

Abstract
Humans are variously and continuously exposed to a wide range of different DNA-damaging agents, some of which are classed as carcinogens. DNA damage can arise from exposure to exogenous agents, but damage from endogenous processes is probably far more prevalent. That said, epidemiological studies of migrant populations from regions of low cancer risk to high cancer risk countries point to a role for environmental and/or lifestyle factors playing a pivotal part in cancer aetiology. One might reasonably surmise from this that carcinogens found in our environment or diet are culpable. Exposure to carcinogens is associated with various forms of DNA damage such as single-stand breaks, double-strand breaks, covalently bound chemical DNA adducts, oxidative-induced lesions and DNA–DNA or DNA–protein cross-links. This review predominantly concentrates on DNA damage induced by the following carcinogens: polycyclic aromatic hydrocarbons, heterocyclic aromatic amines, mycotoxins, ultraviolet light, ionising radiation, aristolochic acid, nitrosamines and particulate matter. Additionally, we allude to some of the cancer types where there is molecular epidemiological evidence that these agents are aetiological risk factors. The complex role that carcinogens play in the pathophysiology of cancer development remains obscure, but DNA damage remains pivotal to this process.
Keywords: bioactivation, cancer, carcinogens, DNA damage
Introduction
DNA damage occurs through exogenous and endogenous processes. Carcinogens, irrespective of their origin, have the ability to evoke the development of DNA damage through a variety of mechanisms. This includes, for instance, covalent binding of carcinogen with DNA or DNA double-strand breaks (DSBs) formed as a result of ionising radiation (IR)-induced free radical generation [1,2]. Carcinogens are categorised as being chemical or physical agents [3], causing DNA damage attributable to their physico-chemical properties, such as DNA molecule distortion or DNA cross-linking [3–6]. Table 1 shows a small subset of environmental and/or dietary carcinogens; however, there are multiple other examples to which humans are potentially exposed (Table 1).
Table 1. Examples of candidate cancer-causing agents.
| Candidate agents | Overview | References |
|---|---|---|
| Heterocyclic aromatic amines (HAAs) | HAAs are activation-dependent, heat-induced mutagenic agents predominantly present in foodstuffs containing nitrogenous and creatine components. Molecular structure of HAAs is dependent on the temperature and level of heat transferred to the food. Can generate SSBs, chromosomal aberrations and DNA adducts in guanine-rich regions. Activated metabolites can attack N2-position of guanine (most common) or C8-atom of guanine (occurs less frequently). | [13–15] |
| Polycyclic aromatic hydrocarbons (PAHs) | Combustion of organic matter results in the generation of PAHs. These are the most abundant indirect-acting carcinogens to which humans are exposed to on a daily basis. Exposure has been associated with the development of breast, skin or lung cancer. Bioactivation of PAHs is required in order for these agents to exhibit mutagenic properties, which is primarily mediated by cytochrome P450 enzymes. Bioactivated metabolites target multiple genomic sites, including guanine and adenine bases via PAH diol epoxides. This results in the generation of bulky BPdG chemical DNA adducts; examples include quinone-mediated cross-linking of N7 position of guanine and N3 of adenine. | [11,16] |
| Ultraviolet (UV) | Direct- and indirect-acting genotoxic cancer-causing agent, primarily absorbed by epidermal components, such as DNA bases (thymine and cytosine) and proteins. This agent is implicated in the causation of skin tumours by targeting pyrimidine bases. Exposure to the epidermis and dermis induces both the up-regulation of cell proliferation and photoproduct generation, including CPDs and (6–4) pyrimidine pyrimidines. | [5,17–19] |
| Aristolochic acid (AA) | Naturally derived acids from Aristolochiaceae plants. Ingestion of these carcinogens shown to be largely associated with nephrotoxicity of the renal cortex and further damage to the bladder and liver; very likely due to the development of bulky chemical DNA adducts. Most abundant and mutagenic form of DNA adduct associated with AA is dA-AA. In exons 2–11 of TP53, bulky chemical DNA adducts result in mutations, primarily of A:T base pairs. | [20–24] |
| Nitrosamines | Metabolism of nitrosamines subsequently induces alkylating DNA damage via the formation of DNA adducts such as O6-alkylguanine, oxidative stress and production of diazonium ions. Humans are exposed to these agents through various foods and tobacco smoke. | [25,26] |
| Mycotoxins | Mycotoxins are fungal-derived metabolites, which primarily contaminate food. The most commonly found mycotoxin is aflatoxin B1, discovered in the early 1960s. These are indirect carcinogens, which require bioactivation via CYP to generate DNA adducts. Adduct formation targeting guanine bases, which induces G → T transversions at codon 249 in TP53. | [27–29] |
| Ionising radiation (IR) | Exposure to ionising radiation induces DNA damage in an indirect or direct manner. The indirect carcinogenic effect is mediated via water radiolysis, which promotes the production of ROS resulting in oxidative damage, which can result in SSBs. The direct effect involves direct interaction of electrons with DNA resulting in molecular distortion and DSBs. | [5,6] |
| Asbestos | Asbestos is highly carcinogenic and used historically in industry and household applications. Exposure to fibres is directly linked to asbestosis, pleural plaques and mesothelioma. Dimension, shape and chemical composition are factors in asbestos pathogenicity. Damage occurs through oxidative stress (may give rise to DNA strand breaks), fibrosis and interaction with the mitotic apparatus of dividing cells. Synergism in the causation of lung cancer is seen with other mutagens, including PAHs, due to asbestos' insoluble core via which adsorbed carcinogens are delivered to target sites where they exert their genotoxic effects. | [30,31] |
| Nanoparticles (NPs) | Nanotechnology engineering has seen increasing usage of nanoparticles in medical, cosmetics and electronic industries. NPs have one dimension <100 nm, aiding cell penetration following inhalation, dermal or oral exposure with consequent ability to cause DNA damage. Damage can be direct and genotoxic effects include DNA adducts resulting from oxidative damage, epigenetic changes and DNA strand breaks. | [32–34] |
Exposure to carcinogens can either directly [7] or indirectly [1,8] induce DNA damage. Subsequent repair mechanisms may result in alterations in DNA sequences, i.e. mutations [2,9]. Induced mutations may be initiating events in cancer causation, when the damage is fixed within oncogenes or tumour suppressor genes [10]. Such risk may also be directly influenced by individual susceptibility and genetic instability [11]. For example, in the inherited genetic disorder Xeroderma Pigmentosum (XP), mutations in the XP proteins disrupt DNA repair resulting in the build-up of sunlight-induced lesions in skin DNA and a high rate of skin cancer [12].
Types of DNA damage: direct- and indirect-acting carcinogens
Carcinogens may fall into two categories: activation-dependent and activation-independent. Those which do not require metabolic activation or any molecular modification in order to induce DNA damage are termed direct-acting carcinogens and examples include nitrosamines, ultraviolet (UV), IR and alkylating agents [5,7,26,35]. These agents have the capability to interact directly with DNA and other cellular components due to their electrophilic groups. These electrophilic groups exhibit an inherent reactivity, allowing them to interact with nitrogen and oxygen atoms within negatively charged cellular macromolecules to induce molecular modifications and distortion [3]. Alteration of DNA bases causes a disarrangement of the genetic material and formation of DNA adducts depending on the type of carcinogen. Failure within DNA repair mechanisms allows DNA lesions to be inherited by daughter cells [7], eventually leading to the accumulation of DNA damage and potentially the development of cancer.
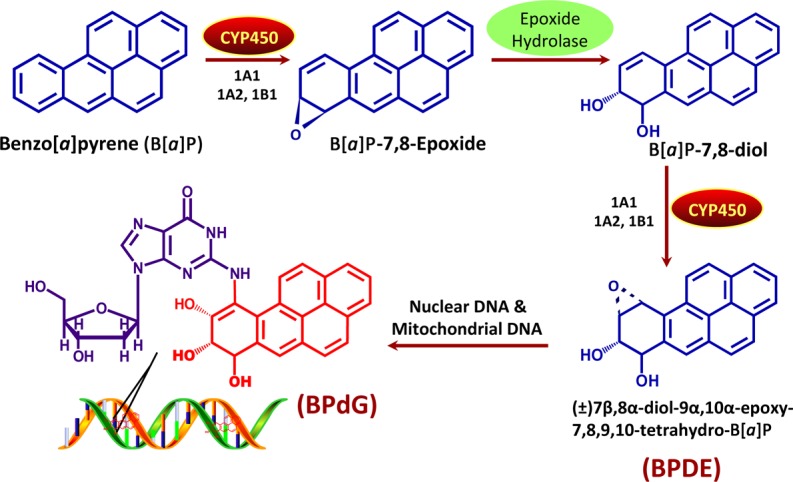
Indirect-acting carcinogens are relatively unreactive parent compounds that include polycyclic aromatic hydrocarbons (PAHs), heterocyclic aromatic amines (HAAs), N-nitrosamines, mycotoxins and aristolochic acid (AA). These typically require bioactivation in host cells to transform them into carcinogenic metabolites or reactive intermediates that are capable of exerting genotoxic effects [1,8,36]. This is often mediated by phase I and/or II metabolic reactions. Phase I reactions include oxidation, reduction or hydrolysis, mainly involving cytochrome P450 (CYP) mixed function oxidase isoforms, commonly referred to as CYPs. These enzymes have the ability to activate carcinogens independently or in conjugation with phase II enzymes such as sulfotransferases and N-acetyltransferase [8,37,38]. A classic example is the bioactivation process of benzo[a]pyrene (B[a]P), which undergoes a multi-step process involving CYP1A1 and epoxide hydrolase-mediated conversion to r7,t8-dihydroxy-t-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene (BPDE) (Figure 1). Bulky chemical adducts are commonly seen as a result of the interaction between activated carcinogens and DNA, e.g. reactive nitrenium ions formed through the reduction and hydrolysation of AA, yield bulky purine DNA adducts at the exocyclic amino group of purines [39]. Nitrosamines encompass a large diverse group of compounds formed by various combinations of amines and nitrogen functional groups. Some nitrosamines are known to be direct-acting carcinogens such as those formed in foodstuffs and are implicated in oesophageal cancer or stomach cancer, while others such as the tobacco-specific lung pro-carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone are bioactivated [40]. Other work shows that nitrosamines can be locally activated within the urothelium [41]. In contrast, α-nitrosoamino aldehydes are highly reactive compounds which are direct-acting mutagens [42]. We see the relevance of this in the high incidence of gastric cancers in certain regions associated with consumption of particular foodstuffs [43].
Figure 1. Benzo[a]pyrene (B[a]P) bioactivation. Major mechanism of DNA binding by B[a]P, a pro-carcinogenic PAH.
Although indirect carcinogens are reliant on activation, a few have the ability to enhance bioactivation by inducing changes in gene expression. PAHs such as B[a]P increase the expression in members of CYP450 family by acting as exogenous ligands of the cytosolic aryl hydrocarbon receptor (AhR)–aromatic receptor nuclear translocator complex [36,44]. Such enzymes are also involved in bioactivation of HAAs, PAHs, AA and aflatoxins, therefore potentially increasing the metabolism and subsequent exposure of DNA to reactive intermediates [1]. Expression of such enzymes has been investigated in tissues possessing the capability of bioactivating carcinogens to reactive electrophiles [45,46].
UV-induced damage
UV-induced lesions promote chemical modification and structural distortion of DNA by forming photoproducts and oxidative stress. Production of photoproducts, such as cyclobutane pyrimidine dimers (CPDs), pyrimidine-(6–4)-pyrimidone photoproducts and their dewar isomers, is achieved through the direct absorption of UVB (290–320 nm) incident photons by DNA bases and methylation of cysteine bases [47,48]. CPDs account for 75% of the mutations, which are induced by UV [47,49]. T-C and C-C CPD lesions are predominant in the tumour suppressor TP53 and in patients with skin cancer. T-T CPDs are less persistent as eradication of these dimers is induced by insertion of adenine bases by DNA repair mechanisms [17]. Lesions that are difficult to remove result in the following: stalling of DNA and RNA polymerase, reduction in DNA replication, protein synthesis and mRNA synthesis [49].
UVA (320–400 nm), a poorly absorbed radiation by DNA [50] with an unknown mutagenic effect, is suggested to be associated with promoting DNA damage by oxidative stress through an activation-independent route [17,50–55]. UVA photons absorbed by photosensitisers promote photo-oxidation reactions largely giving rise to single oxygen molecules or highly reactive electrons, which subsequently target guanine bases for hydration and deprotonation. Hydration of guanine bases promotes production of 8-oxo7,8-dihydroguanyl (8-oxodGuo) radicals which are considered to be a miscoding lesion (a lesion capable of base pairing with either a cytosine or/and adenine residue) in DNA and a marker for oxidative stress [5,51]. UV-derived 8-oxodGuo radicals have not been shown to promote G:T transversions in mammalian cells which are a common hallmark for 8-oxodGuo-induced mutations [51,52]. However, these lesions are known to cause molecular distortion by changing the structure of purine bases within DNA, but other mutagenic factors are likely to be linked with UVA-induced damage [48].
Chemical-induced bulky DNA lesions
Bulky chemical DNA adducts are formed when a reactive electrophilic carcinogen, formed by the metabolism of an indirect carcinogen, binds to a particular nucleophilic moiety in DNA. Nucleophilic targets of the reactive carcinogen include nitrogen and oxygen atoms within the bases and phosphodiester backbone of DNA. The binding of the electrophile to a nucleophile is dependent on the electrophilic strength of the carcinogen [56–58]. Common target sites on DNA include N and O of guanine or N of adenine [56] (see Table 1). Because generation of such adducts leaves the replication process prone to error, the presence of DNA adducts results in replication arrest to facilitate repair mechanisms being recruited to remove the covalently bonded chemical [2]. Unsuccessful repair of the damage often results in transversion or transition mutations. An example of this is seen with a reactive intermediate of the mycotoxin, aflatoxin B1 (AFB1). AFB1–8,9-epoxide interacts with guanine bases in hepatocyte DNA to form adducts [27,28,59]. A common result of replication errors induced by AFB1 is a transversion mutation at codon 249 in exon 7 in TP53, where guanine is substituted by thymine [59].
Oxidative damage
Oxidative-induced DNA damage is formed due to either exogenous or endogenous factors such as UVA, IR or endogenously generated oxygen molecules, which induce intracellular oxidative stress. The most prevalent sources of primarily induced oxidative stress include reactive oxygen species (ROS), such as hydroxyl radicals (OH·), singlet oxygen or reactive nitrogen species such as peroxynitrite [60]. These mutagenic species are known to interact with macromolecules causing defects in DNA synthesis and repair mechanisms, as well as inactivating various key proteins and repair enzymes. Guanine bases are the main target for these species, especially ROS [60–63]. ROS-induced damage forms modified bases, apurinic/apyrimidinic (AP) sites and single-strand breaks (SSBs). The addition of OH· at position C8 within the guanine ring generates the oxidative product, 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG) [60–62]. Similarly, the addition of OH· at position C8 of deoxyadenosine generates the oxidative product 8-oxo-7,8-dihydro-2′-deoxyadenosine (8-oxodA). These radicals are capable of further reduction or oxidation forming 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyGua) or 8-oxo-7,8-dihydroguanine (8-oxoG), in deoxyguanosine or 4,6-diamino-5-formamidopyrimidine (FapyA) or 7,8-dihydro-8-oxoadenine (8-oxoA) in deoxyadenosine. These are non-coding mutagenic DNA bases (cannot be copied by the constitutive replication machinery) [60,63]. Another prevalent oxidative product is thymine glycol, produced by the insertion of OH· at position C5 of thymine rings [63]. Similarly, another oxidation product of cytosine is cytosine glycol, which upon deamination leads to the formation of uracil glycol. These bases are removed by DNA glycosylase enzyme through base excision repair (BER) [60,61,63]. Accumulation of these modified bases over time enhances genomic structure defects and instability. For instance, thymine glycol-induced conformational alterations modify telomeres [64], with 8-oxoDG also playing a role.
Cross-linking damage
Not only do many chemotherapeutic agents (e.g. cisplatin) form DNA cross-links, but also cross-links can result from the endogenous generation of malondialdehyde and acetaldehyde, which can form either in combination or in isolation [4,65]. Cross-linking agents are often clastogens, their exposure resulting in chromosomal aberrations in the form of broken fragments of chromosomes [66]. They generate what can be pre-mutagenic lesions via the formation of covalent bonds between two nucleotide residues (e.g. two exocyclic guanine amino groups) in either the same strand (known as an intrastrand link) or opposite strands (known as interstrand links). Intrastrand links are often repaired via nucleotide excision repair (NER) and homologous recombination (HR). Intrastrand and interstrand links are generated when the cross-linking agent possesses two independent reactive groups, which then react with the DNA base groupings on either the same or the opposing strands. Common targets of the nucleotides for interstrand links include N7 of guanine or the exocyclic N2 amino group of guanine on opposite strands [4]. Interstrand links are toxic DNA lesions that inhibit replication and transcription of the affected DNA, due to the physical obstruction of strand separation [67]. If interstrand link lesions remain unrepaired in the mammalian genome, they can result in catastrophic cell breakdown [4,66,67].
Single- and double-strand breaks
SSBs occur as a consequence of endonuclease enzyme activity during base excision repair [68]. These can be induced by numerous different exogenous and endogenous elements such as UV, IR, B[a]P, mycotoxins and a vast array of intracellular reactions activating the formation of radicals or enzymes including nucleases [68–71]. The most common endogenous source associated with the formation of SSBs is the presence of OH· within cells [68,70]. OH· radicals are induced during oxidative stress and through Fenton's reaction, which involves the intracellular reduction in hydrogen peroxide (H₂O₂) by a transition ion, mostly iron²+ (Fe²+) [68,70]. OH· radicals interact with hydrogen atoms within DNA backbone causing catalysis of phosphodiester bonds resulting in the formation of phosphoglycolate and DNA lesions [70]. Carcinogen-generated radicals also trigger oxidative stress, including a series of events leading to the production of miscoding DNA bases such as thymine glycerol and activation of nucleases [68]. Nuclease activation results in a scenario that resembles the process of apoptosis because of cleavage of the DNA backbone, creating DNA strand lesions [68].
DSBs, if left unrepaired or misrepaired (or misreplicated), can result in cell death, genetic instability and carcinogenesis [72]. The formation of DSBs can be caused endogenously, e.g. during meiosis I DSBs are intentionally induced to ensure chromosomal segregation; they can also be exogenously induced by IR or chemical carcinogens [72,73]. It should be noted that other types of damage can lead to DSBs, and these are often associated with the action of exogenous agents [74]. IR is a significant external agent that induces DSBs directly and indirectly, primarily damage mediated by ROS generated by radiolysis of water. Direct induction of damage occurs when a high-energy particle collides with the phosphodiester backbone of the DNA strands, causing cleavage [56,73]. IR-mediated damage via ROS generation can be targeted by BER, thus generating SSBs. Repair of clustered damage in both strands of DNA can result in closely opposing SSBs, which then present in the form of DSBs [72]. Subsequent repair and processing of DSBs can lead to mutations, loss of heterozygosity and chromosomal translocations resulting in cell death [73].
Human studies of cancers associated with carcinogen-induced DNA damage
There are many human studies implicating environmental- and/or dietary-associated carcinogen exposures in the aetiology of cancer. Carcinogen-DNA damage formed post-combustion of tobacco implicated in the aetiology of lung cancer is a prime example [75]. Studies of migrant populations that demonstrate a generational change in susceptibility to cancers such as breast, prostate, colorectal and stomach cancer also document exposure to DNA-damaging carcinogens [76]. For many of these cancers, there can be compelling evidence, through molecular epidemiology studies, that carcinogen exposure is a pivotal causative factor [77,78]. While the pathophysiology of cancer is undoubtedly complex, many different agents can generate specific forms of DNA damage. Within the scope of this review, only a few typical examples will be highlighted.
Lung cancer and tobacco smoking
Combustion of a cigarette during smoking generates thousands of agents, many of which have the potential to be DNA damaging [79]. It is estimated, in countries where tobacco smoking is common, that 90% of lung cancer cases are directly attributed to smoking [80]; however, it is also well established that smoking-derived carcinogens induce cancers at other tissue sites [81]. Carcinogenic PAHs and tobacco-specific nitrosamines are the major carcinogens found in tobacco smoke, generated in the 900°C combustion environment at a lit cigarette tip induced when a smoker puffs. Following bioactivation of PAHs, primarily via the CYP mixed function oxidase system, their electrophilic metabolites covalently bind to nucleophilic sites on macromolecules, including DNA bases, to form bulky chemical DNA adducts. This is shown in Figure 1 for the pro-carcinogen B[a]P and its major DNA adduct r7,t8,t9-trihydroxy-c-10-(N2-deoxyguanosyl)-7,8,9,10-tetrahydrobenzo[a]pyrene (BPdG). Formation of PAH-DNA adducts can result in the induction of G-T transversions in TP53, due to DNA replication of unrepaired DNA, which produces mutations at sites of DNA adduct formation. At codon 157 in TP53 (a hotspot for mutation induction), G-T transversions are frequently seen in smokers' lung cancers, but not in never-smokers [59].
Prostate cancer and carcinogenic PAH exposure
Residents of countries such as India, China and Japan have typically been at a lower risk of prostate cancer compared with UK residents; however, risk within such ethnic groupings rises in the grandchildren of migrants from India, China or Japan to North/Western Europe, implicating environmental and/or dietary exposures as causative factors [45,46]. Normal human prostate has been shown to possess the extra-hepatic metabolic capacity of phase I and II enzymes able to bioactivate many pro-carcinogens [45]. In fact, some such as CYP1B1 are expressed at a higher level in the cancer-susceptible peripheral zone of human prostate compared with the transition zone [82]. A small cohort study of 12 UK- versus 14 India-resident individuals was undertaken to explore the level of expression of various proteins in benign prostate tissue, including CYP1B1, oestrogen receptor-alpha (ERα) and oestrogen receptor-beta (ERβ). Expression of CYP1B1, an enzyme potentially involved in the metabolic bioactivation of PAHs or HAAs, was markedly higher in UK individuals. Increased ERβ expression correlated with under-expression of CYP1B1, especially in Indian residents [46]. Figure 2 shows an immunostaining of carcinogenic PAH-DNA adducts in the peripheral zone of cancer-free prostates from the U.K. and India stained with antiserum specific for a family of carcinogenic PAHs bound to DNA, and semi-quantified by the Automated Cellular Imaging System (ACIS) [83]. The prostate tissues from India were obtained at a time (2008–2010) when radical retropubic prostatectomy was a rare procedure in this region [84]. While Figure 2B shows a marked interindividual variation in adduct levels, there is a relatively similar range of PAH-DNA adduct levels in the prostates from both groups. One might argue that these groups were very small, and may not be completely representative. Furthermore, India has, in recent years, seen an increasing Westernisation of lifestyle and these tissues would likely have been sourced from individuals from a high socio-economic class. Changes in prostate cancer incidence remain to be seen [76].
Figure 2. PAH-DNA immunostaining of human prostate.

(A) Representative example of PAH-DNA immunostaining in a UK prostate sample stained for carcinogenic PAH-DNA adducts: (left panel) specific PAH-DNA adduct staining is shown by nuclei stained pink and indicated by arrows; (middle panel) the corresponding absorbed serum control shows the same area with no staining and (right panel) haematoxylin staining of the same area shows localisation of nuclei. (B) Values for PAH-DNA adducts/108 nucleotides, for 10 prostate samples from the U.K. and 13 samples from India, were obtained from IHC using ACIS OD/nucleus values (with absorbed serum subtracted) by calculation from a standard curve [85,86].
Urothelial carcinoma and AA
Balkan endemic nephropathy (BEN) and aristolochic acid nephropathy (AAN) [formerly known as Chinese herb nephropathy (CHN)] are conditions with significant evidence of a carcinogen causing DNA damage. BEN and CHN are associated with a high incidence of upper tract urothelial carcinoma and renal failure, both caused by ingestion of AA. Exposure and subsequent metabolism (bioactivation) of AA lead to the formation of aristolactam AL-DNA adducts in urothelial tissue [87,88]. These bulky chemical DNA adducts were shown to be directly linked to A:T to T:A transversion mutations in TP53 in a study conducted by Grollman and colleagues [20,89]. There is significant evidence that AA is both a powerful nephrotoxic and carcinogenic agent with an extremely short latency period, not only in animals but also in humans [90]. In a typical human subject presenting with a urothelial malignancy 6 years post-presentation with AA-associated nephropathy, mutation analysis showed AAG → TAG mutations in codon 139 (Lys → Stop) of exon 5 of TP53 [91].
Measurements of DNA damage
Given the differing forms of DNA damage, a range of techniques measuring different endpoints have been developed. Antisera elicited against DNA adducts or carcinogen-modified DNA samples have been used to detect adducts of specific classes by immunoassay or immunohistochemistry [86]. For bulky chemical DNA adducts, where chemical characterisation is not required, the 32P-postlabelling method based on multi-dimensional thin layer chromatography has been commonly used [83,85,86]. To determine and quantify levels of DNA SSBs or DSBs, alkaline or neutral versions, respectively, of the single cell-gel electrophoresis (‘comet’) assay can be used [92,93]. Post-lysis incorporation of enzymes [formamidopyrimidine DNA glycosylase (Fpg) or 8-oxoGua DNA glycosylase (OGG1) to measure 8-oxoGua in DNA] to cleave bulky lesions or oxidative damage into SSBs can be employed to discriminate an agent's mechanism of DNA damage induction or to enhance the sensitivity of the alkaline version of the comet assay [94]. The cytokinesis-block micronucleus assay determines levels of chromosomal damage, primarily clastogenic or aneuploidy effects [95,96]. Endpoints of oxidative damage such 8-hydroxy-2′-deoxyguanosine (8-OHdG) can be determined using competitive enzyme-linked immunosorbent assay (ELISA) [97]. Highly sensitive variations in the chemical-specific mass spectrometry-based methods have been developed and used very successfully to obtain precise characterisation of DNA adducts in human tissues [98,99].
Repair mechanisms
DNA repair mechanisms [BER, NER, HR and non-homologous end-joining (NHEJ)] maintain genomic stability by eradicating DNA damage induced prior to replication completion [100]. BER is activated upon spontaneous depurination, deamination, methylation and oxidation of DNA bases. It is initiated by hydrolysis of N-glycosyl bond between deoxyribose sugar and DNA base by glycosylase enzyme, creating an abasic site (i.e. AP site) [71,100]. The AP site is cleaved by two enzymes: 5′-AP endonuclease and deoxyribose phosphodiesterase, inducing a nucleotide gap [71,100]. The gap is recovered by DNA polymerase β, which uses a template strand to introduce a new nucleotide, and DNA ligase, which stabilises and seals the phosphate-sugar backbone. An example of BER activation includes the substitution of normal DNA base by oxidative-induced base such as thymine glycol or 8-oxoguanine [67].
NER is a highly conserved process initiated by a multi-subunit complex promoting eradication of UV-induced lesions such as CPDs and bulky chemical-induced DNA adducts (see Figure 1) [71,101]. The mechanism is divided into five stages: recognition, incision, excision, DNA synthesis and DNA ligation [71]. Recognition of DNA damage occurs either due to stalling of RNA polymerase during transcription or by a repair complex, XPC-HHR23B [71,100,101]. Other repair components are recruited such as TFIIH, XPB and XPD, which perform helicase activity and unwind the DNA developing a bubble containing 24–30 nucleotides, followed by the recruitment of pre-incision components (XPA, RPA and XPG). XPF–ERCC1 incision complex removes the oligonucleotide leaving behind a gap within the DNA strand, which is synthesised and sealed by DNA polymerase δ/ε and DNA ligase [71,100,101].
There are two subtypes of NHEJ: classical (C-NHEJ) and alternative (A-NHEJ), both of which require no template for repair of DSB lesions [9]. C-NHEJ encompasses four steps: DNA end recognition, bridging and stabilising of ends, processing and ligation. This process requires heterodimer Ku, containing Ku70/80 subunits. Ku has a high affinity to DNA ends enabling it to localise and bind to the phosphate backbone situated at the break. Once bound, Ku acts as a scaffold recruiting other complexes to allow bridging and to create ligatable ends. DNA ligase IV is activated and stabilised by XRCC4—allowing ligation of the broken ends to repair the DSB [9,102]. Mutation or inhibition of C-NHEJ initiates A-NHEJ, which induces complex indels (insertion/deletion) in the repair junctions affecting genome integrity [9].
HR is predominantly involved in the repair of DSBs and also interstrand cross-links in conjunction with NER. HR utilises sister chromatids as a template ensuring genetic information is retained. Owing to the use of similar/identical nucleotide sequences as a template, the repair can only occur in the S-/G2-phases. HR follows a process of a homology search and DNA strand invasion, which is mediated by RAD51 [103]. DSBs allow for the assembly of RAD51 filaments required for the strand invasion where the invading 3′-end of a template duplex is positioned to initiate repair. The principle of HR allows the exchange of DNA preventing loss of genetic information and providing support for DNA replication in the case of broken or stalled replication forks [103,104]. The distinction between error-prone and error-free DNA repair appears to be an interplay between the DNA repair mechanism and the lesion being repaired; for instance, NHEJ has been regarded as error-prone, but this might have been overestimated, whereas HR, which is typically described as error-free, is increasingly being considered to be highly mutagenic [9].
Conclusion
Cancer is a complex multi-stage process that likely starts with an initiating mutation post-exposure to a DNA-damaging agent, which is followed by mechanisms such as inflammation [105]. However, it may be that not all mutagens are carcinogens. For instance, there are agents that test positive for mutagenicity in Salmonella typhimurium (i.e. the Ames test), but appear to be non-carcinogenic in rodents, e.g. benzene amines and substituted benzene amines [106]; the validity of these observations remains a subject of debate even after several decades. Exposure to carcinogens leads to various forms of DNA damage through indirect and direct pathways. DNA damage can also be implicated in other pathologies, such as neurodegenerative disease [107]. Identification of mutation spectra resulting from carcinogen exposures could give rise to intervention studies resulting in reduced cancer risk in certain cases [108]. For many cancers that may have a dietary and/or lifestyle component [109], there remain enormous gaps in our knowledge regarding candidate causative agents, the interaction between metabolic bioactivation to DNA-damaging species and subsequent repair of the DNA lesion, and the following processes that lead to cancer. Understanding this complex interplay is critical towards understanding the aetiology of this disease.
Acknowledgements
A bursary from UK Environmental Mutagen Society (UKEMS) funded placements for J.L.B. and M.Z. F.L.M. acknowledges invitation to present at the ‘Hydrogen Bonds & DNA: Commemoration of the 70th anniversary of the discovery by J.M. Creeth and colleagues at Nottingham in 1947’ that took place at the University of Nottingham on 10 November 2017, a Biochemical Society and RSC Chemistry Biology Interface Division Focused Meeting. The prostate immunohistochemistry studies were supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH, Bethesda, MD, U.S.A.
Abbreviations
- A-NHEJ
alternative NHEJ
- AA
aristolochic acid
- AAN
aristolochic acid nephropathy
- AFB1
aflatoxin B1
- AP
apurinic/apyrimidinic
- BEN
Balkan endemic nephropathy
- BER
base excision repair
- B[a]P
benzo[a]pyrene
- BPDE
r7,t8-dihydroxy-t-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene
- BPdG
r7,t8,t9-trihydroxy-c-10-(N2-deoxyguanosyl)-7,8,9,10-tetrahydrobenzo[a]pyrene
- C-NHEJ
classical NHEJ
- CHN
Chinese herb nephropathy
- CPDs
cyclobutane pyrimidine dimers
- CYP
cytochrome P450
- DSBs
double-strand breaks
- ERα
oestrogen receptor-alpha
- ERβ
oestrogen receptor-beta
- HAAs
heterocyclic aromatic amines
- HR
homologous recombination
- IR
ionising radiation
- NER
nucleotide excision repair
- NHEJ
non-homologous end-joining
- NP
nanoparticles
- PAHs
polycyclic aromatic hydrocarbons
- ROS
reactive oxygen species
- SSBs
single-strand breaks
- UV
ultraviolet
- XP
Xeroderma pigmentosum
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript.
References
- 1.Smith M.T., Guyton K.Z., Gibbons C.F., Fritz J.M., Portier C.J., Rusyn I. et al. (2016) Key characteristics of carcinogens as a basis for organizing data on mechanisms of carcinogenesis. Environ. Health Perspect. 124, 713–721 10.1289/ehp.1509912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chakarov S., Petkova R., Russev G.C. and Zhelev N. (2014) DNA damage and mutation. Types of DNA damage. BioDiscovery 23, 11 10.7750/BioDiscovery.2014.11.1 [DOI] [Google Scholar]
- 3.Lodish H., Berk A., Zipursky S.L., Matsudaira P., Baltimore D. and Darnell J. (eds.) (2000) DNA damage and repair and their role in carcinogenesis. In Molecular Cell Biology, 4th edn, W. H. Freeman, New York [Google Scholar]
- 4.Huang Y. and Li L. (2013) DNA crosslinking damage and cancer-a tale of friend and foe. Trans. Cancer Res. 2, 144 10.3978/j.issn.2218-676X.2013.03.01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ravanat J.L. and Douki T. (2016) UV and ionizing radiations induced DNA damage, differences and similarities. Radiat. Phys. Chem. 128, 92–102 10.1016/j.radphyschem.2016.07.007 [DOI] [Google Scholar]
- 6.Desouky O., Ding N. and Zhou G. (2015) Targeted and non-targeted effects of ionizing radiation. J. Radiat. Res. Appl. Sci. 8, 247–254 10.1016/j.jrras.2015.03.003 [DOI] [Google Scholar]
- 7.Cohen S.M. and Arnold L.L. (2010) Chemical carcinogenesis. Toxicol. Sci. 120, S76–S92 10.1093/toxsci/kfq365 [DOI] [PubMed] [Google Scholar]
- 8.Wohak L.E., Krais A.M., Kucab J.E., Stertmann J., Ovrebo S., Seidel A. et al. (2016) Carcinogenic polycyclic aromatic hydrocarbons induce CYP1A1 in human cells via a p53-dependent mechanism. Arch. Toxicol. 90, 291–304 10.1007/s00204-014-1409-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodgers K. and McVey M. (2016) Error-prone repair of DNA double-strand breaks. J. Cell Physiol. 231, 15–24 10.1002/jcp.25053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Broustas C.G. and Lieberman H.B. (2014) DNA damage response genes and the development of cancer metastasis. Radiat. Res. 181, 111–130 10.1667/RR13515.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moorthy B., Chu C. and Carlin D.J. (2015) Polycyclic aromatic hydrocarbons: from metabolism to lung cancer. Toxicol. Sci. 145, 5–15 10.1093/toxsci/kfv040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergink S., Toussaint W., Luijsterburg M.S., Dinant C., Alekseev S., Hoeijmakers J.H. et al. (2012) Recognition of DNA damage by XPC coincides with disruption of the XPC–RAD23 complex. J. Cell Biol. 196, 681–688 10.1083/jcb.201107050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ho V., Brunetti V., Peacock S., Massey T.E., Godschalk R.W., Van Schooten F.J. et al. (2017) Exposure to meat-derived carcinogens and bulky DNA adduct levels in normal-appearing colon mucosa. Mutat. Res. Mutagenesis 821, 5–12 10.1016/j.mrgentox.2017.06.005 [DOI] [PubMed] [Google Scholar]
- 14.Fahrer J. and Kaina B. (2017) Impact of DNA repair on the dose-response of colorectal cancer formation induced by dietary carcinogens. Food Chem. Toxicol. 106, 583–594 10.1016/j.fct.2016.09.029 [DOI] [PubMed] [Google Scholar]
- 15.Gibis M. (2016) Heterocyclic aromatic amines in cooked meat products: causes, formation, occurrence, and risk assessment. Compr. Rev. Food Sci. Food Saf. 15, 269–302 10.1111/1541-4337.12186 [DOI] [PubMed] [Google Scholar]
- 16.Luch A. and Baird W.M. (2010) Carcinogenic polycyclic aromatic hydrocarbons. Comp. Toxicol. 2, 850123 10.1016/B978-0-08-046884-6.01407-X [DOI] [Google Scholar]
- 17.Ichihashi M., Ueda M., Budiyanto A., Bito T., Oka M., Fukunaga M. et al. (2003) UV-induced skin damage. Toxicology 189, 21–39 10.1016/S0300-483X(03)00150-1 [DOI] [PubMed] [Google Scholar]
- 18.Kim S.I., Jin S.G. and Pfeifer G.P. (2013) Formation of cyclobutane pyrimidine dimers at dipyrimidines containing 5-hydroxymethylcytosine. Photochem. Photobiol. Sci. 12, 1409 10.1039/c3pp50037c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsumura Y. and Ananthaswamy H.N. (2004) Toxic effects of ultraviolet radiation on the skin. Toxicol. Appl. Pharmacol. 195, 298–308 10.1016/j.taap.2003.08.019 [DOI] [PubMed] [Google Scholar]
- 20.Grollman A.P., Shibutani S., Moriya M., Miller F., Wu L., Moll U. et al. (2007) Aristolochic acid and the aetiology of endemic (Balkan) nephropathy. Proc. Natl Acad. Sci. U.S.A. 104, 12129–12134 10.1073/pnas.0701248104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Attaluri S., Bonala R.R., Yang I.Y., Lukin M.A., Wen Y., Grollman A.P. et al. (2010) DNA adducts of aristolochic acid II: total synthesis and site-specific mutagenesis studies in mammalian cells. Nucleic Acids Res. 38, 339–352 10.1093/nar/gkp815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cosyns J.P. (2003) Aristolochic acid and ‘Chinese herbs nephropathy’. Drug Saf. 26, 33–48 10.2165/00002018-200326010-00004 [DOI] [PubMed] [Google Scholar]
- 23.Lord G.M., Hollstein M., Arlt V.M., Roufosse C., Pusey C.D., Cook T. et al. (2004) DNA adducts and p53 mutations in a patient with aristolochic acid-associated nephropathy. Am. J. Kidney Dis. 43, 1–18 10.1053/j.ajkd.2003.11.024 [DOI] [PubMed] [Google Scholar]
- 24.Debelle F.D., Vanherweghem J.L. and Nortier J.L. (2008) Aristolochic acid nephropathy: a worldwide problem. Kidney Int. 74, 158–169 10.1038/ki.2008.129 [DOI] [PubMed] [Google Scholar]
- 25.Mirvish S.S. (1995) Role of N-nitroso compounds (NOC) and N-nitrosation in etiology of gastric, esophageal, nasopharyngeal and bladder cancer and contribution to cancer of known exposures to NOC. Cancer Lett. 93, 17–48 10.1016/0304-3835(95)03786-V [DOI] [PubMed] [Google Scholar]
- 26.Hebels D.G., Briedé J.J., Khampang R., Kleinjans J.C. and Kok T.M. (2010) Radical mechanisms in nitrosamine-and nitrosamide-induced whole-genome gene expression modulations in Caco-2 cells. Toxicol. Sci. 116, 194–205 10.1093/toxsci/kfq121 [DOI] [PubMed] [Google Scholar]
- 27.Wang J.S. and Groopman J.D. (1999) DNA damage by mycotoxins. Mutat. Res. 424, 167–181 10.1016/S0027-5107(99)00017-2 [DOI] [PubMed] [Google Scholar]
- 28.Hamid A.S., Tesfamariam I.G., Zhang Y. and Zhang Z.G. (2013) Aflatoxin B1-induced hepatocellular carcinoma in developing countries: geographical distribution, mechanism of action and prevention. Oncol. Lett. 5, 1087–1092 10.3892/ol.2013.1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu H.C. and Santella R. (2012) The role of aflatoxins in hepatocellular carcinoma. Hepat. Monthly 12, 10 10.5812/hepatmon.7238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mott F.E. (2012) Mesothelioma: a review. Ochsner J. 12, 70–79 PMID: [PMC free article] [PubMed] [Google Scholar]
- 31.Huang S.X., Jaurand M.C., Kamp D.W., Whysner J. and Hei T.K. (2011) Role of mutagenicity in asbestos fiber-induced carcinogenicity and other diseases. J. Toxicol. Environ. Health B Crit. Rev. 14, 179–245 10.1080/10937404.2011.556051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rim K.T., Song S.W. and Kim H.Y. (2013) Oxidative DNA damage from nanoparticle exposure and its application to workers’ health: a literature review. Saf. Health Work 4, 177–186 10.1016/j.shaw.2013.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wan R., Mo Y., Zhang Z., Jiang M., Tang S. and Zhang Q. (2017) Cobalt nanoparticles induce lung injury, DNA damage and mutations in mice. Part. Fibre Toxicol. 14, 38 10.1186/s12989-017-0219-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh N., Manshian B., Jenkins G.J., Griffiths S.M., Williams P.M., Maffeis T.G. et al. (2009) Nanogenotoxicology: the DNA damaging potential of engineered nanomaterials. Biomaterials 30, 3891–3914 10.1016/j.biomaterials.2009.04.009 [DOI] [PubMed] [Google Scholar]
- 35.Kondo N., Takahashi A., Ono K. and Ohnishi T. (2010) DNA damage induced by alkylating agents and repair pathways. J. Nucleic Acids 2010, 543531 10.4061/2010/543531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walsh A.A., Szklarz G.D. and Scott E.E. (2013) Human cytochrome P450 1A1 structure and utility in understanding drug and xenobiotic metabolism. J. Biol. Chem. 288, 12932–12943 10.1074/jbc.M113.452953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sridhar J., Goyal N., Liu J. and Foroozesh M. (2017) Review of ligand specificity factors for CYP1A subfamily enzymes from molecular modeling studies reported to-date. Molecules 22, 1143 10.3390/molecules22071143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guengerich F.P. (2000) Metabolism of chemical carcinogens. Carcinogenesis 21, 345–351 10.1093/carcin/21.3.345 [DOI] [PubMed] [Google Scholar]
- 39.Turesky R.J. and Le Marchand L. (2011) Metabolism and biomarkers of heterocyclic aromatic amines in molecular epidemiology studies: lessons learned from aromatic amines. Chem. Res. Toxicol. 24, 1169 10.1021/tx200135s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Megaraj V., Zhou X., Xie F., Liu Z., Yang W. and Ding X. (2014) Role of CYP2A13 in the bioactivation and lung tumorigenicity of the tobacco-specific lung procarcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone: in vivo studies using a CYP2A13-humanized mouse model. Carcinogenesis 35, 131–137 10.1093/carcin/bgt269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roos P.H. and Bolt H.M. (2005) Cytochrome P450 interactions in human cancers: new aspects considering CYP1B1. Expert Opin. Drug Metab. Toxicol. 1, 187–202 PMID: [DOI] [PubMed] [Google Scholar]
- 42.Loeppky R.N., Tomasik W., Eisenbrand G. and Denkel E. (1987) Alpha-nitrosaminoaldehydes: highly reactive metabolites. IARC Sci. Publ. 84, 94–99 PMID: [PubMed] [Google Scholar]
- 43.Chen C.S., Pignatelli B., Malaveille C., Bouvier G., Shuker D., Hautefeuille A. et al. (1992) Levels of direct-acting mutagens, total N-nitroso compounds in nitrosated fermented fish products, consumed in a high-risk area for gastric cancer in southern China. Mutat. Res. 265, 211–221 PMID: [DOI] [PubMed] [Google Scholar]
- 44.Wang H., Yamamoto J.F., Caberto C., Saltzman B., Decker R., Vogt T.M. et al. (2010) Genetic variation in the bioactivation pathway for polycyclic hydrocarbons and heterocyclic amines in relation to risk of colorectal neoplasia. Carcinogenesis 32, 203–209 10.1093/carcin/bgq237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin F.L., Patel I.I., Sozeri O., Singh P.B., Ragavan N., Nicholson C.M. et al. (2010) Constitutive expression of bioactivating enzymes in normal human prostate suggests a capability to activate pro-carcinogens to DNA-damaging metabolites. Prostate 70, 1586–1599 10.1002/pros.21194 [DOI] [PubMed] [Google Scholar]
- 46.Singh P.B., Ragavan N., Ashton K.M., Basu P., Nadeem S.M., Nicholson C.M. et al. (2010) Quantified gene expression levels for phase I/II metabolizing enzyme and estrogen receptor levels in benign prostate from cohorts designated as high-risk (UK) versus low-risk (India) for adenocarcinoma at this organ site: a preliminary study. Asian J. Androl. 12, 203 10.1038/aja.2009.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.De Gruijl F.R., Van Kranen H.J. and Mullenders L.H. (2001) UV-induced DNA damage, repair, mutations and oncogenic pathways in skin cancer. J. Photochem. Photobiol. B 63, 19–27 10.1016/S1011-1344(01)00199-3 [DOI] [PubMed] [Google Scholar]
- 48.Bykov V.J., Hemminki K., Sheehan J.M. and Young A.R. (1999) In situ repair of cyclobutane pyrimidine dimers and 6–4 photoproducts in human skin exposed to solar simulating radiation. J. Investig. Dermatol. 112, 326–331 10.1046/j.1523-1747.1999.00523.x [DOI] [PubMed] [Google Scholar]
- 49.Sinha R.P. and Häder D.P. (2002) UV-induced DNA damage and repair: a review. Photochem. Photobiol. Sci. 1, 225–136 10.1039/B201230H [DOI] [PubMed] [Google Scholar]
- 50.Kozmin S., Slezak G., Reynaud-Angelin A., Elie C., de Rycke Y., Boiteux S. et al. (2005) UVA radiation is highly mutagenic in cells that are unable to repair 7,8-dihydro-8-oxoguanine in Saccharomyces cerevisiae. Proc. Natl Acad. Sci. U.S.A. 102, 13538–13543 10.1073/pnas.0504497102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mouret S., Baudouin C., Charveron M., Favier A., Cadet J. and Douki T. (2006) Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc. Natl Acad. Sci. U.S.A. 103, 13765–13770 10.1073/pnas.0604213103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marnett L.J. (2000) Oxyradicals and DNA damage. Carcinogenesis 21, 361–370 10.1093/carcin/21.3.361 [DOI] [PubMed] [Google Scholar]
- 53.Rastogi R.P., Kumar A., Tyagi M.B. and Sinha R.P. (2010) Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J. Nucleic Acids 2010, 592980 10.4061/2010/592980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ravanat J.L., Douki T. and Cadet J. (2001) Direct and indirect effects of UV radiation on DNA and its components. J. Photochem. Photobiol. B 63, 88–102 10.1016/S1011-1344(01)00206-8 [DOI] [PubMed] [Google Scholar]
- 55.Kappes U.P., Luo D., Potter M., Schulmeister K. and Rünger T.M. (2006) Short-and long-wave UV light (UVB and UVA) induce similar mutations in human skin cells. J. Investig. Dermatol. 126, 667–675 10.1038/sj.jid.5700093 [DOI] [PubMed] [Google Scholar]
- 56.Klaunig J.E. and Kamedulis L.M. (2010) 3.09 — Carcinogenicity. Comp. Toxicol. 3, 117–138 10.1016/B978-0-08-046884-6.00315-8 [DOI] [Google Scholar]
- 57.Munnia A., Giese R.W., Polvani S., Galli A., Cellai F. and Peluso M.E. (2017) Bulky DNA adducts, tobacco smoking, genetic susceptibility, and lung cancer risk. Adv. Clin. Chem. 81, 231–277 10.1016/bs.acc.2017.01.006 [DOI] [PubMed] [Google Scholar]
- 58.Rajalakshmi T., AravindhaBabu N., Shanmugam K. and Masthan K.M. (2015) DNA adducts-chemical addons. J. Pharm. Bioallied Sci. 7, 197 10.4103/0975-7406.155901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Robles A.I. and Harris C.C. (2010) Clinical outcomes and correlates of TP53 mutations and cancer. Cold Spring Harb. Perspect. Biol. 2, a001016 10.1101/cshperspect.a001016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kryston T.B., Georgiev A.B., Pissis P. and Georgakilas A.G. (2011) Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 711, 193–201 10.1016/j.mrfmmm.2010.12.016 [DOI] [PubMed] [Google Scholar]
- 61.Cadet J. and Davies K.J. (2017) Oxidative DNA damage and repair: an introduction. Free Radic. Biol. Med. 107, 2–12 10.1016/j.freeradbiomed.2017.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Achanta G. and Huang P. (2004) Role of p53 in sensing oxidative DNA damage in response to reactive oxygen species-generating agents. Cancer Res. 64, 6233–6239 10.1158/0008-5472.CAN-04-0494 [DOI] [PubMed] [Google Scholar]
- 63.Evans M.D., Dizdaroglu M. and Cooke M.S. (2004) Oxidative DNA damage and disease: induction, repair and significance. Mutat. Res. 567, 1–61 10.1016/j.mrrev.2003.11.001 [DOI] [PubMed] [Google Scholar]
- 64.Lee H.T., Bose A., Lee C.Y., Opresko P.L. and Myong S. (2017) Molecular mechanisms by which oxidative DNA damage promotes telomerase activity. Nucleic Acids Res. 45, 11752–11765 10.1093/nar/gkx789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Niedernhofer L.J., Daniels J.S., Rouzer C.A., Greene R.E. and Marnett L.J. (2003) Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J. Biol. Chem. 278, 31426–31433 PMID: [DOI] [PubMed] [Google Scholar]
- 66.Noll D.M., Mason T.M. and Miller P.S. (2006) Formation and repair of interstrand cross-links in DNA. Chem. Rev. 106, 277–301 10.1021/cr040478b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deans A.J. and West S.C. (2011) DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 11, 467–480 10.1038/nrc3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Horváthová E., Slameňová D., Hlinčı´ková L., Mandal T.K., Gábelová A. and Collins A.R. (1998) The nature and origin of DNA single-strand breaks determined with the comet assay. Mutat. Res. 409, 163–171 10.1016/S0921-8777(98)00053-6 [DOI] [PubMed] [Google Scholar]
- 69.Omar H.E. (2013) Mycotoxins-induced oxidative stress and disease. InTech 63–92 10.5772/51806 [DOI] [Google Scholar]
- 70.Dianov G.L. and Parsons J.L. (2007) Co-ordination of DNA single strand break repair. DNA Repair 6, 454–460 10.1016/j.dnarep.2006.10.009 [DOI] [PubMed] [Google Scholar]
- 71.Braithwaite E., Wu X. and Wang Z. (1999) Repair of DNA lesions: mechanisms and relative repair efficiencies. Mutat. Res. 424, 207–219 10.1016/S0027-5107(99)00020-2 [DOI] [PubMed] [Google Scholar]
- 72.Jeggo P.A. and Lobrich M. (2007) DNA double-strand breaks: their cellular and clinical impact. Oncogene 26, 7717–7719 10.1038/sj.onc.1210868 [DOI] [PubMed] [Google Scholar]
- 73.Cannan W.J. and Pederson D.S. (2016) Mechanisms and consequences of double-strand DNA break formation in chromatin. J. Cell. Physiol. 231, 3–14 10.1002/jcp.25048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.De Bont R. and van Larebeke N. (2004) Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 19, 169–185 PMID: [DOI] [PubMed] [Google Scholar]
- 75.Hecht S.S. (2012) Lung carcinogenesis by tobacco smoke. Int. J. Cancer 131, 2724–2732 10.1002/ijc.27816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martin F.L. (2013) Epigenetic influences in the aetiology of cancers arising from breast and prostate: a hypothesised transgenerational evolution in chromatin accessibility. ISRN Oncol. 2013, 624794 10.1155/2013/624794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martin F.L., Cole K.J., Williams J.A., Millar B.C., Harvey D., Weaver G. et al. (2000) Activation of genotoxins to DNA-damaging species in exfoliated breast milk cells. Mutat. Res. 470, 115–124 PMID: [DOI] [PubMed] [Google Scholar]
- 78.Williams J.A., Martin F.L., Muir G.H., Hewer A., Grover P.L. and Phillips D.H. (2000) Metabolic activation of carcinogens and expression of various cytochromes P450 in human prostate tissue. Carcinogenesis 21, 1683–1689 PMID: [DOI] [PubMed] [Google Scholar]
- 79.IARC (2004) Tobacco Smoke and Involuntary Smoking. International Agency for Research on Cancer (IARC) Monographs on the Evaluation of Carcinogenic Risks to Humans Volume 83, IARC, Lyon, France: [PMC free article] [PubMed] [Google Scholar]
- 80.Vargas A.J. and Harris C.C. (2016) Biomarker development in the precision medicine era: lung cancer as a case study. Nat. Rev. Cancer 16, 525–537 10.1038/nrc.2016.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sasco A.J., Secretan M.B. and Straif K (2004) Tobacco smoking and cancer: a brief review of recent epidemiological evidence. Lung Cancer 45, S3–S9 10.1016/j.lungcan.2004.07.998 [DOI] [PubMed] [Google Scholar]
- 82.Ragavan N., Hewitt R., Cooper L.J., Ashton K.M., Hindley A.C., Nicholson C.M. et al. (2004) CYP1B1 expression in prostate is higher in the peripheral than in the transition zone. Cancer Lett. 215, 69–78 PMID: [DOI] [PubMed] [Google Scholar]
- 83.Pratt M.M., John K., MacLean A.B., Afework S., Phillips D.H. and Poirier M.C. (2011) Polycyclic aromatic hydrocarbon (PAH) exposure and DNA adduct semi-quantitation in archived human tissues. Int. J. Environ. Res. Public Health 8, 2675–2691 10.3390/ijerph8072675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sinha S., Siriguri S.R., Kanakmedala S.K. and Bikkasani K. (2011) Prostate biopsy findings in Indian men: a hospital-based study. Indian J. Cancer 48, 175–180 10.4103/0019-509X.82879 [DOI] [PubMed] [Google Scholar]
- 85.John K., Ragavan N., Pratt M.M., Singh P.B., Al-Buheissi S., Matanhelia S.S. et al. (2009) Quantification of phase I/II metabolizing enzyme gene expression and polycyclic aromatic hydrocarbon-DNA adduct levels in human prostate. Prostate 69, 505–519 10.1002/pros.20898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Poirier M.C. (2016) Linking DNA adduct formation and human cancer risk. Environ. Mol. Mutagen. 57, 499–507 10.1002/em.22030 [DOI] [PubMed] [Google Scholar]
- 87.Yang H.Y., Chen P.C. and Wang J.D. (2014) Chinese herbs containing aristolochic acid associated with renal failure and urothelial carcinoma: a review from epidemiologic observations to causal inference. BioMed. Res. Int. 2014, 569325 10.1155/2014/569325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pavlović N.M. (2013) Balkan endemic nephropathy — current status and future perspectives. Clin. Kidney J. 6, 257–265 10.1093/ckj/sft049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kathuria P., Sharma P. and Wetmore S.D. (2015) Adenine versus guanine DNA adducts of aristolochic acids: role of the carcinogen–purine linkage in the differential global genomic repair propensity. Nucleic Acids Res. 43, 7388–7397 10.1093/nar/gkv701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Arlt V.M., Stiborova M. and Schmeiser H.H. (2002) Aristolochic acid as a probable human cancer hazard in herbal remedies: a review. Mutagenesis 17, 265–277 PMID: [DOI] [PubMed] [Google Scholar]
- 91.Lord G.M., Hollstein M., Arlt V.M., Roufosse C., Pusey C.D., Cook T. et al. (2004) DNA adducts and p53 mutations in a patient with aristolochic acid-associated nephropathy. Am. J. Kidney Dis. 43, e11–e17 PMID: [DOI] [PubMed] [Google Scholar]
- 92.Martin F.L., Cole K.J., Orme M.H., Grover P.L., Phillips D.H. and Venitt S. (1999) The DNA repair inhibitors hydroxyurea and cytosine arabinoside enhance the sensitivity of the alkaline single-cell gel electrophoresis (‘comet’) assay in metabolically-competent MCL-5 cells. Mutat. Res. 445, 21–43 PMID: [DOI] [PubMed] [Google Scholar]
- 93.Anderson D. and Laubenthal J. (2013) Analysis of DNA damage via single-cell electrophoresis. Methods Mol. Biol. 1054, 209–218 10.1007/978-1-62703-565-1_14 [DOI] [PubMed] [Google Scholar]
- 94.Collins A.R. (2017) The use of bacterial repair endonucleases in the comet assay. Methods Mol. Biol. 1641, 173–184 10.1007/978-1-4939-7172-5_9 [DOI] [PubMed] [Google Scholar]
- 95.Yared E., McMillan T.J. and Martin F.L. (2002) Genotoxic effects of oestrogens in breast cells detected by the micronucleus assay and the Comet assay. Mutagenesis 17, 345–352 PMID: [DOI] [PubMed] [Google Scholar]
- 96.Fenech M., Kirsch-Volders M., Natarajan A.T., Surralles J., Crott J.W., Parry J. et al. (2011) Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 26, 125–132 10.1093/mutage/geq052 [DOI] [PubMed] [Google Scholar]
- 97.Dąbrowska N. and Wiczkowski A. (2017) Analytics of oxidative stress markers in the early diagnosis of oxygen DNA damage. Adv. Clin. Exp. Med. 26, 155–166 10.17219/acem/43272 [DOI] [PubMed] [Google Scholar]
- 98.Yun B.H., Rosenquist T.A., Nikolić J., Dragičević D., Tomić K., Jelaković B. et al. (2013) Human formalin-fixed paraffin-embedded tissues: an untapped specimen for biomonitoring of carcinogen DNA adducts by mass spectrometry. Anal. Chem. 85, 4251–4258 10.1021/ac400612x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Beland F.A., Churchwell M.I., Doerge D.R., Parkin D.R., Malejka-Giganti D., Hewer A. et al. (2004) Electrospray ionization-tandem mass spectrometry and 32P-postlabeling analyses of tamoxifen-DNA adducts in humans. J. Natl Cancer Inst. 96, 1099–1104 PMID: [DOI] [PubMed] [Google Scholar]
- 100.Abbotts R. and Wilson D.M. (2017) Coordination of DNA single strand break repair. Free Radic. Biol. Med. 107, 228–244 10.1016/j.freeradbiomed.2016.11.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Spivak G. (2015) Nucleotide excision repair in humans. DNA Repair 36, 13–18 10.1016/j.dnarep.2015.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Davis A.J. and Chen D.J. (2013) DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2, 130 10.3978/j.issn.2218-676X.2013.04.02 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li X. and Heyer W.D. (2008) Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 18, 99–113 10.1038/cr.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jasin M. and Rothstein R. (2013) Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 5, a012740 10.1101/cshperspect.a012740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cooks T., Harris C.C. and Oren M. (2014) Caught in the cross fire: p53 in inflammation. Carcinogenesis 35, 1680–1690 10.1093/carcin/bgu134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zeiger E. (2001) Mutagens that are not carcinogens: faulty theory or faulty tests? Mutat. Res. 492, 29–38 PMID: [DOI] [PubMed] [Google Scholar]
- 107.Martin F.L., Williamson S.J., Paleologou K.E., Hewitt R., El-Agnaf O.M. and Allsop D. (2003) Fe(II)-induced DNA damage in α-synuclein-transfected human dopaminergic BE(2)-M17 neuroblastoma cells: detection by the Comet assay. J. Neurochem. 87, 620–630 PMID: [DOI] [PubMed] [Google Scholar]
- 108.Olivier M., Hussain S.P., Caron de Fromentel C., Hainaut P. and Harris C.C. (2004) TP53 mutation spectra and load: a tool for generating hypotheses on the etiology of cancer. IARC Sci. Publ. 157, 247–270 PMID: [PubMed] [Google Scholar]
- 109.Poirier M.C. (2012) Chemical-induced DNA damage and human cancer risk. Discov. Med. 14, 283–288 PMID: [PMC free article] [PubMed] [Google Scholar]