

Abstract
The Acer truncatum Bunge is a particular widespread forest tree species in northern China. VLCFAs are important to eukaryotes survival and play diverse roles throughout the development. So far, there are reports that the Acer truncatum seeds fatty acid (FA) rich in VLCFAs, but little is known about the physiological mechanism responsible for the biosynthesis. A total of approximately 37.07 Gbp was generated, it was comprehensive enough to determine the majority of the regulation VLCFAs biosynthesis genes. The 97,053 different unigenes were assembled and identified, and large numbers of EST-SSRs were determined. The expression profiles of crucial genes (KCS, KCR, HCD and ECR) involved in VLCFAs elongation of fatty acids were also studied. To our knowledge, the present study provides the first comprehensive of the transcriptome of Acer truncatum seeds. This transcriptome dataset have been made publicly available NCBI, we believe that it may provide new resource for future high-throughput gene expression of Acer truncatum seeds growth and development and will provide theoretical basic information for improving the yield of VLCFAs, especially nervonic acid.
Introduction
The Acer truncatum Bunge, a particular widespread forest tree species in northern China, Japan and Korea, and is also found in Europe and Northern America1,2. The seeds fatty acid (FA) rich in nervonic acid (24:1; cis-tetracos-15-enoic acid) and it has been officially admitted as edible oil by the Ministry of Health of China3. Nervonic acid is a very long chain fatty acids (VLCFAs). VLCFAs are fatty acids with an acyl chain of 20–30 carbons and longer4. The chain lenth, the type of polar head, the degree of unsaturation and the associated lipids provide the structural and functional diversity of these fatty acids. VLCFAs are important to eukaryotes survival and play diverse roles throughout the growth and development5. In addition, VLCFAs are important feedstocks for industrial, pharmaceutical and nutraceutical applicatiom6,7. Vegetable oil, as neat fuel, has been to the main source of VLCFAs; therefore, the increase of VLCFA contents in seeds has become an important target for oilseed enhancement.
VLCFAs are synthesized by a microsomal fatty acid extension (FAE) system. Specifically, the C18 fatty acid is first synthesized using the de novo fatty acid synthesis pathway of plastids, and the C2 portion of malonyl-coenzyme A (CoA) is sequentially added to the previously synthesized C18 fatty acids8,9. Analogous to FAS, the FAE is a membrane-bound fatty acid elongation complex, which involves 4 enzymatic reactions: condensation of C18-CoA with malonyl CoA to form a ketoacyl-CoA by ketoacyl-CoA synthase (KCS), reduction of ketoacyl-CoA to a hydroxyacyl-CoA by ketoacyl-CoA reductase (KCR), dehydration of hydroxyacyl-CoA to a enoyl-CoA by hydroxyacyl-CoA dehydratase (HCD), and reduction of enoyl-CoA by enoyl-CoA reductase (ECR)10. The KCS is thought to be the rate-limiting enzyme for the elongation of long-chain fatty acids, since it determines the substrate and tissue specificity of long-chain fatty acid elongation. In addition, regulating the expression of the KCS gene affects the final contents of VLCFAs. In contrast, the other 3 enzymes have no substrate specificity and tissue specificity for VLCFA biosynthesis11–19.
Recently, RNA-Seq has become a very effective and powerful technology in generating comprehensive transcriptome dataset20. Studies in the past have indicated the rapid identification and profiling of differentially expressed genes by de novo transcriptome sequencing in some oil seeds, such as flax, castor bean, olive, peanut, sea buckthorn, tree peony and Camellia oleifera21-27. So far, there are reports that the Acer truncatum seeds FA rich in VLCFAs3, but little is known about the physiological mechanism responsible for VLCFAs biosynthesis.
In this study, we analyzed the transcriptome of Acer truncatum using high-throughput Illumina sequencing technology. In total, more than 37.07 Gbp was generated, it was comprehensive enough to determine the majority of the regulation VLCFAs biosynthesis genes. The 127,791 different transcripts and 97,053 unigenes were assembled and identified, and large numbers of EST-SSRs were determined. To our knowledge, this study provides the first comprehensive of the transcriptome of Acer truncatum seeds. This transcriptome dataset have been made publicly available NCBI, we believe that it may provide new resource for future research on bioengineering breeding and will provide theoretical basic information for improving the yield of VLCFAs.
Results and Discussion
FA profiling in different genotypes of Acer truncatum seeds
Based on the analysis of thirty-six genotypes Acer truncatum seeds FA, we found that different genotypes seeds showed different FA content and composition. There mainly were six kinds of VLCFAs in Acer truncatum, namely arachidic acid (C20:0), eicosenoic acid (C20:1), behenic acid (C22:0), erucic acid (C22:1), lignoceric acid (C24:0), and nervonic acid (C24:1). Although the accumulation of major VLCFAs varied in different genotypes seeds, the three dominant components of VLCFAs composed of eicosenoic acid (C20:1), erucic acid (C22:1) and nervonic acid (C24:1) (Tables S1 and S2).
The results showed that the highest content of VLCFAs genotype was H-11, and the lowest genotype was L-4. Additionally, there were higher content of Nervonic acid (C24:1) in H-11 seeds. However, no obvious differences were observed in several seed morphological traits including seed seed size and dry weight between seeds of H-11 and L-4 plants seeds (Fig. 1 and Table S2). To investigate the biological function on seed VLCFAs accumulation, we performed transcriptome sequencing of these two genotype seeds.
Figure 1.

Characterization of two genotypes of Acer truncatum seeds. (A) The seeds randomly selected from the H-11 and L-4 plants. (B) Quantitative comparisons of seed size between the H-11 and L-4 plants. (C) Quantitative comparison of dry weight of seeds between the H-11 and L-4 plants. (D) Comparison of seeds total FA content between the H-11 and L-4 plants. (E) Comparison of contents of major seeds VLCFAs compositions between the H-11 and L-4 plants. Error bars are standard errors of the mean from three technical replicates. The different letters indicates significant differences at P < 0.05 (Student’s t-test).
Illumina sequencing and de novo assembly
The total of the Acer truncatum seeds mRNA was isolated from a single plant (H-11 or L-4). The sequencing raw data through rigorous quality assessment and data filtering, about 6.23 Gb, 6.21 Gb and 6.08 Gb, as well as 6.13 Gb, 6.21 Gb and 6.21 Gb for H-11 and L-4, respectively. It is presented in Table 1. The high quality sequencing reads has been deposited in the NCBI. Using the Trinity software program, the high quality sequencing reads were de novo assembled28, which produced 127,791 transcripts with an N50 length of 1,122 bp and a mean length of 686.76 bp. The distribution of the transcripts are depicted in Fig. S1 and Table S3. These transcripts were further analyzed for cluster and assembly. We have obtained 97,053 unigenes with an N50 length of 938 bp and a mean length of 598.19 bp for further analysis. It is shown in Fig. 2 and Table 2.
Table 1.
Summary of transcriptome sequencing for two genotypes of Acer truncatum H-11 and L-4 seeds.
| Sample | Read length | No. of reads | Data (bp) | GC% | Q30 (%) |
|---|---|---|---|---|---|
| H-11-1 | 150 + 150 | 20,777,973 | 6,233,391,900 | 53.27 | 88.90 |
| H-11-2 | 150 + 150 | 20,689,010 | 6,206,703,000 | 52.24 | 89.49 |
| H-11-3 | 150 + 150 | 20,278,336 | 6,083,500,800 | 52.34 | 89.18 |
| L-4-1 | 150 + 150 | 20,417,206 | 6,125,161,800 | 49.59 | 89.52 |
| L-4-2 | 150 + 150 | 20,712,428 | 6,213,728,400 | 51.96 | 90.06 |
| L-4-3 | 150 + 150 | 20,705,377 | 6,211,613,100 | 50.44 | 89.61 |
Figure 2.
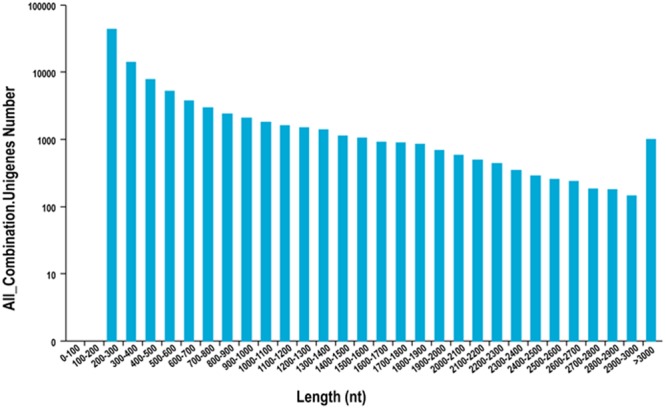
Length distribution of Acer truncatum transcripts.
Table 2.
Overview of the Acer truncatum transcriptome unigenes assembly.
| Unigenes length | Total number | Percentage |
|---|---|---|
| 200-300 | 43,021 | 44.33% |
| 300–500 | 21,857 | 22.52% |
| 500–1000 | 16,338 | 16.83% |
| 1000–2000 | 11,706 | 12.06% |
| 2000+ | 4,131 | 4.26% |
| Total number | 97,053 | |
| Total length | 58,055,717 | |
| N50 length | 938 | |
| Mean length | 598.19 | |
Functional annotation of all unigenes
The assembled unigenes sequences were annotated based on the following databases: NR, SWISS-PROT, GO, COG and KEGG. An overview of functional annotation in Table 3. We used BLASTX to similarity analysis and compared against the NR database. In these assembled unigenes, 71,014 (73.57%) unigenes had significant matches, and about 30% unigenes were showed no significant matches. Previous studies have shown that sequencing of cDNA libraries does not significant hits sequences is about 25% to 35%29–31. Among a wide range of plants with protein sequences, the Acer truncatum assembled unigenes had the highest number of hits against Citrus sinensis at 6,886 hits, followed by Citrus clementina at 4,687 hits, Theobroma cacao at 1,453 hits, Ricinus communis at 1,005 hits, Vitis vinifera at 755 hits, Manihot esculenta at 665 hits, Jatropha curcas at 656 hits and Ziziphus jujuba at 538 hits (Fig. 3). It depicts that the higher similarity of Acer truncatum unigenes and Citrus sinensis genes suggests the possibility we can use Citrus sinensis transcriptomes and genomes as a reference for further analysis.
Table 3.
Functional annotation of the Acer truncatum transcriptome unigenes.
| Database | Unigene | Percentage |
|---|---|---|
| NR | 71,401 | 73.57% |
| Swiss-prot | 26,076 | 26.87% |
| GO | 39,745 | 40.95% |
| COG | 30,605 | 31.53% |
| KEGG | 14,708 | 15.15% |
| All-annotated | 75,499 | 77.79% |
Figure 3.

Species distribution of the top BLAST hits in the NR database.
The SWISS-PROT database is the manually annotated and reviewed protein sequence database. It is a high-quality database of annotated and non-redundant protein sequences, and the results contain experimental results, computational features and scientific conclusions. Among the 97,053 unigenes, 26,076 (26.87%) were similar to the SWISS-PROT database (Table 3). Using the GO database enrichment analysis, the identified assembled unigenes were carried out to classify three independent sets (the cellular component, the molecular function, and the biological process). It depicts that the majority GO terms were assigned to the biological process is 59,615, the molecular function had 27,109 terms assigned, and the cellular component had 60,490 terms assigned (Fig. 4).
Figure 4.

Gene Ontology (GO) classification of assembled unigenes in Acer truncatum. The 39,745 Acer truncatum unigenes were assigned to three main categories: cellular component, molecular function and biological process.
Furthermore, the assembled unigenes were searched against the COG database. A total of 30,605 unigenes have been assigned to the COG classification (Fig. 5). The highest group is the cluster for function prediction only (7,042, 23.01%), and followed by amino acid transport and metabolism (4,110, 13.43%); carbohydrate transport and metabolism (3,613, 11.81%); inorganic ion transport and metabolism (2,651, 8.66%); translation, ribosomal structure and biogenesis (2,579, 8.43%); energy production and conversion (2,545, 8.32%); transcription (2,493, 8.15%); replication, recombination and repair (2,472, 8.08%); posttranslational modification, protein turnover, chaperones (2,304, 7.53%) and signal transduction mechanisms (1,860, 6.08%). However, only 11 and 10 annotations of unigenes are annotated to the nuclear structural and the extracellular structure. The KEGG database was used to analyze the the active biological pathways. The 14,708 assembled unigenes were assigned to 120 biological pathways through this process (Table S4). Among them, the highest metabolic pathway assigned to the unigenes is Ribosome (ko03010, 698 unigenes), followed by Oxidative phosphorylation (ko00190, 566 unigenes), Purine metabolism (ko00230, 563 unigenes), Protein processing in endoplasmic reticulum (ko04141, 526 unigenes), Glycolysis/Gluconeogenesis (ko00010, 491 unigenes), RNA transport (ko03013, 423 unigenes), Spliceosome (ko03040,414 unigenes) and Pyrimidine metabolism (ko00240, 391 unigenes). These results indicate that the growth and development of Acer truncatum seeds is mainly dependent on a large number of substances and energy metabolism.
Figure 5.

Classification of clusters of orthologous groups (COG). The total of 30,605 Acer truncatum unigenes were grouped into 25 classificat.
EST-SSR discovery
As containing highly information molecular marker, SSR markers have become one of the most widely used molecular marker systems for genetics, evolution and breeding research. To explore EST-SSR markers in the assembled unigenes, the 4,039 sequences containing 5,774 EST-SSRs were produced from 15,837 unigenes. Di-nucleotide and tri-nucleotide motifs were the most plentiful with 31.24% (909) and 29.35% (854), respectively (Table 4). The most repeat was AG/CT (1,012), followed by AGC/CTT (400), AT/AT (328), and ACC/GGT (189) (Table S5). The large set of EST-SSR markers identified in this research will help future researchers to better understand the genome-wide adaptive pattern of this species.
Table 4.
Length distribution of EST-SSRs based on the number of repeat units.
| Number of repeat units | Di- | Tri- | Tetra- | Penta- | Hexa- | Total | Percentage |
|---|---|---|---|---|---|---|---|
| 5 | — | 856 | 41 | 8 | 4 | 909 | 31.24% |
| 6 | 454 | 383 | 11 | 2 | 4 | 854 | 29.35% |
| 7 | 297 | 195 | 1 | 1 | 0 | 494 | 16.98% |
| 8 | 224 | 20 | 1 | 0 | 0 | 245 | 8.42% |
| 9 | 200 | 0 | 0 | 0 | 0 | 200 | 6.87% |
| 10 | 189 | 10 | 0 | 0 | 0 | 199 | 6.84% |
| >10 | 7 | 1 | 1 | 0 | 0 | 9 | 0.31% |
Candidate enzymes involved in VLCFAs elongation of fatty acids in Acer truncatum seeds
VLCFAs are synthesized by a microsomal fatty acid extension (FAE) system. Specifically, the C18 fatty acid is first synthesized using the de novo fatty acid synthesis pathway of plastids, and the C2 portion of malonyl-coenzyme A (CoA) is sequentially added to the previously synthesized C18 fatty acids8,9. Analogous to FAS, the elongase complex catalyzes (FAE) is a membrane-bound fatty acid elongation complex, which involves four VLCFAs elongase complex catalyzes: KCS, KCR, HCD, and ECR10.
Based on previous studies, 34 assembled unigenes related to 4 of the enzymes in the VLCFAs FAE were identified in the annotated Acer truncatum seeds transcriptome database (Fig. 6A). In the transcriptome database, KCS is considered to be the rate-limiting enzyme in VLCFAs biosynthesis. Therefore, the isolation and functional analyses of the genes of KCS genes have become the most important thing in the study of VLCFAs biosynthesis. Twenty unigene sequences were annotated as encoding KCS genes.
Figure 6.

Acer truncatum unigenes that may be involved in the fatty acid elongation. (A) The fatty acid elongase complex. (B) The expression level of DEGs related fatty acid elongation in the “H-11” and “L-4”. Error bars are standard errors of the mean from three technical replicates. The different letters indicates significant differences at P < 0.05 (Student’s t-test).
In order to identify the key genes regulating the VLCFAs biosynthesis, DEGs analysis was performed through comparing the expression levels. On the basis of the applied thresholds FDR (False Discovery Rate) <0.01 and log2 (foldchange) ≥2, a total of 3,258 unigenes were identified as DEGs between these two samples (H-11 vs. L-4), which comprised 2,131 up-regulated genes and 1,127 down-regulated unigenes (Fig. S2). Among the 3,258 DEGs, the expression profile of 4 differentially expressed genes in VLCFAs elongation of fatty acids such as KCS, KCR, HCD, and ECR gene were DEGs analysis and confirm the results by RT-qPCR (Fig. 6B). The higher content of VLCFAs genotype group (H-28, H-7, H-14, H-26) and the lower content of VLCFAs genotype group (L-1, L-18, L-19) in 36 genotypes were also DEGs analysis and confirm the results by RT-qPCR (Fig. S3).
The results showed that 16 DEGs analysis exhibited the similar trends with the RT-qPCR transcript levels, among them, 10 DEGs were up-regulated, including c132128 (KCS-like), c121604 (KCS-like), c124374 (KCS-like), c117613 (KCS-like), c126208 (KCS-like), c123713 (KCS-like), c110367 (KCR-like), c70062 (KCR-like), c127044 (HCD-like), c86364 (HCD-like) and 6 DEGs showed no difference between H-11 and L-4, sunch as c119348 (HCD-like), c125484 (ECR-like), c87572 (ECR-like), c128798 (ECR-like), c132697 (ECR-like) and c131607 (ECR-like) (Fig. 6B). These results again confirm that KCS is the rate-limiting enzyme for the elongation of long-chain fatty acids. In addition, regulating the expression of the KCS gene affects the final contents of VLCFAs. However, since VLCFAs biosynthesis are complex processes involving multiple parameters, we only clarifies part of the whole process through the transcriptome sequencing analysis, so it is difficult to determine a precise conclusion. Obviously, additional accurate molecular biology, genomics and proteomic analysis procedures studies are required to verify and validate and further build on our predictions.
Conclusions and Perspectives
To our knowledge, this study provides the first comprehensive of the transcriptome of Acer truncatum seeds. The coverage of the transcriptome, which includes 37.07 Gbp, was comprehensive enough to identify the majority of the regulation VLCFAs biosynthesis genes. A saturation curve for RNA-Seq was provided in Fig. S4. A total of 127,791 different transcripts and 97,053 unigenes were identified in this study. Additionally, we categorized the enzymes involved in the biosynthesis of VLCFAs, such as KCS, KCR, HCD and ECR. Additionally, large numbers of EST-SSRs were determined. This transcriptome dataset have been made publicly available NCBI Short Read Archive (Accession Number: SUB3838977), we believe that it may provide new resource for future high-throughput gene expression of Acer truncatum seeds growth and development as well as its breeding, especially involved in VLCFAs biosynthesis.
Materials and Methods
Plant materials
Thirty-six genotypes of Acer truncatum trees (10-year-old) seeds, were randomly collected from the Fufeng commercial planting base in Boji Country (N34°22′36.62″; E107°53′44.37″; with altitude 550–600 m), Shannxi Province, China on the early September, 2015. The Acer truncatum trees seeds were fully mature in October. The reason for choosing this period is as follows: Our team previously found that VLCFA (especially 24:1 nervonic acid) are converted and accumulated in the middle and late stages of fatty acid synthesis, which is the first of the Acer truncatum seeds rich in VLCFA nervonic acid3.
Fatty acids extraction and composition analysis
The seeds of Acer truncatum, with three biological replicates, were used for extraction of fatty acids. Seeds FA were extracted and analyzed as reported previously in detail3. Briefly, total FA was converted to FA methyl ester at 80 °C in a methanol solution containing 1 M HCl for 2 hours. After extraction, the fatty acid composition of the oil was analyzed by gas chromatography-mass spectrometry (SQ GC-MS, Thermo Fisher). The FAME peaks were identified using the NIST 2014 database and their retention times compared to real standards. Prior to data analysis and statistics, all FAME peaks were quantified by area normalization with a threshold set at 0.1%.
The RNA-seq library construction for sequencing
The seeds were collected from Acer truncatum plants and dissected. After removal of pericarp, they were then immediately frozen and stored in liquid nitrogen prior to further analysis. We extracted the total mRNA using the Plant-RNA Kit (Aidlab -biotech, China). The spectrophotometer and the Agilent 2100 bioanalyzer were used to measure the quality and quantity of purified mRNA. The mRNA-seq library was constructed using the Illumina’s TruSeq RNA Sample Preparation Kit and the library quality was assessed on the Agilent Bioanalyzer 2100 system as previously reported in detail27. The fragment (340 bp ± 25 bp) was purified by gel electrophoresis and then amplified by PCR as a sequencing template. Finally, the mRNA library was sequenced by the HiSeqTM 4000 platform (Illumina Inc., USA).
Illumina sequencing data analysis and assembly
To obtain high quality clean read data for de novo assembly, all adapter sequences and low quality sequences were removed from the raw data. Using the Trinity program (k-mer = 25), the high-quality reads were de novo assembled (http://trinityrnaseq.sourceforge.net/)28. The contigs were clustered and the transcripts were further assembled according to the pairend reads. The longest transcript in the cluster were defined as unigenes. We used EMBOSS Getorf Software to predict the coding area (http://emboss.bioinformatics.nl/cgi-bin/emboss/getorf).
Sequence clustering and functional categorization of unigenes
The assembled unigenes sequences were annotated based on the following databases: NR (NCBI non-redundant protein sequences); NT (nonredundant nucleotide sequence); the Swiss-Prot (Protein sequence database); GO (Gene Ontology); COG (Clusters of Orthologous Groups of proteins) and KEGG (Kyoto Encyclopedia of Genes and Genomes). The best alignment was selected from the matches with an E-value ≤ 10−5. According to the best BLAST comparison (highest score), we give a gene name for each assembly sequence. The ORFs were identified by the “GetORF” (EMBOSS software package)32,33. GO were assigned to the assembled unigene using the “Blast2GO”34,35. The KEGG pathways annotation by the KAAS (KEGG Automatic Annotation Server) (http://www.genome.jp/kegg/kaas/)36.
The EST-SSRs detection
Using the MISA software, the 12,845 unigenes (more than 1 kb) of Acer truncatum were used for the EST-SSRs detection (http://pgrc.ipk-gatersleben.de/misa/). The parameters were adjusted to identify perfect di-, tri-, tetra-, penta- and hexa-nucleotide motifs with a minimum of 6, 5, 4, 4 and 4 repeats espectively as previously described37,38.
Quantitative real-time reverse transcription PCR (RT-qPCR)
The RT-qPCRs were used to examine expression of potential candidate genes in the VLCFAs biosynthetic pathway in Acer truncatum seeds, such as KCS, KCR, HCD, and ECR. The expression of these potential candidate genes were calculated by relative quantification with the actin house keeping gene as a reference. The specific primers are listed in Table S6 for the RT-qPCR reaction according to the candidate genes. The RT-qPCR reactions were performed in a Step One Plus Real-Time PCR System (Applied Biosystems, USA) using a Super Real PreMix kit (SYBR Green) (Tiangen-biotech, China). The RNA relative expression of each gene was calculated as reported previously in detail31.
Electronic supplementary material
Acknowledgements
We want to thank the reviewers for providing constructive comments on the manuscript. We are grateful to Springer Nature Author Services (authorservices.springernature.com) for manuscript editing. This work was supported by the Fundamental Research Funds for the Central Universities (Project No. 2452015042), Natural Science Basic Research Plan in Shaanxi Province of China (Program No. 2017JQ3027), China Postdoctoral Science Foundation (Project No. 2015M572605), Young Talent fund of University Association for Science and Technology in Shaanxi, China (Project No. 20160107) and Yangling Demonstration Zone Science and Technology Program (Project No. 2018NY-26).
Author Contributions
R.K.W. and P.L. performed the experiments, analyzed the data, prepared figures and tables, reviewed drafts of the paper. J.S.F. reviewed drafts of the paper. L.L.L. conceived and designed the experiments, contributed reagents/materials/analysis tools, wrote the paper, reviewed drafts of the paper.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-33999-3.
References
- 1.Guo X, et al. Effects of nitrogen addition on growth and photosynthetic characteristics of Acer truncatum seedlings. Dendrobiology. 2014;72:151–161. doi: 10.12657/denbio.072.013. [DOI] [Google Scholar]
- 2.More, D. & White, J. Cassell’s trees of Britain and Northern Europe. (Cassell, 2003).
- 3.Wang X, Fan J, Wang S, Sun R. A new resource of nervonic acid from purpleblow maple (Acer truncatum) seed oil. Forest products journal. 2006;56:147–150. [Google Scholar]
- 4.Millar AA, Kunst L. Very-long-chain fatty acid biosynthesis is controlled through the expression and specificity of the condensing enzyme. The Plant Journal. 1997;12:121–131. doi: 10.1046/j.1365-313X.1997.12010121.x. [DOI] [PubMed] [Google Scholar]
- 5.Amminger G, et al. Decreased nervonic acid levels in erythrocyte membranes predict psychosis in help-seeking ultra-high-risk individuals. Molecular psychiatry. 2012;17:1150–1152. doi: 10.1038/mp.2011.167. [DOI] [PubMed] [Google Scholar]
- 6.Ntoumani E, Strandvik B, Sabel K. Nervonic acid is much lower in donor milk than in milk from mothers delivering premature infants-Of neglected importance? Prostaglandins, Leukotrienes and Essential Fatty Acids (PLEFA) 2013;89:241–244. doi: 10.1016/j.plefa.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 7.Dingess K, et al. Docosahexaenoic acid, nervonic acid and iso-20 (BCFA) concentrations in human milk from the Global Exploration of Human Milk Project (623.15) The FASEB Journal. 2014;28:1–10. doi: 10.1096/fj.14-0101LTE. [DOI] [PubMed] [Google Scholar]
- 8.Fehling E, Mukherjee KD. Acyl-CoA elongase from a higher plant (Lunaria annua): metabolic intermediates of very-long-chain acyl-CoA products and substrate specificity. Biochimica et Biophysica Acta (BBA)-Lipids and Lipid Metabolism. 1991;1082:239–246. doi: 10.1016/0005-2760(91)90198-Q. [DOI] [PubMed] [Google Scholar]
- 9.Kunst L, Samuels A. Biosynthesis and secretion of plant cuticular wax. Progress in lipid research. 2003;42:51–80. doi: 10.1016/S0163-7827(02)00045-0. [DOI] [PubMed] [Google Scholar]
- 10.Haslam TM, Kunst L. Extending the story of very-long-chain fatty acid elongation. Plant science. 2013;210:93–107. doi: 10.1016/j.plantsci.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 11.Mietkiewska E, et al. Seed-specific heterologous expression of a nasturtium FAE gene in Arabidopsis results in a dramatic increase in the proportion of erucic acid. Plant physiology. 2004;136:2665–2675. doi: 10.1104/pp.104.046839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cahoon EB, et al. Production of fatty acid components of meadowfoam oil in somatic soybean embryos. Plant physiology. 2000;124:243–252. doi: 10.1104/pp.124.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lassner MW, Lardizabal K, Metz JG. A jojoba beta-Ketoacyl-CoA synthase cDNA complements the canola fatty acid elongation mutation in transgenic plants. The Plant Cell. 1996;8:281–292. doi: 10.1105/tpc.8.2.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mietkiewska E, Brost JM, Giblin EM, Barton DL, Taylor DC. Cloning and functional characterization of the fatty acid elongase 1 (FAE1) gene from high erucic Crambe abyssinica cv. Prophet. Plant biotechnology journal. 2007;5:636–645. doi: 10.1111/j.1467-7652.2007.00268.x. [DOI] [PubMed] [Google Scholar]
- 15.Fan Y, Meng HM, Hu GR. & Li FLBiosynthesis of nervonic acid and perspectives for its production by microalgae and other microorganisms. Applied Microbiology & Biotechnology. 2018;102:1–9. doi: 10.1007/s00253-017-8535-7. [DOI] [PubMed] [Google Scholar]
- 16.Hegebarth D, et al. Arabidopsis ketoacyl-CoA synthase 16 forms C36 /C38 acyl precursors for leaf trichome and pavement surface wax. Plant Cell & Environment. 2017;40:1–43. doi: 10.1111/pce.12981. [DOI] [PubMed] [Google Scholar]
- 17.Li D, et al. Regulation of FATTY ACID ELONGATION1 expression and production in Brassica oleracea and Capsella rubella. Planta. 2017;246:1–16. doi: 10.1007/s00425-016-2607-2. [DOI] [PubMed] [Google Scholar]
- 18.Ozseyhan ME, Kang J, Mu X, Lu C. Mutagenesis of the FAE1 genes significantly changes fatty acid composition in seeds of Camelina sativa. Plant Physiology & Biochemistry. 2018;123:1–7. doi: 10.1016/j.plaphy.2017.11.021. [DOI] [PubMed] [Google Scholar]
- 19.Wu X, et al. Chemical composition, crystal morphology and key gene expression of cuticular waxes of Asian pears at harvest and after storage. Postharvest Biology & Technology. 2017;132:71–80. doi: 10.1016/j.postharvbio.2017.05.007. [DOI] [Google Scholar]
- 20.Vijay N, Poelstra JW, Künstner A, Wolf JB. Challenges and strategies in transcriptome assembly and differential gene expression quantification. A comprehensive in silico assessment of RNA‐seq experiments. Molecular ecology. 2013;22:620–634. doi: 10.1111/mec.12014. [DOI] [PubMed] [Google Scholar]
- 21.Venglat P, et al. Gene expression analysis of flax seed development. BMC plant biology. 2011;11:74–88. doi: 10.1186/1471-2229-11-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costa GG, et al. Transcriptome analysis of the oil-rich seed of the bioenergy crop Jatropha curcas L. BMC genomics. 2010;11:462–471. doi: 10.1186/1471-2164-11-462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Munoz-Mérida A, et al. De novo assembly and functional annotation of the olive (Olea europaea) transcriptome. DNA research. 2013;20:93–108. doi: 10.1093/dnares/dss036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yin D, et al. De novo assembly of the peanut (Arachis hypogaea L.) seed transcriptome revealed candidate unigenes for oil accumulation pathways. PLoS One. 2013;8:e73767–e73778. doi: 10.1371/journal.pone.0073767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fatima T, et al. Fatty acid composition of developing sea buckthorn (Hippophae rhamnoides L.) berry and the transcriptome of the mature seed. PloS one. 2012;7:e34099–e34017. doi: 10.1371/journal.pone.0034099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li S-S, et al. Fatty acid composition of developing tree peony (Paeonia section Moutan DC.) seeds and transcriptome analysis during seed development. BMC genomics. 2015;16:208–222. doi: 10.1186/s12864-015-1429-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia E-H, et al. Transcriptome analysis of the oil-rich tea plant, Camellia oleifera, reveals candidate genes related to lipid metabolism. PLoS One. 2014;9:e104150–e104166. doi: 10.1371/journal.pone.0104150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature biotechnology. 2011;29:644–654. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bombarely A, et al. Generation and analysis of ESTs from strawberry (Fragaria xananassa) fruits and evaluation of their utility in genetic and molecular studies. BMC genomics. 2010;11:503–520. doi: 10.1186/1471-2164-11-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park S, Sugimoto N, Larson MD, Beaudry R, van Nocker S. Identification of genes with potential roles in apple fruit development and biochemistry through large-scale statistical analysis of expressed sequence tags. Plant Physiology. 2006;141:811–824. doi: 10.1104/pp.106.080994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L, et al. Comparative transcriptome sequencing and de novo analysis of Vaccinium corymbosum during fruit and color development. BMC plant biology. 2016;16:223–232. doi: 10.1186/s12870-016-0866-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature methods. 2008;5:621. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 33.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends in Genetics. 2000;16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 34.Conesa A, Götz S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. International journal of plant genomics. 2008;5:621–628. doi: 10.1155/2008/619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Conesa A, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 36.Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic acids research. 2004;32:D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei W, et al. Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC genomics. 2011;12:451–464. doi: 10.1186/1471-2164-12-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeng S, et al. Development of a EST dataset and characterization of EST-SSRs in a traditional Chinese medicinal plant, Epimedium sagittatum (Sieb. Et Zucc.) Maxim. BMC genomics. 2010;11:94–105. doi: 10.1186/1471-2164-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.