

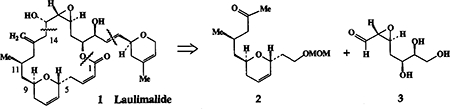
Abstract
The C3-C14 segment of the novel antitumor agent laulimalide has been constructed enantioselectively by utilizing a catalytic asymmetric hetero Diels-Alder reaction of benzyloxyacetaldehyde and Danishefsky’s diene followed by Ferrier rearrangement and asymmetric conjugate reaction as the key steps.
Laulimalide 1 also known as figianolide B, is a 20-membered macrolide isolated from the Indonesian sponge Hyattella Sp.1 More recently, laulimalide has also been isolated from an Okinawan sponge Fasciospongia rimosa.2 Laulimalide represents a new and novel class of macrolide with potent cytotoxicity against the KB cell line with an IC50 value of 15 ng/mL.1b The cytotoxicity of laulimalide against P388, A549, HT29 and MEL28 cell lines is also in the range of 10–50 ng/mL (IC50 values).2b The gross structure of laulimalide was established by NMR studies and more recently its absolute configuration has been elucidated by X-ray crystallographic analysis.2a In view of its limited supply and unique structural features as well as its potential utility as an anticancer agent, synthetic studies of laulimalide became of interest to us. Herein, we report on the asymmetric synthesis of the C3-C14 segment of laulimalide in which a chiral bis(oxazoline)-metal complex catalyzed hetero DielsAlder reaction, a Ferrier type rearrangement of the derived glycal and diastereoselective conjugate addition were utilized to set the C-9, C-5 and C-11 asymmetric centers.
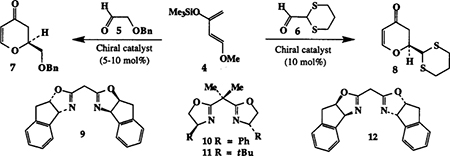
Chiral bis(oxazoline)-metal complex catalyzed cycloaddition reactions have received increasing attention in recent years.3 We recently reported4 constrained chiral bis(oxazoline)-metal complex catalyzed hetero Diels-Alder reactions of Danishefsky′s diene and alkyl glyoxalates to provide dihydropyranone derivatives up to 72% ee. In an effort to further improve the enantioselectivity in this catalytic process, we have. investigated the scope and utility of readily accessible bidentate aldehydes such as benzyloxyacetaldehyde 5 and 1,3-dithianecarboxaldehyde 6. Of particular interest, dihydropyranone derivatives6 resulting from such cyclocondensation reactions are appropriately functionalized for the synthesis of laulimalide segment 2. As shown in Table 1, cyclocondensation of aldehyde 57 and Danishefsky’s diene 4 in the presence of 10% Cu(II)-bis(oxazoline) complex provided good yield (62–76%) of versatile dihydropyranone 78 in high enantiomeric excess. Constrained ligands 9 and 123e are particularly effective providing 2S and 2R-dihydropyranones in 85 and 87% ee’s respectively. In comparison, phenyl and t-butyl based bis(oxazoline) ligands 10 and 11 are less effective (51 and 38% ee). Cyclocondensation of 1,3-dithianecarboxaldehyde 69 with constrained Cu(II)-bis(oxazoline) 12 complex also afforded the dihydropyranone 8 in 46% yield and 81% ee compared to Cu(ll)-bis(oxazoline) ligand 10 which has shown 59% ee 10 and only 20% isolated yield.
Table 1.
Cu(II)-bis(oxazoline) catalyzed Hetero Diels-Alder Reaction at −78°C
| Entry | Aldehyde | Ligand | Time(h) | % Yielda | % eeb | Config.c |
|---|---|---|---|---|---|---|
| 1. | 5 | 10 | 9 | 76 | 51 | 2S |
| 2. | 5 | 11 | 9 | 72 | 38 | 2R |
| 3. | 5 | 9 | 11 | 76 | 85 | 2S |
| 4. | 5 | 12 | 9 | 62 | 87 | 2R |
| 5. | 6 | 12 | 9 | 46 | 81C | 2S |
| 6. | 6 | 10 | 10 | 20 | 59C | 2R |
After silica gel chromatography.
By chiral HPLC and comparison of optical rotation.
By comparison of optical rotation.
For synthesis of the C3-C14 segment of laulimalide, dihydropyranone 7 was prepared on a multigram scale.11 To append the hydroxyethyl side chain and to establish the C-5 stereocenter appropriately, a Ferrier type rearrangement of the corresponding glycal acetate was sought. Thus, dihydropyranone 7 was reduced with 1.5 equiv of DIBAL in benzene at 0° to 5°C for 2 h and the resulting glycal was acetylated with 1.5 equiv of Ac2O and 3 equiv of Et3N in CH2Cl2 in the presence of a catalytic amount of DMAP at 23°C for 6 h to provide 13 in 66% yield (from 7). Ferrier rearrangement of 13 with 2 equiv of tert-butyldimethylsilyl vinyl ether12 and stoichiometric amount of Montmorillonite clay K-1013 as the Lewis acid in CH2C12 at 0°C followed by NaBH4 reduction of the resulting mixture of aldehydes afforded good yield (65–70% from 13) of the dihydropyran 14 (diastereomeric ratio >95:5 by 1H and 13C-NMR). Protection of the alcohol with MOMCl and iPr2NEt furnished the MOM derivative 15 in 88% yield.
To elaborate the C-11 methyl group with appropriate stereochemistry, the benzyl group in 15 was deprotected by exposure to sodium in liquid ammonia (65% yield). Mesylation of the resulting alcohol followed by displacement with tetraethylammonium cyanide provided the cyanide 16 (77% yield). Reduction of 16 with DIBAL resulted in aldehyde which was immediately exposed to a Horner-Emmons olefination reaction to afford the trans α, ß-unsaturated ester 17 in 45% yield. Saponification of 17 with aqueous LiOH afforded the corresponding α, ß-unsaturated acid which was converted to N-enoylsultam 18 (73% yield) utilizing (1S)-(+)-2,10-camphorsultam. Treatment of 18 with 4 equiv of Me2CuLi in Et2O at −78°C for 8 h afforded the conjugate addition product 19 in 68% yield (based on 30% recovery of 18). The 1H-NMR (400 MHz) analysis after chromatography reveals the presence of a mixture of (90:10) diastereomers. Interestingly, reaction of Me2CuLi with the Nenoylsultam derived from (1R)-(+)-2,10-camphorsultam proceeded smoothly in 3 h (no recovery of 18), providing the corresponding diastereomers (mixture ratio 5:95 by 1H-NMR) in 76% isolated yield. The depicted configuration is assigned based on Oppolzer’s model.14 However, either C-11 methyl isomer of laulimalide can be prepared selectively by an appropriate choice of camphorsultam.
Our next synthetic strategy was to convert the sultam 19 to the methyl ketone 2 which would enable us to introduce the C-15 hydroxyl group by an aldol type reaction with an appropriately functionalized epoxyaldehyde 3. Thus, removal of the chiral auxiliary afforded the corresponding acid which was treated with 5 equiv of MeLi at 0°C followed by workup with excess of TMSCI according to Rubottom procedure 15 to furnish 2 (−55.6; c, 0.18, CHC13) in 63 % yield.16
In summary, the C3-C14 segment of antitumor macrolide laulimalide has been synthesized in optically active form utilizing dihydropyranone 7, prepared enantioselectively by a chiral bis(oxazoline)-metal complex catalyzed hetero Diels-alder reaction as the key step. Further synthetic studies of laulimaide are currently under investigation.
Scheme 1:

(a) K-10, OTBS, CH2Cl2, 0°C, 2 h; (b) NaBH4, MeOH, 0°C, 30 min; (c) MOMCl, iPr2NEt, CH2Cl2, 23°C, 8 h; (d) liq. NH3, −78°C, 1 h; (e) MCl, Et3N, DMAP, CH2Cl2, 0°C, 30 min; (f)n-Et4N+CN-, CH3CN-PhH (1:1), reflux,4 h; (g) Diba1-H, Et2O, −78°C, 4h; (h) NaH, THF, 0°C, (EtO)2P(O)CH2CO2Et, 1 h;(i) 1 M LiOH, 23°C, 6 h; (j) Me3CCOC1, Et3N, THF, 0°C then N-lithiosultam, −78°C, 2 h; (k) Me2CuLi, Et2O, −78°C, 8 h; (m) 1M LiOH, 23°C, 5 h; (n) MeLi, THF, 0°C then TMSC1.
Acknowledgments
Acknowledgment:
Financial support of this work by the University of Illinois at Chicago and by the National Institute of Health (GM 53386) is gratefully acknowledged.
References and notes:
- 1.(a) Quinoa E; Kskou Y; Crews P J Org. Chem. 1988,53,3642; [Google Scholar]; (b) Corley DG; Herb R; Moore RE; Scheuer PJ; paul VJ J. Org. Chem. 1988,53, 3644. [Google Scholar]
- 2.(a) Jefford CW;Bernrdinelli G; Tanaka J-I; Higa T Tetrahedron Lett. 1996,37, 159; [Google Scholar]; (b) Tanaka J-I; Higa T;Bernrdinell Gi,.;Jefford CW Chemistry Lett. 1996,255. [Google Scholar]
- 3.(a) Corey EJ; Imai, Zhang H-Y J. Am. Chem. soc 1991, 113, 728; [Google Scholar]; (b) Corey E Isihara K Tetrahedron Lett. 1992, 33, 6807; [Google Scholar]; (c) Evans DA; Miller SJ; Lectka T J. Am. Chem. soc. 1993, 115, 6460; [Google Scholar]; (d) Evans DA; Murry JA; Mat PV; Norcross RD; Miller SJ Angew. Chem. Int. Ed. Engl 1995, 34, 798; [Google Scholar]; (e) Ghosh AK; Mathivanan P; Cappiello J Tetrahedron Lett. 1996, 37, 3815; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Davies IW; Senanayake CH; Larsen RD; Verhoeven TR; Reider PJ Tetrahedron Lett. 1996, 37, 1725; [Google Scholar]; (g) Evans DA; Barnes DM Tetrahedron Lett. 1997, 38, 57; [Google Scholar]; (h) Pfaltz A Acta Chimica Scandinavia 1996, 50, 189 and references cited therein. [Google Scholar]
- 4.Ghosh AK; Mathivanan P; Cappiello J; Krishnan K Tetrahedron: Asymmetry 1996, 7, 2615 For other recent catalytic reports, see ref. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Terada M; Mikami K; Nakai T Tetrahedron Lett. 1991, 32, 935; [Google Scholar]; (b) Corey EJ; Cywin CL; Roper TD Tetrahedron Lett. 1992, 33, 6907; [Google Scholar]; (c) Gao Q; Maruyama T; Mouri M; Yamamoto H J. org. Chem 1992, 57, 1957; [Google Scholar]; (d) Mikmli, Motoyama Y; Terada M J. Am. Chem. soc., 1994, 116, 2811; [Google Scholar]; (e) Motoyama Y; Mikami KJ Chem. soc. Chem. Commun 1994, 1563; [Google Scholar]; (f) Keck G Li X-Y; Krishnamurthy D J. org. Chem 1995, 60, 5998. [Google Scholar]
- 6.For synthetic application of wide variety of dihydropyranones, see, (a) Danishefsky SJ Aldrichim. Acta 1986, 19, 59; [Google Scholar]; (b) Bednarski MD; Lyssikatos JP Comprehensive Organic Synthesis: Selectivity, Strategy and Efficiency in Modern Organic Chemistry, Trost BM Ed., 1991, 2, 661; [Google Scholar]; (c) Meng D; Sorensen E Bertinato, Danishefsky SJ J. org. Chem. 1996, 61, 7998; [DOI] [PubMed] [Google Scholar]; (c) Bertinato P; Sorensen EJ; Meng D; Danishefsky SJ J. Org. Chem. 1996, 61, 8000; [DOI] [PubMed] [Google Scholar]; (d) Paterson I; Cumming JG; Ward RA; Serge Lambley.Tetrahderon 1995, 51, 9393 and references cited therein. [Google Scholar]
- 7.Available from Aldrich. For recent application in catalytic Mukaiyama aldol reaction, see, Evans DA; Murry JA; Kozlowski MC J. Am. Chem. Soc 1996, 118, 5815. [Google Scholar]
- 8.Paterson I; Osborne S Tetrahedron Lett. 1990, 31, 2213 and references cited therein. [Google Scholar]
- 9.Bulman Page PC; Marchington AP; Graham LJ; Harkin SA; Wood WW Tetrahedron 1993, 49, 10369. [Google Scholar]
- 10. Details of the determination of ee’s and absolute configuration of 8 will be reported in due course.
- 11. In a typical procedure, Cu(OTf)2 (Aldrich, 241 mg, 0.66 mmol) and ligand 9 (264 mg, 0.8 mmol) were stirred together in CH2Cl2 (20 mL) at <MI> for 1 h under nitrogen. The resulting deep blue solution (5 mol% catalyst) was cooled to -<MI> and aldehyde 5 (Aldrich, 2 g, 13.3 mmol) in CH2Cl2 (l mL) followed by diene 4 (2.8 m.L, 16.28 mmol) were added. The mixture was stirred for 9 h at - and then quenched with aq. NaHCO3. Standard workup and evaporation of the solvents gave a residue which was stirred with TFA (10 mL) in CH2Cl2 (10 mL) at for 1 h. The reaction was quenched carefully with sat. NaHCO3 solution. Standard workup and chromatography over silica gel afforded 7 (1.8 g, 62% yield) as a colorless oil.
- 12.Sirsi W; Padias AB; Hall HK Jr. J. org. Chem. 1984, 49, 5424. [Google Scholar]
- 13.Toshima K; Miyamoto N; Matsuo G; Nakata M; Matsura S J. Chem. Soc. Chem. Commun. 1996, 1379. [Google Scholar]
- 14.Oppolzer W; Kingma AJ Helv. Chim. Acta 1989, 72, 1337 and references cited therein. [Google Scholar]
- 15.Rubottom GM; Kim C J. org. Chem. 1983, 48, 1550. [Google Scholar]
- 16. All new compounds gave satisfactory spectral data. Ketone 2: 1H-NMR (400 MHz, CDCl3) δ: 5.08 (m, 1H), 5.7 (d, 1H, J −1.2 Hz), 4.63 (s, 2H), 4.3 (br s, 1H), 3.65 (m, 3H), 3.36 (s, 3H), 2.5 (m, 1H), 2.24 (m, 2H), 2.13 (s, 3H), 2.05–1.86 (m, 3H), 1.58–1.23 (m, 3H), 0.92 (d, 3H, J −6.3 Hz); 13C-NMR (100 MHz, CDCl3) d: 223.3, 129.9, 124.1, 96.4, 69.4, 65.2, 64.4, 55.1, 52.2, 42.8, 34.5, 31.1, 29.6, 26.5, 19.3.