

Abstract
Signal transduction systems based on tyrosine phosphorylation are central to cell-cell communication in multicellular organisms. Typically in such a system, the signal is initiated by activating tyrosine kinases associated with transmembrane receptors, which induces tyrosine phosphorylation of the receptor and/or associated proteins. The phosphorylated tyrosines then serve as docking sites for the binding of various downstream effector proteins. It has long been observed that the cooperative association of the receptors and effectors produces higher order protein assemblies (clusters) following signal activation in virtually all phosphotyrosine signal transduction systems. However, mechanistic studies on how such clustering processes affect signal transduction outcomes have only emerged recently. Here we review current progress in decoding the biophysical consequences of clustering on the behavior of the system, and how clustering affects how these receptors process information.
Graphics Abstract
Introduction
Tyrosine phosphorylation regulates a number of biological activities including cell proliferation, differentiation, adhesion, and motility. Indeed, tyrosine phosphorylation evolved around the same time as metazoan organisms, and the new signaling capabilities it afforded may have been important for the transition to multicellular life [1].
Signaling by tyrosine phosphorylation involves the activities of three distinct classes of proteins. Tyrosine kinases phosphorylate tyrosine residues in substrate proteins, while tyrosine phosphatases remove the phosphate, converting phosphotyrosine (pTyr) back to tyrosine. Finally, small modular pTyr binding domains, predominately Src Homology 2 (SH2) domains, bind specifically to tyrosine-phosphorylated peptides, thereby mediating the assembly of multiprotein complexes dependent on tyrosine phosphorylation [2,3]. In most cases, downstream signaling requires the binding of SH2-containing proteins to phosphorylated binding sites. Thus system output depends on phosphorylation and dephosphorylation rates, and on the concentration and affinity of the SH2-containing proteins that bind to the phosphorylated sites.
In most cases, pTyr-mediated signaling is initiated by the activation of tyrosine kinases that are associated with transmembrane receptors. Engagement of the extracellular portion of receptors with specific ligands leads to increased catalytic activity of the associated kinase domain. There are two broad classes of receptors: the receptor tyrosine kinases (RTKs), which have their own intracellular tyrosine kinase domains, and receptors such as cytokine receptors and T-cell receptor, which are non-covalently associated with cytosolic tyrosine kinases. For both classes, it has long been appreciated that dimerization or aggregation of receptors is critical for their activation.
The need for dimerization is easily understood in the case of RTKs. For these receptors, the extracellular ligand-binding domain is linked to the intracellular tyrosine kinase domain by a single helical transmembrane segment. For an isolated receptor molecule, it is difficult to envision how the ligand binding state of the extracellular domain could be communicated through the membrane to the kinase domain. Ligand-induced dimerization, however, provides a ready mechanism to convert ligand binding to a change in state of the catalytic domain. In most cases, it is thought that dimerization leads directly to activation by allowing two closely apposed kinase domains to phosphorylate each other (transphosphorylation). Typically, phosphorylation of critical sites in the so-called activation loop of the kinase locks that kinase in an activated conformation (without activation loop phosphorylation, the active conformation of most protein kinases is poorly populated) [4]. Thus dimerization leads to activation of the dimerized catalytic domains, and also facilitates phosphorylation of other sites on the receptor (or in some cases receptor-associated scaffold proteins) that can then serve as binding sites for SH2 domain-containing effector proteins [5].
Similarly, dimerization or aggregation of receptors non-covalently linked to tyrosine kinases increases the local concentration of such kinases, and thus their likelihood to phosphorylate each other and associated receptor molecules. In some cases, such as the T-cell and B-cell receptors, the activation process is more complicated, involving multiple kinases that are sequentially recruited to receptors and activated, and multiple substrates, including receptor chains, scaffold proteins, and downstream effectors [6].
The binding of SH2 domain-containing effectors to the activated receptor triggers downstream signaling by their relocalization (in the case of receptors, to the plasma membrane) and in some cases by inducing their phosphorylation by receptor-associated kinases. Many of these effectors are themselves enzymes, or associated with enzymes, and relocalization to the plasma membrane brings them into close proximity with their substrates. For example, phospholipase C and phosphatidylinositol 3-kinase both modify plasma membrane inositol lipids, while SOS (which is recruited by the adaptor protein GRB2) and RAS-GAP both act on the small G protein RAS, which is confined to the membrane due to its lipid 4 modification. This proximity effect, combined in some cases with activating phosphorylation by the receptor, increases the rate of reaction for these enzymes, thus propagating downstream signaling.
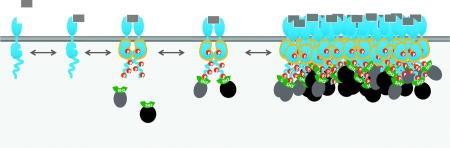
It is now appreciated that in addition to dimerization, in virtually all cases receptors associated with tyrosine kinases aggregate into higher-orders structures (clusters) upon ligand engagement (Fig 1). As noted above, receptor dimerization is needed to facilitate activation of kinases and phosphorylation of other sites on the receptors, but at first glance the effects of higher order clustering are not obvious. Below we consider the biophysical consequences of clustering on the behavior of the system, and how clustering affects information processing by receptors. A list of points of regulation by clustering is shown in Table 1 and its accompanying figure (Fig. 2), and discussed in more detail in the following sections.
Fig. 1.

Signaling by receptor tyrosine kinases. As depicted from the left, binding of ligand (gray rectangle) to the extracellular domain of the receptor stabilizes receptor dimers. The intracellular catalytic domains then phosphorylate their dimer partner, stabilizing the active conformation and promoting phosphorylation of additional sites on the receptor. These phosphosites serve to recruit SH2 domain-containing effectors from the cytosol. Activated receptors typically cluster together into dynamic higher-order assemblies (right). While signaling clusters may be relatively stable, individual phosphorylation and binding events are highly transient, lasting only a few seconds
Table 1.
Points of regulation by clustering
| Point of regulation | Effect and mechanism |
|---|---|
| Receptor kinase activity | Transphosphorylation of activation loop or through asymmetric dimer formation. |
| Recruited kinase activity | Avidity (increased binding). |
| PTP activity (transmembrane) | Potentially reduced activity by spatial exclusion |
| PTP activity (cytosolic) | Potentially reduced activity due to substrate saturation |
| PTP activity (recruited) | Complex effects |
| Binding of effector (monovalent) | Slower turnover kinetics due to high local concentration. Possible effect on Kd through low-affinity interactions (e.g. depletion force). |
| Binding of effector (multivalent) | Increased binding due to avidity. |
| Effector output activity | Kinetic proofreading. May promote threshold-type response. |
Fig. 2.

Points of regulation by clustering discussed in this review. Light color background suggested an upregulation of the underlying molecular interactions (binding, enzymatic activity etc.); dark color suggests a downregulation.
Signaling clusters as liquid protein droplets
The physical properties of signaling clusters had attracted little research attention until recent years. That changed, however, after Li et al. reported the formation of phase-separated protein droplets upon mixing purified signaling proteins that could interact with each other in a multivalent manner [7]. These experiments used the adaptor protein NCK, which contains an SH2 domain and three SH3 domains; a fragment of N-WASP containing multiple SH3 binding sites; and a multiply tyrosine-phosphorylated fragment of Nephrin. In vivo, these components are critical to signaling events that generate actin-rich structures in kidney podocytes [8]. Similar experiments have now been replicated in a two-dimensional environment on a supported lipid bilayer [9] and with other signaling protein systems [10–12]. These results are hugely influential in terms of reshaping the current thinking on the signaling clusters. It is, therefore, worthwhile to discuss a few key concepts arising from these experiments in the context of signaling.
(1) Phase separation
Protein droplet formation in vitro has largely been interpreted in the framework of phase separation – a thermodynamic mechanism that segregates protein molecules from a homogenous solution phase into two new equilibrated phases: the droplets, where the protein concentrations are high, and the surrounding solution, where the protein concentrations are lower. It is plausible that signaling clusters in cells can be understood in the same framework, considering the similarity between the two phenomena, but with one significant caveat: signaling clusters exist in vivo in a state far from equilibrium. In tyrosine phosphorylation signaling, pTyr residues have a very high rate of turnover (on the time-scale of seconds, also see later section on signal initiation), as they are continuously being generated by kinases and removed by phosphatases. It remains to be seen how the phase separation framework fits for systems with this type of energy-consuming turnover dynamics.
(2) Sol-gel transition
There is significant evidence suggesting that the chemical architecture of protein droplets follows that of a gel [13]. Here the tem “gel” refers to its traditional meaning in polymer/colloidal chemistry [14]: biphasic entities containing both a homogenous liquid phase (mostly the water or organic solvent) and a cross-linked chemical network. We know that protein droplets contain large amount of water [7], and the importance of cross-linking can be inferred from the requirement for multivalent components in the protein droplet experiments. Indeed, multivalent binding is a hallmark of protein interactions in cellular signaling, and thus it is likely that the signaling clusters in cells have a similar architecture, although they do also have a few of their own peculiarities. For example, most signaling clusters contain transmembrane proteins, and thus lipid molecules must contribute to the biophysical properties of the resulting structures.
(3) Viscoelasticity
One curious observation on the protein droplets was that they exhibited many “liquid-like” mechanical properties: They are mostly round, indicative of surface tension; they can fuse together into larger droplets; they shear under stress; and one can demonstrate “flow” of materials within a droplet via photobleaching experiments. Nevertheless, a more general understanding of their mechanical properties would involve characterizing their viscoelastic parameters, with the anticipation that they exhibit both solid-like and liquid-like responses in a time-scale dependent manner. In this regard, a critical parameter is the stability of molecular bonds between proteins in the network. When these bonds are sufficiently transient to allow reconfiguration of the network at the time-scale of interest, then the material would appear “liquid”; otherwise it would be more solid-like. In the context of signaling, the physiologically relevant time-scale spans a fairly broad range from milliseconds to many hours or days, and data on the physical properties of signaling clusters are very limited on all timescales.
Dynamics of signaling clusters
The observation of protein droplets in vitro prompted the question of whether the signaling clusters seen so ubiquitously in cells have the same physical nature. Conceptually, creating droplets of signaling molecules that behave like liquid (as opposed to solid) seems to offers advantages in terms of biochemistry: an enzymatic domain within a solid will only be able to access a few substrates that are very close, but in liquid it can diffuse within the droplet to work on more substrate molecules. Experimental investigation of the physical properties of signaling clusters is quite difficult, however, as the small size of many of the clusters in pTyr signaling (sub-micrometer) causes significant technical hurdles. For example, shear-stressed induced flows can be a very effective strategy to probe liquid properties in protein clusters, as was well-demonstrated in relatively large cellular structures such as RNP granules and the nucleolus [11], but this approach has not been used to study receptor clusters. Another strategy is to examine material flow within a droplet by photobleaching half of the droplet, and observing the signal recovery and redistribution from the other half. This test works well for in vitro reconstituted protein droplets, but the signaling clusters in cells are often too small for this approach (Fig 3). A much simpler experiment is to photobleach the whole cluster and measure the overall protein recovery kinetics. This will tell us the turnover rate of the molecular components in the droplet, but it does not directly answer the question whether the droplet liquid-like or solid-like.
Fig 3.

Experimental tests to distinguish protein liquid droplet from gel can be based on either macroscopic properties (left) or microscopic properties (right). Left: FRAP experiment with partial photobleaching of the droplet can test whether the materials within the droplet can flow via diffusion (liquid) or not (gel). Right: In liquid, non-covalent bonds (dashed lines) between protein molecules (represented by the spheres) are broken and reformed frequently, allowing individual molecules to move and rebind to different partners. This type of molecular dynamics is not observed in a gel, where binding is stable (solid lines).
Other experimental strategies probe the molecular dynamics within the signaling clusters by studying individual molecules. Microscopically, the hallmark of liquid is the ability of its molecules to frequently break and reform bonds to different interacting partners nearby, allowing the molecule to shift its position within the bulk material during the process. Evidence of this type of microscopic dynamics can be found in the single-molecule studies by Oh et al. [15] on EGFR signaling clusters. Their data showed a correlation between the apparent diffusion rates of the individual SH2-domain proteins and the strength of binding to their target pTyr sequences. This correlation exists because molecules that have weaker bonds tend to break them more frequently and thus spend more time moving around; this can be quantitatively modeled via standard kinetics theory of the diffusion-reaction system [16]. Another example is from analysis of single-molecule dynamics of the integrin receptor in focal adhesions [17]. The focal adhesion couples the extracellular matrix to the actin cytoskeleton, and is a relatively immobile structure [18]. The experimental data showed, however, that individual integrin molecules within the structure exhibit significantly higher-than-expected lateral diffusional dynamics, suggesting some form of dissociation-rebinding events. Finally, statistical analysis of molecular mobility can also be a powerful tool in deciphering the microscopic interactions a molecule experiences in a droplet, especially when the analyses are performed across multiple time-scales. For example, Huang et al. used such a strategy to deduce the cross-linking architecture of reconstituted LAT:Grb2:SOS droplets [19]. Although their work is based on an in vitro reconstituted system, the experimental strategy is applicable to live cell studies.
Despite many current efforts to characterize the dynamics of signaling clusters, it is probably fair to say that our understanding on this topic is still very limited. Furthermore, few studies have yet addressed more complex questions, such as whether signaling clusters can adopt multiple physical states, and/or undergo transitions between different physical states as a way to regulate signaling outcomes. It is possible that some signaling complexes may transition to a solid state under certain environmental conditions, in which individual components are essentially permanently associated and can no longer move relative to each other [11]. Such a transition could explain the perplexing persistence of some protein-protein interactions, even long after cell lysis, which allows their detection by co-purification or co-immunoprecipitation.
Signaling protein clustering and signal initiation
The prevalence of receptor clusters in pTyr signaling raises the obvious question of whether clustering is necessary for signal initiation. Biochemically, there is no question that dimerized RTKs are perfectly capable of cross-phosphorylating each other, without the need for clustering. In cells, however, it is now increasingly appreciated that receptors face significant dephosphorylation pressure from phosphatases. In the EGFR system, for example, we know that dephosphorylation of an activated receptor occurs within a few seconds [20,21]. Importantly, the dephosphorylation pressure is also present in unactivated cells, as inhibition of tyrosine phosphatases in living cells quickly results in an accumulation of pTyr, indicating that even in resting cells there is rapid turnover of tyrosine phosphorylation [20,22]. The question is thus whether the cluster is needed for signal initiation in the presence of dephosphorylation. Recently, several labs have engineered receptors whose state of oligomerization could be experimentally manipulated (Fig. 4). Data from such experiments indicate that, at least in some signaling systems, the answer seems to be yes.
Fig 4.

Various protein engineering strategies have been used in the literature to exert experimental control of receptor oligmerization states. (Left) DNA-based artificial ligand allows fine tuning of receptor-ligand interaction between a cell and supported lipid bilayer (SLB). (Right) FKBP-based chemical dimerization system has been used to define the cross-linking topology of artificial receptors.
The first system is based on the EphB2 receptor [23]. Unlike many other RTKs, signaling by Eph family (EphA/B) receptors seems to be highly dependent on multivalent interaction between the receptors and their ligands (ephrins) [24]. It was shown two decades ago, using artificial ligands, that monomeric ligand (which induces receptor dimerization) does not produce the downstream signaling effect evoked by oligomeric ligand, which efficiently induces clustering [25]. More recently, researchers have produced engineered EphB2 by inserting copies of the FKBP protein domain into the intracellular portion [26]. This allows them to chemically cross-link the receptor by treating cells with a small-molecule chemical dimerizer (which binds with high affinity the FKBP domain). By varying the number of FKBP domains, they could fine-tune the distribution of oligomers and test how receptor phosphorylation depends on the oligomerization state. They found that dimers were less efficient than higher-order oligomers in inducing receptor phosphorylation, and much less efficient in inducing downstream signaling.
The T cell receptor (TCR) is another system that has been examined [27]. The TCR is a relatively complex receptor in which the ζ chains of the receptor are phosphorylated by cytosolic Src family tyrosine kinases, such as Lck, when the receptor engages MHC-peptide complexes on the surface of antigen-presenting cells. Phosphorylation of TCR ζ chain in turn leads to recruitment and activation of a second cytosolic tyrosine kinase, ZAP70, which phosphorylates downstream effectors and scaffolds such as LAT. In this case, the researchers replaced the extracellular domain of the TCR ζ chain with a short single-stranded DNA strand, allowing them to activate the receptor by presenting complementary DNA strands on a supported lipid bilayer. By tuning the density of the complementary DNA and its affinity to the engineered receptor, and monitoring the cell with single-molecule imaging, they were also able to distinguish receptors in different oligomerization states. Again, their results indicated that the formation of receptor clusters was necessary for efficient receptor phosphorylation and recruitment of ZAP70.
What makes clusters more efficient in generating tyrosine phosphorylation? A simple answer would be that clusters increase the local concentration of reactants (kinase and substrates), which should increase the rate of phosphorylation in a cluster. The simplicity of this statement, however, is extremely misleading. Considering the RTK system, for example, the receptor protein encompasses both the catalytic enzyme (the kinase domain) and the substrate sequence. In a sense, from the RTK point of the view, the substrate is always at (the same) very high local concentration. The concept of “local concentration” is ambiguous here. Therefore, whether clustering increases the phosphorylation rate is far from a trivial question. Further theoretical analysis is needed, as well as experimental data on the accessibility of different substrate sites in cis, in trans in dimers, and in trans in clusters.
Another factor must be considered in the case of receptors that are non-covalently associated with cytosolic tyrosine kinases: most such kinases (for example Src family kinases and ZAP-70) themselves contain SH2 domains that bind to tyrosine-phosphorylated sites [28]. Furthermore, engagement of the SH2 domains of such kinases often stabilizes their catalytically active conformation [29]. This provides a strong positive feedback mechanism, in that the higher the local concentration of tyrosine phosphorylation, the higher the likelihood the kinase will be recruited and will be active, thus leading to even more tyrosine phosphorylation. SH2 domains also promote the processive phosphorylation of substrates by tethering the kinase to substrates [30,31]. Receptor tyrosine kinases have even been reported to recruit and activate soluble nonreceptor tyrosine kinases via SH2-mediated binding, which enhances phosphorylation of the receptor and of receptor-associated proteins [32]. The greater likelihood of nonreceptor tyrosine kinase binding to clustered receptors, via avidity effects and rapid rebinding as discussed below, likely contributes to a more switch-like (“all or nothing”) response for receptor clusters than would be possible in the case of isolated receptors.
In addition to the phosphorylation rates, steady state pTyr levels are also dependent on dephosphorylation rates. Here clustering can have different effects, depending on the type of tyrosine phosphatase. As has been pointed out by several authors recently [33,34], an important factor to consider is the saturation of enzymatic reactions at high concentration. When substrate concentration is sufficiently high, an enzymatic reaction follows zero order kinetics: the reaction rate no longer increases as a function of substrate concentration. It has been recognized more than three decades ago that saturation of competing enzymes, such as kinases and phosphatases, in a network could lead to complex system properties, e.g. switch-like behavior, which was initially called “zero-order ultrasensitivity” [35]. In the specific case of RTK systems, the various tyrosine phosphatases have moderately high Km values (on the order of µM [36]), meaning that they are normally unsaturated when substrates are dispersed, but could reach saturation in a cluster. This makes phosphatases less efficient on a per-substrate basis within the clusters. Because kinase activity is either constant or increased in a cluster (see above), this may be one mechanism whereby clusters promote more efficient phosphorylation.
For larger clusters, another factor that may affect the phosphorylation/dephosphorylation balance is the spatial exclusion of some phosphatases from the interior of the clusters. This mechanism is expected to affect transmembrane, receptor-type phosphatases. This has been most convincingly shown in the TCR system, where one of the major phosphatases is the transmembrane phosphatase CD45 [37]. Single-molecule imaging data showed that CD45 tends to be sterically excluded from the interior of TCR clusters, suggestion that clustering protects internal pTyr sites from dephosphorylation by CD45.
Finally, there is also a small group of PTPs that contain tandem SH2 domains (PTPN6/SHP-1 and PTPN11/SHP-2), which can be actively recruited to phosphorylated/activated receptors. SH2 engagement also increases phosphatase activity. Simplistically, these phosphatases should be recruited more efficiently to clusters with high local concentrations of phosphosites that can engage their SH2 domain (see below). How clustering affects the activity of this group is not well studied and is difficult to predict, due to the presence of multiple counter-acting factors that could potentially play a role. In cells, these phosphatases have been reported to have both positive and negative effects on receptor signaling [38].
Signaling clusters and signal propagation – Thermodynamic considerations
Beyond effects on phosphorylation, cluster formation may also affect how the effector proteins are recruited to the receptors at the cell membrane. For example, in EGFR signaling, the mitogenic effects are largely driven by the recruitment of the RAS activator, SOS, to the plasma membrane. Therefore when considering the impact of EGFR clustering on signaling outcomes, one has to evaluate whether clustering would alter the amount of SOS being recruited and its ability to activate RAS. Unfortunately, the thermodynamics of receptor/effector interaction cannot be easily described by simple affinity parameters such as dissociation constants, which are typically only available for 1:1 binary interactions between specific protein domains. Most, if not all, effector proteins in signaling have multiple binding modes/valencies, due to the presence of multiple modular protein binding domains and/or their binding sites [39]. SOS, for example, has multiple proline-rich SH3 binding sites, each of which can bind to one of the two SH3 domains of GRB2, a component of the signaling complex. At equilibrium, the SOS proteins are partitioned into three groups: ones that are unbound, ones that are bound to receptor clusters via one GRB2 molecule, and ones bound to multiple GRB2 molecules. Yet, it is impossible to compute the values of the partitioning without knowing the exact molecular configurations of the cluster, because the extent of the multivalent binding will depend on the density and orientation of the multivalent partners. Unlike the case of monomer binding in solution, no absolute value can be easily specified without this context. This complexity has long been recognized by the signaling field and is commonly referred to as the “avidity” effect. Avidity decreases the chance of unbinding, since multiple dissociation events must occur within a short time-window for the molecule to escape. While it is generally agreed that the consequence of avidity is higher apparent binding affinity, experimental quantification of the effect, especially in the context of effector recruitment in signaling, is still uncommon in the literature.
One example of such a quantitative study used the reconstituted T cell receptor system, consisting of phosphorylated LAT, GRB2 and SOS proteins [40]. Mixing these components at suitable concentration on supported lipid bilayers can lead to spontaneous phase separation to form protein clusters/droplets. Thus the authors can compare the single-molecule dynamics of two effectors – GRB2 and SOS in this system – between the non-clustered and clustered situation. The results are unambiguous that clustering allows a new population of very stably bound effectors to emerge, presumably corresponding to molecules that are bound with multiple interaction domains. Somewhat surprisingly, this stably bound population represents a fairly small fraction (e.g. 10% for SOS) of total recruited molecules, which seems to suggest that despite potential avidity mechanisms, most molecules interact with the cluster monovalently. The results are also consistent with measurements in cells, e.g. by Oh et al. [15], on the dynamics of tandem-SH2-domain probes that are bound to EGFR clusters. In this case, the “highly stable” population also represents a small fraction, even though the molecule can potentially bind to two pTyr residues simultaneously. Perhaps in most cases the spacing and orientation of sites makes multivalent interaction relatively rare, with the exception of specialized phosphorylation motifs containing precisely spaced sites known to engage effectors with tandem SH2 domains (for example the so-called “ITAM motif” on TCR chains [41], which when phosphorylated binds the tandem SH2 domains of ZAP70).
In considering avidity, another challenge is that when we are dealing with complex macromolecules such as proteins, it is not always obvious what represents a multivalent interaction. Traditional biochemistry is driven primarily by the specific and strong interaction between protein domains. Weak and nonspecific electrostatic, hydrophobic and van der Waal forces between residues outside the specific “interaction surfaces” are generally ignored, albeit justifiably when studying dilute and well-mixed solutions. However, when proteins form clusters, whether or not these weak forces can still be safely ignored is questionable.
Some new experimental evidence indeed suggests that the contribution of these forces should be more carefully examined. For example, in a recent the study of EGFR signaling [22], the authors performed time-resolved measurements of the amount of SH2 domains recruited to activated and phosphorylated EGFR receptor clusters in living cells, and found that the amount continued to increase long after the total tyrosine phosphorylation had reached its peak and started to decline. This effect, which correlated with the rate of clustering, could be observed for many SH2 domains that bind to the EGFR, unless receptor clustering was disrupted using chemical means. This type of clustering-dependent increase in binding is typically an indicator of avidity effects; in this case, however, the protein molecules being measured were isolated (monomeric) SH2 domains taken from various endogenous effectors in the EGFR pathway.
Nominally, a single SH2 domain is a monovalent binder that can only bind to one pTyr peptide. Thus the results suggest that the traditional concept of multivalency may be too narrowly applied when dealing with structures with very high protein density such as the signaling clusters. Part of the effect here is probably kinetic: clustering would make it harder for any cytosolic effector to find its binding target, and thus slow down both the on rate, assuming the on rate is diffusion controlled, as well as the off rate, assuming efficient rebinding [16]. Therefore, a delay is expected between the peak of phosphorylation and the peak of effector binding. However, theoretical calculation predicts that such a delay should be on the order of seconds, and not minutes as was observed in the experiments. Thus it is plausible that the multiple weak nonspecific interactions may play a role here to strengthen the binding in clusters.
One possible driver of interactions in the cell is the so-called “depletion force,” which operates in highly crowded environments such as the cytosol [42]. This force corresponds to the osmotic pressure exerted by other macromolecules that are excluded from the volume between closely adjacent protein molecules. This was recently proposed as a mechanism driving the clustering of transcription factors in response to signaling-induced changes in their phosphorylation [43]. The exact values of the depletion force in signaling clusters will depend on the concentrations of signaling proteins, the concentrations of co-solute (i.e., other small proteins within the aqueous phase of the droplet), as well as the relative size ratios between the two groups. Not all these data are experimentally available yet to permit quantitative modeling of this mechanism, but we are rapidly approaching such a point, as many new studies now focus on quantifying relevant key parameters (copy numbers, stoichiometry etc.) of signaling systems (e.g., [44]).
Signaling clusters and signal propagation – Kinetic considerations
Signaling clusters could also affect downstream signal transduction via a purely kinetic mechanism called kinetic proofreading. The concept of kinetic proofreading was initially used to explain the fidelity of DNA replication [45], although the general principle is now recognized to be applicable in many other biological phenomena [46,47]. In the context of receptor signal transduction, it was shown theoretically by McKeithan [48] that the kinetics of ligand binding to receptors can regulate the efficiency of receptor activation. A more generalized formulism was given by Huang et al. [40] in their discussion of SOS activation in TCR signaling. In all cases, the general theme of kinetic proofreading is that the turnover rate of an upstream binding/dissociation reaction can affect the biochemical outcome of a downstream event. In the example of SOS, RAS activation occurs after it is recruited to T cell receptor complexes at the cell membrane. For each individual molecule, the activation time should be described by a waiting-time distribution function, due to stochasticity of the chemical reactions involved in catalysis (Fig. 5). Importantly, because there are multiple steps in the catalytic pathway, the waiting-time distribution function for SOS activity has both a rise and a decay, i.e. it exhibits a non-zero mode. As a consequence, SOS molecules that exhibit very fast binding/dissociation turnover at the receptor will not activate RAS as efficiently in comparison to the scenario where the turnover is slower. In more general terms, such a kinetic proofreading effect can be seen in any waiting-time distribution function that exhibits a non-zero mode, a condition that is satisfied as long as the process involves more than one sequential chemical reaction, which probably describes most if not all signal transduction events. It is also important to point out that kinetic proofreading is entirely a consequence of kinetics, and require no change in thermodynamic properties such as equilibrium constants in the system.
Fig. 5.

Activation mechanism dictates the waiting time distribution of the activation step in signal transduction (adopted from (Huang et al 2016)). In the example of SOS activation (A), the distribution (B) is exponential if the activation is a single-step reaction (N=0); otherwise, in the more common case of complex, multi-step activation, the distribution has a non-zero mode (N>0). The latter cases would exhibit a kinetic proofreading effect.
How are kinetic proofreading effects related to cluster formation in signaling? Recent experimental evidence indicates that signaling cluster formation can slow down the effector protein turnover at the cluster. Several mechanisms contribute to this effect: First, because high-density clusters promote multivalent binding, the effectors’ dissociation rate is lower in a cluster, because dissociation requires simultaneously breaking multiple protein-protein interactions. This is a consequence related to the avidity effects discussed earlier. Secondly, the high local concentration of binding sites in the cluster necessarily leads to a very high local protein-protein binding rate, which is proportional to concentrations of reactants. Therefore for any newly dissociated protein, there is a high probability of rapid rebinding as opposed to diffusing away, effectively resulting in a slower overall turnover rate [15,49]. Combined, these factors suggest that clusters may activate downstream signaling events much more efficiently than non-clustered receptors. Furthermore, clustering may prevent spurious signal activation due to the random binding of various effectors to the isolated receptors, which may happen at high frequency in unactivated cells considering the high intrinsic turnover rate of pTyr in RTK systems. Such events would lead to only relatively brief binding of effectors, compared to clusters generated by bona fide signals, which would promote more stable effector binding. Therefore, the kinetic proofreading mechanism can confer both noise suppression and threshold-type activation.
Conclusion
Recent studies have prompted a re-evaluation of signaling by receptors linked to tyrosine kinases. Relatively simple, static cartoon models are being replaced by a more sophisticated appreciation of the important roles played by dynamic clusters of receptors and their downstream effectors in signal processing. New approaches from the fields of polymer science, soft-matter physics, and kinetic theory are beginning to provide insight into the sometimes surprising and counterintuitive behaviors of such structures. Given the importance of tyrosine kinase pathways for normal life, and the fact that dysregulation of these pathways drives many human diseases, these new insights are likely to have a major impact on basic and translational research in the coming years.
Highlights.
Receptors associated with tyrosine kinases aggregate into higher-orders structures (clusters) upon ligand engagement.
New approaches from the fields of polymer science, soft-matter physics, and kinetic theory are beginning to provide insight into the biophysical consequences of clustering on the behavior of the system.
Experimental data indicated that receptor clustering impact signal outcomes by altering the balance between phosphorylation and dephosphorylation, and modulating downstream effector activation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lim WA, Pawson T. Phosphotyrosine signaling: evolving a new cellular communication system. Cell. 2010;142:661–667. doi: 10.1016/j.cell.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pawson T. Protein modules and signalling networks. Nature. 1995;373:573–580. doi: 10.1038/373573a0. [DOI] [PubMed] [Google Scholar]
- 3.Schlessinger J, Lemmon MA. SH2 and PTB domains in tyrosine kinase signaling. Sci. STKE Signal Transduct. Knowl. Environ. 2003;2003:RE12. doi: 10.1126/stke.2003.191.re12. [DOI] [PubMed] [Google Scholar]
- 4.Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol. Cell. 2004;15:661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 5.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samelson LE. Immunoreceptor signaling. Cold Spring Harb. Perspect. Biol. 2011;3 doi: 10.1101/cshperspect.a011510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li P, Banjade S, Cheng H-C, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF, Russo PS, Jiang Q-X, Nixon BT, Rosen MK. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012;483:336–340. doi: 10.1038/nature10879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones N, Blasutig IM, Eremina V, Ruston JM, Bladt F, Li H, Huang H, Larose L, Li SS-C, Takano T, Quaggin SE, Pawson T. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature. 2006;440:818–823. doi: 10.1038/nature04662. [DOI] [PubMed] [Google Scholar]
- 9.Banjade S, Rosen MK. Phase transitions of multivalent proteins can promote clustering of membrane receptors. ELife. 2014;3 doi: 10.7554/eLife.04123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017;18:285–298. doi: 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shin Y, Brangwynne CP. Liquid phase condensation in cell physiology and disease. Science. 2017;357 doi: 10.1126/science.aaf4382. [DOI] [PubMed] [Google Scholar]
- 12.Su X, Ditlev JA, Hui E, Xing W, Banjade S, Okrut J, King DS, Taunton J, Rosen MK, Vale RD. Phase separation of signaling molecules promotes T cell receptor signal transduction. Science. 2016;352:595–599. doi: 10.1126/science.aad9964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harmon TS, Holehouse AS, Rosen MK, Pappu RV. Intrinsically disordered linkers determine the interplay between phase separation and gelation in multivalent proteins. ELife. 2017;6 doi: 10.7554/eLife.30294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin J, Douglass A. The Sol-Gel Transition in Chemical Gels. Annu. Rev. Phys. Chem. 1991;42:311–339. doi: 10.1146/annurev.pc.42.100191.001523. [DOI] [Google Scholar]
- 15.Oh D, Ogiue-Ikeda M, Jadwin JA, Machida K, Mayer BJ, Yu J. Fast rebinding increases dwell time of Src homology 2 (SH2)-containing proteins near the plasma membrane. Proc. Natl. Acad. Sci. U. S. A. 2012;109:14024–14029. doi: 10.1073/pnas.1203397109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Berg OG, von Hippel PH. Diffusion-controlled macromolecular interactions. Annu. Rev. Biophys. Biophys. Chem. 1985;14:131–160. doi: 10.1146/annurev.bb.14.060185.001023. [DOI] [PubMed] [Google Scholar]
- 17.Rossier O, Octeau V, Sibarita J-B, Leduc C, Tessier B, Nair D, Gatterdam V, Destaing O, Albigès-Rizo C, Tampé R, Cognet L, Choquet D, Lounis B, Giannone G. Integrins β1 and β3 exhibit distinct dynamic nanoscale organizations inside focal adhesions. Nat. Cell Biol. 2012;14:1057–1067. doi: 10.1038/ncb2588. [DOI] [PubMed] [Google Scholar]
- 18.Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010;11:633–643. doi: 10.1038/nrm2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang WYC, Chiang H-K, Groves JT. Dynamic Scaling Analysis of Molecular Motion within the LAT:Grb2:SOS Protein Network on Membranes. Biophys. J. 2017;113:1807–1813. doi: 10.1016/j.bpj.2017.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jadwin JA, Curran TG, Lafontaine AT, White FM, Mayer BJ. Src homology 2 domains enhance tyrosine phosphorylationin vivoby protecting binding sites in their target proteins from dephosphorylation. J. Biol. Chem. 2018;293:623–637. doi: 10.1074/jbc.M117.794412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kleiman LB, Maiwald T, Conzelmann H, Lauffenburger DA, Sorger PK. Rapid phospho-turnover by receptor tyrosine kinases impacts downstream signaling and drug binding. Mol. Cell. 2011;43:723–737. doi: 10.1016/j.molcel.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jadwin JA, Oh D, Curran TG, Ogiue-Ikeda M, Jia L, White FM, Machida K, Yu J, Mayer BJ. Time-resolved multimodal analysis of Src Homology 2 (SH2) domain binding in signaling by receptor tyrosine kinases. ELife. 2016;5:e11835. doi: 10.7554/eLife.11835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyd AW, Bartlett PF, Lackmann M. Therapeutic targeting of EPH receptors and their ligands. Nat. Rev. Drug Discov. 2014;13:39–62. doi: 10.1038/nrd4175. [DOI] [PubMed] [Google Scholar]
- 24.Himanen JP, Yermekbayeva L, Janes PW, Walker JR, Xu K, Atapattu L, Rajashankar KR, Mensinga A, Lackmann M, Nikolov DB, Dhe-Paganon S. Architecture of Eph receptor clusters. Proc. Natl. Acad. Sci. U. S. A. 2010;107:10860–10865. doi: 10.1073/pnas.1004148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis S, Gale NW, Aldrich TH, Maisonpierre PC, Lhotak V, Pawson T, Goldfarb M, Yancopoulos GD. Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science. 1994;266:816–819. doi: 10.1126/science.7973638. [DOI] [PubMed] [Google Scholar]
- 26.Schaupp A, Sabet O, Dudanova I, Ponserre M, Bastiaens P, Klein R. The composition of EphB2 clusters determines the strength in the cellular repulsion response. J Cell Biol. 2014;204:409–422. doi: 10.1083/jcb.201305037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor MJ, Husain K, Gartner ZJ, Mayor S, Vale RD. A DNA-Based T Cell Receptor Reveals a Role for Receptor Clustering in Ligand Discrimination. Cell. 2017;169:108–119.e20. doi: 10.1016/j.cell.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu. Rev. Biochem. 2000;69:373–398. doi: 10.1146/annurev.biochem.69.1.373. [DOI] [PubMed] [Google Scholar]
- 29.Filippakopoulos P, Müller S, Knapp S. SH2 domains: modulators of nonreceptor tyrosine kinase activity. Curr. Opin. Struct. Biol. 2009;19:643–649. doi: 10.1016/j.sbi.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mayer BJ, Hirai H, Sakai R. Evidence that SH2 domains promote processive phosphorylation by protein-tyrosine kinases. Curr. Biol. CB. 1995;5:296–305. doi: 10.1016/s0960-9822(95)00060-1. [DOI] [PubMed] [Google Scholar]
- 31.Scott MP, Miller WT. A peptide model system for processive phosphorylation by Src family kinases. Biochemistry (Mosc.) 2000;39:14531–14537. doi: 10.1021/bi001850u. [DOI] [PubMed] [Google Scholar]
- 32.Bromann PA, Korkaya H, Courtneidge SA. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 2004;23:7957–7968. doi: 10.1038/sj.onc.1208079. [DOI] [PubMed] [Google Scholar]
- 33.Ferrell JE, Ha SH. Ultrasensitivity part I: Michaelian responses and zero-order ultrasensitivity. Trends Biochem. Sci. 2014;39:496–503. doi: 10.1016/j.tibs.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lemmon MA, Freed DM, Schlessinger J, Kiyatkin A. The Dark Side of Cell Signaling: Positive Roles for Negative Regulators. Cell. 2016;164:1172–1184. doi: 10.1016/j.cell.2016.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldbeter A, Koshland DE. An amplified sensitivity arising from covalent modification in biological systems. Proc. Natl. Acad. Sci. U. S. A. 1981;78:6840–6844. doi: 10.1073/pnas.78.11.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y-L, Yao Z-J, Sarmiento M, Wu L, Burke TR, Zhang Z-Y. Thermodynamic Study of Ligand Binding to Protein-tyrosine Phosphatase 1B and Its Substrate-trapping Mutants. J. Biol. Chem. 2000;275:34205–34212. doi: 10.1074/jbc.M004490200. [DOI] [PubMed] [Google Scholar]
- 37.Douglass AD, Vale RD. Single-Molecule Microscopy Reveals Plasma Membrane Microdomains Created by Protein-Protein Networks that Exclude or Trap Signaling Molecules in T Cells. Cell. 2005;121:937–950. doi: 10.1016/j.cell.2005.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pao LI, Badour K, Siminovitch KA, Neel BG. Nonreceptor protein-tyrosine phosphatases in immune cell signaling. Annu. Rev. Immunol. 2007;25:473–523. doi: 10.1146/annurev.immunol.23.021704.115647. [DOI] [PubMed] [Google Scholar]
- 39.Bhattacharyya RP, Reményi A, Yeh BJ, Lim WA. Domains, motifs, and scaffolds: the role of modular interactions in the evolution and wiring of cell signaling circuits. Annu. Rev. Biochem. 2006;75:655–680. doi: 10.1146/annurev.biochem.75.103004.142710. [DOI] [PubMed] [Google Scholar]
- 40.Huang WYC, Yan Q, Lin W-C, Chung JK, Hansen SD, Christensen SM, Tu H-L, Kuriyan J, Groves JT. Phosphotyrosine-mediated LAT assembly on membranes drives kinetic bifurcation in recruitment dynamics of the Ras activator SOS. Proc. Natl. Acad. Sci. 2016;113:8218–8223. doi: 10.1073/pnas.1602602113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Isakov N. Immunoreceptor tyrosine-based activation motif (ITAM), a unique module linking antigen and Fc receptors to their signaling cascades. J. Leukoc. Biol. 1997;61:6–16. doi: 10.1002/jlb.61.1.6. [DOI] [PubMed] [Google Scholar]
- 42.Marenduzzo D, Finan K, Cook PR. The depletion attraction: an underappreciated force driving cellular organization. J. Cell Biol. 2006;175:681–686. doi: 10.1083/jcb.200609066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wollman AJ, Shashkova S, Hedlund EG, Friemann R, Hohmann S, Leake MC. Transcription factor clusters regulate genes in eukaryotic cells. ELife. 2017;6 doi: 10.7554/eLife.27451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi T, Niepel M, McDermott JE, Gao Y, Nicora CD, Chrisler WB, Markillie LM, Petyuk VA, Smith RD, Rodland KD, Sorger PK, Qian W-J, Wiley HS. Conservation of protein abundance patterns reveals the regulatory architecture of the EGFR-MAPK pathway. Sci. Signal. 2016;9:rs6. doi: 10.1126/scisignal.aaf0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hopfield JJ. Kinetic Proofreading: A New Mechanism for Reducing Errors in Biosynthetic Processes Requiring High Specificity. Proc. Natl. Acad. Sci. U. S. A. 1974;71:4135–4139. doi: 10.1073/pnas.71.10.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ouldridge TE, Rein ten Wolde P. The robustness of proofreading to crowding-induced pseudoprocessivity in the MAPK pathway. Biophys. J. 2014;107:2425–2435. doi: 10.1016/j.bpj.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swain PS, Siggia ED. The role of proofreading in signal transduction specificity. Biophys. J. 2002;82:2928–2933. doi: 10.1016/S0006-3495(02)75633-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McKeithan TW. Kinetic proofreading in T-cell receptor signal transduction. Proc. Natl. Acad. Sci. U. S. A. 1995;92:5042–5046. doi: 10.1073/pnas.92.11.5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lawley SD, Keener JP. Rebinding in biochemical reactions on membranes. Phys. Biol. 2017;14:056002. doi: 10.1088/1478-3975/aa6f93. [DOI] [PubMed] [Google Scholar]