

SUMMARY
Rational design of drug-like small-molecule ligands based on structural information of proteins remains a significant challenge in chemical biology. In particular, designs targeting protein-protein interfaces have met little success given the dynamic nature of the protein surfaces. Herein, we utilized the structure of a small-molecule ligand in complex with toll-like receptor 8 (TLR8) as a model system due to TLR8’s clinical relevance. Over-activation of TLR8 has been suggested to play a prominent role in the pathogenesis of various autoimmune diseases, however there are still few small-molecule antagonists available, our rational designs led to the discovery of six exceptionally potent compounds with ~pM IC50 values. Two X-ray crystallographic structures validated the contacts within the binding pocket. A variety of biological evaluations in cultured cell lines, human peripheral blood mononuclear cells, and splenocytes from human TLR8-transgenic mice further demonstrated these TLR8 inhibitors’ high efficacy, suggesting strong therapeutic potential against autoimmune disorders.
INTRODUCTION
Toll-like receptors (TLRs) play a key role in the innate immune system by recognizing structurally conserved pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) (Hayashi, et al., 2001; Hoshino, et al., 1999; O’Neill, et al., 2013; Takeuchi, et al., 2001; Takeuchi, et al., 2002). Among 10 TLRs in humans, TLR3, 7, 8 and 9 localize in endosomal compartments and detect viral and bacterial signature molecules, including double-stranded RNA (TLR3) (Alexopoulou, et al., 2001), single-stranded RNA (TLR7/8) (Diebold, et al., 2004; Heil, et al., 2004), or unmethylated CpG sequences in DNA (TLR9) (Hemmi, et al., 2000). There is a considerable amount of evidence indicating that excessive activation of TLR8 significantly contributes to the pathogenesis of a variety of autoimmune diseases (Anders, et al., 2005; Farrugia and Baron, 2017), such as rheumatoid arthritis (RA) (Guiducci, et al., 2013; Krieg and Vollmer, 2007; Mullen, et al., 2015; Sacre, et al., 2016; Sacre, et al., 2007; Sacre, et al., 2008), which is one of the most common autoimmune disorders, occurring in 0.5–1.0% of the adult population (Firestein, 2003). However, few small molecules that specifically target TLR8 have been identified (Hu, et al., 2018; Rimbach, et al., 2015), which drove us into the pursuit of TLR8 antagonists. The development of potent and selective small-molecule inhibitors targeting TLR8 could also provide useful tools to further the mechanistic understanding of TLR8 inhibition and regulation, and help in the future development of autoimmune disease therapeutics. (Elshabrawy, et al., 2017)
Few successful developments of small-molecule inhibitors of TLR8 has been reported, suggesting the need for an alternative approach targeting the canonical binding sites of single-stranded RNA. Until recently, our group reported the first well characterized small-molecule TLR8 inhibitor, CU-CPT8m, based on a 7-phenylpyrazolo[1,5-a]pyrimidine scaffold (Zhang, et al., 2018). Interestingly, CU-CPT8m recognized a pocket on the interface of two copies of TLR8 (TLR8 and TLR8*, throughout this paper, asterisks are used to indicate the second TLR8 and its residues. Figure 1A) and prevented them from undergoing conformational changes that are necessary for the proinflammatory signaling (Tanji, et al., 2015). This discovery validated that the unliganded TLR8 dimer is a feasible target for TLR8 modulation, suggesting a novel avenue for anti-autoimmune therapeutic development. Nonetheless, the X-ray crystal structure of TLR8 in complex with CU-CPT8m (Figure 1A) indicated that CU-CPT8m did not fully utilize the functionalities presented at the binding pocket (Zhang, et al., 2018), which might explain its moderate binding affinity.
Figure 1. Structure-based rational design of TLR8 antagonists.
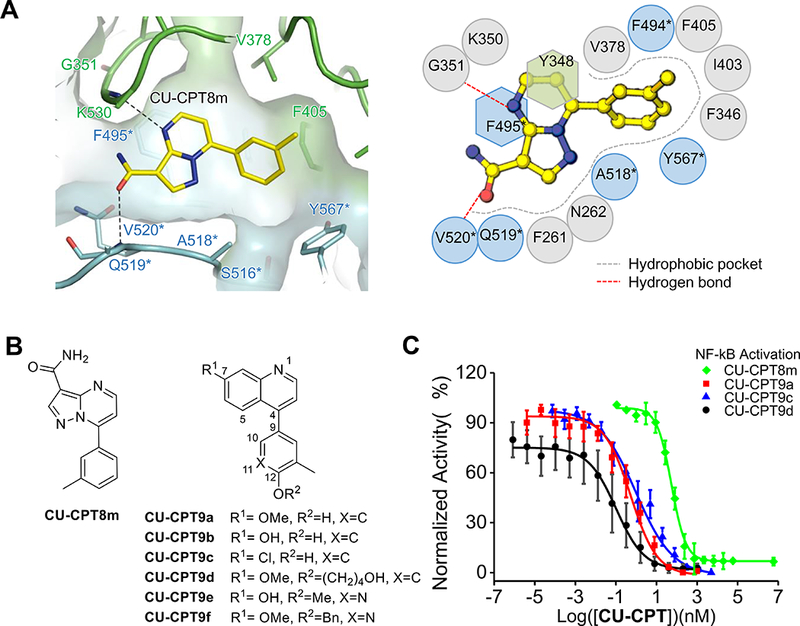
(A) Close-up view of the binding site of CU-CPT8m (left) and the schematic representation of interactions between CU-CPT8m and TLR8 (right). The recognition of previously identified TLR8 inhibitor, CU-CPT8m, is primarily drive by shape complementarity with few specific polar interactions, suggesting room for further optimizations.
(B) Chemical structures of CU-CPT8m and CU-CPT9a - 9f.
(C) Dose-dependent inhibitory effects of TLR8 signaling by CU-CPT8m, CU-CPT9a, CU-CpT9c and CU-CPT9d in hTLR8 HEK-Blue cell using R848 (1 μg/mL) as agonist (data are mean ± SD; n = 3 independent experiments).
Herein we report a series of exceptionally potent TLR8 antagonists through structure based rational design. A second generation of TLR8 inhibitors was identified with IC50 values approaching ~100 pM in various cell lines. Two X-ray crystal structures of CU-CPT9a/9c complexed with the ectodomain of TLR8 confirmed that these compounds also bind to the same pocket of CU-CPT8m. Assays in human peripheral blood mononuclear cells (PBMC) and splenocytes from transgenic mouse showed that CU-CPT9 compounds can effectively and selectively inhibit inflammatory TLR8 signaling. Taken together, these results convincingly demonstrated the feasibility of targeting the TLR dimer interface using small-molecules, laying the groundwork for future drug development.
RESULTS AND DISCUSSION
CU-CPT8m recognized a previously unknown binding pocket on the protein-protein interface of TL8 dimers. It is particular noteworthy that the binding of CU-CPT8m is primarily driven by its size and shape complementarity, with few specific hydrogen binding or electrostatic interactions involved (Zhang, et al., 2018). Based on these observations, we set out to optimize the first generation of TLR8 inhibitors by installing new functionalities, aiming to further utilize additional interactions with residues at the newly identified binding site on the TLR8 dimeric interface.
Multiple synthetic routes were developed to prepare structurally comparable chemical scaffolds (scheme S1). More than 100 different compounds were synthesized to demonstrate a consistent structure-activity relationship (for detailed SAR study, see the supplementary information). In particular, to utilize the hydrogen bond with G351 (Figure 1A), a variety of heterocycles were synthesized. The quinoline core yielded the most potent inhibitors (Figure 1B, and Table S2). In a similar way, the installment of substituents at the 7-position was used to establish interactions with V520* and Q519* (Figure 1A). Next, a hydroxyl group was introduced at the 12-position to explore extra contacts with S516* through a potential H-bond. These modifications resulted in several extremely potent compounds (Figure 1B) with subnanomolar IC50 values (CU-CPT9a - 9c, with IC50 of 0.5 ± 0.1 nM, 0.7 ± 0.2 nM and 1.0 ± 0.2 nM respectively). Given that the binding of CU-CPT8m is primarily driven by size and shape complementarity, larger substitutes on the quinoline and phenol backbones mostly compromised its inhibitory effect (Table S2). However, the relatively open space on the bottom of the hydrophobic pocket recognized by CU-CPT8m suggested the possibility to engineer additional intermolecular contacts (Figure 1A, supplementary movie). Thus, installment of long-chain substituents at 12-position was applied, which yielded several extremely potent inhibitors (Figure 1B). In particular, CU-CPT9d and 9f showed IC50 values of 0.1 ± 0.02nM and 0.8 ± 0.2 nM, respectively. This increased inhibitory effect suggested possible additional hydrophobic interactions with TLR8.
Taken together, six ~pM inhibitors designated CU-CPT9a-9f were identified with IC50 values as low as 0.1 ± 0.02 nM (Figure 1B, 1C) in blocking TLR8 signaling induced by its synthetic agonist, R848 (Jurk, et al., 2002). Moreover, they also showed comparable inhibitory effects toward single-stranded RNA, the native ligand of TLR8 (Figure S1) (Kariko, et al., 2005). Isothermal titration calorimetry (ITC) confirmed the direct binding between CU-CPT9b and the ectodomain of human TLR8 with a dissociation constant (Kd) of 21 nM (Zhang, et al., 2018). As a comparison, the Kd between TLR8 and R848 was determined as 200 nM(Tanji, et al., 2015), highlighting the superior affinity of CU-CPT9b. ITC results also showed that binding of CU-CPT9 derivatives prevented further binding of R848 (Figure S1), supporting the design of targeting an allosteric, competitive binding site. The anti-inflammatory effect of CU-CPTs was further confirmed with the down-regulation of NF-κB, a key downstream target for TLR8 signaling (O’Neill, et al., 2013; Tak and Firestein, 2001). CU-CPT9a showed significant reduction of nuclear NF-κB at concentration of 500 nM (Figure S2). Finally, these compounds all demonstrated minimal cytotoxicity, suggesting promising potential for further therapeutic development (Figure S3).
A major challenge in developing TLR inhibitors is to achieve selectivity among the highly analogous TLR family proteins (Yin and Flynn, 2016). This is especially true for TLR7 and TLR8 since that they both recognize single-stranded RNA as well as non-selective small molecule agonists (e.g. R848) (Tanji, et al., 2013; Zhang, et al., 2016). The specificity of CU-CPT9a and CU-CPT9b was tested using HEK-Blue cells individually overexpressing each human TLR. CU-CPT9a and CU-CPT9b showed good selectivity towards TLR8 over other TLRs (Figure 2A). In particular, TLR7 was not affected below ~μM concentration, showing more than 10,000 folds of difference in selectivity. The specificity of CU-CPT9s against human TLRs was also tested using PBMCs and their subsets that express multiple TLRs (Siednienko and Miggin, 2009). In these cells, CU-CPT9s specifically inhibited cytokine production induced by TLR8 (Cervantes, et al., 2012), but not by TLR2, 4, 5, 7 or 9 (Figure 2B).
Figure 2. Specificity tests of CU-CPT compounds.
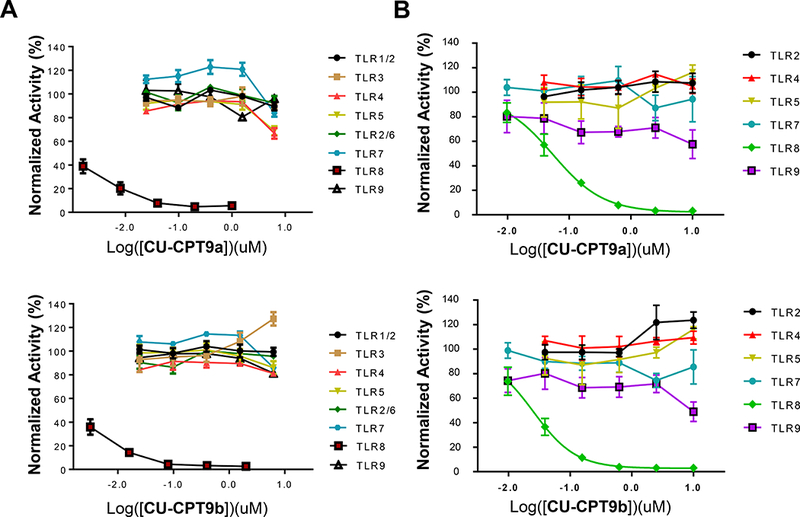
(A) Specificity test of CU-CPT9a (top) and 9b (bottom) in HEK-Blue cells overexpressing individual TLRs. For HEK-Blue hTLR1/2, hTLR2/6, hTLR3, hTLR4, hTLR5, hTLR7/8, and hTLR9 cells, Pam3CSK4 (100 ng/mL), Pam2CSK4 (100 ng/mL), poly(I:C) (5 μg/mL), LPS (lipopolysaccharide) (20 ng/mL), flagellin (50 ng/mL), R848 (1 μg/mL), ODN2006 (0.15 μM) were used as TLR-specific agonists, respectively.
(B) Specificity test of CU-CPT9a (top) and 9b (bottom) in PBMCs (B cells, monocytes, etc.) isolated from human donors. The entire PBMC population was used to assess antagonist activity against TLR2, 4, and 5. Isolated B cells were used to assess antagonist activity against TLR7 and 9, and isolated monocytes were used for TLR8. (data are mean ± SD; n =3 independent experiments).
To validate our structure-based designs and the mechanism of action of these CU-CPT compounds, we successfully solved two structures of TLR8/CU-CPT complexes, TLR8/CU-CPT9a and TLR8/9c (Figure 3), using X-ray crystallography. In an agreement with the previously reported structure of TLR8/CU-CPT9b (Figure 3) (Zhang, et al., 2018), all CU-CPT9s recognize the allosteric pocket identified by CU-CPT8m between two TLR8 protomers, thereby stabilizing the dimer at resting state (Figure 3A). This hydrophobic pocket only exists in the resting state of preformed TLR8 dimer, with several water molecules filling inside. All CU-CPT9s showed similar binding pattern: π-π stacking with Y348 and F495*, van der Waals interactions with hydrophobic residues (F261, F346, V378, I403, F405, F494*, A518*, V520*, and Y567*), H-bonds with G351 and V520*. Also, CU-CPT9 compounds formed water mediated H-bonds with S516*, although only weak electron density corresponding to water molecule was observed in TLR8/CU-CPT9a and 9c complexes due to their lower crystallographic resolutions. Water mediated H-bonds with Q519* was also formed in TLR8/CU-CPT9b, but not in TLR8/CU-CPT9a/9c because of the hindrance of 7-methoxyl group and chlorine atom. The results of co-crystal structures are in accordance with our SAR study and confirmed our rational design: quinoline motif showed optimal inhibition for its π-π stacking with Y348 and F495* and H-bonds with G351; 7-substituents increased inhibitory effect because of their H-bonds with V520* and Q519*; 12-hydroxyl group was critical for its water mediated H-bond with S516* (Figure 3B, 3C).
Figure 3. Crystallography structures of TLR/CU-CPT complexes.
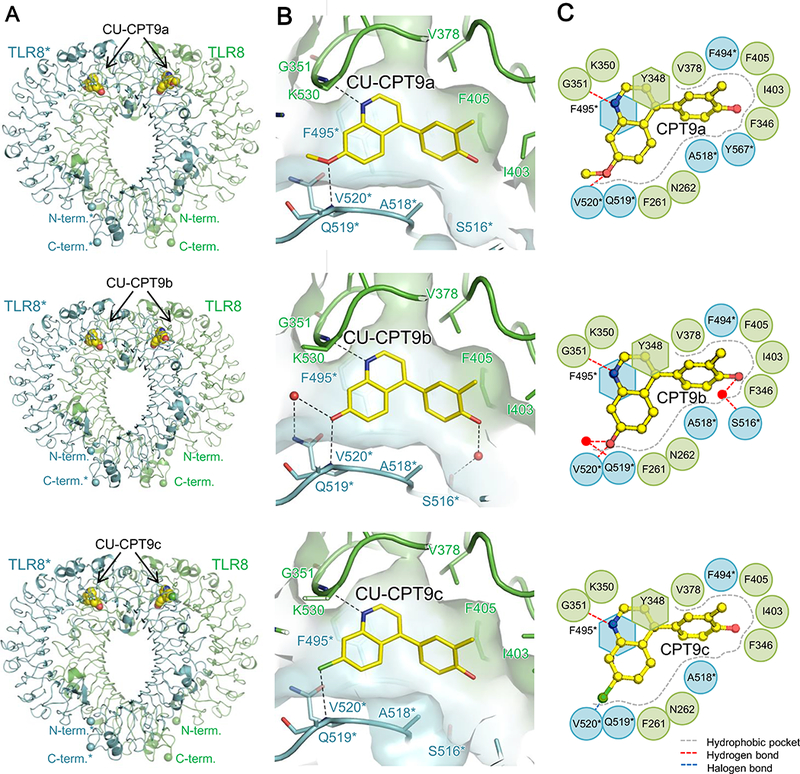
(A) Front views of the TLR8/CU-CPT9a (top, PDB ID: 5Z14), TLR8/CU-CPT9b (middle, PDB ID: 5WYZ) and TLR8/CU-CPT9c (bottom, PDB ID: 5Z15) complexes. TLR8 and its dimerization partner TLR8* are colored green and cyan, respectively. The ligand molecules are illustrated by space-filling representations. The C, O and N atoms of the ligands are colored yellow, red, and blue, respectively.
(B) Close-up view of the binding site of CU-CPT9a (top), CU-CPT9b (middle), and CU-CPT9c (bottom) on the TLR8 surface..
(c) Schematic representations show the interactions within the binding pockets of TLR8/CU-CPT9a (top), TLR8/CU-CPT9b (middle) and TLR8/CU-CPT9c (bottom) complexes.
TLR8 has been shown as a potential therapeutic target for autoimmune diseases (Guiducci, et al., 2013; Sacre, et al., 2008). Although previous works demonstrated the effects of TLR7 and TLR9 in autoimmunity in mice models (Lau, et al., 2005; Leadbetter, et al., 2002), a similar evaluation of TLR8 was not possible since murine TLR8 has been suggested to be nonfunctional (Jurk, et al., 2002). Therefore, we tested the CU-CPT9 compounds on splenocytes isolated from mice harboring a human TLR8 transgene in a TLR7 knock-out background (hTLR8tg/TLR7-KO). The results from mouse splenocytes were consistent with those from human cells, CU-CPT9 compounds demonstrated antagonist activity against the human TLR8 transgene with no activity against mouse TLR9 when using ORN8L (100 ug/mL) (Lan, et al., 2007) and 1018 (7.1 ug/mL) (Friedberg, et al., 2005) as TLR8 and TLR9 agonists, respectively (Figure 4).
Figure 4. Inhibitory effect of CU-CPT compounds in splenocytes.
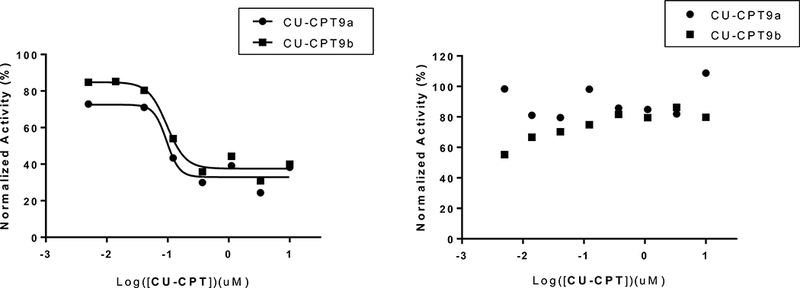
CU-CPT9a and CU-CPT9b demonstrated inhibition of IL-12p40 production as a monitor of TLR8 signaling (left) with no activity against mouse TLR9 (right) in the splenocytes harvested from hTLR8tg/TLR7-KO mice. ORN8L (100 ug/mL) and 1018 (7.1 ug/mL) were used as TLR8 and TLR9 agonists, respectively.
In summary, structure-based rational design has led to the discovery of a second generation of TLR8 inhibitors with ~pM IC50 values. X-ray crystal structures of CU-CPT9 compounds in complex with TLR8 confirmed that these compounds utilized additional interactions at the binding pocket on the TLR8 dimer interface and stabilized the TLR8 dimer at its resting state. Assays in various cell lines, primary human PBMCs, and splenocytes from a transgenic mouse model validated the therapeutic potential of these antagonists. Overall, these results demonstrated remarkable therapeutic potential of these TLR8 antagonists.
SIGNIFICANCE
The development of a new generation of TLR8 antagonist represents a successful demonstration of targeting an unconventional pocket on a protein-protein interface. The structured-based rational design of such a ternary complex is extremely challenging as the binding site is highly dynamic. Our designs showcased the successful application of molecular recognitions in drug development against conventionally undruggable targets. Also, the understanding of TLR8 signaling has lagged behind due to the lack of potent and specific chemical probes. The identification of CU-CPT9 derivatives as TLR8-specific antagonists with exceptionally high potency and selectivity provided useful tools in studying TLR8 signaling. Moreover, the drug-like CU-CPT9 derivatives demonstrated promising activities and toxicities in various cell lines and primary cells, overcoming a significant hurdle to develop small-molecule drug candidates for endosomal TLRs and laying the groundwork for future drug development.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal anti-NF-κB p65 | Cell Signaling Technology |
Cat#8242; RRID: AB_10859369 |
| peroxidase-conjugated AffiniPure Goat Anti-Rabbit IgG (H+L) |
Jackson Immuno Research |
Cat#111–035-144; RRID: AB_2307391 |
| Lamin A/C Antibody | Cell Signaling Technology |
Cat#2032; RRID: AB_2307391 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| For detailed usage of chemicals, please see supplementary information. |
||
| SEAP reporter assay | Invivogen | Cat#rep-qb1 |
| WST-1 cell proliferation assay | Roche | Cat#5015944001 |
| Deposited Data | ||
| TLR8/CU-CPT9a structure | This paper | PDB: 5Z14 |
| TLR8/CU-CPT9b structure | Zhang, et al., 2018 | PDB: 5WYZ |
| TLR8/CU-CPT9c structure | This paper | PDB: 5Z15 |
| TLR8/CU-CPT8m structure | Zhang, et al., 2018 | PDB: 5WYX |
| Experimental Models: Cell Lines | ||
| Human: HEK-Blue-hTLR2 cells | Invovogen | Cat#hkb-htlr2 |
| Human: HEK-Blue-hTLR3 cells | This paper | N/A |
| Human: HEK-Blue-hTLR4 cells | Invovogen | Cat#hkb-htlr4 |
| Human: HEK-Blue-hTLR5 cells | This paper | N/A |
| Human: HEK-Blue-hTLR7 cells | Invovogen | Cat#hkb-htlr7 |
| Human: HEK-Blue-hTLR8 cells | This paper | N/A |
| Human: HEK-Blue-hTLR9 cells | Invovogen | Cat#hkb-htlr9 |
| Human: THP-1 | ATCC | Cat#TIB-202 |
| Experimental Models: Organisms | ||
| Splenocytes from hTLR8tg/TLR7-KO mice | This paper | N/A |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software Inc |
https://www.graphpad.com/ scientific-software/prism/ |
| OriginPro | OriginLab Corporation | https://www.originlab.com/index.aspx?go=Products/Origin |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Hang Hubert Yin (hubert.yin@colorado.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
The human embryonic kidney (HEK)-Blue Null1 (gender: female), HEK-Blue cells (gender: female) with overexpressing of TLR2, TLR4, TLR7, and TLR9 were purchased from Invivogen (California, USA). HEK-Blue cells overexpressing TLR3, TLR5 and TLR8 were generated from HEK-Blue Null1 cells by lentiviral infection. HEK-Blue cells were grown in humidified incubators containing 5% CO2 at 37 °C. HEK-Blue TLR cells were cultured in DMEM media supplemented with 10% fetal bovine serum (FBS), 50U/mL penicillin and 50 μg/mL streptomycin (PenStrep), 100 mg/mL normocin, 5 μg/mL blasticidin and 2 mM L-glutamine. THP-1 cells (gender: male) were purchased from ATCC (Virginia, USA). THP-1 cells were grown in humidified incubators containing 5% CO2 at 37 °C. THP-1 cells were cultured in RPMI 1640 medium supplemented with 10% fetal FBS, 100 U/mL penicillin, 100 μg/mL streptomycin (PenStrep), 2 mM L-glutamine, and 0.05 mM 2-mercaptoethanol. HEK-Blue TLR cells and THP-1 cells are used without further authentication. Cells were checked periodically and were found to be free of mycoplasma contamination. Spleens were isolated from TLR7 knock-out mice (gender: female) harboring a human TLR8 transgene. PBMC cells were collected from anonymous donors.
METHOD DETAILS
SEAP reporter assay
HEK-Blue TLR8 cells were seeded in tissue culture treated 96-well plates with a density of 7.5 × 104 cells/well in DMEM media supplemented with 10% FBS (phosphatases deactivated with heat). These cells were then treated with R848 (1 μg/mL) along with various concentrations of appropriate compounds. Plates are then incubated in humidified incubators containing 5% CO2 at 37 °C for 24 h. After which, 30 μL of culture media was transfer to a new plate and 100 μL of Quanti-Blue (Invivogen, California, USA) was added, the plate was then incubated at 37 °C till color change (30 min-1 h). Readout of absorbance at 620 nm was quantified using Beckman- Coulter DTX 880 Multimode Detector. Readout of only R848 treated cells were normalized as 100% activation, and untreated cells as 0% activation. SEAP assays for each sample were conducted with three biological replicates, each in triplicate.
WST-1 cell proliferation assay
HEK-Blue cells overexpressing TLRs were treated as described in SEAP assay. After incubation of 24 h, supernatant was removed, and 100 µL of 1:10 diluted WST-1 (Roche) was then added to each well. The plate was then incubated at 37 °C till color change (30 min-1 h). Readout of absorbance at 450 nm was quantified using Beckman-Coulter DTX 880 Multimode Detector. Readouts of untreated cells were normalized as 100% survival, and 20% DMSO treated cells as 0% survival.
TLR selectivity assay
The selectivity of sample compound was tested using SEAP reporter assay in HEK-Blue overexpressing various TLRs. Plates were set up in same manner as described in SEAP assay above. Instead of R848, for HEK-Blue hTLR1/2, hTLR2/6, hTLR3, hTLR4, hTLR5, hTLR7, and hTLR9 cells, Pam3CSK4 (100 ng/mL), Pam2CSK4 (100 ng/mL), poly(I:C) (5 µg/mL), LPS (lipopolysaccharide) (20 ng/mL), flagellin (50 ng/mL), R848 (1 µg/mL), ODN2006 (0.15 µM) were used as agonists respectively.
Protein expression, purification and crystallization
The extracellular domain of human Toll-like receptor 8 (hTLR8, residues 27–827) was prepared as described previously (Tanji, et al., 2013) and was concentrated to 16 mg/mL in 10 mM Tris- HCl pH 8.0 and 150 mM NaCl. The protein solutions for the co-crystallization of hTLR8 and inhibitors contained hTLR8 (7.0 mg/mL) and a five-fold excess of inhibitors in a crystallization buffer containing 10 mM Tris-HCl pH 8.0, 150 mM NaCl, and 5% dimethyl sulfoxide (DMSO). Crystallization experiments were performed with sitting-drop vapor-diffusion methods at 293 K. Crystals of hTLR8/CU-CPT were obtained with reservoir solutions containing 12.5 – 13.0% PEG 4000, 0.2 M calcium chloride, 0.1 M Tris-HCl pH 8.0 – 8.3, and 20 – 25% ethylene glycol.
Data collection and structure determination
Diffraction dataset was collected on beamline, PF BL-5A (Ibaraki, Japan), and SPring-8 BL41XU (Hyogo, Japan) under cryogenic condition at 100 K. The wavelength was set to 1.0000 Â. The dataset was processed using the HKL2000 package until the R factor was converged. CU-CPT compounds, N-glycans, and water molecules were modeled into the electron density maps at the latter cycles of the refinement. The quality of the final structure was validated with the PDB validation server (http://wwpdb-validation.wwpdb.org/). The favored and the allowed regions in the Ramachandran plot were 88 % and 11 % for TLR8/CU-CPT9a, and 89 % and 10 % for TLR8/CU-CPT9c. The figures representing structures were prepared with PyMOL (http://www.pymol.org). Coordinates and structure factor have been deposited in the Protein Data Bank with PDB as 5Z14 (TLR8/CU-CPT9a), 5WYZ (TLR8/CU-CPT9b), and 5Z15 (TLR8/CU-CPT9c).
Isothermal titration calorimetry (ITC)
ITC experiments were done in a buffer composed of 25 mM MES pH 5.5, 0.20 M NaCl, and 2.5% DMSO at 298 K using a MicroCal iTC200 (GE Healthcare, Illinois, USA). The titration sequence included a single 0.4 μL injection followed by 18 injections, 2 μL each, with a spacing of 120 seconds between the injections. The titration conditions were as follows: 100 μM inhibitors into 10 μM hTLR8; 100 μM R848 into 10 μM hTLR8/50 μM inhibitors. OrigineLab software (GE Healthcare, Illinois, USA) was used to analyze the raw ITC data.
Immunoblotting
THP-1 cells were seeded in 6-well plates with a density of 2 × 106 cells/well in RPMI 1640 medium supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin (PenStrep), 2 mM L-glutamine, and 0.05 mM 2-mercaptoethanol. THP-1 cells were treated with phorbol-12- myristate-13-acetate (PMA) (100 ng/mL) and incubated in humidified incubators containing 5% CO2 at 37 °C for 24 h. After differentiation, the supernatant was then removed and replaced with unsupplemented RPMI, these cells were then treated with R848 (1 μg/mL) along with various concentrations of appropriate compounds. After incubation of 2 h, THP-1 cells were collected, nuclear protein fraction was extracted using NE-PER Nuclear and Cytoplasmic Extraction kit (Thermo Fisher Scientific, Massachusetts, USA). BCA assay was then used to determine protein concentration. Protein samples were loaded and run in 10% Tris-glycine SDS-PAGE, and then transferred onto a nitro-cellulose membrane (BioRad, California, USA) using electroblotting. P65 (CST; 8242) (1:1000) was then used as primary antibody, and peroxidase-conjugated AffiniPure Goat Anti-Rabbit IgG (H+L) antibody (Jackson Immuno Research; 111–035-144) (1:10000) as secondary antibody, then blots were visualized using Thermo SuperSignal West Pico kit (Thermo Fisher Scientific, Massachusetts, USA). Lamin A/C (CST; 2032) were used as internal controls for nuclear fractions.
Activity on primary human lymphocytes
Buffy coats from human donors were obtained from the Stanford Blood Center and lymphocytes isolated using Ficoll-Paque. For assessing antagonist activity against TLR2, 4, and 5 the entire PBMC population was used. For assessing antagonist activity against TLR7, 8 and 9 specific lymphocyte subsets were isolated using the QuadroMACS Separator System (Miltenyi) and CD19 or CD14 microbeads to isolate B cells (for TLR7 and 9) and monocytes (for TLR8), respectively. Cells were incubated with a dilution series of compounds and a constant amount of agonist. The following agonists were used for each TLR: Pam2CSK4 (0.35mg/mL) for TLR2, lipopolysaccharide (0.35mg/mL) for TLR4, flagellin (0.75mg/mL) for TLR5, R848 (1mg/mL) for TLR7, RNA oligonucleotide ORN8L (100mg/mL) for TLR8, and CpG-containing phosphorothioate DNA oligonucleotide 1018 (1 mM) for TLR9. After 24 to 48 hours, culture supernatants were collected and analyzed for cytokine production indicative of TLR activity (IL- 6 from PBMCs and B cells, TNF-alpha from monocytes). Each activity curve was an average from three individual donors.
Activity on primary mouse splenocytes
To assess TLR8 and TLR9 activity, spleens were isolated from TLR7 knock-out mice harboring a human TLR8 transgene (hTLR8tg/TLR7-KO). Splenocytes were prepared by dissociation through a cell strainer and removal of red blood cells with lysis buffer. Splenocytes were incubated with a dilution series of compounds and a constant amount of a TLR8 agonist (RNA oligonucleotide ORN8L) or a TLR9 agonist (CpG-containing phosphorothioate DNA oligonucleotide 1018). After 48 hours, culture supernatants were collected and analyzed for IL- 12p40 production. Curves represent activity from a single spleen.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical differences were performed using one-way ANOVA with Bonferroni post-test for multiple comparisons. All statistical analyses were performed using GraphPad Prism, version 6.0 for Mac or 7.0 for Windows and OriginPro, a P value of <0.05 was considered statistically significant.
DATA AND SOFTWARE AVAILABILITY
The final atomic coordinates and experimental structure factors were deposited in the Protein Data Bank with accession codes 5Z14 and 5Z15 for TLR8/CU-CPT9a complex, and TLR8/CU-CPT9c complex structures, respectively. All other data supporting the findings of this study are available within the paper and its supplementary information files.
Supplementary Material
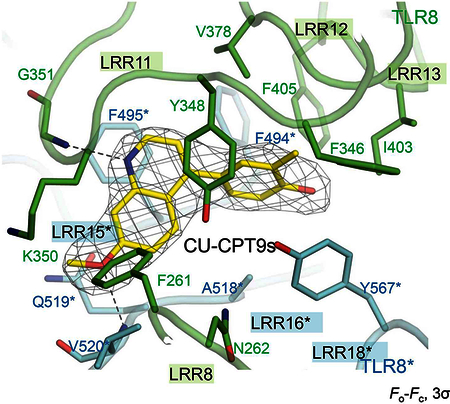
Binding pocket of TLR8 with CU-CPT8m. Related to Figure 1.
Highlight:
1) CU-CPTs successfully target an unconventional pocket on a protein-protein interface.
2) Highly potent and selective CU-CPT provided useful tools in studying TLR8 signaling
3) CU-CPT9 derivatives demonstrated promising therapeutic potentials.
In Brief.
Structure-based rational design led discovery of six exceptionally potent compounds with ~pM IC50 values. Two new X-ray crystallographic structures validated the contacts within the binding pocket. TLR8-transgenic mice further demonstrated these TLR8 inhibitors’ high efficacy, suggesting strong therapeutic potential against autoimmune disorders.
ACKNOWLEDGEMENTS
This work was funded by the National Natural Science Foundation of China (no. 21572114), the National Key R&D Program of China (no. 2017YFA0505200), NIH (R35GM127003), Japanese Ministry of Education, Culture, Sports, Science, and Technology (H.T. and T.S.), CREST JST (T.S.), the Takeda Science Foundation (T.S.), and the Naito Foundation (T.S.). We thank Y. Yamada and A. Shinoda for automated data collection at Photon Factory.
Footnotes
AUTHOR CONTRIBUTIONS
H.Y. designed the project and supervised data analysis. Z.H. performed chemical synthesis, cell culture and cellular inhibition studies. Z.H., S.Z. and S.J. performed immunoblotting experiments. H.T., K.S., U.O. and T.S. expressed protein, solved the crystal structure, performed ITC and gel filtration experiments. J.C. and A.C. performed PBMC and transgenic mice studies. H.Y. and Z.H. wrote the manuscript, all authors edited the manuscript.
DECLARATION OF INTERESTS
H.Y. and Z.H. have filed a patent application based on the technology in this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alexopoulou L, Holt AC, Medzhitov R, and Flavell RA (2001). Recognition of double stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413, 732–738. [DOI] [PubMed] [Google Scholar]
- Anders HJ, Zecher D, Pawar RD, and Patole PS (2005). Molecular mechanisms of autoimmunity triggered by microbial infection. Arthritis Res. Ther. 7, 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battye TG, Kontogiannis L, Johnson O, Powell HR & Leslie AG (2011). iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D. Biol. Crystallogr 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes JL, Weinerman B, Basole C, and Salazar JC (2012). TLR8: the forgotten relative revindicated. Cell Mol. Immunol. 9, 434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold SS, Kaisho T, Hemmi H, Akira S, and Reis e Sousa C (2004). Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303, 1529–1531. [DOI] [PubMed] [Google Scholar]
- Elshabrawy HA, Essani AE, Szekanecz Z, Fox DA, and Shahrara S (2017). TLRs, future potential therapeutic targets for RA. Autoimmun. Rev. 16, 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P & Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Farrugia M, and Baron B (2017). The Role of Toll-Like Receptors in Autoimmune Diseases through Failure of the Self-Recognition Mechanism. Int. J. Inflam. 2017, 8391230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein GS (2003). Evolving concepts of rheumatoid arthritis. Nature 423, 356–361. [DOI] [PubMed] [Google Scholar]
- Friedberg JW, Kim H, McCauley M, Hessel EM, Sims P, Fisher DC, Nadler LM, Coffman RL, and Freedman AS (2005). Combination immunotherapy with a CpG oligonucleotide (1018 ISS) and rituximab in patients with non-Hodgkin lymphoma: increased interferon-alpha/beta-inducible gene expression, without significant toxicity. Blood 105, 489–495. [DOI] [PubMed] [Google Scholar]
- Guiducci C, Gong M, Cepika AM, Xu Z, Tripodo C, Bennett L, Crain C, Quartier P, Cush JJ, Pascual V, et al. (2013). RNA recognition by human TLR8 can lead to autoimmune inflammation. J. Exp. Med. 210, 2903–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, and Aderem A (2001). The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410, 1099–1103. [DOI] [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, and Bauer S (2004). Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303, 1526–1529. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, et al. (2000). A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745. [DOI] [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, and Akira S (1999). Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J. Immunol. 162, 3749–3752. [PubMed] [Google Scholar]
- Hu T, Suter SR, Mumbleau MM, and Beal PA (2018). TLR8 activation and inhibition by guanosine analogs in RNA: Importance of functional groups and chain length. Bioorg. Med. Chem. 26, 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, Lipford G, and Bauer S (2002). Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat. Immunol. 3, 499. [DOI] [PubMed] [Google Scholar]
- Kariko K, Buckstein M, Ni H, and Weissman D (2005). Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175. [DOI] [PubMed] [Google Scholar]
- Krieg AM, and Vollmer J (2007). Toll-like receptors 7, 8, and 9: linking innate immunity to autoimmunity. Immunol. Rev. 220, 251–269. [DOI] [PubMed] [Google Scholar]
- Lan T, Kandimalla ER, Yu D, Bhagat L, Li Y, Wang D, Zhu F, Tang JX, Putta MR, Cong Y, et al. (2007). Stabilized immune modulatory RNA compounds as agonists of Toll-like receptors 7 and 8. Proc. Natl. Acad. Sci. U S A 104, 13750–13755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, et al. (2005). RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J. Exp. Med. 202, 1171–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, and Marshak- Rothstein A (2002). Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 416, 603–607. [DOI] [PubMed] [Google Scholar]
- Mullen L, Ferdjani J, and Sacre S (2015). Simvastatin inhibits TLR8 signaling in primary human monocytes and spontaneous TNF production from rheumatoid synovial membrane cultures. Mol. Med. 21, 726–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA & Dodson EJ (2004). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D. Biol. Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- O’Neill LA, Golenbock D, and Bowie AG (2013). The history of Toll-like receptors - redefining innate immunity. Nat. Rev. Immunol. 13, 453–460. [DOI] [PubMed] [Google Scholar]
- Rimbach K, Kaiser S, Helm M, Dalpke AH, and Eigenbrod T (2015). 2’-O-Methylation within Bacterial RNA Acts as Suppressor of TLR7/TLR8 Activation in Human Innate Immune Cells. J. Innate Immun. 7, 482–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z & Minor W (1997). Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Sacre S, Lo A, Gregory B, Stephens M, Chamberlain G, Stott P, and Brennan F (2016). Oligodeoxynucleotide inhibition of Toll-like receptors 3, 7, 8, and 9 suppresses cytokine production in a human rheumatoid arthritis model. Eur. J. Immunol. 46, 772–781. [DOI] [PubMed] [Google Scholar]
- Sacre SM, Andreakos E, Kiriakidis S, Amjadi P, Lundberg A, Giddins G, Feldmann M, Brennan F, and Foxwell BM (2007). The Toll-like receptor adaptor proteins MyD88 and Mal/TIRAP contribute to the inflammatory and destructive processes in a human model of rheumatoid arthritis. Am. J. Pathol. 170, 518–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacre SM, Lo A, Gregory B, Simmonds RE, Williams L, Feldmann M, Brennan FM, and Foxwell BM (2008). Inhibitors of TLR8 Reduce TNF Production from Human Rheumatoid Synovial Membrane Cultures. J. Immunol. 181, 8002–8009. [DOI] [PubMed] [Google Scholar]
- Siednienko J, and Miggin SM (2009). Expression analysis of the Toll-like receptors in human peripheral blood mononuclear cells. Methods Mol. Biol. 517, 3–14. [DOI] [PubMed] [Google Scholar]
- Tak PP, and Firestein GS (2001). NF-kappaB: a key role in inflammatory diseases. J. Clin. Invest. 107, 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Kawai T, Muhlradt PF, Morr M, Radolf JD, Zychlinsky A, Takeda K, and Akira S (2001). Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 13, 933–940. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, Modlin RL, and Akira S (2002). Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol. 169, 10–14. [DOI] [PubMed] [Google Scholar]
- Tanji H, Ohto U, Shibata T, Miyake K, and Shimizu T (2013). Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science 339, 1426–1429. [DOI] [PubMed] [Google Scholar]
- Tanji H, Ohto U, Shibata T, Taoka M, Yamauchi Y, Isobe T, Miyake K, and Shimizu T (2015). Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat. Struct. Mol. Biol. 22, 109–115. [DOI] [PubMed] [Google Scholar]
- Vagin A & Teplyakov A (2010). Molecular replacement with MOLREP. Acta Crystallogr. D. Biol. Crystallogr 66, 22–25. [DOI] [PubMed] [Google Scholar]
- Yin H, and Flynn AD (2016). Drugging Membrane Protein Interactions. Annu. Rev. Biomed. Eng. 18, 51–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Hu Z, Tanji H, Jiang S, Das N, Li J, Sakaniwa K, Jin J, Bian Y, Ohto U, et al. (2018). Small-molecule inhibition of TLR8 through stabilization of its resting state. Nat. Chem. Biol. 14, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Ohto U, Shibata T, Krayukhina E, Taoka M, Yamauchi Y, Tanji H, Isobe T, Uchiyama S, Miyake K, et al. (2016). Structural Analysis Reveals that Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA. Immunity 45, 737–748. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Binding pocket of TLR8 with CU-CPT8m. Related to Figure 1.