

Abstract
Objective
To identify the mechanism of unexplained hyponatremia and primary polydipsia in schizophrenia and its relationship to the underlying psychiatric illness.
Methods
Briefly review previous studies that led to the conclusion the hyponatremia reflects altered hippocampal inhibition of peripheral neuroendocrine secretion. In greater detail, present the evidence supporting the hypothesis that circuit dysfunction associated with the hyponatremia and the polydipsia contributes to the underlying mental disorder.
Results
Polydipsic patients with and without hyponatremia exhibit enhanced neuroendocrine responses to psychological stress in proportion to structural deformations on their anterior hippocampus, amygdala and anterior hypothalamus. Nonpolydipsic patients exhibit blunted responses and deformations on other hippocampal and amygdala surfaces. The deformations in polydipsic patients are also proportional to diminished peripheral oxytocin levels and impaired facial affect recognition that is reversed by intranasal oxytocin. The anterior hippocampus is at the hub of a circuit that modulates neuroendocrine and other responses to psychological stress and is implicated in schizophrenia. Preliminary data indicate other measures of stress reactivity are also enhanced in polydipsics and that the functional connectivity of the hippocampus with the other structures in this circuitry differs in schizophrenia patients with and without polydipsia.
Conclusion
Polydipsia may identify a subset of schizophrenia patients whose enhanced stress reactivity contributes to their mental illness. Stress reactivity may be a symptom dimension of chronic psychosis that arises from circuit dysfunction that can modeled in animals. Hence polydipsia could be a biomarker that helps to clarify the pathophysiology and heterogeneity of psychosis as well as identify novel therapies. Clinical investigators should consider obtaining indices of water balance, as these may help them unravel and more concisely interpret their findings. Basic researchers should assess if the polydipsic subset is a patient group particularly suitable to test hypotheses arising from their translational studies.
Keywords: hippocampus, stress, neuroendocrine, heterogeneity, oxytocin, vasopressin, polydipsia
1.0 Overview of impaired water excretion, water intoxication and polydipsia
In the first half of the 20th century, unexplained impaired water excretion (Pfister, 1938; Targowla, 1923) and life-threatening hyponatremia (Barahal, 1938) were observed in patients with chronic psychosis and noted to coincide with psychotic exacerbations. Studies at that time also demonstrated that primary polydipsia was the major unexplained physiologic abnormality in those with severe mental illness (Hoskins and Sleeper, 1933; Sleeper and Jellinek, 1936). During the latter half of the 20th century, these unexplained impairments in water excretion and intake were recognized as common causes of morbidity and a frequent cause of death in schizophrenia patients (De Leon et al., 1994; Hawken et al., 2009; Vieweg et al., 1985). More recent studies suggest these patients can be relatively easily distinguished from patients whose hyponatremia is attributable to recognized causes (Atsariyasing and Goldman, 2014; Ittaskul and Goldman, 2014). Primary polydipsia and evidence of impaired water excretion are found in 10–25% and 2–5% of chronic psychotic patients respectively, depending on the criteria used to define water imbalance. All of the patients with unexplained hyponatremia appear to have a primary polydipsia (hence about 1/5th of polydipsic patients exhibit impaired water excretion), though the relationship, if any, between the two disorders was initially unknown. See Supplemental Figure 1 for a primer on water imbalance and Glossary for a definition of terms.
Here, we first briefly summarize studies characterizing the mechanism of the impaired water excretion and its relationship to acute psychosis in hyponatremic polydipsic psychotic patients. Next, we briefly review studies indicating that these patients, and to a somewhat lesser extent those polydipsic patients without impaired water excretion (i.e. normonatremic polydipsics), exhibit diminished anterior hippocampal restraint of peripheral neuroendocrine secretion during psychological stress. This is the opposite of the response seen in nonpolydipsic patients. This work is summarized in greater detail in Goldman, 2009.
The last part of the manuscript reviews evidence these findings are a facet of circuit dysfunction emanating from the anterior hippocampus (AH). Evidence that this dysfunction contributes to the underlying mental disorder, by altering central neuroendocrine activity and/or enhancing stress reactivity, is presented. In addition, we review evidence that nonpolydipsic patients differ markedly on many of these measures suggesting the pathophysiology(ies) of the mental disorders in polydipsic and nonpolydipsic patients likely differ. The mechanism of the polydipsia and the implications of these findings to current efforts to characterize basic dimensions of behavioral functioning and to ‘deconstruct’ schizophrenia are then discussed. Additional background on the pathophysiology of water imbalance for the interested reader is provided in Supplemental Information.
1.1 Reset osmostat and its aggravation by psychosis
Hariprasad (et al., 1980) provided evidence that hyponatremic polydipsic patients have impaired water excretion attributable to a subtle disorder of antidiuretic function called reset osmostat. Several studies subsequently provided further evidence of reset osmostat in this population and indicated it was attributable to elevated activity of the antidiuretic hormone, arginine vasopressin (AVP) (Delva et al., 1990; Kishimoto et al., 1989; Ohsawa et al., 1993; Vieweg et al., 1986, 1990) (see Supplemental Figure 2 for further information about reset osmostat). A concurrently conducted study of osmoregulation, which also carefully measured or controlled recognized influences on AVP function, confirmed the finding and showed resetting could not be attributed to the recognized non-osmotic stimuli for AVP release (Goldman et al., 1988). Subsequent studies further substantiated this conclusion and found that other recognized and putative factors (e.g. oropharyngeal regulation, antipsychotics) could not account for the findings (reviewed in Goldman, 2009). These studies also demonstrated that the resetting appeared to be ameliorated by habituation to the clinical environs (Goldman et al., 1996) and confirmed they were worsened by psychotic exacerbations (Figure 1) (Goldman et al., 1997). Recent trials with highly specific AVP antagonists substantiate the conclusion that the variability in the hyponatremia, and thus the resetting, is attributable to enhanced AVP activity (Jossiasen et al. 2008, 2012). Together these findings provided a plausible physiologic explanation for the original observations linking acute psychosis to impaired water excretion and the life-threatening hyponatremia in hyponatremic polydipsic schizophrenic patients (Goldman, 2009). The mechanism, however, of the resetting and its aggravation by acute psychosis remained unknown.
Fig. 1.

Acute psychosis aggravates reset osmostat in hyponatremic polydipsic patients. Following three weeks of optimal treatment on an inpatient psychiatric research unit, osmoregulation (dotted lines) in six hyponatremic polydipsic (red) and eight matched normonatremic polydipsic (blue) patients was similar and near normal. A transient psychotic exacerbation was then induced with intravenous methylphenidate. While severity of induced psychosis was similar, peak arginine vasopressin (AVP) plasma levels were higher in those subjects with hyponatremia (solid squares) despite their lower sodium levels. Moreover, as the figure illustrates, these peak AVP levels in the hyponatremic subjects (but not the normonatremics half-filled squares) were proportional to concurrent plasma sodium (solid red line: PNa= 10.2PAVP + 114 mEq/L; r = 0.80), consistent with reset osmostat. The resetting appears so severe (i.e. new set point = 114mEq/l) that it would induce water intoxication in the presence of even modest polydipsia. Thus these findings provide a plausible explanation for the observations first made in the early 20th century that acute psychosis in some chronic psychotic patients impairs water excretion and contributes to water intoxication (Data derived from Goldman et al., 1996, 1997). See Supplemental Information for primer on water imbalance and reset osmostat. Normal range of AVP relative to concurrent plasma osmolality is shown in grey.
AVP is a tightly-regulated and measurable neuropeptide released directly from the brain into the peripheral blood stream where it has precise quantifiable actions (increases urine osmolality) reflecting a precise CNS modulated-function (osmoregulation). In reset osmostat, the precision of the system is unaffected (e.g. plasma osmolality accounts for about 80–90% of the variance in AVP levels), just the set point for AVP release is lowered (Figure 1). Many factors (i.e. non-osmotic stimuli) can account for this lowering, some of which are associated with limbic functions (Robertson, 2006). The accessibility and precision of these various components of water balance offer a powerful tool for identifying and characterizing aberrations in CNS function attributable to a non-osmotic stimulus. This is one reason the neurohypophysial system has been described as a “veritable Rosetta stone for neuroendocrinology and neuroscience” (Gainer et al., 2002).
2.0 Reset osmostat and enhanced HPAA and AVP responses to psychologic stress
The association between habituation and acute psychosis to the AVP set point resembled the association between novelty and acute psychosis to variations in other neurohormone levels previously observed in psychiatric patients and attributed to non-specific psychological stress (Coccaro et al., 1984). This observation raised the possibility that psychological stress could be the non-osmotic stimulus that caused the reset osmostat. Consistent with this interpretation, hypothalamic-pituitary-adrenal axis (HPAA) activity (a generally reliable measure of psychological stress) was elevated on admission in polydipsic relative to nonpolydipsic patients (Goldman, et al., 1993).
The problem with this interpretation, however, is that AVP secretion is insensitive to psychological stress in humans (Edelson and Robertson, 1986) and other mammals (Onaka and Yagi,1988). This insensitivity was attributed to neural inhibition by limbic pathways (Onaka and Yagi, 1990) and in this manner resembled the constraint of HPAA secretion by the hippocampus during psychological stress. The HPAA constraint involved a projection from the ventral subiculum (the rodent analogue of the lateral anterior hippocampus (AH) in primates) to the anterior hypothalamus (Herman et al., 1998). Indeed, this same projection enervated adjacent neurons in the anterior hypothalamus responsible for peripheral AVP secretion (Risold and Swanson, 1996; Tasker et al., 1998). Lesion studies demonstrated that lesioning this pathway enhanced both peripheral AVP and HPAA responses to psychological stimuli (Nettles et al., 2000) (see Figure 2 for model). This observation was reproduced in an animal model of schizophrenia that disrupts the neurodevelopment of this hippocampal segment (Chrapusta et al., 2003; Mitchell and Goldman, 2004), substantiating the possibility that this pathway may be altered in persons with schizophrenia.
Fig. 2.
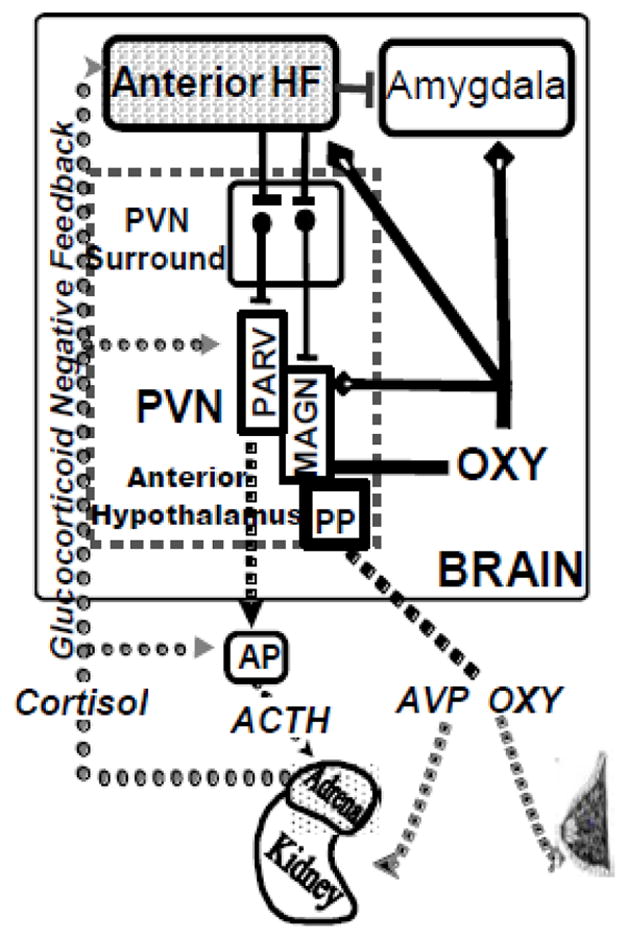
Proposed neurocircuit dysfunction underlying enhanced peripheral neuroendocrine responses to psychologic stress in polydipsic schizophrenia patients. The earlier findings suggested the enhanced AVP release in hyponatremic polydipsic patients might be related to acute psychologic stress, however, psychological, unlike physical, stress does not normally increase AVP release. The figure shows the circuitry which appears to be responsible for inhibiting AVP and HPAA axis release during acute psychological stress.
Neuroendocrine secretion directly into the peripheral circulation occurs from magnocellular neurons (MAGN) which secrete vasopressin (AVP) and oxytocin (OXY). The closely related neuropeptides regulate water excretion and lactation, respectively. ACTH secretagogues are released from adjacent parvacelllular neurons (PVN) into the neurohypophyseal circulation where they modulate HPAA activity by inducing ACTH release from the anterior pituitary. Projections from the lateral anterior hippocampus (AH) relay in the ‘PVN surround’ and terminate in the (paraventricular) PVN and supraoptic nuclei (not shown) of the anterior hypothalamus where they normally inhibit AVP and HPAA axis secretion during psychological stress. Evidence suggests this pathway also may enhance peripheral OXY release. Glucocorticoid negative feedback contributes to the HPAA restraint particularly in the pathway emanating from the AH. The enhanced AVP and HPAA activity observed in polydipsic schizophrenia patients appears to result from disrupted lateral AH function. See Goldman, 2009 for further details.
This lateral AH dysfunction could be responsible for core features of the psychiatric illness if it also disrupts central neuroendocrine activity. Dendrites on MAGN also secrete AVP and OXY directly into the brain where these peptides modulate stress responses and social functions in particular by binding to receptors in the amygdala and hippocampus. In general, the central effects of these peptides oppose each other. The dendritic secretion of OXY parallels peripheral release particularly during psychological stress. This dendritic secretion appears responsible for much of the OXY in the brain. The impact of the hippocampal pathway on central OXY release is unknown but we propose its disruption in polydipsic patients may reduce both peripheral and central oxytocin activity.
The potential relevance of these observations to the impaired water excretion in hyponatremic patients was assessed in a series of studies in four subject groups (matched polydipsic schizophrenic patients with and without hyponatremia, nonpolydipsic schizophrenic patients and healthy controls). The first study compared neuroendocrine responses to physical and psychological stress. Results demonstrated that AVP responses to psychological (but not physical) stress were a) enhanced in hyponatremic polydipsic patients relative to the three other groups, were b) similar in healthy controls and normonatremic polydipsic patients and were c) blunted in nonpolydipsic patients relative to the other three groups (Goldman et al., 2007a). The peripheral HPAA responses were similarly ordered except that they were greater in normonatremic polydipsic patients than health controls. The effect of psychological stress on AVP in the hyponatremic group resembled the effect of acute psychosis, i.e. it further lowered the set point for AVP release.
In the second study, glucocorticoid negative feedback was assessed under conditions sensitive to hippocampal-modulated constraint of HPAA release. Feedback was markedly blunted in the hyponatremic polydipsic patients; intermediately blunted in the normonatremic polydipsic patients; and enhanced in the nonpolydipsic patients relative to healthy controls (Goldman et al., 2007b). These findings complemented those of the first study and both were consistent with diminished hippocampal inhibition of peripheral neuroendocrine secretion (Figure 2).
2.1 Enhanced neuroendocrine responses and lateral anterior hippocampal dysfunction
The third study measured the volume of the anterior and posterior hippocampus in the four groups. Based on the previous HPAA results, we predicted anterior, but not posterior, hippocampal volume would be smaller in the hyponatremic than normonatremic polydipsics, whose anterior volumes, in turn, we predicted would be larger in normonatremic polydipsics than the healthy controls. The results confirmed anterior (but not posterior) hippocampal volumes were smaller in hyponatremic polydipsic patients. Volumes, however, but did not differ in the other three groups and thus were not consistent with the view that the relatively elevated HPAA responses in the polydipsics or the diminished responses in nonpolydipsics were attributable to imageable hippocampal pathology (Goldman et al., 2007c).
A subsequent reanalysis assessing hippocampal shape, however, resolved this disparity and provided even more interesting results. Both polydipsic groups exhibited deformations on the lateral AH surface (more marked in those with hyponatremia) while the nonpolydipsic group exhibited deformations on other hippocampal surfaces relative to healthy controls (Goldman et al., 2011a) (Figure 3). Furthermore, the deformations on the lateral AH were proportional to the enhanced HPAA and AVP responses to stress. Together, the findings offered a plausible mechanism for not only the varying AVP set point in the hyponatremic patients but the enhanced HPAA activity in both polydipsic groups. Furthermore, the finding that nonpolydipsic patients exhibited deformations on other hippocampal surfaces raised the possibility that heterogeneity in schizophrenia might be partly attributable to which hippocampal segments were affected (Goldman, 2009).
Fig. 3.

Lateral anterior hippocampal deformations in polydipsic patients. Panel A shows the average surface map of the bilateral hippocampi in polydipsic hyponatremic schizophrenia patients relative to healthy controls while Panel B shows the map in this group relative to those without polydipsia. Inward deformations on the lateral anterior surface (blue) are apparent in the hyponatremic patients in both panels. Panels C and D focus on the significant groups differences by mapping the extremes of the 5th eigenvector obtained after reducing shape variation to 5 unique patterns (eigenvectors) with principal components analysis. Only the fifth eigenvector differed and the magnitude of the differences was ordered hyponatremic polydipsic> normonatremic polydipsic> healthy control> nonpolydipsic (P<.01). The maximum magnitude reflects findings in polydipsic hyponatremics and the minimum reflects findings in nonpolydipsic patients. Note the blue areas in C reflect prominent inward deformations on the anterior lateral surface representative of the polydipsic hyponatremics, while those in D show inward deformations on other hippocampal surfaces representative of nonpolydipsic patients. The magnitude of these deformations were proportional to those in amygdala and anterior hypothalamic structures which also contribute to neuroendocrine responses to psychological stress. In addition, these deformations predict group differences in peripheral HPAA and AVP responses, as well as basal oxytocin levels providing neural correlates of the peripheral neuroendocrine dysfunction.
2.1 Involvement of other limbic structures implicated in neuroendocrine regulation
The shape analyses also revealed deformations adjacent to the 3rd ventricle and on the right dorsal medial surface of the amygdala in the two polydipsic groups. Again these were more marked in those polydipsic patients with hyponatremia than normonatremia and were completely absent in nonpolydipsic patients (Goldman et al., 2011a). The deformations in the 3rd ventricle were adjacent to the supraoptic (SON) and paraventricular nuclei (PVN) (i.e. hypothalamic nuclei which regulate AVP and HPA activity) (Figure 2). The dorsal medial surface of the amygdala overlies the medial and central nuclei which are interconnected with both the lateral AH and anterior hypothalamic structures adjacent to the SON and PVN (i.e. periventricular surround). Indeed, the extent of the size of the deformations in left AH, right amygdala and 3rd ventricle covaried significantly with each other in the polydipsics, and furthermore the amygdala and 3rd ventricle deformations were correlated with peripheral AVP and HPAA responses to the psychological stressor, respectively. The findings remained significant even when group effects were partialed out (Goldman et al., 2011a). These observations extended the AH dysfunction to other limbic structures involved in modulating neuroendocrine responses to stress (Figure 4).
Fig. 4.

The anterior hippocampus is the at hub of a circuit whose dysfunction could contribute to schizophrenia in the subset of patients with polydipsia. Earlier work demonstrated structural deformations on the anterior hippocampus, amygdala and anterior hypothalamus could underlie the altered peripheral neuroendocrine findings in polydipsic schizophrenics. The findings could also account for core features of the psychiatric illness if they alter central OXY secretion from the anterior hypothalamus or its actions on the hippocampus and amygdala.
In addition, the AH has direct and significant connections with the nucleus accumbens and medial prefrontal cortex to other structures also implicated in and schizophrenia. Together these structures form a circuit which determines stress reactivity by modulating both the impact of stress and the efficacy of coping behaviors.
3.0 Are peripheral findings related to central dysfunction underlying mental disorder?
The findings could explain the elevated neuroendocrine activity in polydipsic patients, but did not establish a relationship to the underlying psychiatric disorder. Possible links between the findings and the mental illness are suggested by the fact the deformations occurred in structures previously implicated in schizophrenia (Heckers, 2001; Rosenfeld et al., 2011; Schobel et al., 2009; Shenton et al., 2001), and that enhanced responses to psychological stress have been implicated in schizophrenia (Myin-Germeys et al. 2003; van Venrooi et al. 2012; Venables, 1977; Walker and Diforio, 1997). Hippocampal function varies markedly along both its medial-lateral as well as its anterior-posterior axes (Faneslow and Dong, 2010; Mitchell and Goldman, 2004). By this reasoning, lateral AH pathology could result in psychosis by disrupting one set of hippocampal-modulated functions, while deformations on other hippocampal surfaces could disrupt other functions. In this model, psychosis is analogous to fever or rash, i.e. a clear but nonspecific indicator of hippopcampal dysfunction (Keshevan et al., 2013).
3.1 Oxytocin secretion from the hypothalamus into the periphery and the brain
Disruption of central neuroendocrine activity is one plausible means by which the hippocampal and other deformations in polydipsics could influence their mental disorder. Disruption of central oxytocin (OXY) is an appealing candidate given its impact on social functioning in healthy controls and at least some patients with schizophrenia (Gumley et al., 2014). Social function is particularly impaired in polydipsic patients (Bralet et al., 2007). OXY is a nonapeptide closely related to AVP. Both are secreted into the periphery as well as directly into the brain from neurons in the anterior hypothalamus (Arakawa, et al., 2010; Legros, 2001) (Figure 2). Anatomic and electrophysiologic studies suggest the anterior hippocampus also influences peripheral OXY secretion (Herman et al., 2002; Risold and Swanson, 1996; Tasker et al., 1998), though this has not been directly studied. The influence of the AH on central OXY release is unknown. Both oxytocin and AVP are active in the brain and influence social functioning and interpersonal behavior as well as stress reactivity (Heinrichs, 2003; Insel, 2010a). The two hormones’ actions generally oppose each other, leading one investigator to call them ‘ying and yang’ hormones (Legros, 2001). The evidence indicates diminished central OXY or increased central AVP could enhance stress reactivity and impair social function (Insel, 2010a).
OXY’s appeal also stems from the fact that, unlike AVP, its peripheral and central release covary (Landgraf and Neumann, 2004) (Figure 2), particularly during psychological stress in laboratory animals (Wotjak et al., 1998). Furthermore, again unlike AVP, the central secretion of OXY from the hypothalamus likely accounts for most of the OXY in the brain (Landgraf and Neumann, 2004). Others note that peripheral OXY levels predict several central oxytocin-modulated social functions in healthy normals (Feldman et al., 2010, 2012) as well as persons with schizophrenia (Kéri et al., 2009), providing additional evidence that the peripheral and central activity are linked.
3.2 Structural deformations and evidence of altered peripheral and central oxytocin activity
To begin to explore the possibility that the structural deformations contribute to the psychiatric disorder by altering central OXY function, plasma OXY levels and an oxytocin-modulated social function (facial affect recognition) were first assessed in the four groups noted above. Impaired facial affect recognition parallels other deficits in social function in schizophrenia and is associated with negative symptoms (Addington et al., 2010; Rosenfeld et al., 2011). As predicted, OXY levels in both polydipsic groups were lower than in either healthy controls or nonpolydipsic patients. Moreover, the drop was proportional to their impaired facial affect recognition (Goldman et al., 2008) as well as their deformations on the left AH and bilateral amygdalae (Goldman et al., 2011a).
3.3 Intranasal oxytocin restores social deficits associated with diminished peripheral levels
To further assess the possible role of diminished central oxytocin in social deficits in polydipsic patients, polydipsic and nonpolydipsic patients were given two doses of intranasal oxytocin as well as placebo on three separate occasions. Intranasal oxytocin crosses into the brain and evidence suggests it increases central oxytocin activity (Neumann et al., 2013). Previous studies indicate intranasal oxytocin improves facial affect recognition (Marsh et al., 2010; Insel, 2010a), particularly for fear (Fischer-Shofty et al., 2010, 2013). As predicted, the results demonstrated that facial affect recognition was more impaired in the polydipsic group, and that they responded more favorably to oxytocin (with the higher dose) (Goldman et al., 2011b). In particular, fear recognition was nearly normalized in those with polydipsia. Together, these observations suggest that the structural deformations also disrupt central neuroendocrine function, which in turn could contribute to the psychiatric disorder(Figure 4).
3.4 Altered central oxytocin receptor activation is also possible
The above findings suggest that the structural deformations diminish central OXY levels, but are not inconsistent with the possibility of diminished receptor response. OXY influences social function and stress reactivity through receptors in both the amygdala (Insel, 2010a; Kirsch et al., 2005; Rosenfeld et al., 2011) and hippocampus (Montag et al., 2012). Impaired fear recognition, which was nearly normalized with supplemental OXY in polydipsics, is proportional to altered amygdala activity in schizophrenia (Gur et al., 2007; Rosenfeld et al., 2011) and variations in the oxytocin receptor have been associated with impaired social functioning in schizophrenia (Cohen et al., 2010). Hence, it is plausible that the deformations reflect impaired action of OXY rather than, or in addition to, diminished secretion (Figure 4).
4.0 Does anterior hippocampal pathology induce a stress diathesis?
For many years, researchers have postulated that chronic psychotic patients are more vulnerable than healthy normals to negative psychological stimuli (Myin-Germeys et al. 2003; van Venrooi et al. 2012; Venables, 1977; Walker and Diforio, 1997). In addition to disrupting central neuroendocrine activity, the neuroendocrine findings could be one component of such a stress diathesis. Measures of autonomic nervous system activity provide preliminary evidence that enhanced stress reactivity extends beyond the neuroendocrine system in polydipsic patients. Autonomic nervous system (ANS) responses to emotional stimuli, particularly skin conductance responses (SCRs), are also modulated by the hippocampus and amygdala (Critchley, 2002; LeDoux, 1992). Previous studies have linked enhanced SCRs to the putative stress diathesis (Ohman et al., 1981). We explored whether SCRs to affective stimuli were increased in polydipsic patients (Goldman et al., 2012).
Results revealed SCRs were elevated in polydipsic patients for negative emotions but were non-significantly diminished for the single positive emotion that was assessed (happy) (Figure 5). In particular, the SCRs to fear were significantly elevated relative to the SCRs to happy in polydipsic compared to nonpolydipsic patients (i.e. group X emotion interaction) (Goldman et al., 2012). This enhanced SCR response to negative emotions in polydipsic patients could reflect enhanced stress reactivity and thus provides preliminary evidence of increased stress reactivity.
Fig. 5.

Preliminary evidence of a more generalized stress diathesis in polydipsic patients. The figure illustrates skin conductance responses to emotional stimuli in five polydipsic and six nonpolydipsic patients. Subjects were asked to assess the intensity of four emotions seen in pictures of faces (10/emotion). SCRs were generally higher in the polydipsics (P <0.10), except for happiness. In particular, the relative intensity of the SCRs to fear and to happiness differed in the two groups (P < 0.05), consistent with the view that these patients have an enhanced response to negative emotional stimuli.
4.1 Significance of blunted stress reactivity in nonpolydipsic patients
Nonpolydipsic patients constitute about 85% of those with schizophrenia, and thus it seems likely the nonpolydipsic subjects included in the studies reviewed above are more representative of the schizophrenia population at large. Unlike those with polydipsia, the nonpolydipsics exhibited blunted responses to psychological stress which is consistent with previous reports of diminished neuroendocrine (Bradley and Dinan, 2010, van Venrooi et al., 2012) and SCR (Ohman, 1981; Venables, 1977) responses to psychological stimuli in schizophrenia. While some argue that this blunting in schizophrenia is abnormal (van Venrooi et al., 2012), the evidence suggests it is more likely an adaptive response to the chronic stress of having schizophrenia. Chronic psychological stress normally blunts HPAA responses to future stresses in humans (Elzinga et al., 2008; MacMillan et al., 2009; Miller et al., 2007). Furthermore, this blunting is correlated with measures of coping ability (Kudielka et al., 2009). A similar relationship of blunted HPAA responses to stress with coping ability, as well as with social functioning, is observed in schizophrenia patients (Brenner et al., 2011; Ikezawa et al., 2012; Straube, 1979). Thus, the blunting in the nonpolydipsic patients may be an indication of the normal response to psychological stress in schizophrenia patients, further underscoring the finding of the enhanced responses in those with polydipsia.
Of particular interest is that Brenner (et al., 2011) did not observe any benefits of coping ability on HPAA response in those schizophrenia patients with enhanced, in contrast to diminished, HPAA responses. Unfortunately, Brenner (et al., 2011) did not report measures of fluid intake in his subjects, as this could provide further evidence of a stress diathesis attributable to AH dysfunction in the polydipsic subset. The ventral subiculum has a projection to the nucleus accumbens which is critical for modulating the salutary benefits of coping on stress reactivity (Aggleton, 2012; Valenti et al., 2011; Gill and Grace, 2011). Hence, the AH may be critical to modulating the salutary benefits of coping behaviors. The accumbens is another limbic structure closely associated with stress reactivity and schizophrenia (Grace, 2012). Coping behaviors, per se, appear to be generated by the medial prefrontal cortex (Deutch et al., 1990; Franklin et al., 2012, Lataster et al., 2014) which also shares direct connections with the AH (Aggleton, 2012) and is associated with stress reactivity and schizophrenia (Goldman and Mitchell, 2004; Grace, 2012). These observations raise the possibility that disrupted interactions between the AH and these other structures could contribute to a stress diathesis (Figure 4).
4.2 Evidence of circuit impairments in hippocampal-modulated stress reactivity
To examine this possibility, we utilized resting state functional magnetic resonance imaging (rs-fMRI) to estimate the connectivity of the hippocampus to the other structures illustrated in Figure 4. Rs-fMRI provides a measure of neural circuit activity (Pettersson-Yeo et al., 2011; Unschuld et al., 2013) which is correlated with task-evoked responses and underlying anatomic connectivity (Fornito et al., 2012; Gu et al., 2010). Functional connection strength is typically estimated by measuring the covariation of the spontaneous changes in blood oxygen level dependent (BOLD) activity between different neural structures.
Subjects in our pilot study were seven polydipsic patients, nine nonpolydipsic patients and nine healthy normals. Preselected ROIs included the four bilateral structures and one midline structure (9 ROIs) illustrated in Figure 4, and four other structures (8 ROIs) (caudate, putamen, pallidum, thalamus) which constitute subcortical components of the cortical-striatal-pallidal-thalamic loops frequently implicated in schizophrenia. BOLD activity in each voxel in these 17 ROIs was correlated with that of every other voxel in that ROI as well as all the voxels in the other 16 ROIs. The r values associated with the resulting 153 connections were Z transformed and then analyzed with mixed model linear regression. The analysis determined if overall connectivity differed; if connections between the hippocampus and other 15 structures differed, and if the connections between the hippocampus and other structures implicated in stress reactivity (Figure 4) differed across groups.
Mean connectivity for all 153 connections between and within the 17 ROIs was lower in the healthy controls compared to the two groups of schizophrenia patients (Z = 5.63, P <0.00001) who did not differ from each other (P = 0.23). Similar results were found when the comparison was limited to the 33 connections between and within the left and right hippocampus and the other 15 ROIs. When restricted to the 17 connections between the hippocampi and structures illustrated in Figure 4, however, polydipsic patients showed greater connectivity than nonpolydipsic patients (nonpolydipsic: Z = 2.26, P <.025). These preliminary results support the view that that hippocampal connectivity with structures which modulate stress reactivity differs in polydipsic and nonpolydipsic patients, which could be a marker of increased stress reactivity.
5.0 Mechanism of primary polydipsia
Little is known about the mechanism of the primary polydipsia, despite it being the most distinguishing feature of this subset of patients. Thirst regulation, per se (Goldman et al., 1988, 1996), appears intact while delusional explanations for drinking are not common. When asked, most polydipsic patients say drinking makes them feel better (May, 1995; Millson et al., 1992). One plausible explanation is that the polydipsia is also a consequence of AH dysfunction, particularly the previously noted influence of the AH on the nucleus accumbens (De Carolis et al., 2010; Hawken and Beninger, 2014; Luchins 1990; Pellon et al., 2011; Wallace et al. 1983). Specifically, increased drinking could be related to the disrupted coping ability (Valenti et al., 2011) and thus represent a more primitive effort to cope with psychological stress (Falk, 1977). Consistent with this interpretation, hippocampal lesions induce a type of polydipsia in laboratory animals (schedule-induced) that is aggravated by psychological stress while drinking reduces the elevated HPAA activity (Brett and Levine, 1979) and other stress markers (Mittleman et al., 1992; Troisi, 2002). Animals with hippocampal lesions also exhibit other odd behaviors that appear analogous to repetitive behaviors (pacing, pica, posturing) (Mittleman et al., 1990) commonly seen in patients with polydipsia (Luchins et al., 1992; Shutty et al., 1995). This issue warrants further study.
6.0 Polydipsia may identify a discrete subset of chronic psychotic patients
Polydipsia identifies a discrete subset of patients with schizophrenia whose peripheral neuroendocrine findings and psychiatric disorder arise from pathologic changes in the anterior hippocampus and its projections to limbic structures implicated in stress modulation. This hypothesis arises from a sequence of studies, reviewed here, assessing the nature and mechanism of the peripheral findings in patients with polydipsia, and subsequently their relationship to the underlying psychiatric illness. Because polydipsic patients are relatively easy to identify and differ from other patients on these measures, assessment of polydipsia may facilitate resolution of discrepant and inconsistent findings in previous research studies. Because the stress and neuroendocrine circuitries are fairly well characterized, further studies may help resolve issues regarding the pathophysiology and treatment of severe mental illness in this subset. Finally, because this pathology can, and to a certain degree has, been reproduced in other mammals, the findings may enhance the ability of translational neuroscientists to create more accurate animal models of psychosis.
By taking a ‘bottom up’ approach, this work draws into question the long held view that animal models cannot capture the core pathology of schizophrenia. Disrupting ventral hippocampal/subicular function (the rodent analogue of the anterior lateral hippocampus) has reproduced findings reported here (Tseng et al., 2009, Valenti et al., 2011) as well as other features of schizophrenia (Lodge and Grace, 2008; Tseng et al., 2009). More significantly, some of these findings can be reversed by deep brain stimulation (Ewing and Grace, 2013), transplantation of GABAergic precursor neurons (Perez and Lodge, 2013) into the ventral hippocampus, or by activation of alpha-5 containing hippocampal GABA receptors (Lodge and Grace, 2011). Hence, translational neuroscience has already revealed potential novel therapies that may be effective in this subset (Grace, 2012).
The strategic approach taken here is unusual. The approach involves careful assessment of peripheral findings (i.e. hyponatremia; reset osmostat; enhanced HPAA responses, diminished plasma oxytocin) that potentially reflect limbic dysfunction. It requires intimate knowledge of neurophysiology with the goal of identifying recognized or putative modulators that are responsible for the findings. In contrast many investigators assume that modulators are not responsible for their findings (i.e. there is a more fundamental disruption of the neurosystem under study), as long as demographic and treatment measures are matched across groups (Goldman, 2009).
While unusual, the approach adheres to the view of mainstream researchers who have concluded that relying on clinical features of mental illness (e.g. DSM-IV), and particularly those of schizophrenia (Insel, 2010b), to guide research into pathophysiology and treatment has been largely unproductive (Hyman, 2007; Kapur et al., 2012). Schizophrenia is increasing viewed as “dysfunction in neural circuits which can be identified with functional neuroimaging and by new methods for quantifying connections in vivo” (Insel, 2010b). In contrast to typical clinical features (e.g. hallucinations, thought disorder, delusions) measures of stress reactivity require assessing how psychological stress modulates different neurosystems. In so doing, it may yield a ‘dimension of observable behavior characterized by neurobiologic measures’ that leads to more productive classification of severe mental illness (NIMH RDoC). This approach also reflects principles associated with the ‘deconstruction’ of schizophrenia : 1) characterize a valid phenotype based on translational neuroscience; 2) elucidate pathophysiological processes that identify intermediate phenotypes; 3) incorporate the heterogeneity of schizophrenia in these efforts; 4) separate causal factors from consequences and adaptive responses; and 5) formulate hypotheses that can be tested in animal models (Karatsoreos and McEwen, 2011; Keshavan et al., 2011; Nava and Röder, 2011).
Other peripheral findings in schizophrenia may similarly reflect altered limbic functioning associated with the psychiatric disorder (Kirkpatrick, 2013). Some possibilities warranting consideration include peripheral features of catatonia (Castillo et al., 1989); inflammatory markers like monocytosis (Bergink et al., 2014; Kirpatrick and Miller, 2013); insulin resistance (Kirkpatrick, 2013) and other neurohormone findings (Bergink et al., 2014; Kudielka et al., 2009).
7.0 Caveats and limitations
At this stage, the idea that ‘discrete pathologic changes in a neurocircuit involved in stress modulation contributes to the psychiatric illness in polydipsic psychotic patients’ is only a hypothesis; albeit one that we believe warrants serious consideration. Nearly all of the studies reviewed here utilized very small samples and many findings have not been reproduced. The first concern is ameliorated somewhat by the fact that identifying discrete subgroups and measuring or controlling recognized modulators greatly diminishes group variance. The latter concern is critical, however, in addition to the concern that some findings have not undergone external review. Furthermore, adequate evidence for a generalized stress diathesis is still missing and other measures of stress-sensitive indices are thus needed.
Part of the hypothesis is predicated on the views that the anterior hippocampus modulates central neuroendocrine activity; that peripheral and central oxytocin secretion covary during psychological stress; and that centrally secreted oxytocin accounts for most of the oxytocin in the brain. Each of these statements is controversial. Furthermore, the other part of the hypothesis, involving the role of the anterior lateral hippocampus in stress reactivity, is not firmly established. The assertion, that the stronger resting state connectivity in polydipsic patients between the hippocampus and the other structures illustrated in Figure 4 reflects dysfunction, may also seem counterintuitive. This is particularly so because diminished temporal-frontal connectivity is a longstanding finding reproduced on a regular basis by schizophrenia researchers. Indeed, task and non-task fMRI measures primarily demonstrate diminished hippocampal connectivity in schizophrenia (but see Salvador et al., 2010; Ford et al., 2014), though these studies, with one exception (Fan et al., 2013), have not examined the connections illustrated in Figure 4 (e.g. Qui et al., 2010; Zhou et al., 2008). One possibility is that the increased hippocampal connectivity in both patient groups reflects the enhanced anterior hippocampal activation which is increasingly recognized as a frequent finding in chronically psychotic patients (Suazo et al., 2013; Tregellas, et al., 2014).
Alternative explanations exist for some findings. Most subjects were receiving antipsychotic therapy during the studies. While patient groups were matched for chlorpromazine equivalents this is not a precise way of controlling for neuroleptic exposure and does not address the possibility of idiosyncratic responses. The fact that most of the peripheral findings appear to have been documented prior to the introduction of antipsychotics somewhat diminishes this concern. Some of the mechanistic interpretations are also open to question. Thus it is possible that hippocampal findings are a consequence, rather than the cause, of the water imbalance or of enhanced HPAA activity (Arango et al., 2001). On the other hand, effects limited to the anterior segment or its lateral surface have not been reported in other populations. Finally, the inability to clearly link the polydipsia to the hippocampal structural and functional findings is concerning. It might seem tempting to instead view it as a primary disorder in fluid intake since the anterior hypothalamus regulates both water intake and excretion. Further studies are needed to determine the mechanism of the polydipsia.
8.0 Next steps
In addition to addressing the issues raised above, we believe the field of research into the pathophysiology and development of novel therapies for psychosis would benefit if investigators obtain estimates of water balance in their clinical studies of psychotic patients. Polydipsia (> 4 liters intake/day) is fairly stable (Schnur et al., 1997; Vieweg et al., 1990) and reliably diagnosed by obtaining two or three spot urine samples (Abassi, et al., 1997). Obtaining both a morning and afternoon urine sample is ideal, though afternoon samples are acceptable. Urine creatinine concentration provides a more reliable estimate than either urine osmolality or specific gravity, though this is not essential (Goldman et al., 1992). Mean urine osmolalities of <200 mOsm/Kg (normal 400 to 1000mOsm/Kg) or urine specific gravities of <1.008 (normal 1.015 to 1.030) are generally accepted as confirming polydipsia. Episodic hyponatremia and reset osmostat can be identified by measuring morning and evening body weight (Vieweg et al., 1988). A 3% or greater increase in body weight is suggestive of reset osmostat, and should be confirmed by obtaining two or three afternoon plasma sodium (<130 me/L; normal 135–150 mEq/L). Identifying which patients are polydipsic may greatly facilitate interpretation of their findings, particularly since the groups are likely to be at opposite ends of the spectrum on other measures besides stress reactivity. Unless polydipsic and nonpolydipsic subjects are distinguished, the results may tend to ‘cancel each other out.’
Supplementary Material
Acknowledgments
Role of Funding Sources
The studies reviewed here were funded by various grants from the Brain Research Foundation; Scottish Rite Foundation; NARSAD and the National Institute of Mental Health.
The author would like to acknowledge the support and encouragement from Daniel J. Luchins, Gary L. Robertson, and Sue Carter. The staffs at the Elgin Mental Health Center, Illinois State Psychiatric Institute and the clinical research centers at the University of Chicago, University of Illinois at Chicago and Northwestern University exhibited both exceptional compassion and technical skills. There have been many students, residents and fellows who have made major contributions to each of these studies. I have also had the pleasure of the knowledge, advice of many capable colleagues. I want to thank the PGY-2 class of psychiatric residents at Northwestern University and Julie Babyar, RN for their helpful comments on the manuscript. These studies were funded by various grants from the Brain Research Foundation; Scottish Rite Foundation; NARSAD and the National Institute of Mental Health (MH43618; MH56525, RR00055 RR13987; MH082295)
Glossary
- Facial Affect Recognition
A component of social cognition that is assessed by having the subject identify the emotion(s) (and in some cases their intensity) displayed on photos of actors. It is consistently impaired in chronically psychotic patients and appears sensitive to variations in central oxytocin activity. Social cognition, in turn, is predictive of negative symptoms and overall functioning in persons with schizophrenia
- Functional connectivity
An index of the extent that neural activity covaries in different brain structures which can provide evidence that the structures constitute a neural circuit
- Glucocorticoid negative feedback
The extent that stress hormone (plasma cortisol) levels influence cortisol regulation. Feedback is generally considered a component of how the hippocampus normally restrains HPAA responses to psychological stress. The dexamethasone suppression test (DST) is an index of general feedback sensitivity. More specific indices (i.e. hippocampal-modulated) can be obtained by varying baseline cortisol levels and the time of day that feedback is assessed
- Hyponatremia
Diminished concentration of sodium ions in the plasma. This is generally interchangeable with hypoosmolemia (i.e. diminished plasma osmolality) which is dilution of all solute in the plasma, which in turn indicates diminished tissue tonicity because water crosses freely across intra- and extra-cellular compartments
- Impaired water excretion
The kidney has a tremendous capacity to retain or excrete water. Normal water excretion is defined relative to concurrent plasma osmolality
- Increased stress reactivity
Defined here as increased sensitivity of affective, cognitive, neuroendocrine, autonomic and behavioral measures to psychologic stress. Generally interchangeable with ‘stress diathesis’ as defined by others
- Non-osmotic stimuli
Modulators of AVP activity independent of changes in tonicity
- Osmoregulation
The neurosystems which regulate and modify water intake and excretion (i.e. water balance) in the service of optimizing tissue tonicity and secondarily maintaining sufficient blood pressure
- Primary Polydipsia
Increased fluid intake not attributable to increased fluid loss. Generally considered to exceed four liters daily
- Psychological stress
Defined here as a stimulus that is interpreted to be threatening to the physical or emotional well-being of the subject; to be distinguished from physical stressors which require a physiologic response to maintain homeostasis. Note that it is difficult to administer standardized stressors to psychotic patients because of their inclination toward idiosyncratic interpretations
- Water intoxication
Impaired brain function due to dilution of tissue tonicity in the brain typically associated with cerebral compression by the skull; potentially life-threatening.
Footnotes
Contributors
Dr. Goldman was solely responsible for the preparation of this manuscript though he received much assistance as detailed in the acknowledgement.
Conflict of Interest
Morris Goldman has received grant funding and consulted for Otsuka Pharmaceuticals.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbasi QA, Carbonell FE, Koczapski AB, Vieweg WV. Measuring and estimating daily urine volume in psychiatric patients: strengths and weaknesses. Schizophr Res. 1997;28(1):87–93. doi: 10.1016/s0920-9964(97)00086-8. [DOI] [PubMed] [Google Scholar]
- Addington J, Girard TA, Christensen BK, Addington D. Social cognition mediates illness-related and cognitive influences on social function in patients with schizophrenia-spectrum disorders. J Psychiatry Neurosci. 2010;35(1):49–54. doi: 10.1503/jpn.080039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggleton JP. Multiple anatomical systems embedded within the primate medial temporal lobe: implications for hippocampal function. Neurosci Biobehav Rev. 2012;36(7):1579–1596. doi: 10.1016/j.neubiorev.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Arango C, Kirkpatrick B, Koenig J. At issue: stress, hippocampal neuronal turnover, and neuropsychiatric disorders. Schizophr Bull. 2001;27(3):477–480. doi: 10.1093/oxfordjournals.schbul.a006888. [DOI] [PubMed] [Google Scholar]
- Arakawa H, Arakawa K, Deak T. Oxytocin and vasopressin in the medial amygdala differentially modulate approach and avoidance behavior toward illness-related social odor. Neuroscience. 2010;171(4):1141–1151. doi: 10.1016/j.neuroscience.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Atsariyasing A, Goldman MB. A systematic review of the ability of urine concentration to distinguish antipsychotic- from psychosis-induced hyponatremia. Psychiatry Res. 2014;217:129–133. doi: 10.1016/j.psychres.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barahal HS. Water intoxication in a mental case. Psychiat Quart. 1938;12(4):767–771. [Google Scholar]
- Bergink V, Gibney SM, Drexhage HA. Autoimmunity, inflammation, and psychosis: a search for peripheral markers. Biol Psychiatry. 2014;75(4):324–331. doi: 10.1016/j.biopsych.2013.09.037. [DOI] [PubMed] [Google Scholar]
- Bradley AJ, Dinan TG. A systematic review of hypothalamic-pituitary-adrenal axis function in schizophrenia: implications for mortality. J Psychopharmacol. 2010;24(4 Suppl):91–118. doi: 10.1177/1359786810385491. [DOI] [PubMed] [Google Scholar]
- Bralet MC, Ton T, Falissard B. Schizophrenic patients with polydipsia and water intoxication more often have a form of schizophrenia first described by Kraepelin. Psychiatry Res. 2007;152(2–3):267–271. doi: 10.1016/j.psychres.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Brenner K, St-Hilaire A, Liu A, Laplante DP, King S. Cortisol response and coping style predict quality of life in schizophrenia. Schizophr Res. 2011;128(1–3):23–29. doi: 10.1016/j.schres.2011.01.016. [DOI] [PubMed] [Google Scholar]
- Brett LP, Levine S. Schedule-induced polydipsia suppresses pituitary-adrenal activity in rats. J Comp Physiol Psychol. 1979;93(5):946–956. doi: 10.1037/h0077619. [DOI] [PubMed] [Google Scholar]
- Castillo E, Rubin RT, Holsboer-Trachsler E. Clinical differentiation between lethal catatonia and neuroleptic malignant syndrome. Am J Psychiatry. 1989;146(3):324–328. doi: 10.1176/ajp.146.3.324. [DOI] [PubMed] [Google Scholar]
- Chrapusta SJ, Egan MF, Wyatt RJ, Weinberger DR, Lipska BK. Neonatal ventral hippocampal damage modifies serum corticosterone and dopamine release responses to acute footshock in adult Sprague-Dawley rats. Synapse. 2003;47(4):270–277. doi: 10.1002/syn.10179. [DOI] [PubMed] [Google Scholar]
- Coccaro EF, Prudic J, Rothpearl A, Nurnberg HG. Effect of hospital admission on DST results. Am J Psychiatry. 1984;141(3):982–985. doi: 10.1176/ajp.141.8.982. [DOI] [PubMed] [Google Scholar]
- Cohen H, Kaplan Z, Kozlovsky N, Gidron Y, Matar MA, Zohar J. Hippocampal microinfusion of oxytocin attenuates the behavioural response to stress by means of dynamic interplay with the glucocorticoid-catecholamine responses. J Neuroendocrinol. 2010;22(8):889–904. doi: 10.1111/j.1365-2826.2010.02003.x. [DOI] [PubMed] [Google Scholar]
- Critchley HD. Electrodermal responses: what happens in the brain. Neuroscientist. 2002;8(2):132–142. doi: 10.1177/107385840200800209. [DOI] [PubMed] [Google Scholar]
- De Carolis L, Stasi MA, Serlupi-Crescenzi O, Borsini F, Nencini P. The effects of clozapine on quinpirole-induced non-regulatory drinking and prepulse inhibition disruption in rats. Psychopharmacology (Berl) 2010;212(1):105–115. doi: 10.1007/s00213-010-1937-1. [DOI] [PubMed] [Google Scholar]
- De Leon J, Verghese C, Tracy JI, Josiassen RC, Simpson GM. Polydipsia and water intoxication in psychiatric patients: a review of the epidemiological literature. Biol Psychiatry. 1994;35(6):408–419. doi: 10.1016/0006-3223(94)90008-6. [DOI] [PubMed] [Google Scholar]
- Delva NJ, Crammer JL, Lawson JS, Lightman SL, Sribne M, Weier BJ. Vasopressin in chronic psychiatric patients with primary polydipsia. Br J Psychiatry. 1990;157(5):703–712. doi: 10.1192/bjp.157.5.703. [DOI] [PubMed] [Google Scholar]
- Deutch AY, Clark WA, Roth RH. Prefrontal cortical dopamine depletion enhances the responsiveness of mesolimbic dopamine neurons to stress. Brain Res. 1990;521(1–2):311–315. doi: 10.1016/0006-8993(90)91557-w. [DOI] [PubMed] [Google Scholar]
- Edelson JT, Robertson GL. The effect of the cold pressor test on vasopressin secretion in man. Psychoneuroendocrinology. 1986;11(3):307–316. doi: 10.1016/0306-4530(86)90016-8. [DOI] [PubMed] [Google Scholar]
- Elzinga BM, Roelofs K, Tollenaar MS, Bakvis P, van Pelt J, Spinhoven P. Diminished cortisol responses to psychosocial stress associated with lifetime adverse events: a study among healthy young subjects. Psychoneuroendocrinology. 2008;33(2):227–237. doi: 10.1016/j.psyneuen.2007.11.004. [DOI] [PubMed] [Google Scholar]
- Ewing SG, Grace A. Deep brain stimulation of the ventral hippocampus restores deficits in processing of auditory evoked potentials in a rodent developmental disruption model of schizophrenia. Schizophren Res. 2013;143(2–3):377–383. doi: 10.1016/j.schres.2012.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk JL. The origin and functions of adjunctive behavior. Anim Learn Behav. 1977;5(4):325–335. [Google Scholar]
- Fan FM, Tan SP, Yang FD, Tan YL, Zhao YL, Chen N, Zuo XN. Ventral medial prefrontal functional connectivity and emotion regulation in chronic schizophrenia: a pilot study. Neurosci Bull. 2013;29(1):59–74. doi: 10.1007/s12264-013-1300-8. d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanselow MS, Dong H-W. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron. 2010;65(1):7–19. doi: 10.1016/j.neuron.2009.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman R, Gordon I, Schneiderman I, Weisman O, Zagoory-Sharon O. Natural variations in maternal and paternal care are associated with systematic changes in oxytocin following parent-infant contact. Psychoneuroendocrinology. 2010;35(8):1133–1141. doi: 10.1016/j.psyneuen.2010.01.013. [DOI] [PubMed] [Google Scholar]
- Feldman R, Zagoory-Sharon O, Weisman O, Schneiderman I, Gordon I, Maoz R, Shalev I, Ebstein RP. Sensitive parenting is associated with plasma oxytocin and polymorphisms in the OXTR and CD38 genes. Biol Psychiatry. 2012;72(3):175–81. doi: 10.1016/j.biopsych.2011.12.025. [DOI] [PubMed] [Google Scholar]
- Fischer-Shofty M, Shamay-Tsoory SG, Harari H, Levkovitz Y. The effect of intranasal administration of oxytocin on fear recognition. Neuropsychologia. 2010;48(1):179–184. doi: 10.1016/j.neuropsychologia.2009.09.003. [DOI] [PubMed] [Google Scholar]
- Fischer-Shofty M, Shamay-Tsoory SG, Levkovitz Y. Characterization of the effects of oxytocin on fear recognition in patients with schizophrenia and in healthy controls. Front Neurosci. 2013;127(7):1–9. doi: 10.3389/fnins.2013.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford JM, Palzes V, Roach BJ, Potkin SG, van Erp TGM, Turner J, Mathalon DH. Visual hallucinations are associated with hyperconnectivity between the amygdala and visual cortex in people with a diagnosis of schizophrenia. Schizophren Bull. 2014:1–10. doi: 10.1093/schbul/sbu031. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornito A, Zalesky A, Pantelis C, Bullmore ET. Schizophrenia, neuroimaging and connectomics. Neuroimage. 2012;62(4):2296–2314. doi: 10.1016/j.neuroimage.2011.12.090. [DOI] [PubMed] [Google Scholar]
- Franklin TB, Saab BJ, Mansuy IM. Neural mechanisms of stress resilience and vulnerability. Neuron. 2012;75(5):747–61. doi: 10.1016/j.neuron.2012.08.016. [DOI] [PubMed] [Google Scholar]
- Gainer H, Yamashita M, Fields RL, Hause SB, Rusnak ML. The magnocellular neuronal phenotype: cell-specific gene expression in the hypothalamo–neurohypophysial system. Prog Brain Res. 2002;139(1):1–14. doi: 10.1016/s0079-6123(02)39003-4. [DOI] [PubMed] [Google Scholar]
- Gill KM, Grace AA. Heterogeneous processing of amygdala and hippocampal inputs in the rostral and caudal subregions of the nucleus accumbens. Intl J Neuropsychopharm. 2011;14(10):1301–1314. doi: 10.1017/S1461145710001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman MB. The mechanism of life-threatening water imbalance in schizophrenia and its relationship to the underlying psychiatric illness. Brain Res Rev. 2009;61(2):210–220. doi: 10.1016/j.brainresrev.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman MB, Blake L, Marks RC. Association of nonsuppression of cortisol on the DST with primary polydipsia in chronic schizophrenia. Am J Psychiatry. 1993;150(4):653–655. doi: 10.1176/ajp.150.4.653. [DOI] [PubMed] [Google Scholar]
- Goldman MB, Gnerlich J, Hussain N. Neuroendocrine responses to a cold pressor stimulus in polydipsic hyponatremic and in matched schizophrenic patients. Neuropsychopharmacology. 2007a;32(7):1611–1621. doi: 10.1038/sj.npp.1301282. [DOI] [PubMed] [Google Scholar]
- Goldman MB, Gomes AM, Carter CS, Lee R. Divergent effects of two different doses of intranasal oxytocin on facial affect discrimination in schizophrenic patients with and without polydipsia. Psychopharmacology (Berl) 2011b;216(1):101–110. doi: 10.1007/s00213-011-2193-8. [DOI] [PubMed] [Google Scholar]
- Goldman MB, Ittaskul P, Gomes AM. Enhanced stress reactivity to emotional stimuli in a discrete subset (polydipsic) of schizophrenia patients. Abstracts Annual Meeting Society for Neuroscience; October 17; New Orleans, La. 2012. p. 772.06. [Google Scholar]
- Goldman MB, Luchins DJ, Robertson GL. Mechanisms of altered water metabolism in psychotic patients with polydipsia and hyponatremia. N Eng J Med. 1988;318(7):397–403. doi: 10.1056/NEJM198802183180702. [DOI] [PubMed] [Google Scholar]
- Goldman MB, Marks RC, Blake L, Petkovic M, Hedeker D, Luchins DJ. Estimating daily urine volume in psychiatric patients: empiric confirmation. Biol Psychiatry. 1992;31(12):1228–1231. doi: 10.1016/0006-3223(92)90343-x. [DOI] [PubMed] [Google Scholar]
- Goldman MB, Marlow-O’Connor M, Torres I, Carter SC. Diminished plasma oxytocin in schizophrenic patients with neuroendocrine dysfunction and emotional deficits. Schizophr Res. 2008;98(1–3):247–255. doi: 10.1016/j.schres.2007.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman MB, Mitchell CP. What is the functional significance of the hippocampal pathology in schizophrenia? Schizophr Bull. 2004;30(2):367–392. doi: 10.1093/oxfordjournals.schbul.a007086. [DOI] [PubMed] [Google Scholar]
- Goldman MB, Robertson GL, Luchins DJ, Hedeker D. The influence of polydipsia on water excretion in hyponatremic, polydipsic, schizophrenic patients. J Clin Endocrinol Metab. 1996;81(4):1465–1470. doi: 10.1210/jcem.81.4.8636352. [DOI] [PubMed] [Google Scholar]
- Goldman MB, Robertson GL, Luchins DJ, Hedeker D, Pandey GN. Psychotic exacerbations and enhanced vasopressin secretion in schizophrenic patients with hyponatremia and polydipsia. Arch Gen Psychiatry. 1997;54(5):443–449. doi: 10.1001/archpsyc.1997.01830170069010. [DOI] [PubMed] [Google Scholar]
- Goldman MB, Torres I, Keedy S, Marlow-O’Connor M, Beenken B, Pilla R. Reduced anterior hippocampal formation volume in hyponatremic schizophrenic patients. Hippocampus. 2007c;17(7):554–562. doi: 10.1002/hipo.20292. [DOI] [PubMed] [Google Scholar]
- Goldman MB, Wang L, Wachi C, Daudi S, Csernansky JG, Marlow-O’Connor M. Structural pathology underlying neuroendocrine dysfunction in schizophrenia. Behav Brain Res. 2011a;218(1):106–113. doi: 10.1016/j.bbr.2010.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman MB, Wood G, Goldman MB, Gavin M, Paul S, Zaheer S. Diminished glucocorticoid negative feedback in polydipsic hyponatremic schizophrenic patients. J Clin Endocrinol Metab. 2007b;92(2):698–704. doi: 10.1210/jc.2006-1131. [DOI] [PubMed] [Google Scholar]
- Grace AA. Dopamine system dysregulation by the hippocampus: implications for the pathophysiology and treatment of schizophrenia. Neuropharmacology. 2012;62(3):1342–1348. doi: 10.1016/j.neuropharm.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Salmeron BJ, Ross TJ, Geng X, Zhan W, Stein Ea Yang Y. Mesocorticolimbic circuits are impaired in chronic cocaine users as demonstrated by resting-state functional connectivity. NeuroImage. 2010;53(2):593–601. doi: 10.1016/j.neuroimage.2010.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumley A, Braehler C, Macbeth A. A meta-analysis and theoretical critique of oxytocin and psychosis: Prospects for attachment and compassion in promoting recovery. Br J Clin Psychol. 2014;53(1):42–61. doi: 10.1111/bjc.12041. [DOI] [PubMed] [Google Scholar]
- Gur RE, Loughead J, Kohler CG, Elliott MA, Lesko K, Ruparel K, Wolf DH. Limbic activation associated with misidentification of fearful faces and flat affect in schizophrenia. Arch Gen Psychiatry. 2007;64(12):1356–1366. doi: 10.1001/archpsyc.64.12.1356. [DOI] [PubMed] [Google Scholar]
- Hariprasad MK, Eisinger RP, Nadler IM, Padmanabhan CS, Nidus BD. Hyponatremia in psychogenic polydipsia. Arch Intern Med. 1980;140(12):1639–1642. [PubMed] [Google Scholar]
- Hawken ER, Beninger RJ. The amphetamine sensitization model of schizophrenia symptoms and its effect on schedule-induced polydipsia in the rat. Psychopharmacology. 2014;231(9):2001–2008. doi: 10.1007/s00213-013-3345-9. [DOI] [PubMed] [Google Scholar]
- Hawken ER, Crookall JM, Reddick D, Millson RC, Milev R, Delva NJ. Mortality over a 20-year period in patients with primary polydipsia associated with schizophrenia: a retrospective study. Schizophren Res. 2009;107(2–3):128–133. doi: 10.1016/j.schres.2008.09.029. [DOI] [PubMed] [Google Scholar]
- Heckers S. Neuroimaging studies of the hippocampus in schizophrenia. Hippocampus. 2001;11(5):520–528. doi: 10.1002/hipo.1068. [DOI] [PubMed] [Google Scholar]
- Heinrichs M. Social support and oxytocin interact to suppress cortisol and subjective responses to psychosocial stress. Biol Psychiatry. 2003;54(12):1389–1398. doi: 10.1016/s0006-3223(03)00465-7. [DOI] [PubMed] [Google Scholar]
- Herman JP, Dolgas CM, Carlson SL. Ventral subiculum regulates hypothalamo-pituitary-adrenocortical and behavioural responses to cognitive stressors. Neuroscience. 1998;86(2):449–459. doi: 10.1016/s0306-4522(98)00055-4. [DOI] [PubMed] [Google Scholar]
- Herman JP, Tasker JG, Ziegler D, Cullinan WE. Local circuit regulation of paraventricular nucleus stress integration: glutamate-GABA connections. Pharm Biochem Behav. 2002;71(3):457–468. doi: 10.1016/s0091-3057(01)00681-5. [DOI] [PubMed] [Google Scholar]
- Hobson JA, English JT. Self-induced water intoxication. Ann Intern Med. 1963;58(2):324–332. doi: 10.7326/0003-4819-58-2-324. [DOI] [PubMed] [Google Scholar]
- Hoskins RG, Sleeper FH. Organic functions in schizophrenia. Arch Neurol Psychiatry. 1933;30(1):123–140. [Google Scholar]
- Hyman SE. Can neuroscience be integrated into the DSM-V? Nat Rev Neurosci. 2007;8(9):725–732. doi: 10.1038/nrn2218. [DOI] [PubMed] [Google Scholar]
- Ikezawa S, Corbera S, Liu J, Wexler BE. Empathy in electrodermal responsive and nonresponsive patients with schizophrenia. Schizophr Res. 2012;142(1–3):71–76. doi: 10.1016/j.schres.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR. The challenge of translation in social neuroscience: a review of oxytocin, vasopressin, and affiliative behavior. Neuron. 2010a;65(6):768–779. doi: 10.1016/j.neuron.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Insel TR. Rethinking schizophrenia. Nature. 2010b;468(7321):187–193. doi: 10.1038/nature09552. [DOI] [PubMed] [Google Scholar]
- Ittasakul P, Goldman MB. Management of water imbalance in schizophrenia. In: Janicak PG, Marder SR, Tandon R, Goldman MB, editors. Schizophrenia: Recent Advances in Diagnosis and Treatment. Springer; New York: 2014. pp. 205–225. [Google Scholar]
- Josiassen RC, Goldman MB, Jessani M, Shaughnessy RA, Albazzaz A, Lee J, et al. Double-blind, placebo-controlled, multicenter trial of a vasopressin V2-receptor antagonist in patients with schizophrenia and hyponatremia. Biol Psychiatry. 2008;64(12):1097–1100. doi: 10.1016/j.biopsych.2008.06.017. [DOI] [PubMed] [Google Scholar]
- Josiassen RC, Curtis JL, Shaughnessy RA, Filmyer DM, Geboy AG, Skuban N, Czerwiec F. Vaptans: a potential new approach for treating chronic hyponatremia in psychotic patients. Clin Schizophren Rel Psychoses. 2012;6(4):21–26. doi: 10.3371/CSRP.6.1.3. [DOI] [PubMed] [Google Scholar]
- Kapur S, Phillips G, Insel TR. Why has it taken so long for biological psychiatry to develop clinical tests and what to do about it? Mol psychiatry. 2012;17(12):1174–1179. doi: 10.1038/mp.2012.105. [DOI] [PubMed] [Google Scholar]
- Karatsoreos IN, McEwen BS. Psychobiological allostasis: resistance, resilience and vulnerability. Trends Cogn Sci. 2011;15(12):576–584. doi: 10.1016/j.tics.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Kéri S, Kiss I, Kelemen O. Sharing secrets: oxytocin and trust in schizophrenia. Social Neurosci. 2009;4(4):287–293. doi: 10.1080/17470910802319710. [DOI] [PubMed] [Google Scholar]
- Keshavan MS, Clementz B, Pearlson GD, Sweeney J, Tamminga C. Reimagining psychoses: an agnostic approach to diagnosis. Schizophren Res. 2013;146(1–3):10–16. doi: 10.1016/j.schres.2013.02.022. [DOI] [PubMed] [Google Scholar]
- Keshavan MS, Nasrallah H, Tandon R. Moving ahead with the schizophrenia concept : From the elephant to the mouse. Schizophren Res. 2011;127(1–3):3–13. doi: 10.1016/j.schres.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick B. Understanding the physiology of schizophrenia. J Clin Psychiatry. 2013;74(3):e05. doi: 10.4088/JCP.12045tx3c. [DOI] [PubMed] [Google Scholar]
- Kirkpatrick B, Miller BJ. Inflammation and schizophrenia. Schizophren Bull. 2013;39(6):1174–1179. doi: 10.1093/schbul/sbt141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch P, Esslinger C, Chen Q, Mier D, Lis S, Siddhanti S, Gruppe H. Oxytocin modulates neural circuitry for social cognition and fear in humans. J Neuroscience. 2005;25(49):11489–11493. doi: 10.1523/JNEUROSCI.3984-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto T, Hirai M, Ohsawa H, Terada M, Matsuoka I, Ikawa I. Manner of arginine vasopressin secretion in schizophrenic patients—with reference to the mechanism of water intoxication. Jpn J Psychiatry Neurol. 1989;43(2):161–169. doi: 10.1111/j.1440-1819.1989.tb02565.x. [DOI] [PubMed] [Google Scholar]
- Kudielka BM, Hellhammer DH, Wüst S. Why do we respond so differently? Reviewing determinants of human salivary cortisol responses to challenge. Psychoneuroendocrinology. 2009;34(1):2–18. doi: 10.1016/j.psyneuen.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Landgraf R, Neumann ID. Vasopressin and oxytocin release within the brain: a dynamic concept of multiple and variable modes of neuropeptide communication. Front Neuroendocrinol. 2004;25(3–4):150–176. doi: 10.1016/j.yfrne.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Lataster J, Collip D, Ceccarini J, Hernaus D, Haas D, Booij L, van Os J, Pruessner J, Van Laere K, Myin-Germeys I. Familial liability to psychosis is associated with attenuated dopamine stress signaling in ventromedial prefrontal cortex. Schizophr Bull. 2014;40(1):66–77. doi: 10.1093/schbul/sbs187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux J. Emotion and the amygdala. In: Aggleton JP, editor. The Amygdala. Wiley Liss; New York: 1992. pp. 339–351. [Google Scholar]
- Legros JJ. Inhibitory effect of oxytocin on corticotrope function in humans: are vasopressin and oxytocin ying-yang neurohormones? Psychoneuroendocrinology. 2001;26(7):649–655. doi: 10.1016/s0306-4530(01)00018-x. [DOI] [PubMed] [Google Scholar]
- Lodge DJ, Grace A. Hippocampal dysfunction and disruption of dopamine system regulation in an animal model of schizophrenia. Neurotox Res. 2008;14(2–3):97–104. doi: 10.1007/BF03033801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Developmental pathology, dopamine, stress and schizophrenia. Neuroscience. 2011;29(3):207–213. doi: 10.1016/j.ijdevneu.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchins DJ. A possible role of hippocampal dysfunction in schizophrenic symptomatology. Biol Psychiatry. 1990;28(2):87–91. doi: 10.1016/0006-3223(90)90625-c. [DOI] [PubMed] [Google Scholar]
- Luchins DJ, Goldman MB, Lieb M, Hanrahan P. Repetitive behaviors in chronically institutionalized schizophrenic patients. Schizophr Res. 1992;8(2):119–123. doi: 10.1016/0920-9964(92)90027-3. [DOI] [PubMed] [Google Scholar]
- MacMillan HL, Georgiades K, Duku EK, Shea A, Steiner M, Niec A. Cortisol response to stress in female youths exposed to childhood maltreatment: Results of the youth mood project. Biol Psychiatry. 2009;66(1):62–68. doi: 10.1016/j.biopsych.2008.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh AA, Yu HH, Pine DS, Blair RJR. Oxytocin improves specific recognition of positive facial expressions. Psychopharmacology. 2010;209(3):225–232. doi: 10.1007/s00213-010-1780-4. [DOI] [PubMed] [Google Scholar]
- May DL. Patient perceptions of self-induced water intoxication. Arch Psychiatr Nurs. 1995;9(5):295–304. doi: 10.1016/s0883-9417(95)80049-2. [DOI] [PubMed] [Google Scholar]
- Miller GE, Chen E, Zhou ES. If it goes up, must it come down? Chronic stress and the hypothalamic—pituitary—adrenocortical axis in humans. Psychol Bull. 2007;133(1):25–45. doi: 10.1037/0033-2909.133.1.25. [DOI] [PubMed] [Google Scholar]
- Millson RC, Koczapski AB, Cook MI, Daszkiewicz M. A survey of patient attitudes toward self-induced water intoxication. Can J Psychiatry. 1992;37(1):46–47. doi: 10.1177/070674379203700110. [DOI] [PubMed] [Google Scholar]
- Mitchell CP, Goldman MB. Neonatal lesions of the ventral hippocampal formation disrupt neuroendocrine responses to auditory stress in the adult rat. Psychoneuroendocrinology. 2004;29(10):1317–1325. doi: 10.1016/j.psyneuen.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Mittleman G, Blaha CD, Phillips AG. Pituitary-adrenal and dopaminergic modulation of schedule-induced polydipsia: behavioral and neurochemical evidence. Behav Neurosci. 1992;106(2):408–420. doi: 10.1037//0735-7044.106.2.408. [DOI] [PubMed] [Google Scholar]
- Mittleman G, Whishaw IQ, Jones GH, Koch M, Robbins TW. Cortical, hippocampal, and striatal mediation of schedule-induced behaviors. Behav Neurosci. 1990;104(3):399–409. doi: 10.1037//0735-7044.104.3.399. [DOI] [PubMed] [Google Scholar]
- Montag C, Brockmann E-M, Lehmann A, Müller DJ, Rujescu D, Gallinat J. Association between oxytocin receptor gene polymorphisms and self-rated “empathic concern” in schizophrenia. PloS One. 2012;7(12):e51882. doi: 10.1371/journal.pone.0051882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myin-Germeys I, Krabbendam L, Delespaul P, van Os J. Do life events have their effect on psychosis by influencing the emotional reactivity to daily life stress? Psychol Med. 2003;33(2):327–333. doi: 10.1017/s0033291702006785. [DOI] [PubMed] [Google Scholar]
- Nava E, Röder B. Adaptation and maladaptation insights from brain plasticity. Prog Brain Res. 2011;191:177–194. doi: 10.1016/B978-0-444-53752-2.00005-9. [DOI] [PubMed] [Google Scholar]
- Nettles KW, Pesold C, Goldman MB. Influence of the ventral hippocampal formation on plasma vasopressin, hypothalamic-pituitary-adrenal axis, and behavioral responses to novel acoustic stress. Brain Res. 2000;858(1):181–190. doi: 10.1016/s0006-8993(99)02281-7. [DOI] [PubMed] [Google Scholar]
- NIMH RDoC, Draft 31. 2011 Jun; http://www.nimh.nih.gov/research-funding/rdoc/nimh-research-domain-criteria-rdoc.shtml.
- NIMH Strategic Plan. 2008 Aug; http://www.nimh.nih.gov/about/strategic-planning-reports/index.shtml.
- Neumann ID, Maloumby R, Beiderbeck DI, Lukas M, Landgraf R. Increased brain and plasma oxytocin after nasal and peripheral administration in rats and mice. Psychoneuroendocrinology. 2013;38(10):1985–1993. doi: 10.1016/j.psyneuen.2013.03.003. [DOI] [PubMed] [Google Scholar]
- Ohman A. Electrodermal activity and vulnerability to schizophrenia: a review. Biol Psychol. 1981;12(2–3):87–145. doi: 10.1016/0301-0511(81)90008-9. [DOI] [PubMed] [Google Scholar]
- Ohsawa H, Kishimoto T, Shimayoshi N, Matsumura K, Tahara K, Kitera K, et al. Atrial natriuretic peptide and arginine vasopressin secretion in schizophrenic patients. Acta Psychiatr Scand. 1993;88(2):130–134. doi: 10.1111/j.1600-0447.1993.tb03426.x. [DOI] [PubMed] [Google Scholar]
- Onaka T, Yagi K. Role of noradrenergic projections to the bed nucleus of the stria terminalis in neuroendocrine and behavioral responses to fear-related stimuli in rats. Brain Res. 1988;788(1–2):287–293. doi: 10.1016/s0006-8993(98)00012-2. [DOI] [PubMed] [Google Scholar]
- Onaka T, Yagi K. Interactions between emotional stress due to fear and hypovolemic stimuli in the control of vasopressin secretion in rats. Neurosci Lett. 1990;120(2):187–190. doi: 10.1016/0304-3940(90)90034-7. [DOI] [PubMed] [Google Scholar]
- Pellón R, Ruíz A, Moreno M, Claro F, Ambrosio E, Flores P. Individual differences in schedule-induced polydipsia: neuroanatomical dopamine divergences. Behav Brain Res. 2011;217(1):195–201. doi: 10.1016/j.bbr.2010.10.010. [DOI] [PubMed] [Google Scholar]
- Perez SM, Lodge DJ. Hippocampal interneuron transplants reverse aberrant dopamine system function and behavior in a rodent model of schizophrenia. Mol Psychiatry. 2013;18(11):1193–1198. doi: 10.1038/mp.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson-Yeo W, Allen P, Benetti S, McGuire P, Mechelli A. Dysconnectivity in schizophrenia: where are we now? Neurosci Biobehav Rev. 2011;35(5):1110–1124. doi: 10.1016/j.neubiorev.2010.11.004. [DOI] [PubMed] [Google Scholar]
- Pfister HO. Disturbances of the autonomic nervous system in schizophrenia and their relationship to the insulin, cardiazol and sleep treatment. Am J Psychiatry. 1938;94:109–118. [Google Scholar]
- Qiu A, Tuan TA, Woon PS, Abdul-Rahman MF, Graham S, Sim K. Hippocampal-cortical structural connectivity disruptions in schizophrenia: an integrated perspective from hippocampal shape, cortical thickness, and integrity of white matter bundles. NeuroImage. 2010;52(4):1181–1189. doi: 10.1016/j.neuroimage.2010.05.046. [DOI] [PubMed] [Google Scholar]
- Risold PY, Swanson LW. Structural evidence for functional domains in the rat hippocampus. Science. 1996;272(5267):1484–1486. doi: 10.1126/science.272.5267.1484. [DOI] [PubMed] [Google Scholar]
- Robertson GL. Regulation of arginine vasopressin in the syndrome of inappropriate antidiuresis. Am J Med. 2006;119(7 Supp1):S36–S42. doi: 10.1016/j.amjmed.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Rosenfeld AJ, Lieberman J, Jarskog LF. Oxytocin, dopamine, and the amygdala: A neurofunctional model of social cognitive deficits in schizophrenia. Schizophr Bull. 2011;37(5):1077–1087. doi: 10.1093/schbul/sbq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador R, Sarró S, Gomar JJ, Ortiz-Gil J, Vila F, Capdevila A, Pomarol-Clotet E. Overall brain connectivity maps show cortico-subcortical abnormalities in schizophrenia. Hum Brain Map. 2010;31(12):2003–2014. doi: 10.1002/hbm.20993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnur DB, Frick S, Smith S. Temporal stability of polydipsia-hyponatremia. Schizophre Res. 1997;26(2–3):199–202. doi: 10.1016/s0920-9964(97)00041-8. [DOI] [PubMed] [Google Scholar]
- Schobel S, Lewandowski NM, Corcoran CM, Moore H, Brown T, Malaspina D, Small SA. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psych. 2009;66(9):938–946. doi: 10.1001/archgenpsychiatry.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenton ME, Dickey CC, Frumin M, McCarley RW. A review of MRI findings in schizophrenia. Schizophr Res. 2001;49(1–2):1–52. doi: 10.1016/s0920-9964(01)00163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutty MS, Jr, McCulley K, Pigott B. Association between stereotypic behavior and polydipsia in chronic schizophrenic patients. J Behav Ther Exp Psychiatry. 1995;26(4):339–343. doi: 10.1016/0005-7916(95)00049-6. [DOI] [PubMed] [Google Scholar]
- Sleeper FH, Jellinek EM. A comparative physiologic psychologic and psychiatric study of polyuric and nonpolyuric schizophrenic patients. J Nerv Ment Dis. 1936;1938:557–563. [Google Scholar]
- Souza RP, Ismail P, Meltzer HY, Kennedy JL. Variants in the oxytocin gene and risk for schizophrenia. Schizophren Res. 2010;121(1–3):279–280. doi: 10.1016/j.schres.2010.04.019. [DOI] [PubMed] [Google Scholar]
- Straube ER. On the meaning of electrodermal nonresponding in schizophrenia. J Nerv Ment Dis. 1979;167(10):601–611. doi: 10.1097/00005053-197910000-00003. [DOI] [PubMed] [Google Scholar]
- Suazo V, Díez Á, Tamayo P, Montes C, Molina V. Limbic hyperactivity associated to verbal memory deficit in schizophrenia. J Psych Res. 2013;47(6):843–850. doi: 10.1016/j.jpsychires.2013.02.007. [DOI] [PubMed] [Google Scholar]
- Tandon R. The evolving nosology of schizophrenia. In: Janicak PG, Marder SR, Tandon R, Goldman MB, editors. Schizophrenia: Recent Advances in Diagnosis and Treatment. Springer; New York: 2014. pp. 13–26. [Google Scholar]
- Tandon R, Goldman MB. Overview of neurobiology. In: Janicak PG, Marder SR, Tandon R, Goldman MB, editors. Schizophrenia: Recent Advances in Diagnosis and Treatment. Springer; New York: 2014. pp. 27–34. [Google Scholar]
- Targowla R. Des troubles fonctionnel du rein dans les maladies mentales. L’excretion del’eau (Kidney malfunction in mental illness: water excretion) Bull Mém Soc Méd Hôp Paris. 1923;47 (9):1711–1715. [Google Scholar]
- Tasker JG, Boudaba C, Schrader LA. Local glutamatergic and GABAergic synaptic circuits and metabotropic glutamate receptors in the hypothalamic paraventricular and supraoptic nuclei. Adv Exp Med Biol. 1998;449:117–121. doi: 10.1007/978-1-4615-4871-3_11. [DOI] [PubMed] [Google Scholar]
- Tregellas JR. Neuroimaging biomarkers for early drug development in schizophrenia. Biol Psychiatry. 2013:1–9. doi: 10.1016/j.biopsych.2013.08.025. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tregellas JR, Smucny J, Harris JG, Olincy A, Maharajh K, Kronberg E, Eichman LC. Intrinsic hippocampal activity as a biomarker for cognition and symptoms in schizophrenia. Am J Psychiatry. 2014;171(5):549–556. doi: 10.1176/appi.ajp.2013.13070981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troisi A. Displacement activities as a behavioral measure of stress in nonhuman primates and human subjects. Stress. 2002;5(1):47–54. doi: 10.1080/102538902900012378. [DOI] [PubMed] [Google Scholar]
- Tseng KY, Chambers RA, Lipska BK. The neonatal ventral hippocampal lesion as a heuristic neurodevelopmental model of schizophrenia. Behav Brain Res. 2009;204(2):295–305. doi: 10.1016/j.bbr.2008.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unschuld PG, Buchholz AS, Varvaris M, van Zijl PC, Ross CA, Pekar JJ, Hock C, Sweeney JA, Tamminga CA, Keshavan MS, Pearlson GD, Thaker GK, Schretlen DJ. Prefrontal brain network connectivity Indicates degree of both schizophrenia risk and cognitive dysfunction. Schizophr Bull. 2014;40(3):653–664. doi: 10.1093/schbul/sbt077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Venrooi JM, Fluitman SB, Lijmer JG, Kavelaars A, Heijnen CJ, Westenberg HGM. Impaired neuroendocrine and immune response to acute stress in medication-naive patients with a first episode of psychosis. Schizophr Bull. 2012;38(2):272–279. doi: 10.1093/schbul/sbq062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti O, Lodge DJ, Grace AA. Aversive stimuli alter ventral tegmental area dopamine neuron activity via a common action in the ventral hippocampus. J Neurosci. 2011;31(11):4280–4289. doi: 10.1523/JNEUROSCI.5310-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables PH. The electrodermal psychophysiology of schizophrenics and children at risk for schizophrenia: controversies and developments. Schizophr Bull. 1977;3(1):28–48. doi: 10.1093/schbul/3.1.28. [DOI] [PubMed] [Google Scholar]
- Vieweg WV, David JJ, Rowe WT, Peach MJ, Veldhuis JD, Spradlin WW. Correlation of cigarette-induced increase in serum nicotine levels with arginine vasopressin concentrations in the syndrome of self-induced water intoxication and psychosis (SIWIP) Can J Psychiatry. 1986;31(2):108–111. doi: 10.1177/070674378603100206. [DOI] [PubMed] [Google Scholar]
- Vieweg WV, David JJ, Rowe WT, Wampler GJ, Burns WJ, Spradlin WW. Death from self-induced water intoxication among patients with schizophrenic disorders. J Nerv Ment Dis. 1985;1173(3):161–165. doi: 10.1097/00005053-198503000-00005. [DOI] [PubMed] [Google Scholar]
- Vieweg WV, Harrington DP, Westerman PS, McKelway RB, Hundley PL, Yank GR. Seasonal stability of water balance among schizophrenic patients subject to water intoxication. Prog Neuropsychopharmacol Biol Psychiatry. 1990;14 (2):215–222. doi: 10.1016/0278-5846(90)90102-m. [DOI] [PubMed] [Google Scholar]
- Vieweg WV, Hundley PL, Godleski LS, Tisdelle DA, Pruzinsky T, Yank GR. Diurnal weight gain as a predictor of serum sodium concentration in patients with psychosis, intermittent hyponatremia, and polydipsia (PIP syndrome) Psychiatry Res. 1988;26(3):305–312. doi: 10.1016/0165-1781(88)90125-4. [DOI] [PubMed] [Google Scholar]
- Walker EF, Diforio D. Schizophrenia: a neural diathesis-stress model. Psychol Rev. 1997;104(4):667–685. doi: 10.1037/0033-295x.104.4.667. [DOI] [PubMed] [Google Scholar]
- Wallace M, Singer G, Finlay J, Gibson S. The effect of 6-OHDA lesions of the nucleus accumbens septum on schedule-induced drinking, wheel running and corticosterone levels in the rat. Pharmacol Biochem Behav. 1983;18(1):129–136. doi: 10.1016/0091-3057(83)90262-9. [DOI] [PubMed] [Google Scholar]
- Wotjak CT, Ganster J, Kohl G, Holsboer F, Landgraf R, Engelmann M. Dissociated central and peripheral release of vasopressin, but not oxytocin, in response to repeated swim stress: new insights into the secretory capacities of peptidergic neurons. Neuroscience. 1998;85(4):1209–1222. doi: 10.1016/s0306-4522(97)00683-0. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Shu N, Liu Y, Song M, Hao Y, Liu H, Jiang T. Altered resting-state functional connectivity and anatomical connectivity of hippocampus in schizophrenia. Schizophren Res. 2008;100(1–3):120–132. doi: 10.1016/j.schres.2007.11.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.