NPs and role of cytokines in DC cross talk:
Prior studies targeting NPs to DCs have focused on single DC type, and optimal DC subset for vaccine targeting has not been defined (25). We hypothesized that simultaneously targeting multiple DC subsets may yield superior T cell responses. To this end, we compared NPs targeting either DC-SIGN+ cells, BDCA3+ cells alone or in combination. Combination of NPs targeting both DC-SIGN and BDCA3 led to significantly greater activation of antigen-specific T cells compared to targeting either subset of DCs alone (Figure 5d-5e). To extend the combinatorial targeting to a cancer antigen, PBMCs from cancer patients (n=4, with a diagnosis of either melanoma or myeloma) were stimulated with blank NPs or SOX2 loaded NPs decorated with anti-BDCA3 and anti-DCSIGN antibodies. Combinatorial targeting with BDCA3 and DCSIGN antibody labeled SOX2NPs led to stimulation of SOX2 specific T cells in two out of the four patients tested (Figure 5f).
We hypothesized that the mechanism underlying this potentiation of immune stimulation may involve cross-talk between two subsets of DCs and mediated in part by cytokines. We examined secretion of IFNα, β, γ, and λ, IL1α, IL4, IL5, IL6, IL8, IL10, IL3, IL15, IL23, IL12p70 and TNFα by NP loaded DCs. Exposure of BDCA3+ DCs to NPs leads to enhanced secretion of IFNλ, IL15, IL6, IL8 and TNFα (Figure 6a). Exposure to Poly-IC carrying particles did not further enhance these cytokine responses (data not shown). Exposure of monocyte derived DCSIGN+ DCs to NPs leads enhanced secretion of IL15, IL6, IL8 and TNFα (Figure 6a). Addition of LPS decorated particles led to further increase in IL6 (218 vs 614pg/ml p=0.0001), TNFα (164 vs 1003pg/ml; p=0.017), IL13(10 vs 100pg/ml; p=0.02) and IL23 (19 vs 64pg/ml; p=0.02) when compared to NPs alone. As the proportion of BDCA3+ MDCs is much lower than other myeloid subsets, we reasoned that this subpopulation may be an important component of the cross-talk. We were particularly interested in examining the role of IL15, IFNλ and IL6 since these cytokines have been shown to be important for T cell activation. Antibody-mediated blockade of IL-15 inhibited the potentiation of T cell activation seen with combinatorial DC targeting. In contrast, blockade of interferon-lambda or IL-6 did not lead to significant inhibition of T cell activation (Figure 6b). Together these data demonstrate that IL-15 signaling during combinatorial DC targeting plays an important role in the enhanced activation of T cell responses observed with combinatorial targeting of DC-SIGN+ and BDCA3+ DCs.
Fig. 6. Combinatorial targeting to BDCA3+ and DC-SIGN+ dendritic cell subsets potentiates immune response through an IL-15 dependent mechanism.
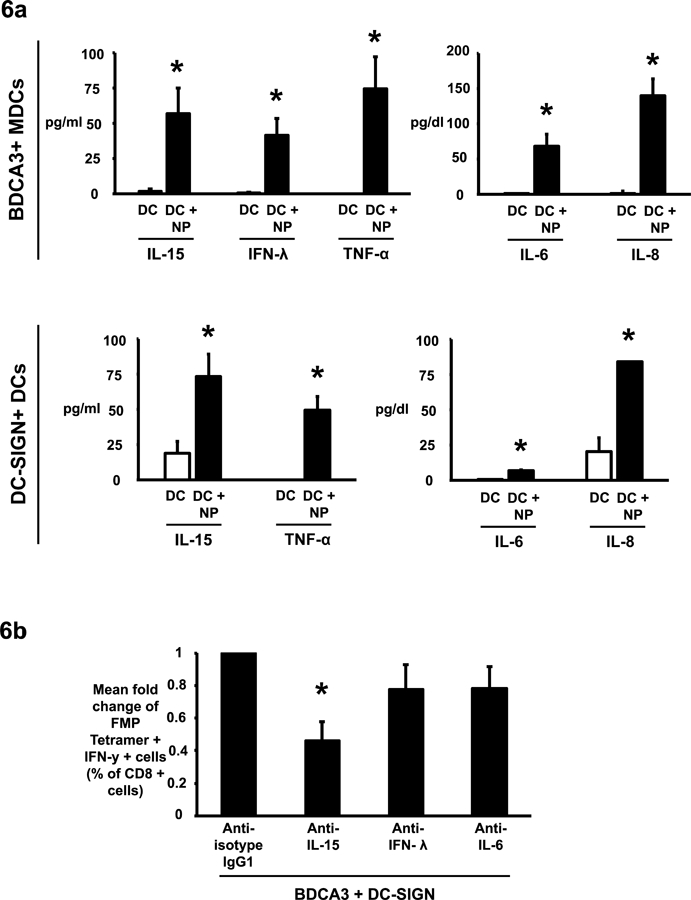
(6a). The effects of nanoparticles on isolated BDCA3+ myeloid dendritic cells and DC SIGN positive monocyte DCs was studied by incubating them overnight with NP-FMP (peptide concentration 0.05μg/ml) at 37°C. The supernatants were then collected and analyzed by VeriPlex™ Human Cytokine ELISA Kit. BDCA3+ MDCs and DC-SIGN DCs incubated alone were used as negative controls. The graph shows mean cytokine expression levels (IL-15, IFN-lambda, TNFα, IL-6 and IL8) per 30,000 APCs ± standard error of the mean for cells obtained from 3 different healthy donors. * p<0.05
(6b). PBMCs were targeted with BDCA3 and DC-SIGN coated NP-FMP either in the presence of IL-15 blocking antibody, IFN-λ blocking antibody, IL-6 blocking antibody or isotype control antibody. After 10–12 days the cultures were analyzed for the presence of FMP tetramer positive, IFN-γ secreting CD8 T cells. Bar graph shows fold decrease in FMP tetramer positive IFN-γ producing CD8 T cells in cultures with blocking antibodies compared to cultures with isotype control antibody (n=4 different donors). * p=0.038
Discussion:
In this study, we have shown that simultaneously targeting antigen-bearing NPs to multiple human DC subsets leads to superior immune activation. Therefore these data suggest that the optimal strategy for targeting DCs may not lie in targeting a specific subset as currently being pursued (5), but rather concurrently targeting multiple DC subsets. The concept that optimal activation of immune responses may involve simultaneous activation of multiple DC subsets has also emerged from studies of protective immunity to pathogens (21, 33). It is notable that the Yellow Fever vaccine, which is one of the most potent vaccines in humans does indeed target multiple DC subsets in vivo (34). Similarly Kastenmuller et al demonstrated that protective immunity following a protein-TLR agonist conjugate vaccine required the engagement of multiple DC subsets (35). These data therefore have clear implications for design of DC-targeting NP (or other) vaccines as they suggest a need to consider simultaneous targeting of different DC subsets.
Integrative synergy with recruitment of diverse DC subsets may in principle be due to multiple mechanisms including differences in their functional properties (2). Recent studies suggest that in contrast to murine DCs, most human DC subsets may be quite similar in terms of antigen cross-presentation and phagocytic function (23). Therefore enhanced immune activation with combinatorial targeting may instead be due to cross-talk such as that mediated by cytokines released by these DC subsets. BDCA3+ MDCs (or their murine equivalent) are known to secrete interferon-lambda as well as IL-15 upon activation (36–41). Our data suggest that IL-15-mediated signaling but not interferon-lambda released in the DC cultures is important for enhanced immune activation that we observed following combinatorial targeting involving BDCA3+ MDCs. Trans-presentation of IL15 by CD8α+ DCs (murine equivalents of BDCA3+ DCs) was shown to play a role in expansion of memory T cells (39, 42). IL-15 is already well known to activate myeloid DCs and enhance their APC function (38, 43). Further studies are needed to better define the optimal combinations of DCs to target. One of the challenges in the studies of human DC subsets in vivo relates to differences in the biology of murine versus human DCs (23). It is not clear that this biology is faithfully reproduced yet in humanized mice in vivo (44) and therefore formal evaluation of DC-cross talk in humans in vivo may require careful human studies to improve vaccine design.
The novel NP platform described here has clear implications for translation to the clinic as the safety of PLGA nanoparticles is already well established (8). The decoration of NPs with avidin as utilized here, provides a simple yet effective strategy to generate NPs targeting different cell types using biotinylated antibodies (27). In this study we have also shown that NPs can be easily encapsulated with complex peptide mixtures and therefore effectively serve as poly-epitope vaccines. Generation of immunity against multiple epitopes may be particularly important in the context of immunity to pathogens and tumors. The demonstration that NPs loaded with overlapping peptide libraries (as shown here for SOX2) (45) can induce immunity provides a novel platform for clinical evaluation of NP-based vaccines irrespective of the HLA type. Furthermore, the ability to load multiple peptides also makes this technology readily amenable to personalized cancer vaccines, such as those against oncogenic mutation-derived peptides in tumor cells. Recent advances in engineering of these materials including nanogels can also be adapted towards combinatorial targeting of multiple DC subsets with poly-epitope vaccines against pathogens and tumors (46).
Supplementary Material
Acknowledgments
This work is supported by RO1AI079222 and the Dana Foundation grant to Kavita M. Dhodapkar, CA106802, CA135110, Multiple Myeloma Research Foundation and Bunker Professorship to Madhav V. Dhodapkar, and R01AR 064350 and U19 AI082713 Pilot grant to Tarek M. Fahmy.
Footnotes
Footnotes:
References:
- 1.Steinman RM, and Banchereau J. 2007. Taking dendritic cells into medicine. Nature 449: 419–426. [DOI] [PubMed] [Google Scholar]
- 2.Palucka K, and Banchereau J. 2013. Human dendritic cell subsets in vaccination. Current opinion in immunology 25: 396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palucka K, and Banchereau J. 2013. Dendritic-cell-based therapeutic cancer vaccines. Immun 39: 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bandyopadhyay A, Fine RL, Demento S, Bockenstedt LK, and Fahmy TM. 2011. The impact of nanoparticle ligand density on dendritic-cell targeted vaccines. Biomaterials 32: 3094–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dhodapkar M, Zhao B, Wang D, Carvajal RD, Keohan M, Chuang E, Sanborn R, Lutzky J, Powderly JD, Kluger H, Sznol M, Tejwani S, Crocker A, Vitale L, Ramakrishna V, Yellin M, Davis T, and Keler T. 2012. A Phase I trial of a novel vaccine targeting NY-ESO1 to the dendritic cell receptor DEC-205 in combination with Toll-like receptor agonists (meeting abstract). J Immunother 35: 740–740. [Google Scholar]
- 6.Paulis LE, Mandal S, Kreutz M, and Figdor CG. 2013. Dendritic cell-based nanovaccines for cancer immunotherapy. Curr Opin Immunol 25: 389–395. [DOI] [PubMed] [Google Scholar]
- 7.Cruz LJ, Tacken PJ, Rueda F, Domingo JC, Albericio F, and Figdor CG. 2012. Targeting nanoparticles to dendritic cells for immunotherapy. Methods in enzymology 509: 143–163. [DOI] [PubMed] [Google Scholar]
- 8.Metcalfe SM, and Fahmy TM. 2012. Targeted nanotherapy for induction of therapeutic immune responses. Trends Mol Med 18: 72–80. [DOI] [PubMed] [Google Scholar]
- 9.Ali OA, Emerich D, Dranoff G, and Mooney DJ. 2009. In situ regulation of DC subsets and T cells mediates tumor regression in mice. Sci Transl Med 1: 8ra19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, Ravindran R, Stewart S, Alam M, Kwissa M, Villinger F, Murthy N, Steel J, Jacob J, Hogan RJ, Garcia-Sastre A, Compans R, and Pulendran B. 2011. Programming the magnitude and persistence of antibody responses with innate immunity. Nature 470: 543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Demento SL, Eisenbarth SC, Foellmer HG, Platt C, Caplan MJ, Mark Saltzman W, Mellman I, Ledizet M, Fikrig E, Flavell RA, and Fahmy TM. 2009. Inflammasome-activating nanoparticles as modular systems for optimizing vaccine efficacy. Vaccine 27: 3013–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goldinger SM, Dummer R, Baumgaertner P, Mihic-Probst D, Schwarz K, Hammann-Haenni A, Willers J, Geldhof C, Prior JO, Kundig TM, Michielin O, Bachmann MF, and Speiser DE. 2012. Nano-particle vaccination combined with TLR-7 and −9 ligands triggers memory and effector CD8(+) T-cell responses in melanoma patients. Eur J Immunol 42: 3049–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schreibelt G, Klinkenberg LJ, Cruz LJ, Tacken PJ, Tel J, Kreutz M, Adema GJ, Brown GD, Figdor CG, and de Vries IJ. 2012. The C-type lectin receptor CLEC9A mediates antigen uptake and (cross-)presentation by human blood BDCA3+ myeloid dendritic cells. Blood 119: 2284–2292. [DOI] [PubMed] [Google Scholar]
- 14.Tacken PJ, Ginter W, Berod L, Cruz LJ, Joosten B, Sparwasser T, Figdor CG, and Cambi A. 2011. Targeting DC-SIGN via its neck region leads to prolonged antigen residence in early endosomes, delayed lysosomal degradation, and cross-presentation. Blood 118: 4111–4119. [DOI] [PubMed] [Google Scholar]
- 15.Tacken PJ, Zeelenberg IS, Cruz LJ, van Hout-Kuijer MA, van de Glind G, Fokkink RG, Lambeck AJ, and Figdor CG. 2011. Targeted delivery of TLR ligands to human and mouse dendritic cells strongly enhances adjuvanticity. Blood 118: 6836–6844. [DOI] [PubMed] [Google Scholar]
- 16.Cruz LJ, Tacken PJ, Fokkink R, Joosten B, Stuart MC, Albericio F, Torensma R, and Figdor CG. 2010. Targeted PLGA nano- but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro. Journal of controlled release : official journal of the Controlled Release Society 144: 118–126. [DOI] [PubMed] [Google Scholar]
- 17.Tel J, Sittig SP, Blom RA, Cruz LJ, Schreibelt G, Figdor CG, and de Vries IJ. 2013. Targeting Uptake Receptors on Human Plasmacytoid Dendritic Cells Triggers Antigen Cross-Presentation and Robust Type I IFN Secretion. J Immunol [DOI] [PubMed]
- 18.Cruz LJ, Tacken PJ, Pots JM, Torensma R, Buschow SI, and Figdor CG. 2012. Comparison of antibodies and carbohydrates to target vaccines to human dendritic cells via DC-SIGN. Biomaterials 33: 4229–4239. [DOI] [PubMed] [Google Scholar]
- 19.Demento SL, Siefert AL, Bandyopadhyay A, Sharp FA, and Fahmy TM. 2011. Pathogen-associated molecular patterns on biomaterials: a paradigm for engineering new vaccines. Trends in biotechnology 29: 294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, Cheong C, Liu K, Lee HW, Park CG, Steinman RM, and Nussenzweig MC. 2007. Differential antigen processing by dendritic cell subsets in vivo. Science 315: 107–111. [DOI] [PubMed] [Google Scholar]
- 21.Pulendran B, Tang H, and Denning TL. 2008. Division of labor, plasticity, and crosstalk between dendritic cell subsets. Current opinion in immunology 20: 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luci C, and Anjuere F. 2011. IFN-lambdas and BDCA3+/CD8alpha+ dendritic cells: towards the design of novel vaccine adjuvants? Expert Rev Vaccines 10: 159–161. [DOI] [PubMed] [Google Scholar]
- 23.Segura E, Durand M, and Amigorena S. 2013. Similar antigen cross-presentation capacity and phagocytic functions in all freshly isolated human lymphoid organ-resident dendritic cells. J Exp Med 210: 1035–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohn L, Chatterjee B, Esselborn F, Smed-Sorensen A, Nakamura N, Chalouni C, Lee BC, Vandlen R, Keler T, Lauer P, Brockstedt D, Mellman I, and Delamarre L. 2013. Antigen delivery to early endosomes eliminates the superiority of human blood BDCA3+ dendritic cells at cross presentation. J Exp Med 210: 1049–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kreutz M, Tacken PJ, and Figdor CG. 2013. Targeting dendritic cells--why bother? Blood 121: 2836–2844. [DOI] [PubMed] [Google Scholar]
- 26.Fahmy TM, Demento SL, Caplan MJ, Mellman I, and Saltzman WM. 2008. Design opportunities for actively targeted nanoparticle vaccines. Nanomedicine (Lond) 3: 343–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park J, Mattessich T, Jay SM, Agawu A, Saltzman WM, and Fahmy TM. 2011. Enhancement of surface ligand display on PLGA nanoparticles with amphiphilic ligand conjugates. Journal of controlled release : official journal of the Controlled Release Society 156: 109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fahmy TM, Samstein RM, Harness CC, and Mark Saltzman W. 2005. Surface modification of biodegradable polyesters with fatty acid conjugates for improved drug targeting. Biomaterials 26: 5727–5736. [DOI] [PubMed] [Google Scholar]
- 29.Dhodapkar KM, Kaufman JL, Ehlers M, Banerjee DK, Bonvini E, Koenig S, Steinman RM, Ravetch JV, and Dhodapkar MV. 2005. Selective blockade of inhibitory Fc gamma receptor enables human dendritic cell maturation with IL-12p70 production and immunity to antibody-coated tumor cells. Proc Natl Acad Sci U S A 102: 2910–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, Ramos AH, Woo MS, Weir BA, Getz G, Beroukhim R, O’Kelly M, Dutt A, Rozenblatt-Rosen O, Dziunycz P, Komisarof J, Chirieac LR, Lafargue CJ, Scheble V, Wilbertz T, Ma C, Rao S, Nakagawa H, Stairs DB, Lin L, Giordano TJ, Wagner P, Minna JD, Gazdar AF, Zhu CQ, Brose MS, Cecconello I, Jr UR, Marie SK, Dahl O, Shivdasani RA, Tsao MS, Rubin MA, Wong KK, Regev A, Hahn WC, Beer DG, Rustgi AK, and Meyerson M. 2009. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nature genetics 41: 1238–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dhodapkar KM, Gettinger SN, Das R, Zebroski H, and Dhodapkar MV. 2013. SOX2-specific adaptive immunity and response to immunotherapy in non-small cell lung cancer. Oncoimmunology 2: e25205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spisek R, Kukreja A, Chen LC, Matthews P, Mazumder A, Vesole D, Jagannath S, Zebroski HA, Simpson AJ, Ritter G, Durie B, Crowley J, Shaughnessy JD Jr., Scanlan MJ, Gure AO, Barlogie B, and Dhodapkar MV. 2007. Frequent and specific immunity to the embryonal stem cell-associated antigen SOX2 in patients with monoclonal gammopathy. The Journal of experimental medicine 204: 831–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pulendran B, Oh JZ, Nakaya HI, Ravindran R, and Kazmin DA. 2013. Immunity to viruses: learning from successful human vaccines. Immunol Rev 255: 243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Querec T, Bennouna S, Alkan S, Laouar Y, Gorden K, Flavell R, Akira S, Ahmed R, and Pulendran B. 2006. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. The Journal of experimental medicine 203: 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kastenmuller K, Wille-Reece U, Lindsay RW, Trager LR, Darrah PA, Flynn BJ, Becker MR, Udey MC, Clausen BE, Igyarto BZ, Kaplan DH, Kastenmuller W, Germain RN, and Seder RA. 2011. Protective T cell immunity in mice following protein-TLR7/8 agonist-conjugate immunization requires aggregation, type I IFN, and multiple DC subsets. J Clin Invest 121: 1782–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lauterbach H, Bathke B, Gilles S, Traidl-Hoffmann C, Luber CA, Fejer G, Freudenberg MA, Davey GM, Vremec D, Kallies A, Wu L, Shortman K, Chaplin P, Suter M, O’Keeffe M, and Hochrein H. 2010. Mouse CD8alpha+ DCs and human BDCA3+ DCs are major producers of IFN-lambda in response to poly IC. The Journal of experimental medicine 207: 2703–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, Keller AM, Joffre O, Zelenay S, Nye E, Le Moine A, Faure F, Donckier V, Sancho D, Cerundolo V, Bonnet D, and Reis e Sousa C. 2010. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. The Journal of experimental medicine 207: 1261–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dubsky P, Saito H, Leogier M, Dantin C, Connolly JE, Banchereau J, and Palucka AK. 2007. IL-15-induced human DC efficiently prime melanoma-specific naive CD8+ T cells to differentiate into CTL. Eur J Immunol 37: 1678–1690. [DOI] [PubMed] [Google Scholar]
- 39.Sosinowski T, White JT, Cross EW, Haluszczak C, Marrack P, Gapin L, and Kedl RM. 2013. CD8alpha+ dendritic cell trans presentation of IL-15 to naive CD8+ T cells produces antigen-inexperienced T cells in the periphery with memory phenotype and function. J Immunol 190: 1936–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anguille S, Lion E, Tel J, de Vries IJ, Coudere K, Fromm PD, Van Tendeloo VF, Smits EL, and Berneman ZN. 2012. Interleukin-15-induced CD56(+) myeloid dendritic cells combine potent tumor antigen presentation with direct tumoricidal potential. PloS one 7: e51851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Colpitts SL, Stoklasek TA, Plumlee CR, Obar JJ, Guo C, and Lefrancois L. 2012. Cutting edge: the role of IFN-alpha receptor and MyD88 signaling in induction of IL-15 expression in vivo. J Immunol 188: 2483–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stonier SW, and Schluns KS. 2010. Trans-presentation: a novel mechanism regulating IL-15 delivery and responses. Immunol Lett 127: 85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perera PY, Lichy JH, Waldmann TA, and Perera LP. 2012. The role of interleukin-15 in inflammation and immune responses to infection: implications for its therapeutic use. Microbes Infect 14: 247–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Akkina R 2013. Human immune responses and potential for vaccine assessment in humanized mice. Curr Opin Immunol 25: 403–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dhodapkar MV, and Dhodapkar KM. 2011. Vaccines targeting cancer stem cells: are they within reach? Cancer J 17: 397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Look M, Stern E, Wang QA, DiPlacido LD, Kashgarian M, Craft J, and Fahmy TM. 2013. Nanogel-based delivery of mycophenolic acid ameliorates systemic lupus erythematosus in mice. J Clin Invest 123: 1741–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.