

Summary
We have uncovered the existence of extracellular vesicle (EV)-mediated signaling between cell types within the adipose tissue (AT) proper. This phenomenon became evident in our attempts at generating an adipocyte-specific knock out of caveolin 1 (cav1) protein. While we effectively ablated the CAV1 gene in adipocytes, cav1 protein remained abundant. With the use of newly generated mouse models, we show that neighboring endothelial cells (ECs) transfer cav1-containing EVs to adipocytes in vivo, which reciprocate by releasing EVs to ECs. AT-derived EVs contain proteins and lipids capable of modulating cellular signaling pathways. Furthermore, this mechanism facilitates transfer of plasma constituents from ECs to the adipocyte. The transfer event is physiologically regulated by fasting/refeeding and obesity, suggesting EVs participate in the tissue response to changes in the systemic nutrient state. This work offers new insights into the complex signaling mechanisms that exist between adipocytes, stromal vascular cells and potentially distal organs.
ETOC
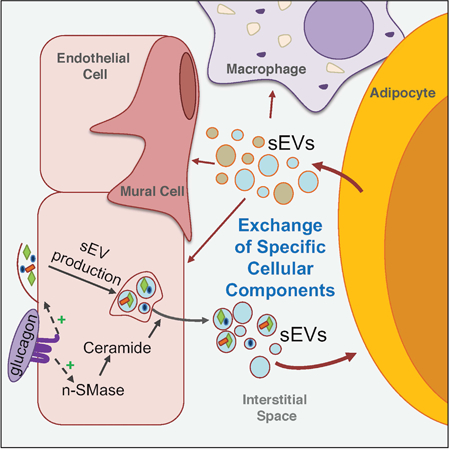
Extracellular vesicles exchange protein and lipid signals between endothelial cells and adipocytes conveying information about nutrient state changes from the blood in adipose tissues
Graphical Abstract
INTRODUCTION
The communication of adipose tissue (AT) with other organs through secreted hormones is indispensable for the maintenance of systemic metabolic health (Crewe et al., 2017; Kusminski et al., 2016). While well established in oncology, studies have only recently recognized the important role that extracellular vesicles (EV) play in organ-to-organ communication of metabolic signals (Thomou et al., 2017; Zhao et al., 2018). Several types of EVs exist, but in the context of signaling, exosomes and microvesicles display marked potential (van Niel et al., 2018). Exosomes generally have the distinguishing features of a nano-size range (30–150 nm), and contain specific biochemical markers, such as the enrichment of tumor susceptibility gene 101 (TSG101) and ALG-2-interacting protein (ALIX) (Lotvall et al., 2014; Willms et al., 2016) and exosomes are of endosomal origin. Microvesicles range in size from 50– 1000nm and originate by budding of the plasma membrane (Kowal et al., 2016). Given the overlap in size and similarity in biochemical properties, it is often difficult to experimentally distinguish exosomes from small microvesicles (Kowal et al., 2016)). Thus, the general term small EV (sEV) is often used to describe this highly heterogenous population of EVs isolated by a 100 000xg spin.
Many cell types secrete protein- and miRNA-rich sEVs that have intricate effects on recipient cells. sEVs produced by adipocytes induce an aggressive metabolic phenotype in melanoma cells, promote metastasis of lung cancer cells and alter TGF-β signaling in hepatocytes (Conlan et al., 2017; Koeck et al., 2014; Lazar et al., 2016). sEVs secreted from other cells in AT, such as macrophages or stem cells, can also have striking systemic effects when injected into mice, such as altering whole body insulin sensitivity, glucose tolerance, and inflammation, which provides a new therapeutic avenue for the treatment of obesity and type 2 diabetes (Deng et al., 2009; Ying et al., 2017; Zhao et al., 2018). Although sEVs can potentially be engineered to provide a pharmacological payload, it is surprising that the most promising therapeutic application is through harnessing the characteristics of native EVs, complete with the cell-prepared cocktail of proteins, lipids, miRNAs and cell-targeting mechanisms (Conlan et al., 2017).
These studies have laid an important foundation for our continued understanding of how cell-specific sEVs modulate whole body metabolism. However, it is not clear if tissue-derived EVs also participate in cell-to-cell communication within the tissue proper. In tumor microenvironment studies, EVs have emerged as an essential signaling mechanism between tumor cells and stromal cells to modulate proliferation, survival and immunity (Lazar et al., 2016; Zhao et al., 2016). We provide evidence in this study that, in the context of metabolic regulation, sEVs mediate a form of cell-to-cell communication within AT, contributing to the multifaceted crosstalk between adipocytes and stromal vascular cells.
Here, we report the presence of a pool of EVs that reside in white AT and allow for a sizeable exchange of cellular material between cell types. This phenomenon became apparent in our adipocyte-specific caveolin 1 (cav1) knockout mouse model. Cav1 is an important membrane-bound structural and signaling protein and is highly abundant in adipocytes and endothelial cells (ECs). Despite successful ablation of the cav1 gene, the cav1 protein remained readily detectable in adipocytes. This was found to be the result of sEV-mediated trafficking of cav1 from neighboring ECs to the adipocytes. We used cav1 as a marker for AT EVs and developed a novel mouse model of cell-specific miniSOG-tagged cav1 overexpression. This model, combined with our cell-specific membrane-halo mouse, allowed us for the first time to track the movement of EVs between cell types in vivo. These AT sEVs are rich in protein and sphingolipids conferring a range of signaling capabilities. We show here that AT sEV production is increased in the fasted state, partially due to glucagon-stimulated sEV secretion from ECs. This response is absent in ECs isolated from mouse models of genetic and diet-induced obesity. Furthermore, we provide evidence that extracellular molecules are taken up by ECs and packaged into sEVs, marking a new mechanism by which blood-borne signals are integrated and supplied to AT. Overall, this EV-mediated signaling mechanism may be important for the appropriate response of AT to changes in systemic energy balance.
RESULTS
Adipocyte-Specific Elimination of Cav1 in vivo
The cav1 gene was specifically ablated in mature adipocytes using the loxP system combined with either the constitutively expressed adiponectin-cre (aCre) or an adipocyte- specific TET-ON inducible cre (iCre) transgene (Fig. S1A and B). Gene ablation was confirmed in in vitro differentiated adipocytes expressing aCre by PCR amplification of the recombined allele (Fig. 1A). In addition, immunofluorescent staining for cav1 showed depletion of cav1 protein in perilipin (PLIN)-positive differentiated adipocytes expressing aCre, although cav1 was still detected in the surrounding stromal vascular cells (SVF; Fig. 1B). A dramatic reduction in cav1 mRNA was observed in mature adipocytes directly isolated and analyzed from the sWAT of mice expressing either cre system (Fig.1C and D). However, in vivo, cav1 protein remained readily detectable in adipocytes isolated from sWAT in the inducible model, being reduced by just 40 % (Fig. 1E and G). Even in the model of constitutive cre expression, where cav1 protein half-life is essentially irrelevant as the cre is active from birth, we observed only a 50–60% reduction in cav1 protein (Fig. 1F and G). This is in contrast to our previously published work where we successfully depleted cav1 protein in hepatocytes using the same floxed cav1 mouse (Asterholm et al., 2012). This data raises the intriguing possibility that the cav1 protein detected in these KO adipocytes was not endogenously produced, but instead originated from a source outside of the adipocyte. Intracellular localization of cav1 provides further evidence. Cav1 protein in aCre-expressing mice was still enriched at the plasma membrane in sWAT adipocytes, although at lower abundance than the control mice (Fig. 1H). The greatest fold reduction in cav1 content was found in the microsomal fraction corresponding to the processing of nascent cav1 peptides in the ER and the vesicular trafficking of cav1 between compartments (Fig. 1H). Although cav1 was abundantly detected at the plasma membrane in aCre mice, characteristic flask-shaped caveolae were not formed (Fig. 1I). These data suggest that, if there is an exogenous source of cav1 protein, it does not localize and retain its function in the adipocyte as the endogenously produced protein.
Figure 1: Efficient Depletion of Adipocyte Cav1 mRNA Results in a Partial Knockout of Cav1 Protein.
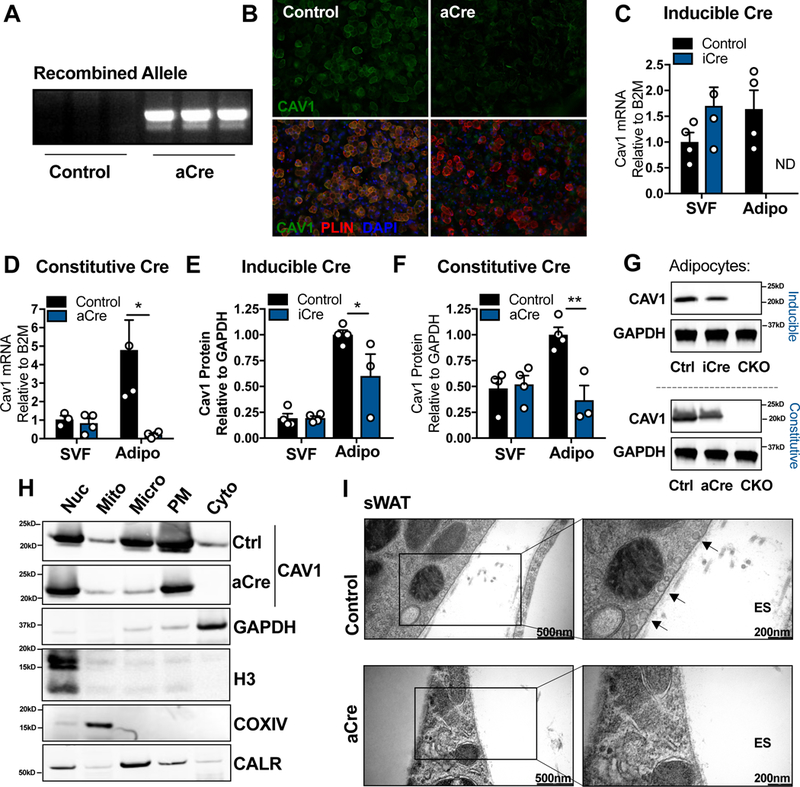
SVF cells were isolated from sWAT of control or adiponectin promoter-cre (aCre)-expressing mice (A–B; n = 3). SVF cells were differentiated and (A) analyzed for cre-driven recombination and (B) stained for cav1 and the adipocyte marker, perilipin (PLIN). Adipocyte and SVF fractions from the sWAT of control (Ctrl), adipocyte-specific constitutively active cre (aCre), adipocyte-specific inducible cre (iCre) or whole body cav1 knockout (CKO) mice were analyzed for cav1 mRNA (C–D; n = 3–4 mice per group; “ND” is “Not Detected”) or cav1 protein (E–G; n = 3–4 mice per group) in 15 wk old mice for aCre and 3 wk doxycycline treatment for iCre mice. (H) Mature adipocytes were isolated from the sWAT of control or aCre-expressing mice and fractionated into subcellular fractions: nucleus (Nuc), mitochondria (Mito), Microsomes (Micro), plasma membrane (PM) and cytoplasm (Cyto). Control and aCre cav1 samples were run on the same gel but the images were separated for easy comparison. (I) Electron micrograph of control or aCre-expressing sWAT (representative of 2 mice per group). Arrows indicate examples of caveolar structures. Data is presented as mean ± SEM
Cav1 is Trafficked from Endothelial Cells to Adipocytes in vitro via EVs.
To determine if different cell types within the sWAT depot share membrane components, we transplanted sWAT pieces from mice in which all cells were constitutively labeled with membrane-bound red fluorescent protein (RFP) into the dorsal subcutaneous fat pad of mice where only mature adipocytes were labeled with plasma membrane halo-tag (Fig. 2A and Fig. S2). The transplanted sWAT pieces were allowed to integrate into the host fat pad for 3 wk. Both RFP and halo signals were detected in the same adipocytes at the site of transplantation, suggesting that adipocytes exchange membrane components with other cell types in the tissue (Fig. 2B).
Figure 2: Endothelial Cell-Derived Cav1 is Transferred to Adipocytes in vitro.
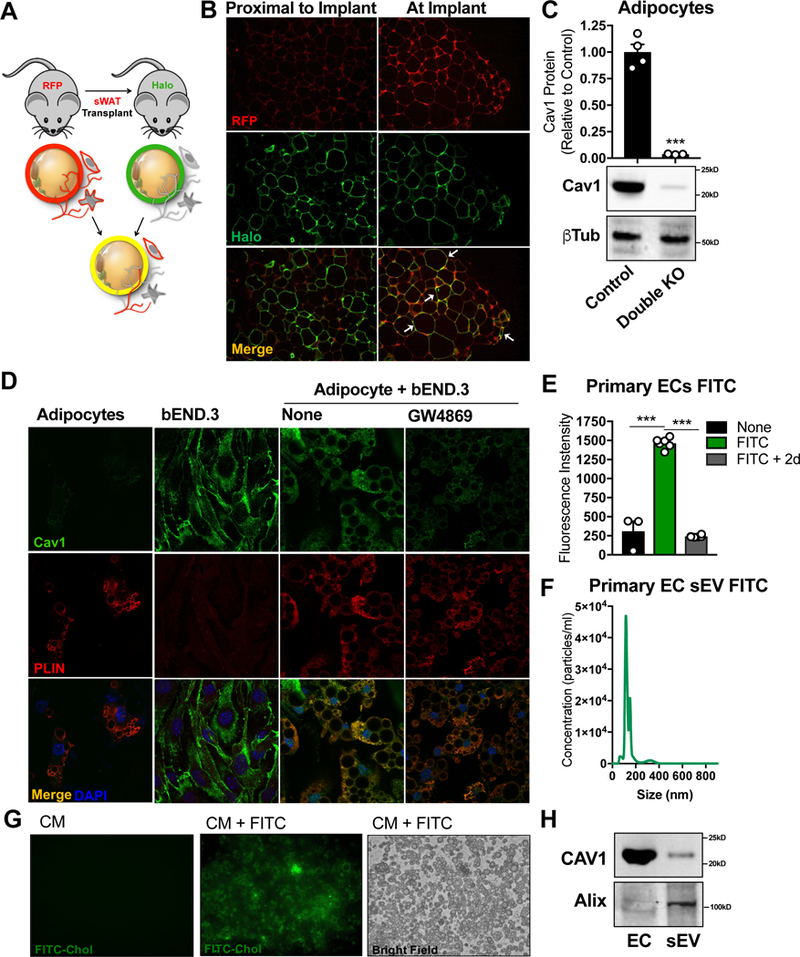
(A) Transplant schematic: pieces of sWAT from an MTMG mouse (where every cell is labeled with RFP) were implanted in the dorsal sWAT fat pad of a mouse expressing an adipocyte- specific halo tag at the plasma membrane. (B) Confocal images of RFP and Halo in tissue sections (n = 3 independent transplants). Arrows indicate examples of RFP and halo colocalization. (C) Western blot densitometry and representative image of cav1 expression in isolated mature adipocytes from the sWAT of control or adipocyte and EC double cav1 KO mice (n = 3–4). (D) sWAT-derived SVF from whole body cav1 KO mice was differentiated into adipocytes and co-cultured with bEND.3 ECs for 2 d ± GW4869 (representative immunofluorescent confocal image of n = 3). (E–H) Primary CD31+ EC were labeled with FITC- PEG-Cholesterol to tag cell membranes (E; “FITC”), washed thoroughly and given fresh media for 2 d (E; “FITC + 2d”). (F) Appearance of FITC-labeled exosomal particles in the EC conditioned media following 2 d incubation. (G) WT differentiated adipocytes were treated with conditioned media from ECs alone (CM) with or ECs pre-treated with FITC (CM + FITC). (H) sEVs were isolated from EC conditioned media and analyzed by Western blot for the presence of cav1 and the exosomal marker Alix, compared to whole EC lysate. All cell culture experiments are representative of 3 independent experiments. Data is presented as mean ± SEM
Previous studies have shown that cav1 is secreted by select cell types and has been identified in the interstitial fluid of AT (Celis et al., 2005; Chang et al., 2017), establishing inter- cellular trafficking of cav1 as a viable theory. However, exchange of cav1 between cells has not been directly demonstrated. Endothelial cells (ECs) are the most likely candidate for a potential source of external cav1 transferred to adipocytes, as ECs have a pronounced presence in AT and highly express cav1 protein (Lisanti et al., 1994). In support of this hypothesis, co-deletion of cav1 from adipocytes and ECs in vivo using a combination of adiponectin-cre and VECAD- cre respectively, resulted in efficient knockout of cav1 protein in isolated adipocytes from the sWAT depot (Fig. 2C and Fig. S1 A and C). We pursued further mechanistic evidence for this phenomenon of EC to adipocyte trafficking in vitro. The SVF fraction from the sWAT of whole body cav1 KO mice was differentiated into mature adipocytes and co-cultured with immortalized mouse ECs (bEnd.3), which have wildtype levels of cav1 protein. The cells were fixed and stained for cav1 and the adipocyte marker PLIN. Cav1 signal was robustly detected in the adipocytes, suggesting that cav1 can be trafficked from ECs to adipocytes in vitro (Fig. 2D). This likely occurs through a vesicular mechanism, as cav1 is a membrane-bound protein and most adipocytes in the co-culture are not in direct physical contact with an EC. WT ECs were co-cultured with cav1 KO adipocytes in the presence of GW4869, an inhibitor of neutral sphingomyelinase (n-SMase) and commonly used to suppress exosome biogenesis. Despite its specificity, it may also inhibit the secretion of other types of EVs. GW4869 treatment resulted in diminished adipocyte cav1 signal (Fig. 2D). This suggests cav1 can be transferred by an EV-based mechanism. Adipocytes also readily take up sEVs produced from primary AT ECs. Primary CD31+ ECs were treated with FITC-PEG-cholesterol to label the plasma membrane. ECs were then washed thoroughly to remove excess dye (Fig. S3) and given fresh media for 2 days. Over this period, the ECs became completely depleted of FITC signal (Fig. 2E). This corresponded with the appearance of FITC-labeled exosome-sized vesicles in the EC media as determined by nanoparticle tracking analysis (NTA; Fig. 2F). Furthermore, isolated sEVs from EC-conditioned media (CM) contained cav1 protein (Fig. 2H). This CM was then transferred to differentiated WT adipocytes and incubated under normal culture conditions for 6 hrs. Adipocytes robustly took up labeled EC sEVs resulting in a dramatic increase in adipocyte FITC signal (Fig. 2G). These results, taken together, suggest that ECs secrete cav1-containing EVs that are taken up by neighboring adipocytes in culture. Although we focused on EC sEVs, cav1 could also be trafficked on other forms of EVs, such as larger microvesicles, a process we cannot formally exclude (Fig. S6).
Cav1 is Trafficked from Endothelial Cells to Adipocytes in vivo
To determine if this trafficking event occurs in vivo, we generated a mouse that allows us to over-express a mini-SOG-tagged cav1 fusion protein (cav1SOG; mini Singlet Oxygen Generator) under the control of the TRE promoter in a TET-OFF system (Fig. S1C). Using a VECAD-tTA transgene, we accomplished constitutive overexpression of cav1SOG specifically in ECs in vivo. In this way, we could track the movement of cav1 produced in ECs within the AT. We validated the newly generated anti-miniSOG antibody via Western blot and IF by demonstrating that cav1SOG was only detected in the mice carrying the TRE-cav1SOG transgene (Fig. 3A and B). In addition, the miniSOG signal was identified in vascular structures (Fig. 3A) and the EC-containing SVF of sWAT (Fig. 3B). Remarkably, cav1SOG was also readily detected in the purified adipocyte fraction (Fig. 3B). This data indicates that cav1SOG is transferred from ECs to adipocytes in vivo.
Figure 3: Cav1 is Trafficked from Endothelial Cells to Adipocytes in vivo.
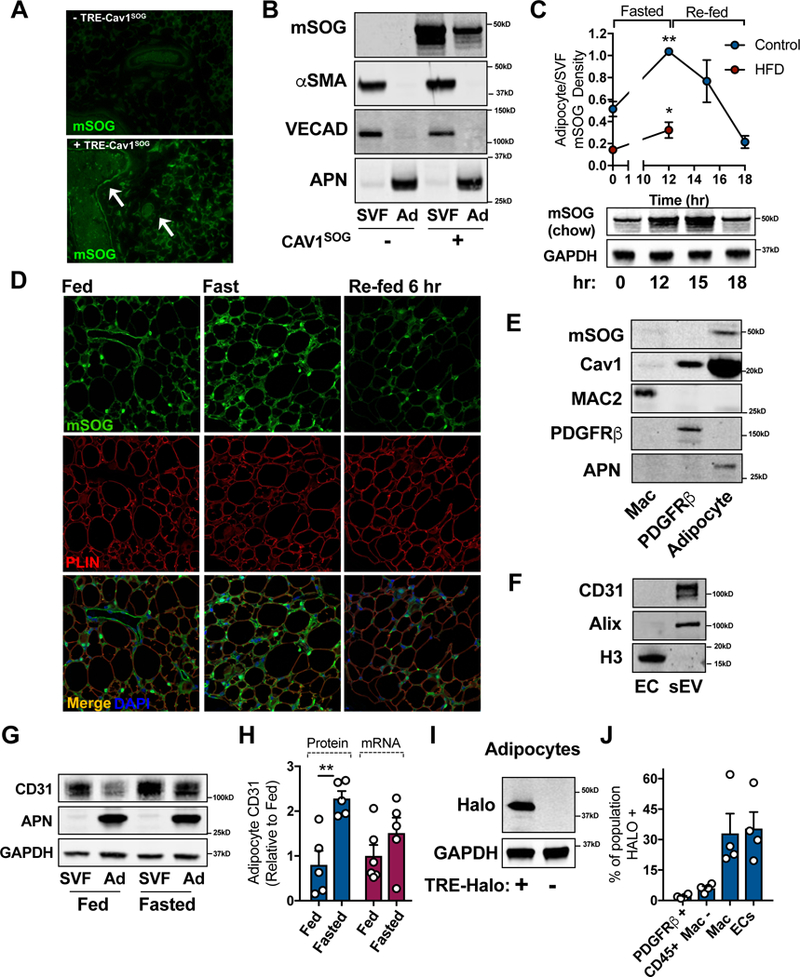
(A) Immunohistochemical analysis of the mini-SOG (mSOG)-tagged cav1 protein. Arrows indicate examples of positively stained vascular structures. (B) Western blot analysis of miniSOG in purified SVF and adipocyte (Ad) fractions from sWAT. (C) Western blot densitometry of the miniSOG tag in purified adipocyte fraction from the sWAT of mice with the specified diet relative. Mice were fed a high fat diet (HFD) for 12 wk. Data is presented as the ratio of adipocyte miniSOG/SVF miniSOG (n = 4). Western blot image is representative of adipocyte mSOG signal over time. (D) Immunohistochemistry for miniSOG and PLIN as detected by confocal microscopy (n = 4). (E) The presence of cav1SOG in FACS-sorted macrophages (MAC2), preadipocytes (PDGFRβ) and isolated adipocytes (APN) from sWAT. (F) Western blot for CD31 in EC lysate and EC-derived sEVs. (G and H) CD31 protein levels in SVF and Adipocyte (Ad) fractions purified under fed or fasting conditions. (H) Densitometry of CD31 protein levels and mRNA content in isolated adipocytes (n = 5–6). (I) Validation of adipocyte-specific membrane-halo mouse by Western blot expression of the Halo tag in isolated mature adipocytes. (J) Flow Cytometric analysis of Halo tag in sWAT cell populations: preadipocytes (PDGFRβ), hematopoietic cells excluding macrophages (CD45), macrophages (Mac) and endothelial cells (EC; n = 4). Data is presented as mean ± SEM
Cav1 Transfer Between Endothelial Cells and Adipocytes is Regulated by Fasting and Feeding
To establish if the transfer of cav1 to adipocytes is physiologically relevant, we monitored the behavior of this exchange under different physiological conditions. A major function of AT is to maintain systemic metabolic homeostasis despite dramatic changes in exogenous nutrient availability. Upon expressing cav1SOG in ECs, we found that the amount of cav1SOG that is trafficked from ECs to adipocytes is substantially increased in the fasted state and progressively returns to baseline levels when mice are re-fed 3 and 6 hrs (Fig. 3C). We can also demonstrate this by IF where, in the fasted state, considerably more cav1SOG was identified in the extracellular space and at the level of the adipocyte than in the fed state (Fig. 3D). Upon re-feeding for 6 hr, the amount of cav1SOG detected in the interstitium and the adipocyte was similar to that observed in the fed state. In contrast, fasting-induced cav1SOG transfer to adipocytes is severely blunted in mice first fed a high fat diet for 12 wk (Fig. 3C). This data suggests that the extent of cav1 trafficking is dictated by the metabolic requirements of the organism. Furthermore, cav1-containing EVs from ECs are likely targeted largely to adipocytes, compared to other cells in the adipose tissue, as cav1sog was not detected in preadipocytes/mural cells and minimally detected in macrophages (Fig. 3E).
We next determined if cav1 was selectively transferred to adipocytes or if other common plasma membrane proteins were also trafficked, as would be expected with an EV-based mechanism. Proteomics analysis of sEVs isolated from primary EC culture media identified CD31, an EC-enriched plasma membrane protein, as an abundant protein in sEVs compared to others like VE-cadherin. We confirmed the presence of CD31 in EC-derived sEVs by Western blot (Fig. 3F). Interestingly, per µg of protein, EC-derived sEVs were highly enriched for CD31 compared to an EC cell lysate (Fig. 3F). Furthermore, we detected CD31 in purified mature adipocytes from the sWAT and found that CD31 protein accumulated in adipocytes following a 16 hr fast (Fig. 3G and H). This was not due to endogenous expression, as CD31 mRNA was almost undetectable in adipocytes and did not increase fasting (Fig. 3H). Therefore, the trafficking of membrane bound proteins from ECs to adipocytes likely extends far beyond cav1. This “plasma membrane sharing” may explain why is has historically been challenging to find an adipocyte-specific plasma membrane maker.
As illustrated in Fig. 3C, the influx of cav1SOG to the adipocyte is reversed upon re- feeding, indicating there is also a mechanism for the shedding of cav1 and other plasma membrane proteins from the adipocyte. To test this, we used the TRE-plasma membrane-halo mouse and inducibly labeled the adipocyte plasma membranes with a halo tag by treating mice with doxycycline for 2 wk (Fig. S2). As demonstrated in Fig. S2 and Fig. 3I, the halo tag is only expressed in the AT of mice carrying the transgene and specifically labeled adipocyte plasma membranes. Following the labeling period, the presence of the halo tag in other adipose tissue cells was assessed by Flow Cytometry using a florescent halo tag ligand. We found a striking 35% of the total EC population contained the halo tag (Fig. 3J). In addition, other populations of sWAT SVF also contained the halo tag, such as macrophages, CD45+ hematopoietic cells (excluding macrophages) and pdgfrβ+ preadipocytes/mural cells (Fig. 3J). The percentage of the macrophage population that was positive for halo was similar to that of ECs, whereas CD45+ and pdgfrb+ cells contained less than 10% halo positive signal. This data suggests that there is considerable membrane-based communication between the adipocytes, ECs and other SVF cells.
White Adipose Tissue Produces Cav1-containing sEVs
We next pursued evidence for the presence of a tissue EV pool that may transport cav1, and other signaling molecules, between cells in the AT. Immunofluorescent staining of wild type AT revealed areas of the interstitial space that were positive for the exosomal marker CD63, which was not cell-associated (DAPI stain; Fig 4A). Furthermore, mice overexpressing cav1SOG in ECs displayed the presence and co-localization of CD63 and cav1SOG in the intercellular space (Fig. 4A). This data indicates that cav1SOG may be packaged into sEVs in ECs and transferred to neighboring adipocytes.
Figure 4: Validation of Tissue sEV Isolation Protocol.
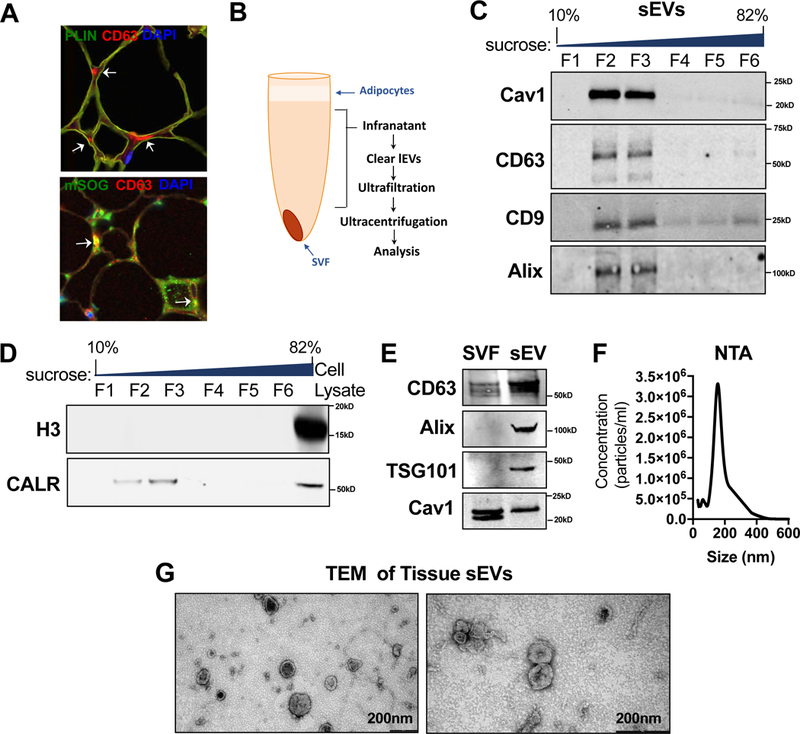
(A) Immunohistochemical staining for the exosomal marker CD63, the adipocyte maker PLIN and miniSOG. Arrows indicate regions of extracellular CD63 staining (red) or CD63-mSOG colocalization (yellow) by confocal imaging. (B) sWAT tissue sEV isolation schematic. (C–D) Western blot of recovered proteins in a sucrose gradient. (E) Western blot detection of cav1 or exosomal makers: CD63, Alix and TSG101 in sWAT SVF or tissue sEV fractions. (F) Nanoparticle tracking analysis (NTA) of purified sWAT tissue sEVs. The experiment was repeated three times. (G) Transmission electron micrograph (TEM) of purified sWAT tissue sEVs.
We developed a protocol to isolate sEV and large EV (lEV) fractions from white AT (Fig. 4B). As illustrated in Fig. 4C, the sEV fraction from sWAT equilibrated in a sucrose gradient at a density range constant with that published for exosomes (1.13–1.19g/ml; F2–3;(Thery et al., 2006), but likely also contains other forms of sEVs. The sEV pellet contained proteins that are characteristic of exosomes, such as tetraspanins and ALIX, which had little to no presence at densities corresponding to endoplasmic reticulum microsomes (F4–5) and Golgi membranes (F1; (Thery et al., 2006) Fig. 4C). Cav1 was strongly detected only within the exosome-containing density range (Fig. 4C). Furthermore, the sEV fraction was devoid of histone 3 and contained calreticulin at lower abundance than an SVF cell lysate (Fig. 4D). These data suggest that the sEV pellet isolated from AT with this protocol contained little to no contamination with cellular debris. The lEV fraction contained a robust signal for cav1 (Fig. S6 A), suggesting this form of EV could participate in inter-cellular transfer of cav1. These EVs likely also contained apoptotic bodies, a type of EV that is rich in calreticulin and histones and floats at a density range of 1.16–1.28 g/ml (F4–5; Fig. S6 B;(Shin et al., 2017; Thery et al., 2009). Here, we focused our efforts mainly on characterizing the properties and signaling capacities of the sEV fraction.
The sEV fraction was enriched for established exosome proteins when compared to a SVF cell lysate, such as CD63, Alix and TSG101 (Fig. 4E). Other common exosomal proteins were identified by an unbiased mass spectrometry approach, such as the Flotillins and CD81 (Table S1). The presence of Cav1 was also identified in the AT sEVs by mass spectrometry (12 peptides, 61.8% coverage) and confirmed by Western blot analysis (Fig. 4E). Interestingly, amongst the two previously described cav1 isoforms (α and β), which arise from differential translational start sites at two distinct methionine residues 32 amino acids apart (Scherer et al., 1995), the α isoform was selectively packaged into sEVs (Fig. 4E) and is the form transferred to adipocytes (Fig. 3B). This further underlines the highly selective enrichment process in tissue EVs. However, as previously mentioned, we do not believe the isoform that is transferred to adipocytes is fully functional with regards to forming caveolar structures (Fig. 1I). By nanoparticle tracking analysis (NTA) and electron microscopy, we show that the isolated sEVs fall within the appropriate size distribution expected of a sEV/exosomal fraction (Fig. 4F and G). These EVs were morphologically heterogeneous, although the majority exhibited a cup-like structure (Fig. 4G).
Tissue sEVs Contain Proteins and Lipids with Signaling Potential
Serum EVs contain an array of proteins, metabolites and RNA species that modulate signaling between various organs. Likewise, identification of a tissue EV population has exciting implications for cell-cell signaling mechanisms within the AT itself. Proteomics analysis was employed to determine differences in sEV protein cargo as a function of fed/fasted conditions. Fig. 5A and Table S2 illustrates the range of signaling pathways in AT sEVs that are enhanced from the fed to the fasted state. For example, proteins involved in Wnt and MAPK signaling were enriched during fasting. Other pathways that were significantly enriched in tissue sEVs during the fasted state included polyamine metabolism, antioxidant response and transport of small molecules (Fig. 5A and Table S2). Interestingly, mitochondrial respiratory chain components were also significantly represented in AT sEV under fasting conditions, a subcellular compartment that is generally considered to be excluded from exosomal cargo (Lotvall et al., 2014). In contrast, pathways that were found to be significantly reduced in tissue sEVs isolated under fasting conditions included metabolism of lipids and metabolism of amino acids (Fig. 5B and Table S3).
Figure 5: Tissue sEVs Contain Various Signaling Proteins and Lipids.
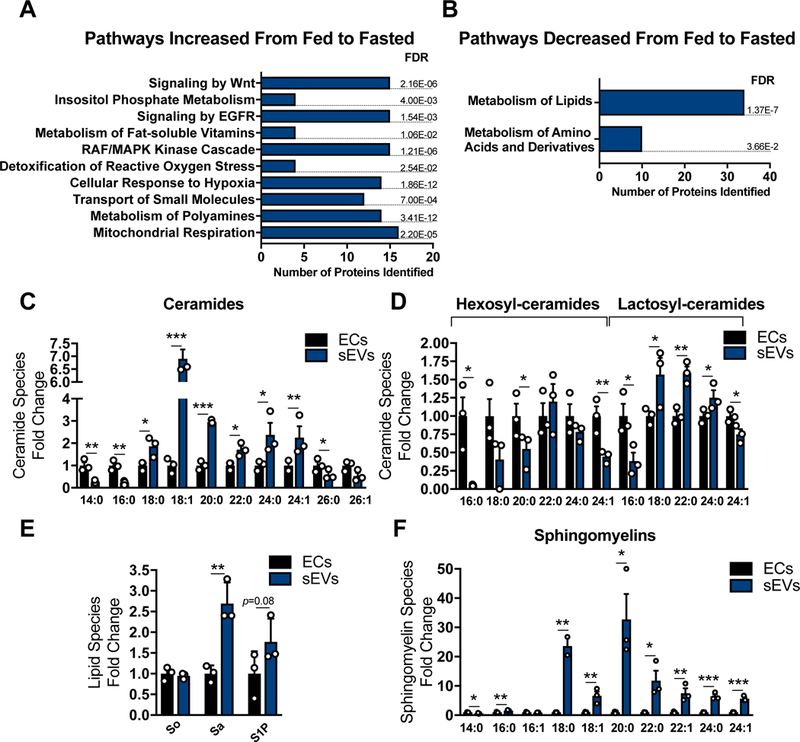
Proteomics pathway analysis of sWAT tissue sEVs isolated from mice under fed or fasting conditions. Pathway analyses was conducted on the subset of proteins identified to be significantly higher (A) or lower (B) in the fasted vs fed states. (C–F) Sphingolipid quantification in AT ECs or AT sEVs (AT EVs were pooled from 5 mice for each condition; n = 3). Abbreviations: So: sphingosine; Sa: sphinganine; S1P: sphingosine 1-phosphate. Data is presented as mean ± SEM
In addition to signaling proteins, we determined if these tissue sEVs contained a specific ceramide profile, as these lipid species have a well-documented spectrum of signaling capacities particularly in the context of stress. The shortest chain length (C14 and C16), and the longest chain length ceramides (C26) were significantly lower in AT sEVs compared to whole EC lysate (Fig. 5C). In contrast, intermediate chain length ceramides (C18, C20, C22 and C24) were highly enriched in sEVs when compared to ECs (Fig. 5C). Furthermore, hexosyl- ceramides were generally lower in tissue sEVs compared to the cell lysates, whereas lactosyl- ceramides were higher in the AT sEVs (Fig. 5D). Lastly, AT sEVs displayed higher levels of sphinganine, sphingosine-1phosphate (S1P) and all sphingomyelin species (Fig. 5E and F). These data indicate that AT sEVs have a unique macromolecular composition with the potential to alter a wide range of signaling pathways within the tissue.
We directly tested whether cav1SOG was transported in sEVs from ECs to adipocytes in vivo. AT sEVs were isolated from mice overexpressing cav1SOG in ECs and were analyzed for the presence of the miniSOG-tag in the fed and fasted states. Cav1SOG was detected in AT sEVs, as was endogenous cav1 (Fig. 6A). Interestingly sEVs from fasting conditions contained less endogenous cav1 but higher phospho-cav1 (Fig. 6A). Thus, phopho-cav1 is enriched by 4.2- fold in sEVs. This suggests that a large fraction of cav1 that enters sEVs is in an active signaling state, particularly under fasting conditions. The overall abundance of endogenous cav1 or cav1SOG in sEVs was not different under the fed relative to the fasted conditions and thus, cannot explain the observation that more cav1 is trafficked to adipocytes in the fasted state. However, the total yield of sEVs from the tissue is significantly increased in the fasted state, suggesting an overall enhancement of sEV production and release (Fig. 6B).
Figure 6: Glucagon Induces EC sEV Secretion.
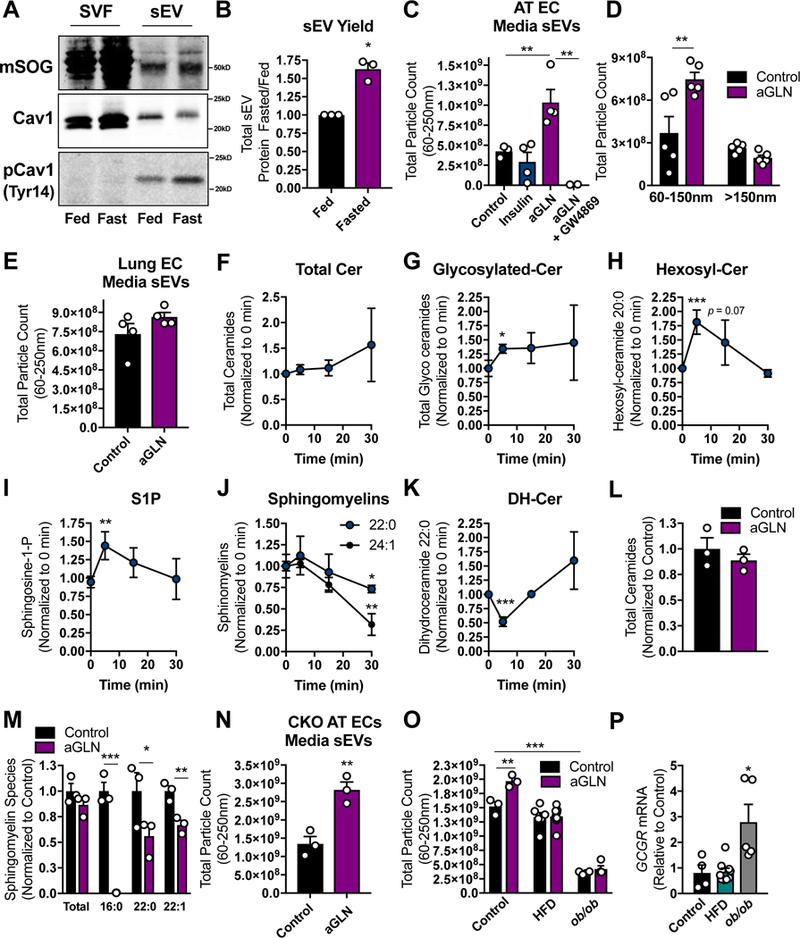
(A) Mouse model of cav1SOG overexpression specifically in ECs. Cav1SOG, endogenous cav1 and endogenous p-cav1Tyr14 were detected by Western blot in SVF and sEV fractions of sWAT tissue (representative of n = 4). (B) AT sEV yield as determined by protein concentration in a fixed volume of re-suspended sEVs. (C–E and N–O) Quantification of media EVs by NTA. The parent cells were ECs isolated from AT (C and D) or Lung (E) treated with insulin (5 µg/ml) or acyl-glucagon (aGLN; 15 nM) as indicated. (F–K) Sphingolipid quantification by mass spectrometry in AT ECs during GLN stimulation (20 nM) at the time-points shown (“cer” was used to abbreviate ceramides). Total ceramide (L) and sphingomyelin (M) content of ECs treated with acyl-glucagon (15 nM) for 2 d. (N) NTA quantification of media EVs from AT ECs isolated from whole body cav1KO mouse sWAT or (O) WT sWAT from mice under the indicated dietary conditions. Mice in the HFD condition were fed for 12 wk before AT EC isolation. (P) Glucagon receptor mRNA expression in culture primary AT ECs isolated from mice fed under the specified conditions. Data is presented as mean ± SEM
Glucagon Enhances AT EC sEV Production Through Ceramide Generation
The proteomics data showed a general trend toward an increase in EC-specific membrane markers identified in the tissue sEVs in the fasted state (data not shown), reflecting a greater contribution of ECs to the tissue sEV pool under fasting conditions. However, AT sEVs likely originate from multiple cell types. To better understand the specific communication between ECs and adipocytes in the fed and fasted states, we cultured primary AT ECs in the presence of either insulin (fed state) or a stabilized form of glucagon (acylated-glucagon; fasted state) for 2 days and quantified the sEVs released into the media. Insulin had no effect on sEV release; however, glucagon treatment resulted in a striking increase sEV production (Fig. 6C). This effect was completely blocked by the addition GW4869 during the incubation (Fig. 6C). We determined that glucagon selectively increased the release of sEV populations and not lEVs from primary ECs (Fig. 6D). Interestingly, glucagon-stimulated sEV release was not observed in primary ECs isolated from lung tissue (Fig. 6E), suggesting that this glucagon-mediated effect is not present in all CD31+ cells. Rather, it is specific for ECs in AT.
It is well established that endosomal-derived exosome production and release is stimulated by n-SMase-dependent ceramide synthesis from sphingomyelin (Essandoh et al., 2015; Li et al., 2013b; Trajkovic et al., 2008). GW4869 is a potent inhibitor of neutral n-SMase. It therefore inhibits exosome production, but may also affect secretion of other forms of sEVs due to the resulting alterations in cellular sphingolipid composition. LC-MS lipidomics analysis revealed that glucagon stimulation of ECs resulted in a trend toward increased total cellular ceramide synthesis (Fig. 6F), but significantly induced the production of glycosylated ceramides (Fig. 6G) within 5 minutes of treatment. Hexosyl-ceramides displayed a particularly robust response to glucagon (Fig. 6H). In addition, the well described signaling sphingolipid species (and product of ceramide degradation) S1P was also induced within 5 min of glucagon stimulation (Fig. 6I). This data indicates that the ceramides produced in response to glucagon are also rapidly turned over. Consistent with a sMAse-specific mechanism of rapid ceramide generation, the level of EC sphingomyelin species decreased over the 30 min time-course of glucagon treatment (Fig. 6J). Interestingly, we also detected a significant drop in dihydroceramide C22:0 at 5 min glucagon treatment (Fig. 6K). This suggests some participation or compensation of the de novo ceramide biosynthetic pathway; however, based on experiments with GW4869, n-sMAse is likely the major pathway for ceramide-induced sEV production in response to glucagon (Fig. 6C). Treatment of ECs with acyl-glucagon for the time frame in which we harvest sEVs (2 d), did not result in high cellular ceramide levels (Fig. 6L), a likely effect of high rates of ceramide turnover, although there was a significant depletion of sphingomyelins (Fig. 6M).
Glucagon-mediated sEV production remained intact in ECs isolated from Cav1KO AT, suggesting Cav1 itself does not participate in this process (Fig. 6N). Furthermore, glucagon did not enhance sEV secretion in ECs isolated from the AT of mice on a high fat diet for 12 wk compared to age-matched mice on a control diet (Fig. 6O). Similarly, ECs from the AT of ob/ob mice, a genetic mouse model of obesity, displayed a severe defect in basal and glucagon- stimulated sEV production (Fig. 6O). These differences in glucagon response could not be attributed to differential expression of the glucagon receptor as ECs from mice on a high fat diet expressed GCGR to the same level as the control cells and those from ob/ob mice displayed higher GCGR expression (Fig. 6P).
Glucagon Signals to Enhance Extracellular Cargo Uptake and Packaging into sEVs
We have shown that glucagon increases sEV production from AT ECs. It is important to elucidate the physiological significance of this event. Proteomics analysis of cultured AT EC sEVs revealed the presence of a number of bovine serum proteins despite extensive washing of the sEV pellet to remove FBS components (Fig. 7A). In fact, many FBS-derived bovine proteins were found to have abundance equal to or greater than the mouse proteins originating from the ECs themselves (Fig. 7A). This data suggests that ECs engulf extracellular components, which are subsequently packaged into sEVs. Furthermore, there was some specificity to the media components that were incorporated into the sEVs, as bovine immunoglobulins were not detected in the sEV preparation. This is consistent with the major function of ECs, which is to relay messages from circulation to the underlying tissue, a process that is critical for metabolically important tissues, such as AT. ECs do this through well-defined pathways of transcytosis of soluble or albumin-bound nutrients and hormones. We tested whether BSA- cargo is packaged into sEVs as a mechanism to transfer blood-borne signals to AT cells. Glucagon and, to a lesser extent insulin, significantly enhanced BSA uptake in primary AT ECs (Fig. 7B and C). Overall BSA uptake was diminished in AT ECs isolated from cav1KO mice (Fig. 7C), which is consistent with previous studies that report uptake of BSA-bound cargo is largely reliant on phosphorylation-dependent activation of cav1 (Tyr14)(Li et al., 2013a; Zimnicka et al., 2016). However, the extent to which glucagon or insulin enhanced BSA uptake was unaffected by the absence of cav1 (Fig. 7C). Accordingly, glucagon treatment did not augment BSA-mediated phosphorylation of cav1 (Fig. 7D), compared to both C2 ceramide and H2O2, which were used as positive controls (Wehinger et al., 2015). This data suggests that insulin and glucagon enhance BSA uptake by a cav1-independent mechanism. It is likely occurring through an endocytosis or macropinocytosis-dependent mechanism (Li et al., 2013a). In addition, glucagon-induced BSA uptake and sEV production occur through distinct pathways, as GW4869 only suppressed sEV production (Fig. 6C) but not BSA uptake (Fig. 7E). Furthermore, basal BSA uptake was enhanced in AT ECs isolated from mice fed a high fat diet for 12 wk, however no further simulation of BSA uptake by glucagon was observed (Fig. 7F). This data indicates dysregulation of this pathway by dietary nutrient excess.
Figure 7: Glucagon Stimulates ECs to Take up Extracellular Components Which are Subsequently Incorporated into sEVs.
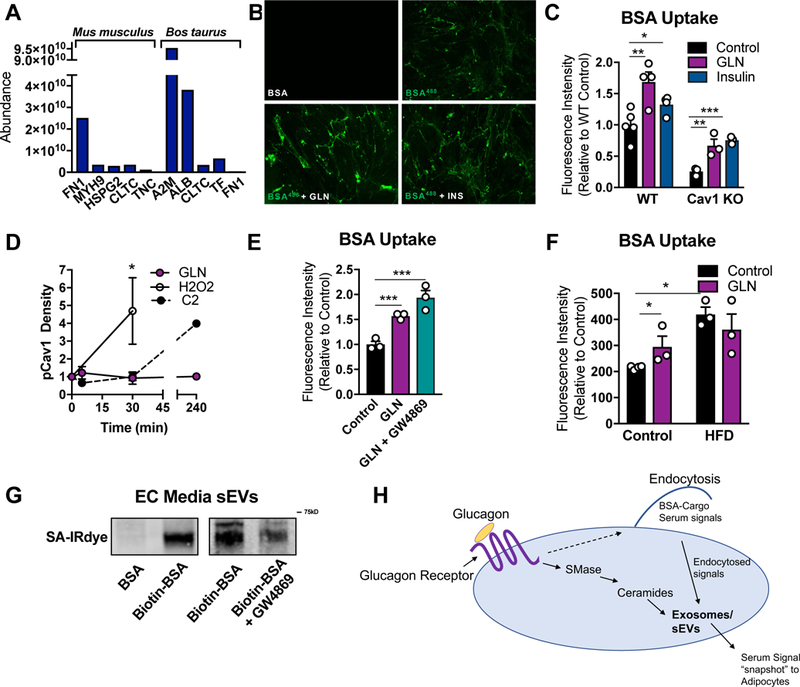
(A) mus musculus- and Bos taurus- specific proteins identified by Mass Spectroscopy in AT EC- derived sEVs. (B and C) BSA-conjugated Alexa Flour488 uptake assay was conducted on AT ECs stimulated with either insulin (5 µg/ml) or glucagon (GLN; 10nM). (B) A representative fluorescence image following BSA-Alexa Flour488 uptake. (C) Quantification of Alexa Flour488 intensity (n = 3–4). (D) Western blot densitometry of p-cav1Tyr14 under the indicated conditions. (E and F) BSA-Alexa Flour488 uptake as described in B–C. ECs in E were isolated from mice on a chow or high fat diet (HFD) for 12 wk. (G) ECs were treated with biotinylated-BSA, washed and provided fresh media. Media sEVs were isolated, resuspended in a fixed volume, and analyzed for biotin-BSA by SDS PAGE and streptavidin-IRdye (SA-IRdye) reactivity. (H) Model for the physiological significance of glucagon-stimulated BSA uptake and sEV production. Data is presented as mean ± SEM
We determined if extracellular BSA taken up by ECs is packaged into sEVs, as observed with FBS-derived proteins (Fig. 7A). AT ECs were serum-starved for 4 hours, treated with normal BSA or biotinylated-BSA for 30 min. Following BSA-treatment cells were washed thoroughly and provided with fresh media in the presence or absence of GW4869. After 2 d, sEVs from the media were isolated by ultracentrifugation and analyzed for the presence of biotin by SDS PAGE and streptavidin reactivity. A 65kD biotinylated protein, corresponding to biotinylated BSA, was observed in the sEV prep (Fig. 7G). Biotin incorporation into EVs was reduced with GW4869 treatment (Fig. 7G), a reflection of overall suppression of sEV production. Taken together these results indicate that AT ECs take up extracellular molecules, which are packaged into sEV and secreted (Fig. 7H).
DISCUSSION
We have isolated and characterized a pool of AT tissue-derived sEVs that traffic between cells within the tissue to communicate metabolic signals. Previous studies have suggested the existence of AT tissue EVs, as AT interstitial fluid contains signaling proteins, such as those involved in the MAPK and nitric oxide pathways (Celis et al., 2005). Others have shown that cultured AT pieces secrete EVs (Deng et al., 2009). In this study, we have developed and refined a protocol to isolate sEVs directly from AT in vivo, with minimal contamination by ruptured cells, and with sufficient yield for downstream applications. Initial proteomic characterization of these tissue sEVs has revealed that the protein profile is altered upon fasting (Fig. 5A and B). Of particular interest, mitochondrial electron transport chain proteins were highly represented in tissue sEVs isolated under fasting conditions. Mitochondria are generally not considered a source for exosomal cargo (Lotvall et al., 2014). However, a recent study reported that mitochondrial fatty acid oxidation proteins were identified in sEVs secreted from cultured 3T3-F442A-differentiated adipocytes (Lazar et al., 2016), although the physiological relevance of these findings in vivo were not clear. The incorporation of mitochondrial proteins into AT sEVs seems to be selective, as many electron transport chain proteins are significantly increased in the fasted state, whereas mitochondrial fatty acid oxidation proteins are decreased (Table S2 and 3). We suspect that the ratio of mitochondrial components that are packaged into sEVs may affect the metabolic profile of recipient cells. In fact, we have strong evidence for such a mechanism in a transgenic model of altered mitochondrial function selectively in adipocytes (Clair Crewe and Christine Kusminski, unpublished observations).
sEVs isolated from AT, were found to be enriched in sphingolipids, a class of lipids that have important signaling and structural properties (Hla and Dannenberg, 2012). Depending on the species produced, ceramides can be potent second messengers for stress-related pathways (Ruvolo, 2001). This includes pathological signaling in the context of type 2 diabetes (Chavez and Summers, 2012). Enhanced integration of ceramides into tissue sEVs may reflect activation of nutrient stress responses in parent and recipient cells alike (Fig. 5C). In addition, products of ceramide metabolism, hexosyl- and lactosyl- ceramides, also displayed differential enrichment in AT sEVs compared to total cellular content of these lipids. Hexosyl-ceramides were lower and lactosyl-ceramides were higher in the sEV fraction compared to whole cell lysates (Fig. 5D). This is particularly interesting because these carbohydrate-ceramide derivatives act as precursors for the ganglioside family of lipids, which are highly abundant on the outer leaflet of the plasma membrane, and complex with cholesterol and sphingomyelin to form membrane microdomains (Krengel and Bousquet, 2014). A property of gangliosides that is potentially important for sEV biology is their role in cell-cell recognition and adhesion through the hydrophilic glycan head group, which can interact with proteins and other carbohydrate moieties on the surface of recipient cells (Krengel and Bousquet, 2014). Differential incorporation of glycosylated-ceramides into sEV membranes may target sEVs to specific cell types.
Although the tissue EV pool is likely the product of EV secretion from multiple cell types, including adipocytes and AT ECs, we focused here on EV-mediated communication between ECs and adipocytes. ECs constitute the interface between the plasma and the interstitial space where tissue cells reside. Bi-directional movement of nutrients and signaling molecules to and from interstitial fluid occur through well characterized mechanisms of EC transcytosis, including caveolae- and clathrin- mediated transcytosis, fused membrane channels or paracellular transport of small molecules (Simionescu et al., 2009; Tuma and Hubbard, 2003). Albumin, specifically and almost exclusively, traverses the endothelial membrane through caveolin- mediated transcytosis. Molecules that would otherwise have little to no solubility in plasma bind and travel with albumin into the tissue. These molecules include fatty acids, amino acids, hormones, xenobiotics and cations. In keeping with previous reports, we have shown that cav1KO AT ECs display severe deficits in BSA uptake (Li et al., 2013a). However, glucagon- and insulin-stimulated BSA uptake is not dependent on cav1 (Fig. 6C). Instead this transport occurs through another pathway, likely a form of non-discriminant endocytosis of plasma components. Furthermore, BSA and bound cargo that is taken up by AT ECs is packaged into sEVs and secreted (Fig.7 A and G). This process is accelerated by glucagon, which, as we report here, enhances sEV biogenesis. We propose that this process is a way for ECs to “sample” the plasma, integrate all blood-borne signals into a single package and deliver this “snapshot” of the systemic metabolic state to tissue cells. In particular, adipocytes seem to be the primary target cell for EC-derived sEVs (Fig. 3E). This process relays more complex information than other forms of transcytosis, like information about the relative concentration of plasma components. Additionally, delivery of signals in this way would facilitate synergistic activation of multiple cellular pathways, as all signals enter the cell in concert. Because EC sEVs contain extracellular/serum signaling molecules, we can speculate that the effect of EC sEVs on adipocyte physiology is dependent on the type of signals in the blood at any given time or condition. If so, it follows that EC sEVs would have the ability to customize the functional response of adipocytes to systemic changes in metabolism. Future work will be required to understand the potentially wide-ranging implications of the EC-adipocyte communication axis for adipocyte function and, consequently, whole-body metabolism.
Glucagon-stimulated EV secretion is not a phenomenon that occurs in all ECs. Instead, the presence of this mechanism is dependent on the tissue from which the ECs were isolated. ECs isolated from different tissues differ in morphology and functionality, a result of both genetic and environmental influences (Conway and Carmeliet, 2004; Craig et al., 1998). AT ECs respond robustly to glucagon whereas lung ECs do not (Fig. 6E). This could be due to a number of differences, including the expression of the glucagon receptor or downstream signaling components like sMAse. ECs in AT are essential for the integration and coordination of AT function with system-wide metabolic demands, which requires the movement of innumerable signaling molecules in and out of the tissue. In contrast, although transcytosis in lung ECs is also important, the major function of lung capillaries is to facilitate the diffusion of oxygen and CO2 molecules to and from circulation. These functional differences provide a physiological explanation for the divergent responses of AT and lung ECs observed in this study.
The discovery and characterization of circulating EVs such as microvesicles and exosomes has offered remarkable insight into inter-organ communication, and the therapeutic potential of targeting these EVs. Likewise, understanding the role that EVs may play in cell-to- cell communication within a given tissue microenvironment may be just as fruitful, particularly for those tissues where the local interaction of tissue cells can have a dramatic effect on whole body metabolic homeostasis. A prominent example of this is white AT, where an appropriate, coordinated response of adipocytes, endothelial cells, immune cells and fibroblasts is required to expand or contract the fat pad depending on nutrient availability (Crewe et al., 2017). The efficiency at which AT completes this task directly reflects the difference between systemic metabolic health and pathological responses. Further efforts to understand tissue EV biology will aid in development of novel therapies with broad applications for the treatment of diabetes.
STAR Methods
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Adiponectin | Covance, rabbit anti- serum |
N/A |
| Alix | Santa Cruz | Cat#: 53540 |
| CALR | Cell Signaling Technology |
Cat#: 12238 |
| Cav1 | Cell Signaling Technology |
Cat#: 3267 |
| Beta tubulin | Abcam | Cat#: ab6046 |
| CD9 | BD Biosciences | Cat#: 553758 |
| CD31 for Western Blot | Cell Signaling Technology |
Cat#: 77699 |
| CD31 for primary endothelial cell sorting | Pharmingen | Cat#: 553370 |
| CD63 | Santa Cruz | Cat#: 5275 |
| COXIV | Abcam | Cat#: ab5694 |
| GAPDH | Cell Signaling Technology |
Cat#: 2118 |
| Halo | Promega | Cat#: G9281 |
| H3 | Cell Signaling Technology |
Cat#: 4499 |
| MAC2 (Galectin-3) Clone M3/38 | BioLegend | Cat#: 125401 |
| miniSOG | Covance, rabbit anti- serum |
N/A |
| pCav1 Tyr14 | Cell Signaling Technology |
Cat#: 3251 |
| PDGFRβ | Santa Cruz | Cat#: sc-432 |
| Perilipin | Fitzgerald | Cat#: 20R-PP004 |
| RFP (Biotin) | Abcam | Cat#: ab34771 |
| SMA | Abcam | Cat#: 5694 |
| TSG101 | Santa Cruz | Cat#: 7964 |
| VECAD | Abcam | Cat#: ab33168 |
| Donkey anti-rabbit IRDye 680RD | Li-cor | Cat#: 926–68073 |
| Donkey anti-mouse IRDye 680RD | Li-cor | Cat#: 926–68072 |
| Goat anti-rat IRDye 680RD | Li-cor | Cat#: 925–68076 |
| Goat anti-rabbit IRDye 800CW | Li-cor | Cat#: 925–32211 |
| Goat anti-mouse IRDye 800CW | Li-cor | Cat#: 926–32210 |
| Chicken anti-mouse Alexa Fluor 594 | Invitrogen | Cat#: A-21201 |
| Goat anti-guinea pig Alexa Fluor 647 | Invitrogen | Cat#: A-21450 |
| Goat anti-rabbit Alexa Fluor 488 | Invitrogen | Cat#: A-11008 |
| APC Anti-CD140b clone APB5 | BD Biosciences | Cat#: 136008 |
| BB700 Anti-CD11b clone M1/70 | BD Biosciences | Cat#: 566417 |
| BV421 Anti-CD45 clone 30-F11 | BD Biosciences | Cat#: 563890 |
| FITC Anti-CD31 clone 390 | BD Biosciences | Cat#: 102405 |
| PECF594 Anti-F4/80 clone T45–2342 | BD Biosciences | Cat#: 565613 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 3-isobutyl-1-methylxanthine (IBMX) | Sigma | Cat#: I7018 |
| Amicon Ultra-15 centrifugal filter units (100KD) | Millipore | Cat#: UFC910024 |
| BSA (Fatty acid free, low endotoxin) | Sigma | Cat#: A8806 |
| BSA (Alexa Flour488- Conjugated) | ThermoFisher | Cat#: A13100 |
| CaCl2 | Sigma | Cat#: C7902 |
| Collagenase D | Roche | Cat#: 11088882001 |
| Collagenase Type I | Worthington | Cat#: X7H9763A |
| C2-Ceramide | Enzo | Cat#: BML-SL100– 0005 |
| Dapi | Sigma | Cat#: D9542 |
| Dexamethasone | Sigma | Cat#: D4902 |
| Dispase II | Roche | Cat#: 04942078001 |
| DMEM high glucose | ThermoFisher | Cat#: 11965092 |
| DMEM F12 with GlutaMAX | ThermoFisher | Cat#: 10565–042 |
| Doxycycline diet | Bio Serve | Cat#: S4107 |
| Dynabeads | Invitrogen | Cat#: 110.35 |
| EDTA | ChemCruz | Cat#: Sc-203932 |
| EGTA | Sigma | Cat#: E3889 |
| Endothelial Cell Growth Supplement (ECGS) | Alfa Aesar | Cat#: J64516 |
| EZ-Link NHS-Biotin | ThermoFisher | Cat#: 20217 |
| Fc Block | BD Biosciences | Cat#: 553142 |
| Fetal Bovine Serum (FBS) | Fisher Scientific | Cat#: 03–600-511 |
| Fetal Bovine Serum (FBS) Exosome-Free | System Biosciences | Cat#: EXO-FBSHI- 50A-1 |
| FITC-PEG-Cholesterol | Nanocs | Cat#: PG2-CSFC-5k |
| Gelatin (2%) | Sigma | Cat#: G1393 |
| Gentamycin | Gibco | Cat#: 15750–060 |
| Glucagon | Sigma | Cat#: G2044 |
| Glucagon (Acylated) | A generous gift from William Holland (Utah) |
N/A |
| Glucose | Gibco | Cat#: 15023–021 |
| GW4869 | Sigma | Cat#: 567715 |
| Halo Tag TMRDirect | Promega | Cat#: G299A |
| HEPES | Gibco | Cat#: 15630–080 |
| Insulin | Sigma | Cat#: I6634 |
| IRDye 800CW Streptavidin | Li-COR | Cat#: 926–32230 |
| iScript cDNA Synthesis Kit | BIORAD | Cat#: 1708890 |
| NaF | Sigma | Cat#: S6776 |
| Na3VO4 (Orthovanadate) | Sigma | Cat#: S6508 |
| NTA 488nm Fluorescence Standard Beads | Malvern | Cat#: NTA4095 |
| NuPAGE 4–12% Bis-Tris Gel | ThermoFisher | Cat#: NP0335BOX |
| MatTek Glass Bottom Microwell Dishes | MatTek | Cat#: P35G-1.5–10- C |
| MgCl2 | Fisher Scientific | Cat#: M35–500 |
| Penicillin-Streptomycin (10, 000U/mL) | ThermoFisher | Cat#: 15140122 |
| Sucrose | Fisher Scientific | Cat#: BP220–212 |
| SYBR Green PCR Master Mix | Applied Biosystems | Cat#: 4309155 |
| Syringe-driven filter (0.22 µm) | Millipore | Cat#: SLGP033NS |
| Trehalose | Sigma | Cat#: T0167 |
| Triton-X100 | Sigma | Cat#: T9284 |
| TRIzol reagent | Fisher Scientific | Cat#: 12034977 |
| Red Blood Cell Lysis Buffer | Sigma | Cat#: R7757 |
| Rosiglitazone | Sigma | Cat#: R2408 |
| Experimental Models: Cell Lines | ||
| bEND.3 Cells | A generous gift from Michael Dellinger, UT Southwestern |
N/A |
| Experimental Models: Organisms/Strains | ||
| mouse: B6;FVB-Tg floxed cav1 | (Asterholm et al., 2012) |
N/A |
| mouse: B6-Tg Membrane halo | This paper | N/A |
| mouse: B6-Tg TRE-miniSOG | This paper | N/A |
| mouse: B6-Tg adiponectinP-rtTA | (Wang et al., 2010) | N/A |
| mouse: Cav1tm1Mls/J (Cav1 KO) | Jackson Laboratory | No. 004585 |
| mouse: B6.Cg-Tg(tet0-cre)1Jaw/J | Jackson Laboratory | No. 006234 |
| mouse: B6;FVB-Tg (Adipoq-cre)1Evdr/J | Jackson Laboratory | No. 028020 |
| mouse: WT C57BL6/J | Jackson Laboratory | No. 000664 |
| mouse: B6. FVB-Tg (Cdh5-cre)7Mlia/J (VECAD-cre) | Jackson Laboratory | No. 006137 |
| mouse: FVB-Tg (Cdh5-tTA) D5Lbjn/J (VECAD-tTA) | Jackson Laboratory | No.013585 |
| Oligonucleotides | ||
|
Mus Cav1 qPCR Forward Primer: 5’-ATGTCTGGGGGCAAATACGTG-3 |
Sigma | N/A |
|
Mus Cav1 qPCR Reverse Primer: 5’-CGCGTCATACACTTGCTTCT-3 |
Sigma | N/A |
|
Mus GgR qPCR Forward Primer: 5’-CCGAAACTACATCCATGGGAACCTGT-3 |
Sigma | N/A |
|
Mus GgR qPCR Reverse Primer: 5’-ATCTTCTGGCTGTACCGTGTCTTCAG-3’ |
Sigma | N/A |
| beta-2-microglobulin (B2M) qPCR Forward Primer: 5’-TTCTGGTGCTTGTCTCACTGA-3’ |
Sigma | N/A |
| beta-2-microglobulin (B2M) qPCR Reverse Primer: 5’-CAGTATGTTCGGCTTCCCATTC-3’ |
Sigma | N/A |
| Recombinant DNA | ||
| pDisplay-Halo vector | ThermoFisher | V66020 |
| pBAD vector | A kind gift from Roger Tsien) |
N/A |
| pTRE-promoter vector | Clontech | Cat#: 631059 |
| Software and Algorithms | ||
| Excel | Microsoft | N/A |
| Fiji (ImageJ) | Fiji | https://fiji.sc/ |
| FlowJo | FlowJo | https://www.flowjo.com/ |
| Image J | NIH | https://imagej.nih.gov/ij/ |
| LabSolutions V 5.82 | Shimadzu Scientific Instruments |
N/A |
| LabSolutions Insight V 2.0 program packages | Shimadzu Scientific Instruments |
N/A |
| Prism | GraphPad Software | GraphPad Software |
| Reactome | Fabregat et al., 2016 | N/A |
| Other | ||
| Zeiss LSM8800 Airyscan Confocal Microscope | Zeiss | N/A |
| Keyence BZ-X700 Fluorescence Microscope | Keyence | N/A |
| 1200EX Transmission Electron Microscope | JEOL | N/A |
| LSRFortessa SORP | BD Bioscience | N/A |
| FACS Aria Fusion | BD Bioscience | N/A |
| MagNA Lyser | Roche | N/A |
| Odyssey Infrared Imager | Li-cor | N/A |
| Nanosight NS300 | Malvern Instruments | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Information and requests for reagents and mice should be directed to the Lead Contact, Philipp Scherer (Philipp.Scherer@UTSouthwestern.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Models
All animal experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of Texas Southwestern (UTSW) Medical Center (Dallas, TX). The Cav1 gene was specifically ablated in mature adipocytes using the loxP system combined with the constitutively active adiponectin-cre (aCre). An inducible mouse model was also generated with a tetracycline response element (TRE)- driven cre recombinase (iCre). Specificity for the adipocyte in this model was accomplished by using the reverse tetracycline transactivator (rtTA) under the control of the adiponectin (APN) promoter (Fig. S1).
To generate tissue-specific doxycycline-inducible Cav1-mSOG mice, the coding sequence for full length murine caveolin-1 was first cloned from murine subcutaneous adipose tissue cDNA; a Kozak sequence of 5′-GCCGCCACC-3′ was placed in front of the caveolin-1 start codon. This cDNA was cloned into a pBAD vector containing sites for insertion of a C-terminal double miniSOG. After amplification, the Cav1-miniSOG fusion protein sequence was removed by NheI and XbaI and cloned into the “tight” pTRE-promoter vector in which we had previously placed a rabbit β-globin 3′UTR at the 3’ end. Following linearization and purification, TRE-Cav1- miniSOG DNA was injected into C57Bl6/N embryos by the Transgenic Core at UT Southwestern Medical Center. Positive founder pups were screened for both tight repression at baseline and doxycycline inducibility in adipose tissue when crossed with the adiponectin-rtTA mouse line.
Generation of the inducible, plasma membrane-targeted halo mouse was accomplished using a pDisplay-Halo vector. HindIII and NotI were used to excise the IgK-HA-Halo-myc- PDGFR fragment, which was cloned into the TRE- promotor vector and processed for submission to the Transgenic Core as described above. Induction of gene expression in all inducible models was achieved by 600 mg/kg doxycycline in the diet for the indicated durations. Both control and transgenic mice received doxycycline. All mice used in this study were males.
In all cases where doxycycline was required to induce gene expression, it was supplied in the mouse diet. All mice received doxycycline including controls without the inducible transgenes (TRE-gene). Reference Fig. S1 and Fig. S2 for mouse model breeding strategies and control mice genotypes.
bEND.3 Cell Line
The immortalized mouse endothelial cell line bEND.3 was cultured in DMEM (4.5g/L glucose) supplemented with 10% FBS and penicillin-streptomycin. Documentation of these cells does not include information about their sex origin.
Stromal Vascular Cells and Adipocyte Differentiation
SVF cells were isolated from male mice and cultured in DMEM/F12 media containing 10% FBS, glutamax, penicillin-streptomycin and gentamicin at 5% CO2 in 37˚C. For in vitro differentiation experiments SVF cells were allowed to grow to ~95% confluency. Adipogenesis was induced by culture media supplemented with 0.5 mM 3-isobutyl-1-methylxanthine, 1µM dexamethasone, 5µg ml-1 Insulin and 1 µm rosiglitazone for 48hr. Following the 48hr induction cells were cultured in media containing only 5µg ml-1 Insulin.
Primary Adipose Tissue Endothelial Cells
Endothelial cells from young male mice were plated on gelatin-coated plates (0.2% gelatin) and used for experiments at, or before passage 3. Endothelial cells were cultured in DMEM (4.5 g/L glucose) supplemented with 20% FBS, penicillin-streptomycin, and 100 µg/ml ECGS. For all sEV-related experiments, exosome-free FBS was used.
METHOD DETAILS
Isolation of Primary Adipocytes, Stromal Vascular Cells (SVF) and Endothelial Cells
The inguinal fat pads were dissected from male mice between the ages of 4–6 wk. Fat tissues were minced and digested for 1 hr at 37˚C in buffer containing 100 mM HEPES, 1.5% BSA, 5 mM glucose, 1 mM calcium and 1mg/ml collagenase D. For adipocyte isolation, 2.4U/ml dispase II was included in the digestion buffer. Collagenase I was substituted for collagenase D for lung endothelial cell isolation. The dispersed tissue was then filtered through a 100 µm cell strainer, and centrifuged at 600 g for 5 min at 4˚C. The floating adipocytes were removed and used for biochemical analysis where indicated. The pelleted SVF cells were resuspended in culture media (DMEM/F12) and filtered through a 45 µm cell strainer. Cells were centrifuged again as described above. The pelleted cells that were used for Flow Cytometry or FACS were incubated in Red Cell Lysis buffer and stained according to the Flow Cytometry section. Otherwise isolated SVF cells were either plated for adipocyte differentiation experiments or bead-sorted with anti-CD31-conjugated Dynabeads for endothelial cell isolation.
Adipocyte Subcellular Fractionation
Mature adipocytes were isolated from the sWAT of 5 control and 8 aCre mice as described above. Adipocytes were pooled by respective genotype and homogenized in ice-cold buffer (10mM HEPES pH 7.4, 250mM sucrose, 10mM KCl, 2mM MgCl2, 1mM EDTA and 1mM EGTA) with a motor-driven Teflon/glass homogenizer. The nucleus and plasma membrane were pelleted at 720 g for 5 min, and washed in homogenization buffer. The supernatant was poured through cheesecloth to separate the floating fat cake from the cleared homogenate. The supernatant was then centrifuged at 16 000xg for 20min to pellet the mitochondria and residual plasma membrane. The supernatant was centrifuged at 30 000xg to recover the microsomal fraction. The resulting supernatant was kept as the cytoplasmic fraction. Both the nuclear and mitochondrial pellets were resuspended in buffer containing 10 mM Tris pH 7.4, 1 mM EDTA and each was layered on a 1.12 M sucrose cushion with a 22G needle. The samples were then centrifuged at 100 000 x g for 60 min. The pellets contained the mitochondrial and nuclear fractions respectively, which were resuspended in an appropriate volume of 10 mM Tris pH 7.4, 1 mM EDTA supplemented with protease inhibitor cocktail. The supernatants from this spin contained floated plasma membranes. These plasma membrane bands were combined, diluted ~8 times the volume with homogenization buffer and spun 80 000xg to pellet the plasma membranes. The plasma membrane fraction was resuspended in the same buffer as the other fractions.
Flow Cytometry
Flow cytometry experiments were carried out on a BD Bioscience LSRFortessa SORP flow cytometer (Children’s Medical Center Research institute flow Core at UT Southwestern). Cells were sorted on a BD Bioscience FACS Aria Fusion (Children’s Medical Center Research institute flow Core at UT Southwestern). Antibodies used for flow cytometry analysis can be found in the Key Resources Table. For flow cytometry: samples of SVF cells from inguinal WAT depots (isolated as described in Isolation of Primary Adipocytes, Stromal Vascular Cells (SVF) and Endothelial Cells section) were first incubated at 4°C for 10 min in 10 uL of PBS containing 2% FBS with anti-mouse CD16/CD32 Fc Block (clone 2.4G2) (1:200). Cells were then incubated rocking at 4°C for 1 hour with primary an tibodies (FITC anti-CD31 1:200, BV421 anti- CD45 1:200, PE or APC anti-CD140b (Pdgfb) 1:200, PE-CF594 anti F4/80 1:200, BB700 anti CD11b 1:200) and TMRDirect Halo Tag fluorescent ligand. The cells were then washed once with 200 uL PBS/2%FBS. Cells were centrifuged 5min at 4°C at 600 g and re-suspended in 200uL of PBS/2%FBS.
Flow Cytometry Gating Strategy
Cells were initially selected by size, on the basis of forward scatter (FSC) and side scatter (SSC). Live cells were then gated on both SSC and FSCWidth singlets, ensuring that only individual cells were recorded. Unstained SVF cells isolated from wild-type mice were used to determine background fluorescence levels. The populations were gated as shown in Fig. S4 and 5. Anti-F4/80 PE-CF594 and anti-CD11b BB700 antibodies were used to gate the macrophage population (F4/80+;CD11b+, Mac) and distinguish the non-macrophage population (F4/80-;CD11b-, No Mac). From F4/80+;CD11b+ population, cells containing anti-halo PE signal (Mac Halo) were gated. From the F4/80-;CD11b- population, cells were gated for 3 subpopulations: endothelial cells, hematopoietic and preadipocytes/mural cells. An anti-CD31 antibody was used to select for the endothelial cell population (CD31+). From this population, cells containing anti-halo PE signal (CD31 Halo) was gated. An anti-CD45 antibody to select the hematopoietic population (CD45+). From CD45+ population, cells containing anti-halo PE signal (CD45 Halo) was selected. Finally, the PDGFb+ preadipocyte cells were isolated from the CD31-;CD45- population using an anti-PDGFb antibody. Preadipocytes containing anti-halo PE were gated. The WT sample was used as a negative control to assure the specificity of the PE signal. All Flow Cytometry data was analyzed with FlowJo.
Cell Labeling Techniques
Where indicated cells were labeled with 5 µM FITC-PEG-Cholesterol (Nanocs, PG2-CSFC-3K). This dye was chosen because the cholesterol is naturally occurring, high-affinity membrane anchor that would likely be incorporated into EVs of all origins either by direct entry into plasma membrane-derived EVs or by entering the endocytic pathway with other components of the plasma membrane. PEGylation of cholesterol allowed for the compound to be easily solubilized in the aqueous media and excess dye easily washed away (Fig. S3). Cells were then washed 5 times with PBS (Fig. S3) and given fresh media for 2 d to allow for the accumulation of FITC- PEG-Cholestrol- labeled EVs in the media. Cell debris and large EVs were cleared from the media (as described in the Isolation of sEVs From Cell Culture Conditioned Medium section) and NTA analysis was used to determine the presence of FITC containing particles. SVF- differentiated adipocytes were then treated with the FITC-sEV containing conditioned media for 6 hr, washed, and analyzed for adipocyte-specific FITC signal indicative of adipocyte sEV uptake. Biotinylation of BSA was accomplished by the treatment of fatty acid- and endotoxin- free BSA with N-Hydroxysuccinimidobiotin per manufacturer’s instruction (EZ-Link NHS-Biotin).Biotinylated BSA was dialyzed against sterile PBS to remove all unreacted reagent. Cells were treated with biotin-BSA as indicated.
BSA-Alexa Flour 488 and Biotin-BSA Uptake Assay
Primary ECs were grown to ~95% confluency on MatTek glass bottom microwell dishes. The cells were washed in PBS and serum-starved in HBSS for 3 hr. The cells were then treated with 0.1 mg/ml Alexa Flour 488- conjugated BSA or 0.5 mg/ml biotinylated BSA in HBSS supplemented with 0.5 mg/ml unlabeled BSA for 30 min. Where indicated, cells were also treated with either insulin (5 µg/ml), glucagon (20 nM) or GW4869 (10 µM) during the BSA incubation period. Post-treatment ECs were washed thoroughly with PBS and either fixed in 4% paraformaldehyde for visualization of Alexa Flour 488-BSA or given fresh media for assessment of biotin-BSA incorporation into sEVs. Cells were visualized by a Keyence BZ-X700 microscope. Fluorescence intensity was determined by ImageJ software.
Transplant Studies
Male membrane halo mice were treated with doxycycline (600 mg/kg) in the diet for 2 wk to induce plasma membrane-targeted halo expression specifically in adipocytes. After the labeling period subcutaneous adipose tissue was excised from 6 wk old male MTMG mice, cut into ~ 100 mg pieces and surgically implanted into the dorsal subcutaneous adipose region of the halo mice. Recipient halo mice were maintained on doxycycline diet during the 3 wk period of implant integration, following which, the implants and surrounding inguinal adipose tissue were removed and processed for immunofluorescent staining.
Immunofluorescence
Adipose tissue was harvested, fixed overnight in 10% PBS-buffered formalin, and washed with 50% ethanol. Tissues were embedded in paraffin blocks by the University of Texas Southwestern Medical Center Molecular Pathology Core and sectioned (5 µm). For cells, following experimental procedures cells were washed with PBS and fixed for 20 min with 4% paraformaldehyde. For BSA-Alexa Flour-488 uptake assays cells were imaged after fixation. For detection of cav1 and perilipin, cells were permeabilized post fixation with 0.1% triton X-100. Primary antibodies used for both tissue sections and fixed cells were: perilipin, RFP, CD63, cav1, Halo, or affinity-purified anti-miniSOG (Covance). Fluorescence secondary antibodies used included: goat anti-rabbit Alexa Fluor 488, goat anti-guinea pig Alexa Fluor 647 and chicken anti-mouse Alexa Fluor 594. Where indicated, confocal images were acquired on a Zeiss LSM8800 Airyscan. Other fluorescence pictures were taken with a Keyence BZ-X700 microscope.
Isolation of sEVs from Adipose Tissue
Mice to be used for sEV isolation were perfused through the heart at the left ventricle with PBS, 5 mM EDTA at a rate of 1 ml/min for 2 min to removed blood from the tissue. EDTA was washed out with a second perfusion of PBS alone. The inguinal fat pads from 5 mice were combined and digested as described above for the isolation of primary cells from adipose tissue. Special care was taken not to mince the tissue too finely as to avoid ruptured cells. Samples were diluted 1:1 in PBS and 2 mM EGTA to inhibit collagenase activity. Following centrifugation at 600 g for 5 min at room temperature the floating adipocytes were removed and discarded. The supernatant containing the sEVs was transferred into a glass 30 ml centrifuge tube and centrifuged at 1200 g at 4˚C for 15 min. The supernatant was centrifuged again at 10 000 g at 4˚C for 15 min. The resulting supernatant was then filtered through a 0.22 µm syringe- driven filter to remove any contaminating microvesicles and any remaining cellular debris. Clarified samples were then concentrated to 250 ul in 100 KD MWCO Amicon Ultra-15 centrifugal filter units and diluted to 10 ml in PBS containing 25 mM trehalose. Samples were then centrifuged in a fixed angle rotor at 100 000 g for 1.5 hr to pellet sEVs. The pellet was resuspended in PBS-trehalose buffer and centrifuged again at 100 000 g for 1.5 hr. The final pellet was resuspended in PBS and 2 mM trehalose.
Isolation of sEVs From Cell Culture Conditioned Media
Endothelial cells were seeded equally between conditions and incubated in growth media containing exosome-free FBS for 48 hr. The media was recovered and centrifuged at 600 g at 4˚C for 5 min to remove whole cells. The supernatant was then centrifuged at 1200 g for at 4˚C for 15 min to remove cell debris and apoptotic bodies. The resulting supernatant was centrifuged at 10 000 g for 15 min at 4˚C 30 min to remove lEVs. The final supernatant was centrifuged in a fixed angle rotor at 100 000 g for 1.5 hr to pellet sEVs. The pellet was resuspended in PBS and centrifuged again at 100 000 g for 1.5 hr.
Nanoparticle Tracking Analysis
EV concentration and size distribution was determined by the dynamic light scattering technology of the Nanosight NS300. The NS300 was fitted with a flow-cell top plate, sample pump and 488 nm laser. The script was programed to take 5 videos for 30 seconds each for every sample. The final size and intensity values were averages between the 5 runs. For use of the Nanosight in fluorescence mode: the filter was engaged at the filter position for the blue laser (488 nm) to detect FITC signal. The thresholding pixels were optimized with the NTA 488 nm Fluorescence Standard Beads which contain fluorescent beads of known concentration and size distribution. Videos were acquired at the highest camera level under the same script conditions for unlabeled particles.
Proteomics and Pathway Analysis
Samples were run into an SDS-PAGE gel. Proteins were excised from the gel and submitted to the UT Southwestern Proteomics Core. Samples were processed as previously described (Kalantari et al., 2016). Pathway Analysis was conducted with Reactome software.
Sucrose Gradient Fractionation Analysis of EVs
Isolated EVs were analyzed by sucrose gradient centrifugation according to previously published protocol (Chiou and Ansel, 2016). Crude EV preparations were diluted in sucrose to a final concentration of 82%. The EVs were layered at the bottom of a sucrose gradient ranging from 82–10% (1.002–1.34g/ml calculated density). Samples were centrifuged at 100 000xg for 16 hr following which fractions were collected as follows: 10–16% (F1), 22–28% (F2), 34–40% (F3), 46–52% (F4), 58–64% (F5), and 70–82% (F6). The fractions were diluted 1:100 in PBS and centrifuged at 100 000xg for 1.5 hr to pellet any EVs. Pellets were resuspended in PBS and prepped for Western Blot. Samples were loaded into SDS-PAGE gels by volume normalization.
Sphingolipid Quantification
Sphingolipids were quantified by LC/ESI/MS/MS using a Shimadzu Nexera X2 UHPLC system couple to a Shimadzu LCMS-8050 triple quadrupole mass spectrometer. Lipid species were identified and quantified based on their molecular mass and fragmentation patterns. All species were verified with lipid standards. Data were processed using the LabSolutions V 5.82 and LabSolutions Insight V 2.0 program packages.
Western Blot
Protein was extracted from cells or tissue by homogenization in RIPA buffer supplemented with 1 mM EDTA, 20 mM NaF, 2 mM Na3VO4, and protease inhibitor cocktail. Proteins were separated by SDS-PAGE and transferred to either nitrocellulose or PVDF membrane. For Western blots the gel lanes were loaded with equal protein: 20 µg/well for adipocytes and SVF cells and 30 µg/well for EV preparations unless indicated otherwise. The sucrose gradient experiments were analyzed by equal volume loading of SDS PAGE gels. The blots were then incubated overnight at 4˚C with primary antibodies. The Odyssey Infrared Imager was used to visualized Western blots with Li-cor IRdye secondary antibodies.
Transmission Electron Microscopy (TEM)
Electron microscopy samples were prepared by UT Southwestern Electron Microscopy Core. Briefly, sWAT pieces were fixed by perfusion with 4% paraformaldehyde and 1% glutaraldehyde in 0.1 mM sodium cacodylate buffer. The fixed tissue was then transferred to 2.5% glutaraldehyde in 0.1 mM sodium cacodylate buffer, post fixed in buffered 1% osmium tetroxide, en bloc stained in 4% uranyl acetate in 50% ethanol. Thin sections were cut on a Leica Ultracut UCT ultramicrotome and poststained with 2% uranyl acetate and lead citrate. Images were acquired on the JEOL 1200EX transmission electron microscope. For visualization of EVs by TEM, 5 µl of the sEV sample was fixed overnight in 4% paraformaldehyde. 5 µl of the fixed sEV solution was transferred to each Formvar-carbon coated (200µ Cu) electron microscopy grids. The grids were covered in incubated for 10 min. The grids were washed once with distilled water and stained with 2% uranyl acetate before visualization.
qPCR
Isolated adipocytes and SVF were homogenized in TRIzol (Invitrogen) using a TissueLyser. RNA was isolated per the manufacturer’s protocol. RNA quality and yield were determined by absorbance at 260nm, 280nm and 230nm. cDNA was prepared by reverse transcribing 1 µg of RNA with the iScript cDNA Synthesis Kit. The results were calculated by the standard threshold cycle method and was used for normalization
QUANTIFICATION AND STATISTICAL ANALYSIS
Data is presented as mean ± SEM. *p<0.05, ** p<0.01 and ***p<0.001 by two-tailed Student’s t test or one-way ANOVA. A p value < 0.05 was determined to be statistically significant. All cell culture and qPCR data points were the average of technical duplicates. For primary cell culture experiments, the n value corresponds to a cell preparation from separate mice. For immortalized bEND.3 cell culture experiments the n value corresponds to an experiment conducted on separate passage number. For all mouse studies, the n value corresponds to individual mice of a given treatment. Data was analyzed used Prism GraphPad Software.
Supplementary Material
Highlights.
-
o
A network of EV-mediated exchange of cellular material exists in adipose tissue
-
o
This exchange can replace protein levels in adipocytes despite a gene disruption
-
o
EV trafficking in adipose tissue is regulated by the systemic metabolic state
-
o
Endothelial cells pack extracellular molecules in EVs for release to adipose tissue
Acknowledgements
We thank the following UT Southwestern Core Units: Histology Core for assistance in embedding and processing of tissue samples, the Metabolic Core for sphingolipidomics, the Proteomics Core for protein identification and the Transgenic Core for their help in the generation of mice. This study was supported by US National Institutes of Health (NIH) grants R01-DK55758, P01-DK088761, R01-DK099110 and P01-AG051459, as well as by an unrestricted grant from the Novo Nordisk Research Foundation (P.E.S.). C.C is supported by F32-DK113704.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Asterholm IW, Mundy DI, Weng J, Anderson RG, and Scherer PE (2012). Altered mitochondrial function and metabolic inflexibility associated with loss of caveolin-1. Cell Metab 15, 171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celis JE, Moreira JM, Cabezon T, Gromov P, Friis E, Rank F, and Gromova I (2005). Identification of extracellular and intracellular signaling components of the mammary adipose tissue and its interstitial fluid in high risk breast cancer patients: toward dissecting the molecular circuitry of epithelial-adipocyte stromal cell interactions. Mol Cell Proteomics 4, 492–522. [DOI] [PubMed] [Google Scholar]
- Chang CC, Chen CY, Wen HC, Huang CY, Hung MS, Lu HC, Chen WL, and Chang CH (2017). Caveolin-1 Secreted from Adipose Tissues and Adipocytes Functions as an Adipogenesis Enhancer. Obesity (Silver Spring) 25, 1932–1940. [DOI] [PubMed] [Google Scholar]
- Chavez JA, and Summers SA (2012). A ceramide-centric view of insulin resistance. Cell Metab 15, 585–594. [DOI] [PubMed] [Google Scholar]
- Chiou N-T, and Ansel KM (2016). Improved exosome isolation by sucrose gradient fractionation of ultracentrifuged crude exosome pellets
- Conlan RS, Pisano S, Oliveira MI, Ferrari M, and Mendes Pinto I (2017). Exosomes as Reconfigurable Therapeutic Systems. Trends Mol Med 23, 636–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway EM, and Carmeliet P (2004). The diversity of endothelial cells: a challenge for therapeutic angiogenesis. Genome Biol 5, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig LE, Spelman JP, Strandberg JD, and Zink MC (1998). Endothelial cells from diverse tissues exhibit differences in growth and morphology. Microvasc Res 55, 65–76. [DOI] [PubMed] [Google Scholar]
- Crewe C, An YA, and Scherer PE (2017). The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis. J Clin Invest 127, 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng ZB, Poliakov A, Hardy RW, Clements R, Liu C, Liu Y, Wang J, Xiang X, Zhang S, Zhuang X , et al. (2009). Adipose tissue exosome-like vesicles mediate activation of macrophage-induced insulin resistance. Diabetes 58, 2498–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essandoh K, Yang L, Wang X, Huang W, Qin D, Hao J, Wang Y, Zingarelli B, Peng T, and Fan GC (2015). Blockade of exosome generation with GW4869 dampens the sepsis-induced inflammation and cardiac dysfunction. Biochim Biophys Acta 1852, 2362– 2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabregat A, Sidiropoulos K, Garapati P, Gillespie M, Hausmann K, Haw R, Jassal B, Jupe S, Korninger F, McKay S , et al. (2016). The Reactome pathway Knowledgebase. Nucleic Acids Res 44, D481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hla T, and Dannenberg AJ (2012). Sphingolipid signaling in metabolic disorders. Cell Metab 16, 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalantari R, Hicks JA, Li L, Gagnon KT, Sridhara V, Lemoff A, Mirzaei H, and Corey DR (2016). Stable association of RNAi machinery is conserved between the cytoplasm and nucleus of human cells. RNA 22, 1085–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeck ES, Iordanskaia T, Sevilla S, Ferrante SC, Hubal MJ, Freishtat RJ, and Nadler EP (2014). Adipocyte exosomes induce transforming growth factor beta pathway dysregulation in hepatocytes: a novel paradigm for obesity-related liver disease. J Surg Res 192, 268–275. [DOI] [PubMed] [Google Scholar]
- Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, Dingli F, Loew D, Tkach M, and Thery C (2016). Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A 113, E968–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krengel U, and Bousquet PA (2014). Molecular recognition of gangliosides and their potential for cancer immunotherapies. Front Immunol 5, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusminski CM, Bickel PE, and Scherer PE (2016). Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat Rev Drug Discov 15, 639–660. [DOI] [PubMed] [Google Scholar]
- Lazar I, Clement E, Dauvillier S, Milhas D, Ducoux-Petit M, LeGonidec S, Moro C, Soldan V, Dalle S, Balor S , et al. (2016). Adipocyte Exosomes Promote Melanoma Aggressiveness through Fatty Acid Oxidation: A Novel Mechanism Linking Obesity and Cancer. Cancer Res 76, 4051–4057. [DOI] [PubMed] [Google Scholar]
- Li HH, Li J, Wasserloos KJ, Wallace C, Sullivan MG, Bauer PM, Stolz DB, Lee JS, Watkins SC, St Croix CM , et al. (2013a). Caveolae-dependent and -independent uptake of albumin in cultured rodent pulmonary endothelial cells. PLoS One 8, e81903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Liu K, Liu Y, Xu Y, Zhang F, Yang H, Liu J, Pan T, Chen J, Wu M , et al. (2013b). Exosomes mediate the cell-to-cell transmission of IFN-alpha-induced antiviral activity. Nat Immunol 14, 793–803. [DOI] [PubMed] [Google Scholar]
- Lisanti MP, Scherer PE, Vidugiriene J, Tang Z, Hermanowski-Vosatka A, Tu YH, Cook RF, and Sargiacomo M (1994). Characterization of caveolin-rich membrane domains isolated from an endothelial-rich source: implications for human disease. J Cell Biol 126, 111–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotvall J, Hill AF, Hochberg F, Buzas EI, Di Vizio D, Gardiner C, Gho YS, Kurochkin IV, Mathivanan S, Quesenberry P , et al. (2014). Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles 3, 26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvolo PP (2001). Ceramide regulates cellular homeostasis via diverse stress signaling pathways. Leukemia 15, 1153–1160. [DOI] [PubMed] [Google Scholar]
- Scherer PE, Tang Z, Chun M, Sargiacomo M, Lodish HF, and Lisanti MP (1995). Caveolin isoforms differ in their N-terminal protein sequence and subcellular distribution. Identification and epitope mapping of an isoform-specific monoclonal antibody probe. J Biol Chem 270, 16395–16401. [DOI] [PubMed] [Google Scholar]
- Shin S, Han D, Park MC, Mun JY, Choi J, Chun H, Kim S, and Hong JW (2017). Separation of extracellular nanovesicles and apoptotic bodies from cancer cell culture broth using tunable microfluidic systems. Sci Rep 7, 9907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simionescu M, Popov D, and Sima A (2009). Endothelial transcytosis in health and disease. Cell Tissue Res 335, 27–40. [DOI] [PubMed] [Google Scholar]
- Thery C, Amigorena S, Raposo G, and Clayton A (2006). Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol Chapter 3, Unit 3 22. [DOI] [PubMed] [Google Scholar]
- Thery C, Ostrowski M, and Segura E (2009). Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 9, 581–593. [DOI] [PubMed] [Google Scholar]
- Thomou T, Mori MA, Dreyfuss JM, Konishi M, Sakaguchi M, Wolfrum C, Rao TN, Winnay JN, Garcia-Martin R, Grinspoon SK , et al. (2017). Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 542, 450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B, and Simons M (2008). Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319, 1244–1247. [DOI] [PubMed] [Google Scholar]
- Tuma P, and Hubbard AL (2003). Transcytosis: crossing cellular barriers. Physiol Rev 83, 871–932. [DOI] [PubMed] [Google Scholar]
- van Niel G, D’Angelo G, and Raposo G (2018). Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 19, 213–228. [DOI] [PubMed] [Google Scholar]
- Wang ZV, Deng Y, Wang QA, Sun K, and Scherer PE (2010). Identification and characterization of a promoter cassette conferring adipocyte-specific gene expression. Endocrinology 151, 2933–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehinger S, Ortiz R, Diaz MI, Aguirre A, Valenzuela M, Llanos P, Mc Master C, Leyton L, and Quest AF (2015). Phosphorylation of caveolin-1 on tyrosine-14 induced by ROS enhances palmitate-induced death of beta-pancreatic cells. Biochim Biophys Acta 1852, 693–708. [DOI] [PubMed] [Google Scholar]
- Willms E, Johansson HJ, Mager I, Lee Y, Blomberg KE, Sadik M, Alaarg A, Smith CI, Lehtio J, El Andaloussi S , et al. (2016). Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci Rep 6, 22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Riopel M, Bandyopadhyay G, Dong Y, Birmingham A, Seo JB, Ofrecio JM, Wollam J, Hernandez-Carretero A, Fu W , et al. (2017). Adipose Tissue Macrophage- Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 171, 372–384 e312. [DOI] [PubMed] [Google Scholar]
- Zhao H, Shang Q, Pan Z, Bai Y, Li Z, Zhang H, Zhang Q, Guo C, Zhang L, and Wang Q (2018). Exosomes From Adipose-Derived Stem Cells Attenuate Adipose Inflammation and Obesity Through Polarizing M2 Macrophages and Beiging in White Adipose Tissue. Diabetes 67, 235–247. [DOI] [PubMed] [Google Scholar]
- Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T, Marini JC, Tudawe T, Seviour EG, San Lucas FA , et al. (2016). Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. Elife 5, e10250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimnicka AM, Husain YS, Shajahan AN, Sverdlov M, Chaga O, Chen Z, Toth PT, Klomp J, Karginov AV, Tiruppathi C , et al. (2016). Src-dependent phosphorylation of caveolin-1 Tyr-14 promotes swelling and release of caveolae. Mol Biol Cell 27, 2090–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.