

Abstract
The arterial blockage in patients with peripheral arterial disease (PAD) restricts oxygen delivery to skeletal muscles distal to the blockage. In advanced-stage PAD patients, this creates a chronic ischemic condition in the affected muscles. However, in the majority of PAD patients, the muscles distal to the blockage only become ischemic during physical activity when the oxygen demands of these muscles are increased. Therefore, the skeletal muscle of most PAD patients undergoes repeated cycles of low-grade ischemia-reperfusion each time the patient is active and then rests. This has been speculated to contribute to the biochemical and morphological myopathies observed in PAD patients. The current study aimed to determine, using a rodent model, whether repeated hind limb muscle contractions during blood flow restriction to the hind limb muscles increases NF-κB activity. We, subsequently, determined whether an increase in NF-κB activity during this condition is required for the increased transcription of specific atrophy-related genes and muscle fiber atrophy. We found that hind limb muscle contractions during blood flow restriction to the limb increased NF-κB activity, the transcription of specific atrophy-related genes, and caused a 35% decrease in muscle fiber cross-sectional area. We further found that inhibition of NF-κB activity, via gene transfer of a dominant-negative inhibitor of κBα (d.n. IκBα), prevented the increase in atrophy gene expression and muscle fiber atrophy. These findings demonstrate that when blood flow to skeletal muscle is restricted, repeated cycles of muscle contraction can cause muscle fiber atrophy that requires NF-κB-IκBα signaling.
Keywords: muscle wasting, nuclear factor κB, peripheral arterial disease, ischemia
exercise claudication is a condition associated with peripheral arterial disease (PAD) that describes pain, or discomfort, in the lower limb muscles during physical activity, which subsides during rest. The pain is caused by a relative ischemia, induced by an inability to deliver the required oxygen needed to meet the demands of the working muscles due to arterial narrowing associated with PAD. During recovery, the substrate demands of the muscle are gradually reduced, and the ischemia and pain subside. Exercise claudication, then, causes low-grade ischemia-reperfusion (I-R).
Several studies show that in PAD patients I-R induced by a single bout of exercise increases plasma malondialdehyde levels and thiobarbituric acid-reactive substances—both markers of oxidative modification of lipids (18, 51). In addition, using a rodent model of exercise claudication, we have previously shown an increase in markers of protein oxidation and lipid peroxidation within skeletal muscle (24, 25). Furthermore, circulating levels of TNF-α, IL-6, IL-8, and tumor necrosis factor receptor 1 (TNFR1) are significantly higher in PAD patients compared with control subjects under resting conditions, with TNF-α and IL-6 further increasing in PAD patients, but not control subjects, following exercise (6, 13, 28, 50). Moreover, IL-6 mRNA and interleukin-1β (IL-1β) mRNA expression is increased ∼23-fold and ∼8-fold, respectively, in hypoperfused skeletal muscle of PAD patients compared with normoperfused muscle (53). Combined, these studies highlight that exercise claudication increases both circulating and muscle levels of oxidants and cytokines. Given that oxidants and cytokines can each promote muscle catabolism (3, 4, 36, 54), exercise claudication could lead to significant skeletal muscle atrophy. This is potentially very important since the skeletal muscle fiber cross-sectional area of PAD patients is significantly reduced compared with control subjects (16, 37, 43). Determining whether exercise claudication contributes to this muscle fiber atrophy is important in advancing our understanding of the causes of atrophy in PAD patients.
One major signaling pathway that is activated by both oxidants and cytokines, and is involved in the regulation of skeletal muscle mass is the nuclear factor-κB (NF-κB) pathway (15, 33, 34). The NF-κB Rel family of transcription factors consists of five members (p65, c-Rel, RelB, p50, and p52) that dimerize, and are bound and retained in the cytosol by the inhibitory factor of κBα (IκBα) (56). Activation of the classic NF-κB pathway leads to phosphorylation and degradation of IκBα, nuclear translocation of Rel dimers, and induction or repression of NF-κB target gene transcription (40). Importantly, an increase in NF-κB activity is sufficient to cause skeletal muscle atrophy (7) and is required for muscle fiber atrophy during muscle disuse (7, 21, 26) and cancer (7). NF-κB activity is known to increase in skeletal muscle during traditional I-R, in which blood flow is completely occluded and then restored (1, 35, 44). However, to our knowledge, there are no published data to suggest whether NF-κB activity is increased following the low-grade I-R associated with exercise claudication. Therefore, the current study determined, using a rat model, whether exercise claudication increases NF-κB transcriptional activity and leads to skeletal muscle fiber atrophy.
As stated above, claudication refers to the pain associated with ischemia, and the current study does not measure pain. However, we use the word claudication in the current study since it is a term specific to exercise-induced ischemic pain in PAD patients, and the model used here is a model of exercise-induced ischemia in PAD patients. Further, since this model uses electrical stimulation, rather than exercise, to cause repeated muscle contraction-induced claudication, we refer to this rodent model as contractile claudication rather than exercise claudication. We hypothesized that a single bout of contractile claudication increases NF-κB transactivation and increases the expression of specific atrophy-related genes, and that repeated bouts of contractile claudication further increases these variables leading to skeletal muscle fiber atrophy.
MATERIALS AND METHODS
Animals
Male Sprague-Dawley rats (225 g) were purchased from Charles River Laboratories (Wilmington, MA, USA) and used for all subsequent experiments. All animal procedures were approved by the University of Florida Institutional Animal Care and Use Committee.
Expression and Reporter Plasmids
The enhanced green fluorescent protein (EGFP)-c1 plasmid was obtained from Clontech (Palo Alto, CA, USA) and the dominant-negative inhibitor of κBα-EGFP [dominant-negative (d.n.) IκBα-EGFP, also referred to as IκBαΔN-EGFP] plasmid was created as previously described (26). The protein produced from this construct lacks amino acids 1–36 and, therefore, the serine 32 and serine 36 phosphorylation sites that are necessary for its subsequent degradation. The NF-κB-GL3 reporter plasmid contains a trimerized NF-κB site from the IgK light-chain enhancer inserted into the IL-2 minimal promoter, driving expression of luciferase (gift from Dr. Steffan Ho). Plasmid DNA was prepared and isolated using an endotoxin-free maxi or mega prep kit (Qiagen, Valencia, CA).
Plasmid Injection
Plasmid injection and sequential transfection of skeletal muscle have been detailed previously (38, 49). Briefly, 10 μg of EGFP or d.n. IκBα-EGFP and 40 μg of the NF-κB reporter plasmid were injected into the soleus muscle in a total volume of 50 μl in 1 × PBS. Following injection, electric pulses were delivered using an electric pulse generator (Electro square porator ECM 830; BTX) by placing two paddle-like electrodes on each side of the muscle. Five pulses were delivered in 200-ms interpulse intervals, each with an effective intensity of 100 V/cm and 20-ms duration.
Contractile Claudication Model
The limbs of animals were assigned to a control (Con) group that underwent sham surgery, a ligated-only group that had the femoral artery ligated (Lig), a stimulated group that underwent in vivo electrical stimulation, to cause repeated muscle contractions and mimic exercise (Stim), and a ligated + stimulation group that had the femoral artery ligated followed, 2 days later, by in vivo electrical stimulation (Lig + Stim). We injected the soleus muscle of each of these groups with the EGFP expression plasmid 5 days prior to ligation or sham surgery. In an additional group of animals, the solei were injected with a d.n. IκBα-EGFP plasmid, and the limbs were assigned to a Lig + Stim group.
To perform the ligation surgery, animals were anesthetized with isoflurane (5% for induction, 1.5–2.5% for maintenance, delivered via O2), a small incision was made directly above the inguinal fold, and the femoral artery was exposed and isolated by blunt dissection. For limbs in the Lig and Lig + Stim groups, two ligatures were placed tightly around the vessel, and the vessel was cut between the ties. The same procedure was performed on the sham limbs, with the exception being that the femoral artery was neither tied, nor cut.
Two days following the ligation or sham surgery, in vivo muscle contractions began using a stimulation protocol previously optimized to cause a significant decrease in force production from limbs in which the femoral artery is ligated, but not in control limbs (24, 25, 27). The decrease in force production indicates insufficient oxygen delivery to the muscles. For this procedure, animals were again anesthetized with isoflurane (5% for induction, 1.5–2.5% for maintenance, delivered via O2), placed in the prone position, and secured in a reproducible position with limited mobility of the lower leg except at the tibiotarsal joint. A stainless-steel stimulating electrode (cathode) was placed transcutaneously near the sciatic nerve midway between the posterior ischeal spine and the greater femoral trochanter. Another electrode (anode) was inserted 3 mm subdermally in the midline of the lower back. Stimulations were administered at a frequency of 2 Hz at 100 V using a Grass 48 stimulator (Grass Techonologies, West Warwick, RI).
In preliminary experiments stimulations were administered for one 30-min bout, and the soleus muscle was removed at various time points for measurement of NF-κB activity (0 min, 30 min, 60 min, 120 min, and 240 min poststimulation). In the subsequent “single bout of muscle contraction” experiments, stimulations were again administered for one 30-min bout, and the soleus muscle was removed 60 min poststimulation for measurement of NF-κB activity and 4 h following the bout for mRNA expression measurements. In the “repeated bouts of muscle contraction” experiments, stimulations were administered for 15 min, 4 times per day with 3 h between stimulation bouts, for 5 days. The soleus muscles were removed 60 min following the final bout for measurement of NF-κB activity and 4 h following the final bout for mRNA expression and muscle fiber cross-sectional area measurements.
The procedure of femoral artery ligation in the rat does not induce ischemia at rest due to the number of collateral and re-entrant vessels (48). However, during repeated muscle contractions, such as induced here via electrical stimulation, the increase in muscle blood flow is only ∼40% of the expected increase in control muscles (2). This is comparable to the increase in muscle blood flow in exercising PAD patients, which is ∼50% of the increase in exercising control subjects (41). For these reasons, the model used here has previously been used by our group, and others, to mimic exercise claudication (17, 24, 25, 27). We have previously shown, using this model, that force production from the triceps surae muscle group decreases by ∼10% in control limbs and ∼50% in ligated limbs after 15 min of stimulation (24), and by ∼15% and 70% in control and ligated limbs, respectively, after 30 mins of stimulation (24, 25, 27). The small decrease in control limbs demonstrates that the stimulation protocol is not intense.
Muscle Removal
Soleus muscles were removed under anesthesia (70 mg/kg pentobarbital sodium) and either processed immediately for RNA isolation—placed at a consistent length in OCT on a tongue depressor, which was immediately frozen in dry-ice-cooled isopentane and stored at −80°C for histochemical analysis— or frozen in liquid nitrogen and stored at −80°C for subsequent biochemical analyses.
NF-κB Reporter Activity
Following homogenization in a passive lysis buffer (Promega, Madison, WI) and centrifugation for 20 min at 5,000 g, 20 μl of the supernatant was added to 100 μl of luciferase reagent (Promega) for determination of total muscle luciferase activity, using an LMax II microplate luminometer (Molecular Devices, Sunnyvale, CA).
RNA Isolation, cDNA Synthesis, and RT-PCR
Total RNA was isolated from soleus muscles using the TRIzol-based method and cDNA synthesized using the RETROscript first-strand synthesis kit (Ambion, Austin, TX), as previously described (49). The concentration and quality of RNA were measured via the ratio of absorbance at 260 nm and 280 nm, and via agarose gel electrophoresis and staining to check for the integrity of 18 and 28S RNA. The 260/280 ratio in all samples, which ranged between 1.8 and 2.0. cDNA (5 μl), was then used as a template for real-time qPCR using the following primer sets (Applied Biosystems, Carlsbad, CA): atrogin-1 (GenBank accession no. NM_133521), Muscle RING-finger protein-1 (MuRF1; GenBank accession no. NM_080903), neuronal precursor cell-expressed developmentally downregulated 4 (Nedd4; GenBank accession no. NM_012986.1), cathepsin L (GenBank accession no. NM_013156.1), and 18S (GenBank accession no. X03205.1). The sequences that Applied Biosystems use in designing these primers is proprietary and, therefore, not available to report. TaqMan probe-based chemistry (Applied Biosystems) was used to allow detection of PCR products on a 7300 ABI instrument, and quantitation of gene expression was performed using the relative standard curve method and normalized to 18S gene expression.
Immunohistochemistry
Cross sections (10 μm) were cut with a cryostat microtome (Microm HM 550; Microm International, Walldorf, Germany) from the midbelly of the soleus muscle and fixed in 4% paraformaldehyde. The muscle sections were incubated with wheat germ agglutinin Texas Red-X conjugate (Invitrogen, Carlsbad, CA) for visualization of muscle fibers under fluorescence microscopy, and images were captured with an Olympus IX50 camera. The muscle fiber area of ∼250 fibers from each muscle was traced and measured using Image Pro Discovery software (Media Cybernetics, Bethesda, MD).
Western blot analysis
Protein concentrations of muscle homogenates were determined using a detergent-compatible assay (Bio-Rad, Hercules, CA, USA). Samples were diluted in loading buffer (Bio-Rad) containing 5% β-mercaptoethanol to achieve a protein concentration of 2 mg/ml and were heat denatured. Equal amounts of protein were loaded onto 4–15% linear gradient gels (Bio-Rad) and separated using SDS- PAGE. Proteins were transferred for 90 min at 100 V onto an Immobilon-FL polyvinylidene fluoride membrane (Millipore, Bedford, MA). The blots were blocked in PBS containing 5% milk and 0.05% Tween (blocking buffer) for 1 h at room temperature, and incubated overnight at 4°C with primary antibody diluted in blocking buffer. The following primary antibodies were used: anti-GFP (sc-8334; Santa Cruz Biotechnology, Santa Cruz, CA); anti-IκBα, 1:1,000 (sc-371; Santa Cruz Biotechnology); anti-MuRF1, 1:500 (MP3401; ECM Biosciences, Versailles, KY); anti-atrogin-1, 1:500 (AP2041; ECM Biosciences); anti-tubulin, 1:1,000 (T6074; Sigma-Aldrich, St. Louis, MO). Following a series of washes, the membranes were incubated with Alexa Fluor 680 or IRDye800 (LI-COR Biosciences, Lincoln, NE) fluorescent dye-conjugated secondary antibodies and visualized using the Odyssey infrared imaging system (LI-COR Biosciences). Relative quantification of proteins was determined by measuring the fluorescence of each lane at the appropriate molecular weight.
Statistics
All data were analyzed using a one-way ANOVA for comparisons between each group, followed by Bonferroni corrections for multiple comparisons when appropriate (GraphPad Software, San Diego, CA). All data are expressed as means ± SE, and significance was established at the P < 0.05 level.
RESULTS
Single Bout of Muscle Contractions
NF-κB activity.
Preliminary experiments were conducted to determine the optimal time point to measure NF-κB-dependent luciferase reporter activity following a 30-min single bout of contractile claudication (Fig. 1A). On the basis of these data, subsequent experiments on NF-κB-dependent reporter activity were conducted 1 h following the stimulation period. In these subsequent experiments, NF-κB activity was unchanged in muscles from the Lig only group, and muscles from the Stim only group compared with control, but increased >3-fold (P < 0.05) in muscles from the Lig + Stim group (Fig. 1B). Since IκBα is an endogenous inhibitory factor of NF-κB activity, we further sought to determine whether a decrease in IκBα expression is required for the Lig + Stim-induced NF-κB transactivation. To do this, we injected and electrotransferred a d.n. IκBα expression plasmid into the soleus muscle of a subset of rats prior to the Lig + Stim. The increased NF-κB activity in Lig + Stim muscles was completely abolished in Lig + Stim muscles expressing the d.n. IκBα. Furthermore, IκBα protein levels significantly decreased 30% in Lig + Stim muscles compared with all other groups (P < 0.05; Fig. 1C).
Fig. 1.
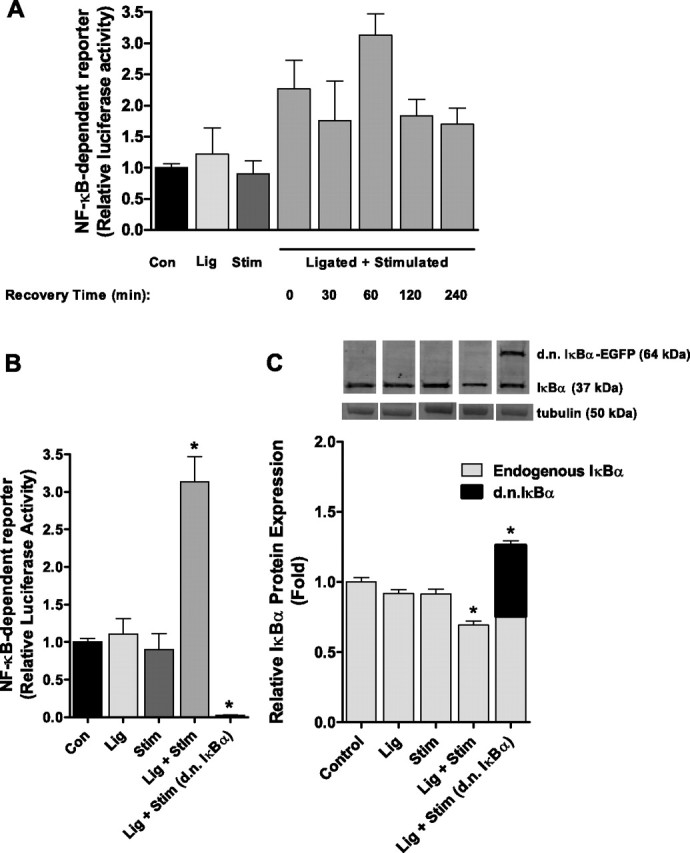
NF-κB transcriptional activity following a single bout of contractile claudication. A: Preliminary data (bars represent means ± SE of 3 muscles per group) to determine the time course of NF-κB transactivation following a single bout of contractile claudication. B: NF-κB activity is significantly increased in the soleus muscle of Lig + Stim animals 1 h following the stimulation. Expression of the dominant-negative (d.n.) IκBα in Lig + Stim muscles abolished NF-κB activity. C: Endogenous IκBα protein expression and ectopically expressed d.n. IκBα protein expression, showing a 30% decrease in endogenous IκBα protein levels in the Lig + Stim group. Data for Lig + Stim (d.n. IκBα) are presented as a stacked bar to show endogenous IκBα and d.n. IκBα-EGFP expression. Bars for B and C represent means ± SE of six muscles per group. *P < 0.05 vs. all other groups.
mRNA expression of selected genes.
Quantitative RT-PCR was used to determine whether a single bout of contractile claudication increased the expression of specific genes known to be increased in multiple models of muscle atrophy, and to determine whether the d.n. IκBα affects the expression of these genes. We measured the mRNA expression of the E3 ubiquitin ligase genes, atrogin-1/MAFbx, MuRF1, Nedd4, and the lysosomal protease cathepsin L, 4 h following the stimulation bout. Atrogin-1 mRNA expression was unchanged across all groups (Fig. 2A). However, the mRNA expression of MuRF1, Nedd4, and cathepsin L was significantly increased in the Lig + Stim group 2.3-fold, 2-fold, and 1.9-fold, respectively, compared with control (Fig. 2, B–D). The increase in MuRF1 and cathepsin L in the Lig + Stim group was also significantly different to the Lig group and to the Stim group (P < 0.05). The increase in Nedd4 in the Lig + Stim group was significantly different to the Lig group (P < 0.05), but it did not reach significance compared with the Stim group (P = 0.09). Importantly, the increases in the mRNA expression of MuRF1, Nedd4, and cathepsin L in the Lig + Stim muscles were abolished in Lig + Stim muscles expressing the d.n. IκBα (P < 0.05). This suggests that MuRF1, Nedd4, and cathepsin L are NF-κB target genes (either direct or indirect).
Fig. 2.
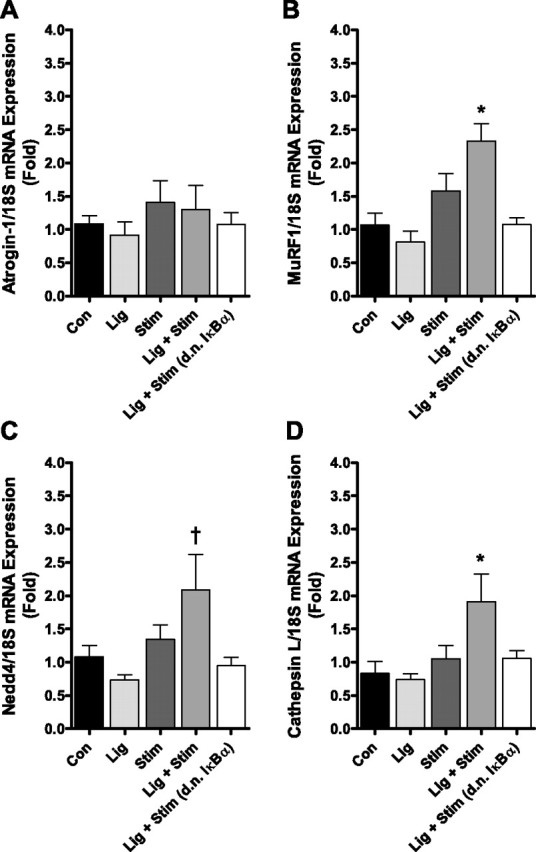
mRNA levels of atrogin-1 (A), MuRF1 (B), Nedd4 (C), and cathepsin L (D) following a single bout of contractile claudication. MuRF1, cathepsin L, and Nedd4 mRNA expression is significantly increased in the Lig + Stim group, and this increase is abolished in Lig + Stim muscles expressing the d.n. IκBα. Atrogin-1 mRNA expression is unchanged across all groups. Bars represent means ± SE of six muscles per group. *P < 0.05 vs. all other groups. †P < 0.05 vs. Con, Lig, and Lig + Stim (d.n. IκBα).
Repeated Bouts of Muscle Contraction
NF-κB activity.
Following repeated bouts of muscle contraction, NF-κB activity was unchanged in the Lig group compared with the control, but increased 6.5-fold in the Stim group and 18-fold in the Lig + Stim groups compared with control (Fig. 3A). However, this increase in NF-κB transcriptional activity in the Lig + Stim group was completely abolished in Lig + Stim muscles expressing the d.n. IκBα (Fig. 3A). These findings suggest, as with the single bout of muscle contraction, that IκBα degradation is necessary for NF-κB activation in this model of exercise claudication. Indeed, IκBα protein levels were decreased 67% in Lig + Stim muscles compared with controls (Fig. 3B).
Fig. 3.

NF-κB transcriptional activity following repeated bouts of contractile claudication. A: NF-κB activity is significantly increased in the stim group and in the Lig + Stim group, compared with all groups. The increase in NF-κB activity in the Lig + Stim muscles is abolished in Lig + Stim muscles expressing the d.n. IκBα. B: representative Western blot with one sample from each group and bar graph of mean changes from six muscles per group of endogenous IκBα protein expression (37 kDa) and ectopically expressed d.n. IκBα-EGFP protein expression (64 kDa). Endogenous IκBα protein expression is down-regulated in the Lig + Stim group. Data for Lig + Stim (d.n. IκBα) are presented as a stacked bar to show endogenous IκBα and d.n. IκBα-EGFP expression. *P < 0.05 vs. all other groups; **P < 0.01 vs. all other groups.
mRNA expression of selected genes.
Atrogin-1, MuRF1, Nedd4, and cathepsin L were significantly increased by 3.2-fold, 3.2-fold, 2.2-fold, and 3.2-fold, respectively, in the Lig + Stim group compared with control. However, the increased expression of each of these genes was abolished in Lig + Stim muscles expressing the d.n. IκBα (Fig. 4). Neither the Lig group nor the Stim group showed any significant increase in the mRNA expression of any of these genes. Furthermore, we measured the protein level of both atrogin-1 and MuRF1 and found that both were increased in the Lig + Stim group compared with control and that this increase was abolished in Lig + Stim muscles expressing the d.n. IκBα (Fig. 5). Equal protein loading was verified by probing blots for tubulin.
Fig. 4.
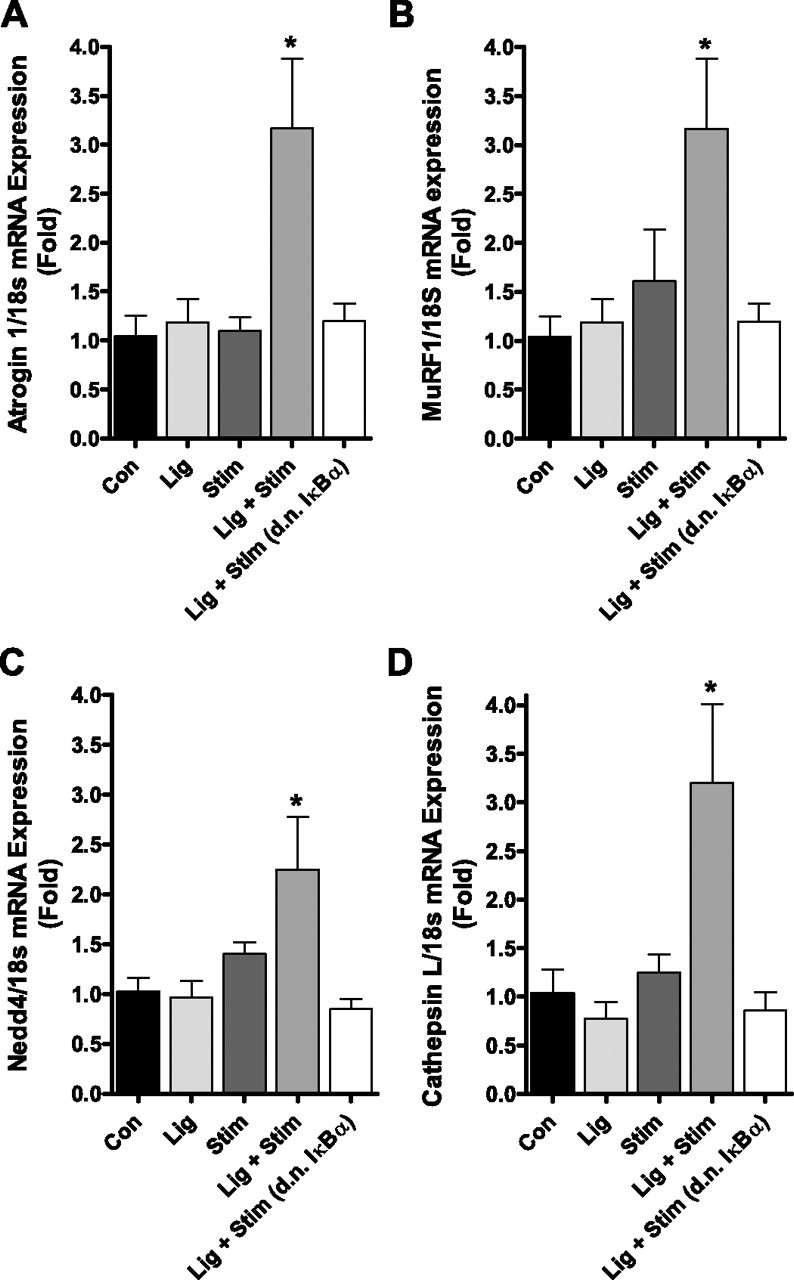
mRNA levels of atrogin-1 (A), MuRF1 (B), Nedd4 (C), and cathepsin L (D) following repeated bouts of contractile claudication. Each gene is significantly increased in the Lig + Stim group, and this increase is abolished in Lig + Stim muscles expressing the d.n. IκBα. Bars represent means ± SE of six muscles per group. *P < 0.05 vs. all other groups.
Fig. 5.
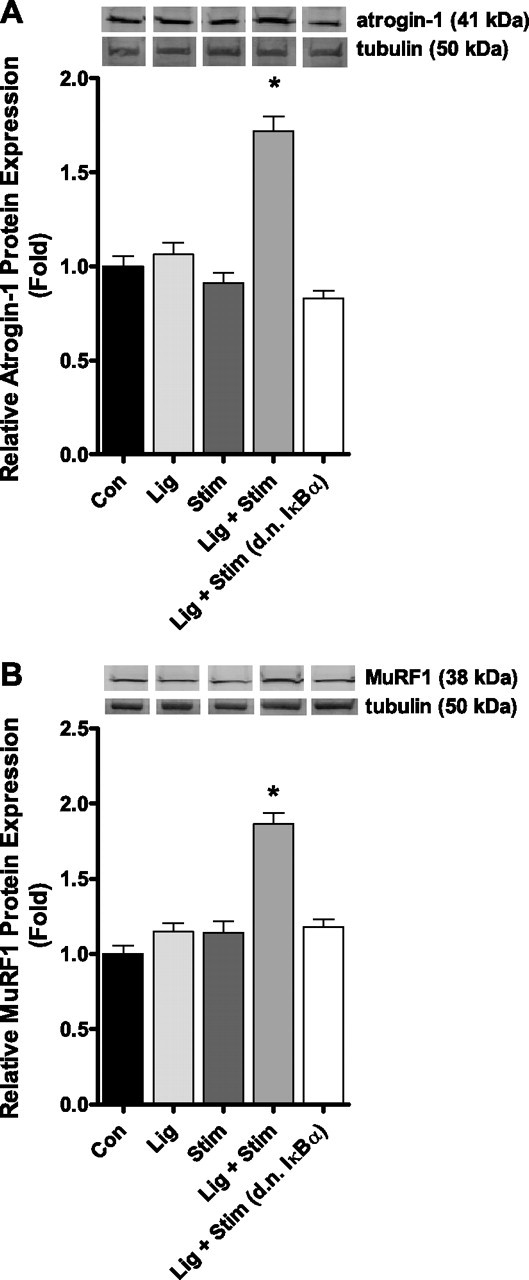
Representative Western blot with one sample from each group and bar graph of atrogin-1 (A) and MuRF1 (B) protein expression following repeated bouts of contractile claudication. Both atrogin-1 and MuRF1 protein levels are significantly increased in the Lig + Stim group, and this increase is abolished in Lig + Stim muscles expressing the d.n. IκBα. Bars represent means ± SE of six muscles per group. *P < 0.05 vs. all other groups.
Muscle fiber atrophy.
Since NF-κB is sufficient to cause skeletal muscle atrophy in vivo (7) and required for skeletal muscle atrophy associated with cancer cachexia (7) and disuse (7, 26, 55), we next sought to determine whether 1) repeated bouts of contractile claudication causes skeletal muscle atrophy, and 2) if so, whether NF-κB is required for this atrophy. Muscles in the Con, Lig, and Stim groups were injected with the EGFP-c1 plasmid, and the cross-sectional area of both green fluorescent fibers (i.e., those expressing EGFP) and black fibers (i.e., those fibers not transduced) was measured. Muscles in the Lig + Stim group were injected with the d.n. IκBα-EGFP plasmid and again the cross-sectional area of green fluorescent fibers (i.e., those expressing the d.n. IκBα-EGFP) and black fibers (i.e., those fibers not transduced) were measured. There were no differences between the mean cross-sectional area of transduced (green) or nontransduced (black) fibers in the Con, Lig, or Stim groups (Con black = 2,837 ± 176.2 μm2, Con green = 2,749 ± 90.67 μm2; Lig black = 2,872 ± 245.6 μm2, Lig green = 2,748 ± 152.3 μm2; Stim black = 2,755 ± 144.5 μm2, Stim green 2,834 ± 132.5 μm2). Five days of repeated bouts (15 min × 4 times/day) of contractile claudication (Lig + Stim group) decreased the mean fiber cross-sectional area by 35% in nontransduced fibers (i.e., black fibers, 1,834 ± 219.9 μm2). However, this decrease in muscle cross-sectional area was prevented in fibers from the Lig + Stim group expressing the d.n. IκBα-EGFP (2,942 ± 136.60 μm2). These findings clearly demonstrate that expression of EGFP alone has no effect on muscle fiber cross-sectional area, and suggest that NF-κB-IκBα signaling is required for the muscle atrophy associated with contractile claudication. No distinct evidence of muscle damage was observed in any group (Fig. 6, A–D)
Fig. 6.
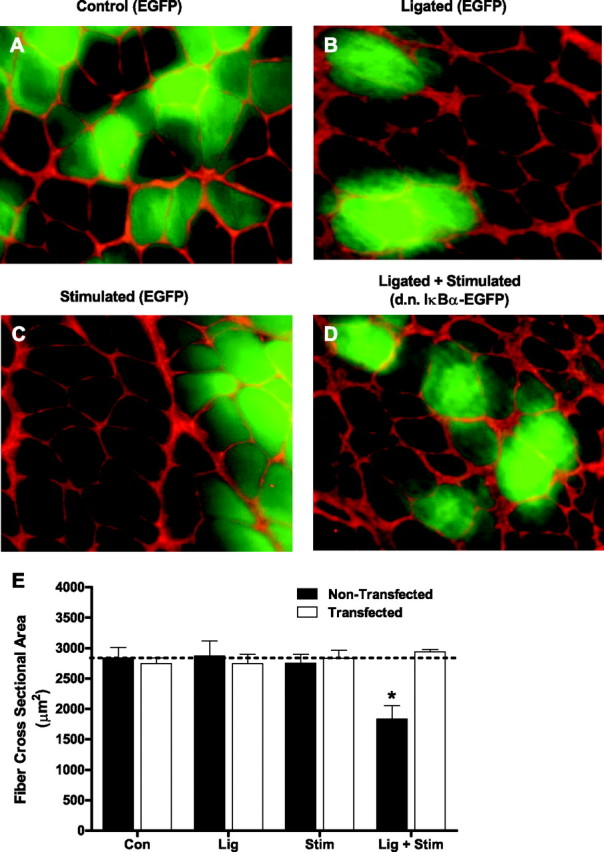
Representative cross sections of the soleus muscle from control (A), ligated (B), stimulated (C), and Lig + Stim (D) groups. Muscle fibers fluorescing green in control, ligated, and stimulated sections are expressing EGFP, whereas muscle fibers fluorescing green in Lig + Stim sections are expressing the d.n. IκBα-EGFP fusion protein. E: Muscle-fiber cross-sectional area of ∼250 fibers per muscle (including both transfected and nontransfected fibers) from six muscles per group. *P < 0.05 vs. all other groups.
DISCUSSION
Skeletal muscle mass and fiber cross-sectional area are significantly reduced in PAD patients compared with age-matched controls, which may be due to a variety of factors, including hypoxia, inactivity, hormonal imbalances, or alterations in the nutritional status of patients (10, 37, 45). In addition to these factors, there is evidence that plasma and muscle levels of cytokines and oxidants are chronically elevated in PAD patients, and further increased following exercise (6, 13, 18, 24, 28, 42, 50, 51, 53). Since both cytokines and oxidants can induce muscle protein loss through activation of various catabolic signaling pathways, these factors may also play a prominent role in the muscle atrophy associated with PAD. One such signaling pathway that is redox sensitive and potently activated by specific cytokines is the NF-κB signaling pathway (15, 33, 34), and NF-κB signaling is directly involved in the regulation of skeletal muscle mass (7, 21, 26, 55). However, to our knowledge, there are currently no data to suggest whether NF-κB activity is increased in the skeletal muscle of resting or exercising PAD patients. In the present study, we restricted blood supply to the hind limb of rodents and then superimposed repeated muscle contractions to mimic the condition of exercise in PAD patients, which we refer to here as contractile claudication. We show that NF-κB transcriptional activity is increased ∼3-fold in the soleus muscle after a single bout of contractile claudication, and increased ∼18-fold following repeated bouts of contractile claudication over 5 days. We further show, using in vivo gene transfer of a dominant-negative IκBα protein, that IκBα degradation and subsequent NF-κB activation are necessary for skeletal muscle atrophy associated with contractile claudication in our model.
NF-κB activity.
The increase in NF-κB activity following contractile claudication was expected since acute and chronic exercise (19, 20, 22, 23), and ischemia-reperfusion (1, 35, 39, 44) all increase NF-κB activity in skeletal muscle, and exercise claudication is characterized by exercise-induced ischemia followed by recovery. However, our data not only demonstrate that contractile claudication significantly increases NF-κB transcriptional activity but that the magnitude of the increase in NF-κB-dependent reporter activity is greater with repeated bouts of contractile claudication over 5 days. Furthermore, overexpression of d.n. IκBα prevented the contractile claudication-induced increase in NF-κB activity, suggesting that IκBα degradation is necessary for contractile claudication-induced NF-κB activity. The lack of an increase in NF-κB activity following a single stimulation-only bout but an increase following repeated stimulation-only bouts is presumably a reflection of the mild stimulation protocol used that only generates sufficient stress to the muscle to activate NF-κB when applied repeatedly.
Atrophy-related genes.
We also show in the current study that a single bout and repeated bouts of contractile claudication significantly increase the mRNA expression of specific genes that have been implicated in the regulation of muscle mass during various physiological conditions (26, 32, 52). However, the increased expression level of these atrophy genes is abolished in contractile claudicant muscles expressing the d.n. IκBα. This confirms that, as previously shown (26), these genes are either direct or indirect NF-κB target genes. Atrogin-1, MuRF1, and Nedd4 are all E3 ubiquitin ligases that ubiquitinate specific protein substrates that are subsequently degraded by the lysosome or proteasome (8). Although the identification of targets of the various ubiquitin ligases in skeletal muscle is still in its infancy, some targets of atrogin-1, MuRF1, and Nedd4 have been identified.
Atrogin-1 and MuRF1 have received significant attention since the discovery that their independent absence in skeletal muscle significantly spares muscle mass during denervation (5). Atrogin-1 is now known to ubiquitinate and promote the degradation of eukaryotic initiation factor 3 subunit 5 (eIF3-f) (31) and suppresses MyoD transcriptional activity (30), both of which could lead to decreased muscle protein.
MuRF1 may play a significant role in promoting the degradation of myofibrillar proteins since myosin heavy chain (9), myosin-binding protein C (MyBP-C) (11), and myosin light chains 1 and 2 each undergo MuRF1-dependent degradation (11). Given that these myofibrillar proteins are involved in muscle contraction, their loss could affect muscle function. Therefore, the contractile claudication-induced increase in atrogin-1 and MuRF1 mRNA levels could be responsible, in part, for the decreased muscle fiber size discovered in the present study.
Nedd4 is associated with muscle atrophy, during which it ubiquitinates and down-regulates Notch1 (29). While the physiological implications of Notch1 down-regulation in whole muscle during an atrophy condition are not clear, in adult skeletal muscle, Notch1 is required for satellite cell activation and proliferation and muscle regeneration (12). Given that the skeletal muscle of PAD patients are characterized by centralized nuclei (an indicator of muscle regeneration) (43), it is likely that regeneration is ongoing in the muscles of PAD patients and an increase in Nedd4, and subsequent decrease in Notch1, could negatively affect this. Cathepsin L is involved in the degradation of proteins in the lysosomes and is increased in multiple models of skeletal muscle atrophy, including disuse, cancer, sepsis, diabetes, and starvation (14, 26, 32, 46). However, the specific role of cathepsin L in the regulation of skeletal muscle mass is as yet unidentified.
The three-fold increase in NF-κB activity following a single bout of exercise claudication appears to be sufficient to increase MuRF1, Nedd4, and cathepsin L mRNA levels since the d.n. IκBα abolished NF-κB activity and the increased transcription of these genes. However, the 6.5-fold increase in NF-κB activity following repeated stimulations only was not associated with the increased mRNA level of these genes. This discrepancy could be due to the specific NF-κB Rel protein dimers and/or cofactors that MuRF1, Nedd4, and cathepsin L require for their transcription. Contractile claudication, but not contraction (stimulation) alone, may preferentially form dimers of Rel proteins and/or increase the presence or availability of cofactors that these genes require for their increased transcription. Alternatively, differential epigenetic modification of histone proteins could explain the discrepancy. For example, acetylation of histones by acetyltransferase proteins promotes the unwinding of DNA and increased accessibility of gene promoters, whereas deacetylation of histones by deacetylases promotes the rewinding of DNA and gene silencing (47). If the cellular acetylation:deacetylation balance were tipped toward acetylation during contractile claudication, but toward deacetylation during contraction alone, this could promote chromatin relaxation at the MuRF1, Nedd4, and cathepsin L promoters only during contractile claudication. Under this scenario, despite comparable increases in NF-κB reporter activity, increased accessibility of these atrophy gene promoters to Rel proteins would only occur during contractile claudication. Clearly, this speculation warrants much further investigation.
Muscle fiber atrophy.
The significant decrease in skeletal muscle fiber cross-sectional area found in the current study demonstrates that skeletal muscle fiber atrophy can occur following 5 days of repeated contractile claudication, as a model of exercise claudication. Therefore, although the presence of PAD and/or its associated comorbidities may contribute to the muscle atrophy in PAD patients, our findings, here, suggest that repeated bouts of exercise claudication may also be a major factor in the progression of muscle wasting in PAD patients. These data are in agreement with our previous findings, using the same model, that showed contractile claudication increases markers of oxidative damage in skeletal muscle (24, 25, 27). Our results are also in conjunction with data from exercising PAD patients demonstrating that exercise increases plasma cytokine and oxidant levels (6, 13, 18, 28, 50, 51). On the basis of these findings, our results suggest that exercise claudication can have, at least short term, detrimental effects. Therefore, factors such as duration, frequency, and intensity of exercise should be considered when prescribing exercise to PAD patients to maximize the cardiovascular benefits of exercise, while minimizing the potentially detrimental systemic and muscular effects.
We also found that IκBα degradation and NF-κB activation are necessary for the muscle fiber atrophy associated with contractile claudication in our rodent model. Although we hypothesized that inhibition of NF-κB activity would attenuate muscle fiber atrophy because NF-κB is required for muscle fiber atrophy during muscle disuse (7, 21, 26, 55) and during cancer cachexia (7), the complete prevention of muscle fiber atrophy was unexpected. This suggests that either NF-κB is the sole activated signaling pathway leading to muscle fiber atrophy in our model or that inhibition of NF-κB activation subsequently represses the activation of an additional atrophy-inducing signaling pathway.
In conclusion, contractile claudication, in a rodent model, significantly increases NF-κB transcriptional activity and the expression level of specific atrophy-related genes in skeletal muscle, and causes skeletal muscle fiber atrophy. Furthermore, gene transfer of d.n. IκBα abolished the increase in NF-κB activity, the increased transcription of atrophy genes and muscle fiber atrophy associated with contractile claudication, demonstrating that IκBα degradation is necessary for these processes.
Perspectives and Significance
The findings of the current study, using a rodent model, suggest that repeated cycles of exercise claudication may play a role in the progression of muscle atrophy. This has potentially important implications for PAD patients who undergo repeated cycles of physical activity-induced claudication each day, possibly for years. However, since exercise provides important overall cardiovascular benefits, it is important to better understand the influence of exercise on the skeletal muscle pathophysiology in PAD patients. Therefore, future work using both animal models and human tissue will further delineate the combined influence of exercise and blood flow restriction on skeletal muscle pathophysiology, so that novel and appropriate therapies may be developed.
GRANTS
This work was supported by a James & Esther King Biomedical Research Program, Florida Department of Health Grant 08KN-07 (to A. R. Judge).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
ACKNOWLEDGMENTS
We thank Brittany Gagnon and Jason Liounakos for technical assistance and Sarah Senf and Dr. Sarah Reed for editorial comments.
REFERENCES
- 1. Andrade-Silva AR , Ramalho FS , Ramalho LN , Saavedra-Lopes M , Jordao AA , Vanucchi H , Piccinato CE , Zucoloto S. Effect of NFκB inhibition by CAPE on skeletal muscle ischemia-reperfusion injury. J Surg Res : 254–262, 2009. [DOI] [PubMed] [Google Scholar]
- 2. Angersbach D , Jukna JJ , Nicholson CD , Ochlich P , Wilke R. The effect of short-term and long-term femoral artery ligation on rat calf muscle oxygen tension, blood flow, metabolism and function. Int J Microcirc Clin Exp : 15–30, 1988. [PubMed] [Google Scholar]
- 3. Anker SD , Chua TP , Ponikowski P , Harrington D , Swan JW , Kox WJ , Poole-Wilson PA , Coats AJ. Hormonal changes and catabolic/anabolic imbalance in chronic heart failure and their importance for cardiac cachexia. Circulation : 526–534, 1997. [DOI] [PubMed] [Google Scholar]
- 4. Argiles JM , Busquets S , Lopez-Soriano FJ. The pivotal role of cytokines in muscle wasting during cancer. Int J Biochem Cell Biol : 2036–2046, 2005. [DOI] [PubMed] [Google Scholar]
- 5. Bodine SC , Latres E , Baumhueter S , Lai VK , Nunez L , Clarke BA , Poueymirou WT , Panaro FJ , Na E , Dharmarajan K , Pan ZQ , Valenzuela DM , DeChiara TM , Stitt TN , Yancopoulos GD , Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science : 1704–1708, 2001. [DOI] [PubMed] [Google Scholar]
- 6. Brevetti G , De Caterina M , Martone VD , Ungaro B , Corrado F , Silvestro A , de Cristofaro T , Scopacasa F. Exercise increases soluble adhesion molecules ICAM-1 and VCAM-1 in patients with intermittent claudication. Clin Hemorheol Microcirc : 193–199, 2001. [PubMed] [Google Scholar]
- 7. Cai D , Frantz JD , Tawa NE , Melendez PA , Oh BC , Lidov HG , Hasselgren PO , Frontera WR , Lee J , Glass DJ , Shoelson SE. IKKβ/NF-κB activation causes severe muscle wasting in mice. Cell : 285–298, 2004. [DOI] [PubMed] [Google Scholar]
- 8. Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J : 7151–7160, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clarke BA , Drujan D , Willis MS , Murphy LO , Corpina RA , Burova E , Rakhilin SV , Stitt TN , Patterson C , Latres E , Glass DJ. The E3 Ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab : 376–385, 2007. [DOI] [PubMed] [Google Scholar]
- 10. Clyne CA , Mears H , Weller RO , O'Donnell TF. Calf muscle adaptation to peripheral vascular disease. Cardiovasc Res : 507–512, 1985. [DOI] [PubMed] [Google Scholar]
- 11. Cohen S , Brault JJ , Gygi SP , Glass DJ , Valenzuela DM , Gartner C , Latres E , Goldberg AL. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J Cell Biol : 1083–1095, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Conboy IM , Rando TA. The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev Cell : 397–409, 2002. [DOI] [PubMed] [Google Scholar]
- 13. DePalma RG , Hayes VW , May PE , Cafferata HT , Mohammadpour HA , Brigg LA , Chow BK , Shamayeva G , Zacharski LR. Statins and biomarkers in claudicants with peripheral arterial disease: cross-sectional study. Vascular : 193–200, 2006. [DOI] [PubMed] [Google Scholar]
- 14. Deval C , Mordier S , Obled C , Bechet D , Combaret L , Attaix D , Ferrara M. Identification of cathepsin L as a differentially expressed message associated with skeletal muscle wasting. Biochem J : 143–150, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dodd SL , Gagnon BJ , Senf SM , Hain BA , Judge AR. ROS-mediated activation of NF-κB and Foxo during muscle disuse. Muscle Nerve : 110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hedberg B , Angquist KA , Henriksson-Larsen K , Sjostrom M. Fibre loss and distribution in skeletal muscle from patients with severe peripheral arterial insufficiency. Eur J Vasc Surg : 315–322, 1989. [DOI] [PubMed] [Google Scholar]
- 17. Hickey NC , Hudlicka O , Gosling P , Shearman CP , Simms MH. Intermittent claudication incites systemic neutrophil activation and increased vascular permeability. Br J Surg : 181–184, 1993. [DOI] [PubMed] [Google Scholar]
- 18. Hickman P , Harrison DK , Hill A , McLaren M , Tamei H , McCollum PT , Belch JJ. Exercise in patients with intermittent claudication results in the generation of oxygen derived free radicals and endothelial damage. Adv Exp Med Biol : 565–570, 1994. [DOI] [PubMed] [Google Scholar]
- 19. Ho RC , Hirshman MF , Li Y , Cai D , Farmer JR , Aschenbach WG , Witczak CA , Shoelson SE , Goodyear LJ. Regulation of IκB kinase and NF-κB in contracting adult rat skeletal muscle. Am J Physiol Cell Physiol : C794–C801, 2005. [DOI] [PubMed] [Google Scholar]
- 20. Hollander J , Fiebig R , Gore M , Ookawara T , Ohno H , Ji LL. Superoxide dismutase gene expression is activated by a single bout of exercise in rat skeletal muscle. Pflügers Arch : 426–434, 2001. [DOI] [PubMed] [Google Scholar]
- 21. Hunter RB , Kandarian SC. Disruption of either the Nfkb1 or the Bcl3 gene inhibits skeletal muscle atrophy. J Clin Invest : 1504–1511, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ji LL , Gomez-Cabrera MC , Steinhafel N , Vina J. Acute exercise activates nuclear factor (NF)-κB signaling pathway in rat skeletal muscle. FASEB J : 1499–1506, 2004. [DOI] [PubMed] [Google Scholar]
- 23. Ji LL , Gomez-Cabrera MC , Vina J. Exercise and hormesis: activation of cellular antioxidant signaling pathway. Ann NY Acad Sci : 425–435, 2006. [DOI] [PubMed] [Google Scholar]
- 24. Judge AR , Dodd SL. Oxidative damage to skeletal muscle following an acute bout of contractile claudication. Atherosclerosis : 219–224, 2003. [DOI] [PubMed] [Google Scholar]
- 25. Judge AR , Dodd SL. Xanthine oxidase and activated neutrophils cause oxidative damage to skeletal muscle after contractile claudication. Am J Physiol Heart Circ Physiol : H252–H256, 2004. [DOI] [PubMed] [Google Scholar]
- 26. Judge AR , Koncarevic A , Hunter RB , Liou HC , Jackman RW , Kandarian SC. Role for IκBα, but not c-Rel, in skeletal muscle atrophy. Am J Physiol Cell Physiol : C372–C382, 2007. [DOI] [PubMed] [Google Scholar]
- 27. Judge AR , Selsby JT , Dodd SL. Antioxidants attenuate oxidative damage in rat skeletal muscle during mild ischaemia. Exp Physiol : 479–485, 2008. [DOI] [PubMed] [Google Scholar]
- 28. Kirk G , Hickman P , McLaren M , Stonebridge PA , Belch JJ. Interleukin-8 (IL-8) may contribute to the activation of neutrophils in patients with peripheral arterial occlusive disease (PAOD). Eur J Vasc Endovasc Surg : 434–438, 1999. [DOI] [PubMed] [Google Scholar]
- 29. Koncarevic A , Jackman RW , Kandarian SC. The ubiquitin-protein ligase Nedd4 targets Notch1 in skeletal muscle and distinguishes the subset of atrophies caused by reduced muscle tension. FASEB J : 427–437, 2007. [DOI] [PubMed] [Google Scholar]
- 30. Lagirand-Cantaloube J , Cornille K , Csibi A , Batonnet-Pichon S , Leibovitch MP , Leibovitch SA. Inhibition of atrogin-1/MAFbx mediated MyoD proteolysis prevents skeletal muscle atrophy in vivo. PLoS One : e4973, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lagirand-Cantaloube J , Offner N , Csibi A , Leibovitch MP , Batonnet-Pichon S , Tintignac LA , Segura CT , Leibovitch SA. The initiation factor eIF3-f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J : 1266–1276, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lecker SH , Jagoe RT , Gilbert A , Gomes M , Baracos V , Bailey J , Price SR , Mitch WE , Goldberg AL. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J : 39–51, 2004. [DOI] [PubMed] [Google Scholar]
- 33. Li YP , Reid MB. NF-κB mediates the protein loss induced by TNF-α in differentiated skeletal muscle myotubes. Am J Physiol Regul Integr Comp Physiol : R1165–R1170, 2000. [DOI] [PubMed] [Google Scholar]
- 34. Li YP , Schwartz RJ , Waddell ID , Holloway BR , Reid MB. Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-κB activation in response to tumor necrosis factor-α. FASEB J : 871–880, 1998. [DOI] [PubMed] [Google Scholar]
- 35. Lille ST , Lefler SR , Mowlavi A , Suchy H , Boyle EM , Farr AL , Su CY , Frank N , Mulligan DC. Inhibition of the initial wave of NF-κB activity in rat muscle reduces ischemia/reperfusion injury. Muscle Nerve : 534–541, 2001. [DOI] [PubMed] [Google Scholar]
- 36. McClung JM , Judge AR , Talbert EE , Powers SK. Calpain-1 is required for hydrogen peroxide-induced myotube atrophy. Am J Physiol Cell Physiol : C363–C371, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McDermott MM , Hoff F , Ferrucci L , Pearce WH , Guralnik JM , Tian L , Liu K , Schneider JR , Sharma L , Tan J , Criqui MH. Lower extremity ischemia, calf skeletal muscle characteristics, and functional impairment in peripheral arterial disease. J Am Geriatr Soc : 400–406, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mitchell-Felton H , Kandarian SC. Normalization of muscle plasmid uptake by Southern blot: application to SERCA1 promoter analysis. Am J Physiol Cell Physiol : C1269–C1276, 1999. [DOI] [PubMed] [Google Scholar]
- 39. Park JW , Qi WN , Cai Y , Urbaniak JR , Chen LE. Proteasome inhibitor attenuates skeletal muscle reperfusion injury by blocking the pathway of nuclear factor-κB activation. Plast Reconstr Surg : 1808–1818, 2007. [DOI] [PubMed] [Google Scholar]
- 40. Pereira SG , Oakley F. Nuclear factor-κB1: regulation and function. Int J Biochem Cell Biol : 1425–1430, 2008. [DOI] [PubMed] [Google Scholar]
- 41. Pernow B , Saltin B , Wahren J , Cronestrand R , Ekestroom S. Leg blood flow and muscle metabolism in occlusive arterial disease of the leg before and after reconstructive surgery. Clin Sci Mol Med : 265–275, 1975. [DOI] [PubMed] [Google Scholar]
- 42. Pipinos II , Judge AR , Zhu Z , Selsby JT , Swanson SA , Johanning JM , Baxter BT , Lynch TG , Dodd SL. Mitochondrial defects and oxidative damage in patients with peripheral arterial disease. Free Radic Biol Med : 262–269, 2006. [DOI] [PubMed] [Google Scholar]
- 43. Pipinos II , Swanson SA , Zhu Z , Nella AA , Weiss DJ , Gutti TL , McComb RD , Baxter BT , Lynch TG , Casale GP. Chronically ischemic mouse skeletal muscle exhibits myopathy in association with mitochondrial dysfunction and oxidative damage. Am J Physiol Regul Integr Comp Physiol : R290–R296, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Qi WN , Chaiyakit P , Cai Y , Allen DM , Chen LE , Seaber AV , Urbaniak JR. NF-κB p65 involves in reperfusion injury and iNOS gene regulation in skeletal muscle. Microsurgery : 316–323, 2004. [DOI] [PubMed] [Google Scholar]
- 45. Regensteiner JG , Wolfel EE , Brass EP , Carry MR , Ringel SP , Hargarten ME , Stamm ER , Hiatt WR. Chronic changes in skeletal muscle histology and function in peripheral arterial disease. Circulation : 413–421, 1993. [DOI] [PubMed] [Google Scholar]
- 46. Sacheck JM , Hyatt JP , Raffaello A , Jagoe RT , Roy RR , Edgerton VR , Lecker SH , Goldberg AL. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J : 140–155, 2007. [DOI] [PubMed] [Google Scholar]
- 47. Saha RN , Pahan K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ : 539–550, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Seifert FC , Banker M , Lane B , Bagge U , Anagnostopoulos CE. An evaluation of resting arterial ischemia models in the rat hind limb. J Cardiovasc Surg (Torino) : 502–508, 1985. [PubMed] [Google Scholar]
- 49. Senf SM , Dodd SL , McClung JM , Judge AR. Hsp70 overexpression inhibits NF-κB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J : 3836–3845, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Signorelli SS , Mazzarino MC , Di Pino L , Malaponte G , Porto C , Pennisi G , Marchese G , Costa MP , Digrandi D , Celotta G , Virgilio V. High circulating levels of cytokines (IL-6 and TNFα), adhesion molecules (VCAM-1 and ICAM-1) and selectins in patients with peripheral arterial disease at rest and after a treadmill test. Vasc Med : 15–19, 2003. [DOI] [PubMed] [Google Scholar]
- 51. Silvestro A , Scopacasa F , Oliva G , de Cristofaro T , Iuliano L , Brevetti G. Vitamin C prevents endothelial dysfunction induced by acute exercise in patients with intermittent claudication. Atherosclerosis : 277–283, 2002. [DOI] [PubMed] [Google Scholar]
- 52. Stevenson EJ , Giresi PG , Koncarevic A , Kandarian SC. Global analysis of gene expression patterns during disuse atrophy in rat skeletal muscle. J Physiol : 33–48, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Testa M , De Ruvo E , Russo A , Citterio F , Serino F , Mangoni A , Capogrossi MC , Sperti G. Induction of interleukin-1β and interleukin-6 gene expression in hypoperfused skeletal muscle of patients with peripheral arterial disease. Ital Heart J : 64–67, 2000. [PubMed] [Google Scholar]
- 54. Valdez H , Lederman MM. Cytokines and cytokine therapies in HIV infection. AIDS Clin Rev 187–228, 1997. [PubMed] [Google Scholar]
- 55. Van Gammeren D , Damrauer JS , Jackman RW , Kandarian SC. The IκB kinases IKKα and IKKβ are necessary and sufficient for skeletal muscle atrophy. FASEB J : 362–370, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zandi E , Rothwarf DM , Delhase M , Hayakawa M , Karin M. The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell : 243–252, 1997. [DOI] [PubMed] [Google Scholar]