

Abstract
miR-30 is a microRNA frequently overexpressed in human cancers. However, the biological consequence of miR-30 overexpression in cancer has been unclear. In a genetic screen, miR-30 was found to abrogate oncogenic-induced senescence, a key tumor-suppressing mechanism that involves DNA damage responses, activation of p53 and induction of p16INK4A. In cells and mouse models, miR-30 disrupts senescence and promotes cancer by suppressing 2 targets, CHD7 and TNRC6A. We show that while CHD7 is a transcriptional coactivator essential for induction of p16INK4A in senescent cells, TNRC6A, a miRNA machinery component, is required for expression and functionality of DNA damage response RNAs (DDRNAs) that mediate DNA damage responses and p53 activation by orchestrating histone modifications, chromatin remodeling and recruitment of DNA damage factors at damaged sites. Thus, miR-30 inhibits both p16INK4A and p53, 2 key senescence effectors, leading to efficient senescence disruption. These findings have identified novel signaling pathways mediating oncogene-induced senescence and tumor-suppression, and revealed the molecular and cellular mechanisms underlying the oncogenic activity of miR-30. Thus, the miR-30/CHD7/TNRC6A pathway is potentially a novel diagnostic biomarker and therapeutic target for cancer.
Keywords: oncogene-induced senescence, microRNA, DNA damage responses, p16INK4A, p53
INTRODUCTION
MicroRNAs (miRNA) regulate physiological and pathological conditions such as cancer (1). After processing by the RNase III Drosha/DGCR8 and Dicer/TRBP complexes, the leading strand of a mature miRNA is incorporated into the miRNA-induced Silencing Complex (miRISC) that suppresses the expression of target mRNA (2). miRISC contains a miRNA-binding endonuclease Ago2, helicases, and mRNA-binding proteins including TNRC6A, B, and C (3).
Senescence is a stable proliferative arrest associated with exhaustion of replicative potential of primary cells. Activation of oncogenes such as ras in early-passage cells causes a senescence-like phenotype known as oncogene-induced senescence (4), mediated by DNA damage responses triggered by DNA hyper-replication (5;6). Induction of senescence depends on sequential activation of the ERK and p38 MAPK pathways (7–9), and is marked by induction of p16INK4A, p53 and p21WAF1 (10). Disruption of senescence accelerates cancer development (11;12), indicating that oncogene-induced senescence suppresses tumorigenesis in vivo. The role of miRNAs in senescence has begun to unveil. Senescent cells display distinct miRNA expression profiles; some miRNAs indeed are involved in senescence (13–16).
In addition to its ability to mediate miRNA production and functionality, the miRNA machinery was recently implicated in DNA damage responses (DDRs) (17–19), a signaling pathway that mediates proliferative arrest and DNA damage repair in response to DNA lesions. In multiple organisms, DNA double strand breaks (DSBs) induces the production of 21-nucleotide small RNA (DNA damage response RNA, DDRNA) from the sequences near the DSB, which are critical for DDRs (17;18). Components of the miRNA pathway, Drosha, Dicer and Ago2, are essential for the biogenesis of DDRNA and DSB-induced DDRs, including focus formation and homologous recombination (HR)-mediated DNA repair, thus highlighting a novel function of the miRNA pathway in DDRs. While the exact role of DDRNAs in DDR is currently unknown, they may promote recruitment of methyltransferases and acetyltransferases to DSB, and mediate histone modifications and chromatin remodeling that is critical for the assembly of DDR factors at damage sites and DNA repair (19).
In this study, we identified miR-30, a microRNA with increased expression in human cancers, as an inhibitor of oncogene-induced senescence, and demonstrated that miR-30 disrupts senescence and accelerates tumorigenesis both in cells and mouse by targeting CHD7, a transcriptional coactivator of p16INK4A, and TNRC6A, a miRNA pathway component essential for ras-induced DDRNA expression and functionality, DDRs and subsequent activation of p53. miR-30 thus inhibits activation of both p16INK4A and p53, leading to senescence disruption. These findings reveal CHD7 and TNRC6A as novel components in signaling pathways mediating oncogene-induced senescence, and identify miR-30 as a novel cancer driver, which may serve as a new diagnostic marker and therapeutic target for cancer.
RESULTS
miR-30 disrupts oncogenic ras-induced senescence
We screened a lentiviral expression library containing 524 miRNAs (Biosettia) to search for miRNAs that, when overexpressed, inhibit oncogenic ras-induced senescence in BJ primary human fibroblasts. We identified 47 miRNAs in the primary screen, and miR-30 was identified as one of the 8 hits in retests (Fig. S1A). Increased expression of each of the 5 miR-30 family members abrogated induction of proliferative arrest and SA-β-gal by Ha-rasV12 in BJ cells (Fig. 1A–C, S1B). Mutant miR-30c with a scrambled seed sequence failed to do so (Fig. S1C–D). Same results were reproduced in primary WI-38 human fibroblasts and human mammary epithelial cells (HMECs) (Fig. S1E–F).
Fig. 1.
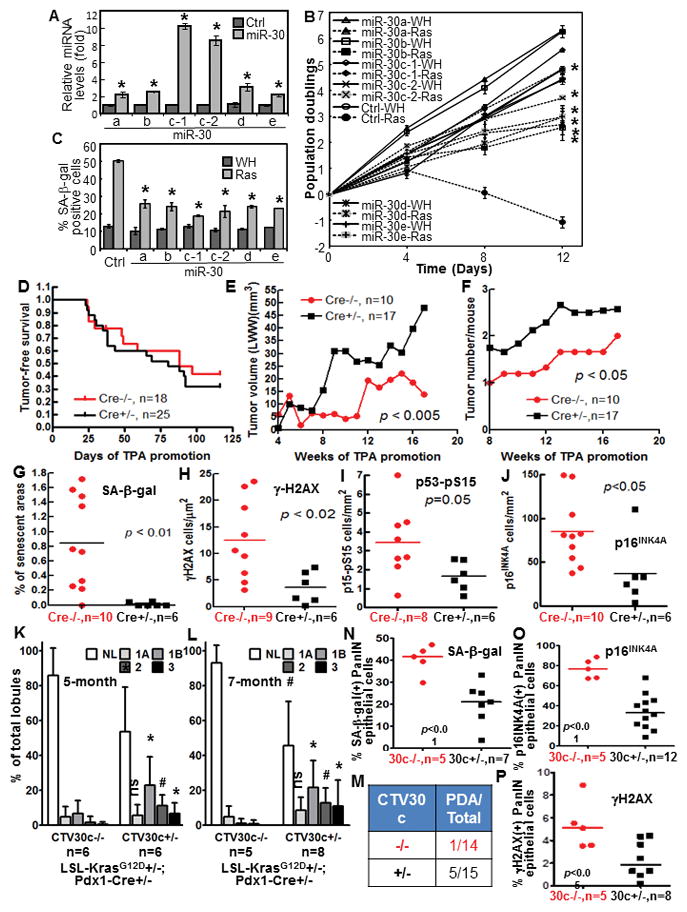
miR-30 disrupts ras-induced senescence, and miR-30c-1 promotes cancer development and disrupts senescence, DDR and p53 and p16INK4A induction in mouse models.
(A) miR-30 levels in BJ cells transduced with miR-30 family members (miR-30) or vector (Ctrl) measured by qRT-PCR.
(B) Growth curves of cells transduced with miR-30 members or vector (Ctrl) and ras (Ras) or vector (WH).
(C) % of SA-β-gal-positive cells on day-10 in cells in (B).
(A–C) Values are mean±SD for triplicates. * p < 0.05 vs Ctrl (A) or Ctrl-Ras (B, C) by Student t test.
(D) Tumor-free survival in miR-30-1 transgenic (Cre+/−) and non-transgenic (Cre−/−) littermates in 2-stage DMBA carcinogenesis.
(E–F) Average tumor growth rate (E) and number of tumors/mice (F) in miR-30-1 transgenic (Cre+/−) and non-transgenic (Cre−/−) littermates in 2-stage carcinogenesis.
(G–J) Quantification of cells positive for SA-β-gal (G), γ-H2AX (H), p53-pS15 (I) and p16INK4A (J) in DMBA-induced papilloma from miR-30-1 transgenic (Cre+/−) and non-transgenic (Cre−/−) littermates, normalized to tumor areas. p values determined by unpaired t test.
(K–L) % of pancreatic lobules containing normal (NL) and neoplastic ducts by grade (1A, 1B, 2 and 3) in 5-month (K) or 7-month (L) miR-30c-1 transgenic (CTV30c+/−) or control (CTV30c−/−) littermates. Values are mean±SD. *, p < 0.05; #, p < 0.01; ns, not significant for CTV30c+/− vs CTV30c−/− in unpaired t test.
(M) Number of miR-30c-1 transgenic (CTV30c+/−) and control (CTV30c−/−) littermates that developed PDA by month-7.
(N–P) % of SA-β-gal- (N), 16INK4A- (O) and γH2AX- (P) positive epithelial cells in PanIN lesions in miR-30c-1 transgenic (30c+/−) or control (30c−/−) pancreata. p values determined by unpaired t test.
miR-30c disrupts senescence and enhances cancer development in murine cancer models
We generated inducible miR-30c-1 transgenic lines (20) (Fig. S2A–C) and analyzed the role of miR-30 in DMBA-induced skin carcinogenesis in which senescence suppresses tumorigenesis (12). Treatment of mouse skin with DMBA followed by TPA induces skin papillomas, the majority of which are driven by activating ras mutations (21;22). We created a strain specifically overexpressing miR-30c in skin epithelium, the origin of DMBA-induced skin papillomas, but not in other tissues (Fig. S2A, S2D). While there was no difference in the incidence or latency of tumor induction (Fig. 1D), DMBA/TPA induced faster growing (Fig. 1E) and more (Fig. 1F) tumors in the miR-30c transgenic than in the control littermates. SA-β-gal-positive senescent cells were detected in skin tumors from the wild type mice, but barely in miR-30c transgenic tumors (Fig. 1G, S2E). Thus, miR-30c accelerates skin carcinogenesis and disrupt senescence induction in vivo.
We further analyzed the effect of miR-30 in a ductal pancreatic cancer model driven by KrasG12D (LSL-KrasG12D; Pdx1-Cre) (23;24). When crossed into this model, miR-30c expression was increased specifically in pancreas but not in other tissues of miR-30c-1 transgenic mice as compared to control littermates (Fig. S2F). Pancreatic cancer development in this model is characterized by histological progression from normal ductal morphology to low-grade (1A and 1B) pancreatic intraepithelial neoplasias (PanINs), high-grade (2 and 3) PanINs, and eventually invasive pancreatic ductal adenocarcinoma (PDA)(23). At both 5- and 7-month of age, the pancreas from the miR-30c-1 transgenic mice contained less normal lobules and more lobules containing grade 1B, 2 and 3 PanIN ducts, than that from nontransgenic littermates (Fig. 1K, L, S2J). Moreover, 5 of the 15 miR-30c-1 transgenic mice developed PDA at 4-, 5-, 6-, 6.5-, 7-month, respectively, while only 1 of the 14 nontransgenic mice developed PDA at 7-month (Fig. 1M). Thus, miR-30c accelerates KrasG12D-driven pancreatic cancer progression. The development of PanIN lesions is accompanied by senescence induction by KrasG12D in PanINs epithelial cells (24). We detected less SA-β-gal positive epithelial cells in PanIN lesions from the miR-30c-1 transgenic than the control pancreas (Fig. 1N, S2G). Thus, miR-30c promotes pancreatic cancer development and disrupts senescence induction by oncogenic Kras.
miR-30 disrupts ras-induced senescence by targeting CHD7 and TNRC6A
To identify senescence-related miR-30 targets, we made 3′UTR reporters for the top 40 targets predicted by TargetScan and miRBase. 15 of these reporters (Table S1), including those for CHD7 and TNRC6A (Fig. 2A, Table S2) were suppressed by cotransfected miR-30. miR-30 had no effect on the reporters containing mutant miR-30-binding sites (Fig. 2A, Table S2). All 5 miR-30 members inhibited the expression of endogenous CHD7 and TNRC6A mRNA in BJ cells (Fig. 2B). Thus, CHD7 and TNRC6A are direct miR-30 targets. We generated lentiviral shRNAs for these 15 miR-30 targets, and those for CHD7 and TNRC6A abrogated ras-induced senescence in BJ (Fig 2C–H), WI-38 and HMEC cells (Fig. S1G–J). Ras-induced senescence was restored strongly by cotransfected TNRC6A and CHD7 in BJ-miR-30 cells, but only slightly by TNRC6A or CHD7 alone (Fig. 2I, J). Thus, miR-30 disrupts ras-induced senescence by targeting CHD7 and TNRC6A, which are essential for senescence induction.
Fig. 2.
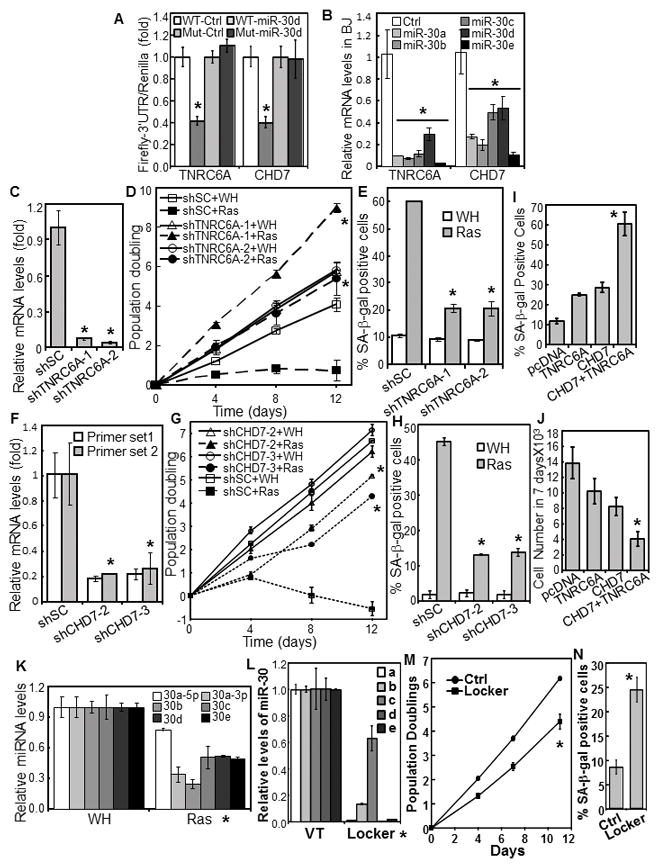
miR-30 disrupts senescence by targeting TNRC6A and CHD7.
(A) miR-30 suppresses the 3′UTR-reporters of TNRC6A and CHD7. The 3′UTR Firefly luciferase reporters of TNRC6A and CHD7 containing wild type (WT) or mutant (Mut) miR-30-binding sites were cotransfected into 293 cells with CMV-Renilla Luciferase and miR-30d or vector (Ctrl). Firefly luciferase activity was determined 24 hrs later and normalized to Renilla luciferase activity.
(B) TNRC6A and CHD7 mRNA were determined in BJ cells transduced with miR-30 members or vector (Ctrl) by qRT-PCR.
(C, F) TNRC6A (C) or CHD7 (F) mRNA were measured in BJ cells transduced with shRNA for TNRC6A (C) or CHD7 (F) or a scrambled shRNA (shSC) by qRT-PCR.
(D, G) Growth curves of BJ cells transduced with shRNA for TNRC6A (D) or CHD7 (G) or scrambled shRNA (shSC) and HaRasV12 (Ras) or vector (WH).
(E, H) % of SA-β-gal-positive cells in populations in (D) and (G), respectively, on day-9 of the growth curves.
(I–J) BJ cells transduced with miR-30c and HaRasV12 were transfected with TNRC6A, CHD7, both or vector (pcDNA3) together with pcDNA3-GFP. GFP-positive cells were sorted and stained for SA-β-gal (I) and numbers counted after 7 days (J).
(K) Levels of miR-30 family members were measured in BJ cells transduced with HaRasV12 (Ras) or vector (WH) by qRT-PCR.
(L–M) Inhibition of miR-30 induces senescence. BJ cells transduced with a miR-30-Locker that inhibits the expression of all miR-30 isoforms (Locker, Biosettia) or vector control (Ctrl) were analyzed for the expression levels of miR-30a–e by qRT-PCR (L), and oncogenic ras-induced senescence by growth curve analysis (M) and SA-β-gal staining (N).
(A–N) Values are mean±SD for triplicates. * p < 0.05 vs Ctrl, shSC, WH, or VT by Student t test.
Levels of the miR-30 isoforms were downregulated by oncogenic ras (Fig. 2K). In addition, downregulation of all miR-30 isoforms by a lentiviral vector expressing sponges and hairpin inhibitors for miR-30a–e (Biosettia, Inc) (Fig. 2L) induced senescence in BJ cells (Fig. 2M–N). Thus, miR-30 is an integral component of the senescence-inducing pathway.
CHD7 mediates ras-induced p16INK4A expression as a transcriptional coactivator
CHD7 is an ATP-dependent helicase and a transcriptional coactivator(25–29). In a survey of expression of senescence effectors, ectopic expression of miR-30 or CHD7 shRNA reduced ras-induced p16INK4A at both protein (Fig. 3A) and mRNA (Fig. 3B) levels. Cotransfected CHD7 stimulated transcription from a 3 kb (−3570 ~ +306) and a 1.2 kb (−891 ~ +306) p16INK4A promoter in 293T cells (Fig. 3C). Transcription from these 2 reporters was also induced by oncogenic ras in BJ cells (Fig. 3D). Thus, CHD7 is essential for ras-induced p16INK4A transcription.
Fig. 3.
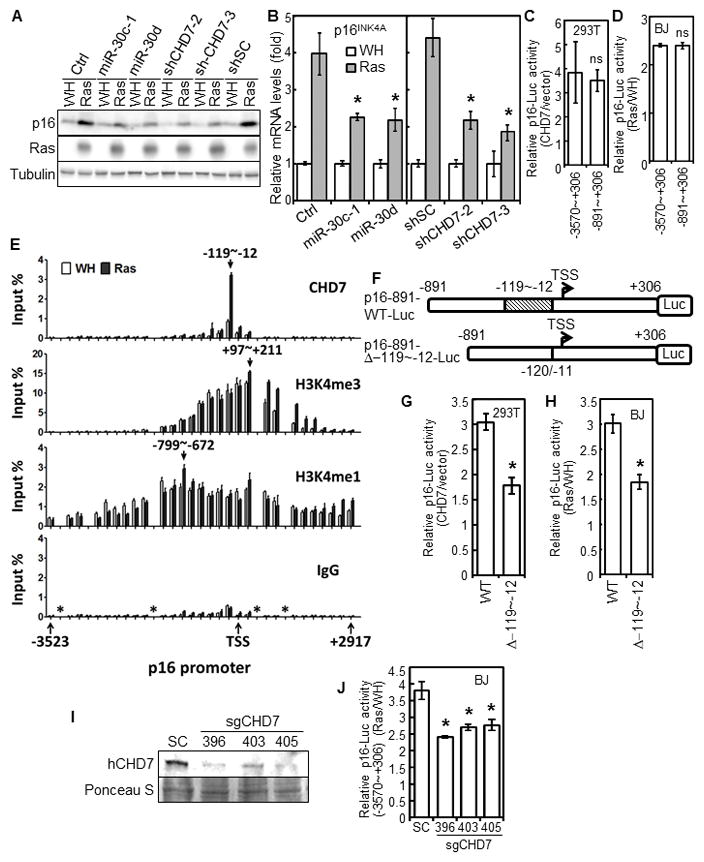
CHD7 mediates ras-induced p16INK4A expression as a transcriptional coactivator.
(A–B) p16INK4A protein (A) and mRNA (B) levels were measured in BJ cells transduced with miR-30 or vector (Ctrl) or CHD7 shRNA or a scrambled shRNA (shSC) and HaRasV12 (Ras) or vector (WH). * p < 0.05 vs Ctrl or shSC by Student t test.
(C) Luciferase reporters for the 3kb (−3570~+306) and 1.2kb (−891~+306) p16INK4A promoters were transfected into 293T cells with pcDNA3-CHD7 or vector.
(D) BJ cells containing stable luciferase reporters for the 3kb- and 1.2kb-p16INK4A promoters were transduced with HaRasV12 (Ras) or vector (WH).
(C, D) ns, no statistical significance by Student t test.
(E) ChIP analysis of the p16INK4A promoter. Abundance of DNA between −3523~+2917 of p16INK4A promoter immnuoprecipitated from BJ cells transduced with HaRasV12 (Ras) or vector (WH) by CHD7, H3K4me3, H3K4me1 antibodies or IgG was determined by qRT-PCR, and normalized to input. Arrows, positions of the DNA binding peaks. *, regions lacking efficient primers.
(F) Diagrams of the p16INK4A promoter luciferase reporters used in (G) and (H), showing position of the deleted CHD7-binding site and transcriptional start site (TSS).
(G) The p16INK4A promoter luciferase reporter with (WT) or without the CHD7-binding site were transfected into 293T cells with pcDNA3-CHD7 or vector.
(H) BJ cells containing a stable p16INK4A promoter luciferase reporter with (WT) or without the CHD7-binding site (Δ−119~−12) were transduced with HaRasV12 or vector.
(G, H) * p < 0.05 vs WT by Student t test.
(I–J) BJ cells containing a stable luciferase reporter for the 3kb (−3570~+306) p16INK4A promoters were transduced with CRISPR/Cas9 for CHD7 or a scrambled sequence (SC), and then with HaRasV12 (Ras) or vector (WH). CHD7 protein (I) and luciferase activity (J) were determined. * p < 0.05 vs SC by Student t test.
(C–D, G–H, J) Luciferase activity was determined 48 hrs (C, G) or 7 days (D, H, J) later and normalized to protein concentrations.
(B, C–E, G–H, J) Values are mean±SD for triplicates.
Using Chromatin immunoprecipitation (ChIP) assay, we mapped the CHD7 binding site to −119 ~ −12 of the p16INK4A promoter (Fig. 3E). Binding of CHD7 to this region was greatly enhanced by oncogenic ras (Fig. 3E). Consistent with previous reports (26;27), the CHD7 binding site is located in an open chromosomal region marked by H3K4me3 and H3K4me1. However, the H3K4me3- and H3K4me1-binding sites were much broader than that of CHD7 and not dependent on ras (Fig. 3E), suggesting that although CHD7 binds to open chromatin regions containing H3K4me, H3K4me may not be the only determinant for CHD7 binding. Further demonstrating the functional significance of CHD7 binding, deletion of the CHD7 binding site (−119 ~ −12) (Fig. 3F) reduced induction of the p16INK4A transcription by cotransfected CHD7 in 293T cells (Fig. 3G), and by oncogenic ras in BJ cells (Fig. 3H). Deletion of CHD7 from BJ cells by the CRISPR/Cas9 (Fig. 3I) also abrogated ras-induced p16INK4A transcription (Fig. 3J). Thus, CHD7 mediates ras-induced p16INK4A expression as a transcriptional coactivator.
Inactivation of miRNA machinery disrupts oncogenic ras-induced senescence
Requirement of TNRC6A in senescence led us to examine the role of other components of the miRNA pathway. shRNAs that inhibited the expression of TNRC6B, a homolog of TNRC6A, abrogated oncogenic ras-induced growth arrest and accumulation of SA-β-gal (Fig. S3A–C). Silencing of Dicer to modest levels (shDicer-1 and -4) disrupted senescence; while a more potent Dicer shRNA (shDicer-2), although reduced proliferation of both control and ras-expressing cells, also prevented induction of SA-β-gal by ras (Fig. S3D–F). Ago2 knockdown moderately reduced cell proliferation, but essentially blocked ras-induced senescence (Fig. S3G–I). Thus, multiple components of the miRNA pathway are essential for oncogene-induced senescence, consistent with prior reports that downregulation of miRNA machinery increases tumorigenesis(30–33). Our results have thus suggested an important role of the miRNA pathway in oncogene-induced senescence.
miR-30 and TNRC6A shRNA disrupt oncogenic ras-induced DNA damage responses
Since the miRNA pathway plays an essential role in DDR by regulating the biogenesis of small non-coding DDRNAs that orchestrate the assembly of DNA damage foci and chromatin remodeling around the damage sites (17–19;34), we investigated whether miR-30, through inhibition of a miRNA pathway component TNRC6A, disrupts senescence by impairing oncogene-induced DDR. Ras induced formation of 53BP1 and γH2AX foci, consistent with previous reports(5;6), which was impaired by miR-30 or TNRC6A shRNAs, and by Dicer shRNA (Fig. 4A–C). miR-30 (Fig. 4D) and TNRC6A shRNA (Fig. 4E) abrogated ras-induced DDR signaling, including phosphorylation of γH2AX, Chk2-T68, p38 MAPK and p53-S15, and induction of a p53 transcriptional target p21WAF1, and abolished ras-induced p53 transcriptional activity on a stably integrated reporter (PG-Luc)(8) (Fig. 4F). Thus, miR-30 and TNRC6A shRNA disrupt ras-induced DDR, an intermediate step toward subsequent p53 activation and senescence induction.
Fig. 4.
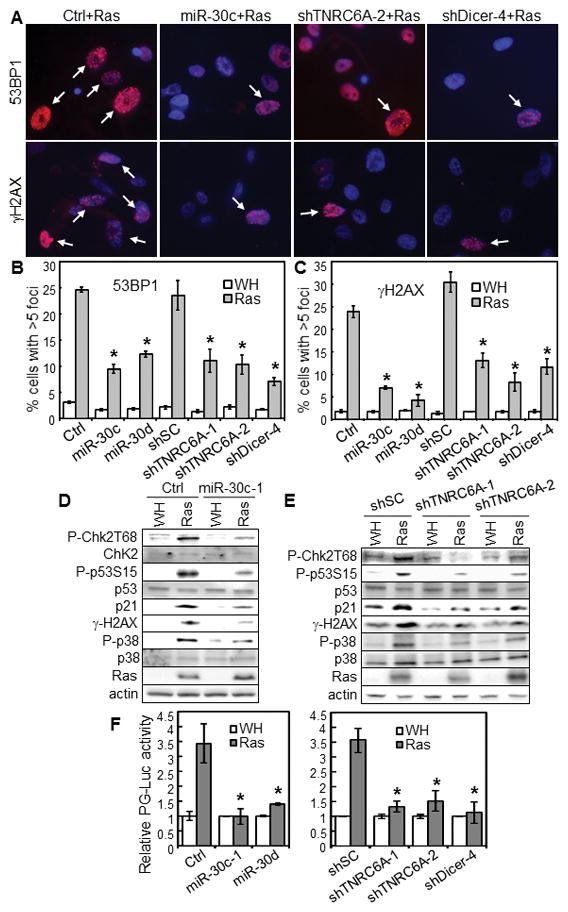
TNRC6A is essential for ras-induced DDR and p53 activation.
(A–C) BJ cells transduced with miR-30 or vector (Ctrl) or shRNA for TNRC6A or Dicer or a scrambled sequence (shSC) and HaRasV12 (Ras) or vector (WH) were stained for 53BP1- or γ-H2AX-foci and photographed (A). % of cells with > 5 53BP1- (B) or γ-H2AX (C)-foci (arrows) were quantified.
(D–E) Western blot of BJ cells transduced with miR-30c-1 or vector (Ctrl) (D) or shRNA for TNRC6A or a scrambled sequence (shSC) (E) and then with HaRasV12 (Ras) or vector (WH) on day 8 post Ras-transduction.
(F) BJ cells with a stable p53-dependent luciferase reporter (PG-Luc) were transduced with miR-30 or vector (Ctrl) (left) or shRNA for TNRC6A or Dicer or a scrambled shRNA (shSC) (right) and then with HaRasV12 (Ras) or vector (WH). Luciferase activity was determined 7 days later and normalized to protein concentrations.
(B–C, F) Values are mean±SD for triplicates. * p < 0.05 vs Ctrl or shSC by Student t test.
miR-30 and TNRC6A shRNA disrupt γ-radiation-induced DNA damage responses and p53 activation
We tested the impact of miR-30 and TNRC6A on DDR induced by genotoxic agents. Ectopic expression of miR-30 and TNRC6A knockdown greatly reduced formation of γ-radiation-induced 53BP1 foci (Fig. 5A, S4A), but only modestly (although with statistical significance) reduced γ-radiation-induced γH2AX foci (Fig. S4A, B). miR-30 or TNRC6A shRNA also disrupted γ-radiation-induced DDR signaling, including phosphorylation of γH2AX, Chk2-T68 and p53-S15, induction of p21WAF1 (Fig. S4C), and γ-radiation-induced p53 transcriptional activity (Fig. S4D). Thus, by suppressing TNRC6A, miR-30 interferes with γ-radiation induced DDR and p53 activation.
Fig. 5.
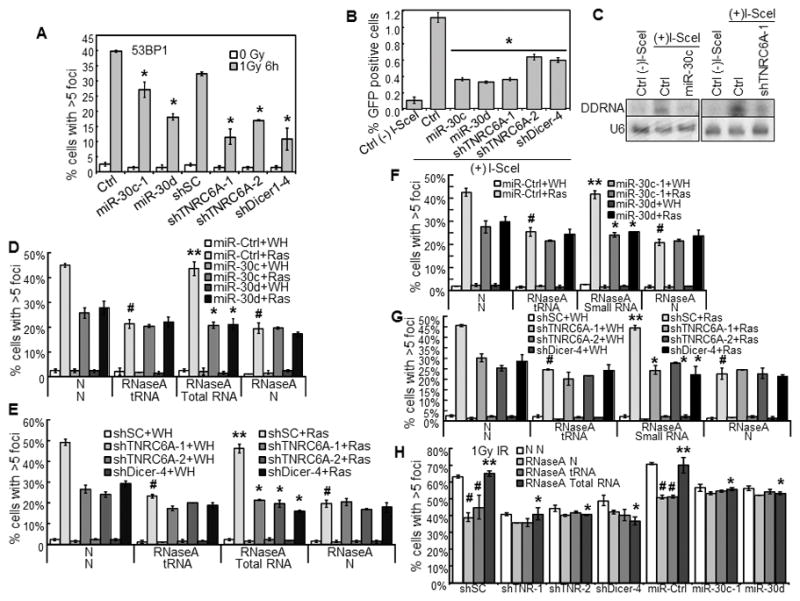
miR-30 and TNRC6A shRNA disrupt γ-radiation induced 53BP1-foci, HR-mediated repair, and expression and functionality of DDRNAs.
(A) % of cells with 53BP1-foci in BJ cells transduced with miR-30 or vector (Ctrl) or shRNA for TNRC6A or Dicer or a scrambled sequence (shSC) and treated with 0 or 1Gy γ-radiation.
(B) miR-30 and TNRC6A shRNA disrupt HR-mediated repair. I-SceI-EGFP-U2OS cells were transduced with miR-30, shRNA for TNRC6A or Dicer or vector control (Ctrl), and then transduced with I-SceI [(+)I-SceI] or control [(−)I-SceI]. % of GPF positive cells was determined by flow cytometry 7 days later.
(A, B) * p < 0.05 vs Ctrl or shSC by Student t test.
(C) miR-30 and TNRC6A shRNA reduce the DDRNA levels. Autoradiogram of Northern blot analysis of DDRNAs in I-SceI-EGFP-U2OS cells after transduction with miR-30c-1, shRNA for TNRC6A or vector (Ctrl) and I-SceI [(+)I-SceI] or control [(−)I-SceI]. A radio-labeled DNA fragment from the region surrounding the I-SceI site was used as probe for DDRNAs. U6 RNA was used as loading control.
(D–H) miR-30 and TNRC6A shRNA abrogate the functionality of DDRNA. BJ cells transduced with miR-30 or vector (miR-Ctrl) (D, F) or shRNA for TNRC6A or Dicer or a scrambled sequence (shSC) (E, G) and then with HaRasV12 (Ras) or vector (WH) were permeabilized, treated with RNaseA or not (N), and after washing and incubation with RNasin, incubated with total (D–E) or small (≤200 nucleotides) (F–G) RNA isolated from senescent cells, yeast total RNA (tRNA) or vehicle (N). Cells were stained for 53BP1-foci and quantified.
(H) BJ cells transduced with shRNA for TNRC6A (shTNR-1, 2) or Dicer or a scrambled sequence (shSC) or miR-30 or vector (miR-Ctrl) were treated with 1Gy γ-radiation, permeabilized after 24 hrs, treated with RNaseA or not (N), and after washing and incubation with RNasin, incubated with total RNA isolated from 1Gy γ-radiation-treated BJ cells (total RNA), yeast total RNA (tRNA) or vehicle (N). Cells were stained for 53BP1-foci and quantified.
(A–B, D–H) Values are mean±SD for triplicates.
(D–H) # p < 0.05 vs N/N by Student t test. ** p < 0.05 vs RNaseA/tRNA by Student t test. * p < 0.05 vs miR-Ctrl+Ras (D, F) or shSC+Ras (E, G) or miR-Ctrl/shSC+IR (H) by Student t test.
One important component of DDR is checkpoint activation, which results in proliferative arrest, senescence and/or apoptosis, to prevent the transmission of damaged DNA. Different doses of γ-radiation inhibited the amount of BrdU incorporating cells in control BJ cells, indicating a S phase checkpoint arrest, which was alleviated by miR-30 or TNRC6A shRNA (Fig. S4E). Thus, miR-30 and TNRC6A shRNA impair γ-radiation-induced S phase checkpoint arrest. Consistent with the disruption of proliferative arrest, miR-30 and TNRC6A shRNA, as well as Dicer shRNA, greatly reduced the percentage of SA-β-gal-positive cells in γ-radiated BJ populations (Fig. S4F), indicating that miR-30 and TNRC6A shRNA abrogate γ-radiation-induced senescence. γ-radiation barely induced any apoptosis in BJ cells (Fig. S4G), suggesting that senescence, rather than apoptosis, is the major response to γ-radiation in these cells.
miR-30 and TNRC6A shRNA disrupt homologous recombination (HR)-mediated DNA repair, and reduce the level and abrogate the functionality of DDRNA
Another component of DDR is repair of damaged DNA. To investigate the role of miR-30 and TNRC6A in repair, we used U2OS cells carrying a single integrated copy of the I-SceI-EGFP reporter for HR(35). Expression of I-SceI by lentivirus in the control U2OS/I-SceI-EGFP reporter cells results in a cut at the I-SceI site, and HR-mediated repair of the cut restores a functional EGFP, leading to accumulation of 1.1% of GFP-positive cells indicative of HR repair efficiency. miR-30 or TNRC6A shRNA reduced the efficiency of HR-mediated DNA damage repair by 2–3-fold as compared to the control cells (Fig. 5B). As reported previously(17;18), Dicer shRNA also reduced HR-mediated repair. These suggest that by suppressing TNRC6A, miR-30 disrupts both checkpoint activation and repair after DNA damage.
Drosha, Dicer and Ago2 are essential for the biogenesis of DDRNA from the sequences near DSB sites, which are critical for DDR (17;18). To investigate the role of TNRC6A and miR-30 on the expression and functionality of DDRNA, we took advantage of the I-SceI-EGFP system in which I-SceI induces a single DSB at a defined genomic locus. In Northern blotting analysis using a probe derived from the region surrounding the I-SceI site, we detected accumulation of DDRNA upon expression of I-SceI, which was essentially abolished by miR-30 or TNRC6A shRNA (Fig. 5C). Thus, miR-30 and TNRC6A shRNA either disrupt the biogenesis or reduces the stability of DDRNA.
Drosha and Dicer are responsible for the processing, while TNRC6A is required for functionality, of miRNA. We reason that TNRC6A may also be required for the functionality of DDRNA in DDR. It was previously shown that transient membrane permeabilization and RNase A treatment leads to degradation of all RNAs including DDRNAs without affecting protein levels, and impairs IR-induced DDR focus formation. This defect in DDR foci can be rescued by incubating RNase A-treated cells with total or gel-purified small RNA isolated from IR-treated cells, which contains DDRNA(18). As in IR-treated cells, RNase A treatment greatly reduced ras-induced formation of 53BP1 foci in BJ cells. Total or purified small (< 200 nucleotides) RNA isolated from ras-expressing BJ cells, but not yeast total RNA (tRNA), restored focus formation in control cells, but not in cells expressing miR-30 or TNRC6A shRNA (Fig. 5D–G). Exogenous total RNA isolated from IR-treated BJ cells also restored foci formation in control BJ cells first treated with IR and then with RNase A, but not in cells expressing miR-30 or TNRC6A shRNA (Fig. 5H). Therefore, TNRC6A is required for the functionality of DDRNA in DDR, and by suppressing TNRC6A, miR-30 abrogates the functionality of DDRNA, thus disrupting DDR. Interestingly, exogenous total and small RNA isolated from senescent cells or IR-treated cells did not restore focus formation in BJ cells with Dicer knockdown, either (Fig. 5E, G, H), suggesting that the processing and functionality of DDRNA might be coupled.
TNRC6A is recruited to DNA damage sites and mediates histone modifications and chromatin remodeling and recruitment of DDR factors on these sites
To further investigate the mechanism by which TNRC6A regulates DDR, we examined the role of TNRC6A in DNA damage-induced chromatin remodeling, an essential step in DDR in which DDRNAs may act as guiding molecules(19). Histones including γ-H2AX, H2AX and H3 are released from chromatin adjacent to DNA damage sites, when chromatin is extracted in 1.0M NaCl(36). ras-induced NaCl solubility of these 3 histones was abrogated by miR-30 and TNRC6A shRNA (Fig. 6A), indicating that miR-30 attenuates TNRC6A-mediated chromatin remodeling resulted from ras-induced DNA damage. DDRNAs recruit acetyltransferases and methyltransferases to DSBs, resulting in local histone H4K16Ac and H4K20me modifications, respectively, which are essential for chromatin remodeling after DNA damage(19;37). In ChIP assays in U2OS cells expressing ER-I-PpoI, a 15bp-cutter endonuclease that cleaves DNA at defined sites(19;38), 4-hydroxytamoxifen (4-OHT) induction of ER-I-PpoI led to increased H4K16Ac and H4K20me3 near DSB at a defined locus. These modifications were abrogated by miR-30 or TNRC6A shRNA (Fig. 6B). ChIP assays also revealed that TNRC6A is localized to the I-PpoI cut site, along with 53BP1, γH2AX, ATM and Ago2 (Fig. 6C). TNRC6A is required at least for recruitment of γH2AX and Ago2, because ectopic expression of miR-30 or TNRC6A shRNA reduced γH2AX and Ago2 binding to the cut sites (Fig. 6D). Thus, TNRC6A is recruited to DNA damage sites, where it mediates generation and functionality of DDRNAs, histone modifications and chromatin remodeling, and recruitment of DDR factors.
Fig. 6.
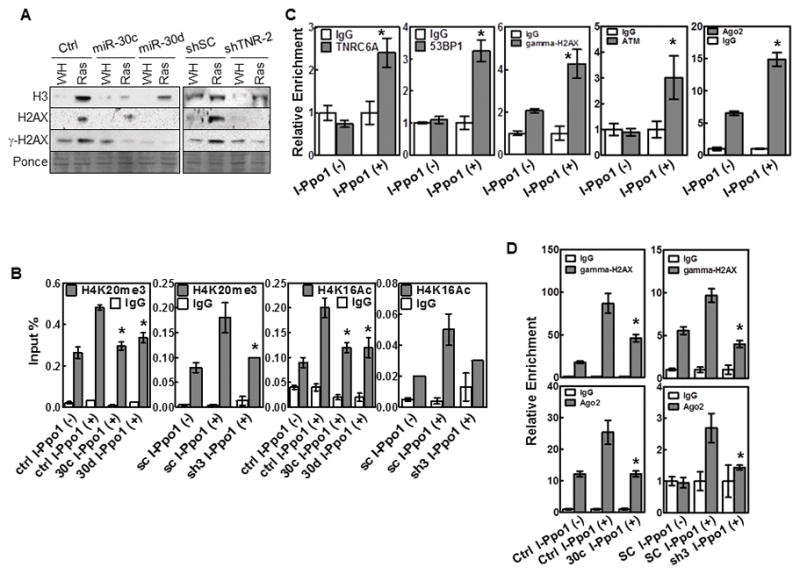
TNRC6A is localized at DNA damage sites and mediates histone modifications and chromatin remodeling and recruitment of DDR factors after DNA damage.
(A) Western blot of supernatants from NaCl extraction of BJ cells transduced with miR-30c or vector control (Ctrl), TNRC6A shRNA (shTNR-2) or scrambled shRNA (shSC), and HaRasV12 (Ras) or vector (WH).
(B) ChIP analysis of U2OS cells transduced with miR-30 (30c, 30d) or vector (Ctrl), or TNRC6A shRNA (sh3) or scrambled shRNA (sc), and ER-I-PpoI with [I-PpoI(+)] or without [I-PpoI(−)] 4-OHT induction, using anti-H4K20me3 or anti-H4K16Ac antibodies or IgG.
(C) TNRC6A, 53BP1, γH2AX, ATM and Ago2 are co-localized at DNA damage sites. ChIP analysis of U2OS-ER-I-PpoI cells with [I-PpoI(+)] or without [I-PpoI(−)] 4-OHT induction.
(D) TNRC6A is required for binding of γH2AX and Ago2 to DNA damage sites. ChIP analysis of U2OS cells transduced with miR-30c (30c) or vector (Ctrl), or TNRC6A shRNA (sh3) or a scrambled sequence (SC), and ER-I-PpoI with [I-PpoI(+)] or without [I-PpoI(−)]4-OHT induction.
(B–D) Abundance of immunoprecipitated DNA near the I-PpoI site on chromosome 1 was measured by qRT-PCR and normalized to input. Values are mean±SD for triplicates. * p < 0.05 vs Ctrl I-PpoI(+) or SC I-PopI(+) (B, D) or I-PpoI(−) (C) by Student t test.
(C–D) Relative enrichment was calculated.
miR-30 suppresses CHD7 and TNRC6A expression, disrupts DNA damage response and inhibits activation of p16INK4A and p53 in mouse cancel models
We investigated the impact of miR-30 on p16INK4A, DDR and p53 activation in vivo. Consistent with the specific expression of miR-30c-1 in skin keratinocytes in the skin carcinogenesis model and in pancreas in the pancreatic cancer model, expression of both CHD7 and TNRC6A was suppressed specifically in respective tissues in miR-30c-1 transgenic, as compared to those from wild type littermates (Fig. S2D, F). Thus, CHD7 and TNRC6A are miR-30 targets in mouse models.
Corresponding to reduced senescence in the skin tumors from miR-30c transgenic mice (Fig. 1G, S2E), these tumors contained less cells positive for γH2AX or activated p53 (p53-pS15), indicating abrogation of DDR, and less p16INK4A-positive cells, as compared to those from wild type littermates (Fig. 1H–J, S2E). Thus, miR-30 disrupts senescence and promotes skin carcinogenesis while suppressing its targets CHD7 and TNRC6A and inhibiting p16INK4A induction, DDR and p53 activation. In the pancreatic model, enhanced cancer progression and disrupted senescence was also accompanied by reduction in the γH2AX- and p16INK4A-positive epithelial cells in PanIN lesions from the miR-30c-1 transgenic as compared to the control pancreas (Fig. 1O–P, S2G). This supports a notion that miR-30 abrogates senescence and promotes cancer development by disrupting p16INK4A induction and DDR through suppression of CHD7 and TNRC6A, respectively. The number of cells positive for p53-pS15 or p53 was very low in these lesions, suggesting that p53 may not mediate DDR in this model. Expression of Cre itself did not induce DDR in normal skin and pancreas in these models (Fig. S2H).
DISCUSSION
Although several studies indicated that miR-30 is overexpressed in human cancers, the biological significance of increased expression of miR-30 in cancer had been unclear. Here, we demonstrate that miR-30 disrupts oncogene-induced senescence, a key tumor-suppressing mechanism, and promotes tumorigenesis in cell culture and mouse models, by targeting CHD7 and TNRC6A. We are currently analyzing the role of the miR-30/CHD7/TNRC6A pathway in human cancers, and our preliminary results indicate that suppression of CHD7 and TNRC6A expression and senescence by miR-30 also occurs in human cancers and contributes to poor patient survival. These findings establish miR-30 as a cancer driver and reveal new molecular and cellular mechanisms underlying the oncogenic activity of miR-30.
We found that the 2 miR-30 targets involved in senescence, CHD7 and TNRC6A, are essential for the induction of 2 key senescence effectors, p16INK4A and p53, respectively. CHD7 mediates oncogenic ras-induced p16INK4A expression as a transcriptional coactivator. TNRC6A, when recruited to DNA damage sites, mediates the expression and functionality of DDRNAs derived from DNA damage sites, which promote histone modifications and chromatin remodeling, and recruitment of DDR factors at the damage sites, thus triggering DDR and subsequently p53 activation. These results provide the molecular mechanism for senescence disruption by miR-30 and suggest that a microRNA can disrupt a phenotype by simultaneously suppressing multiple targets that synergistically confer this phenotype.
Our studies have identified CHD7 and TNRC6A as novel components in the signaling pathway mediating ras-induced senescence, and as novel tumor suppressors. CHD7 deficiency is embryonic lethal in mouse(39), and causes CHARGE syndrome in human, a genetic disease characterized by complex birth defects(40). CHD7 is a transcriptional coactivator mapped to DNase I-hypersensitive open chromatin regions containing H3K4me(26;27), and recruited to enhancers by H3K4me or transcriptional activators/coactivators including Oct4, Sox2, Sox9, Twist, p300 and PBAF(27–29). We found that the CHD7 binding site on the p16INK4A promoter is located in a H3K4me-enriched region, but does not overlap with H3K4me peaks, suggesting that H3K4me is not the only determinant for CHD7 binding, and that CHD7 may be recruited to an H3K4me-enriched, open chromatin region on the p16INK4A promoter by a transcription factor.
TNRC6A, TNRC6B and TNRC6C comprise the trinucleotide repeat-containing (TNRC)-6 protein family(41). By interacting with Ago proteins in the miRISC complex, TNRC6A-C play an important role in miRNA-mediated gene silencing(3;42). Our data demonstrate that TNRC6A is recruited to DNA damage sites along with other DDR factors and is required for ras- and IR-induced DDR focus formation, DDR signaling and senescence, for IR-induced checkpoint activation, for HR-mediated repair, for histone modifications and chromatin remodeling at damage sites, and for recruitment of DDR factors to damage sites. These identify TNRC6A as a new component in the recently discovered DDR pathway involving the miRNA machinery. The essential role of TNRC6A in this pathway indicates that not only the miRNA machinery components responsible for miRNA processing (Drosha and Dicer), but also those for miRNA functionality (TNRC6A and TNRC6B), participate in the DDRNA-mediated DDR and ras-induced senescence. It is therefore likely that the biogenesis and functionality of miRNAs and DDRNAs are mediated by the miRNA machinery in a similar fashion, in that Drosha and Dicer process precursors of DDRNAs into mature DDRNAs. Mature DDRNAs are then incorporated into a RISC-like complex containing Ago2 and TNRC6 proteins, which is targeted to DNA damage sites by the DDRNAs to orchestra the assembly of DNA damage foci and induction of DDR.
Interestingly, exogenous RNA isolated from senescent cells were also unable to restore focus formation in BJ cells with Dicer knockdown, indicating that Dicer is also required for the functionality of DDRNAs. Meanwhile, TNRC6A knockdown also reduced the levels of DDRNAs. These findings suggest that the processing or the level of DDRNAs and their functionality are coupled. Indeed, it was suggested that miRNA processing is coupled to their functionality. The association of Dicer and Ago2 is required for loading of mature miRNAs to Ago2, and thus the formation of a functional RISC complex(43); the stability of miRNAs depends on Ago2(44). It is likely that similar to miRNAs, DDRNAs are also stabilized upon binding to Ago2 and TNRC6A.
MATERIALS AND METHODS
Cell culture, antibodies and plasmids
BJ cells were maintained in MEM, and WI38, U2OS-HR reporter cells, 293T and LinX-A cells in DMEM, with 10% FCS. Primary antibodies were from Sigma (actin), Santa Cruz (Ras C-20, p53 FL-393, p21WAF1 C-19), Cell Signaling (phospho-p53-Ser15, phospho-Chk2T68, γ-H2AX, phosphor-p38, p38), Abcam (CHD7 ab176807, TNRC6A ab84403) and Novus Biologicals (53BP1). HRP-conjugated goat anti-rabbit secondary antibody was from Jackson Laboratories. Alexa Fluor® 488 Goat Anti-Rabbit IgG was from ThermoFisher Scientific. Plasmid construction is described in Supplemental Materials.
Retrovirus- and lentivirus-based gene transduction
Analysis of senescence
Senescence was analyzed by growth and SA-β-Gal staining as described (7). For analysis of restoration of ras-induced senescence, BJ cells transduced with miR-30 and HaRasV12 were transfected with 22.5 μg of pcDNA3-CHD7, pcDNA3-TNRC6A, both pcDNA3-CHD7 and pcDNA3-TNRC6A, or vector control (pcDNA3), together with 7.5 μg of pcDNA3-GFP in 45 μl of X-tremeGENE™ HP DNA Transfection Reagent (Roche) in 10-cm plates. GFP positive cells were sorted by flow cytometry 2 days later, and 5000 cells/well were seeded into 12-well plates. Cells were stained for SA-β-gal 2 days later and cell numbers were counted 5 days later.
Western blot analysis
Western blot was performed as described(7).
RNA isolation and quantitative Real-Time PCR
RNA was isolated from cells using TRIzol and quantified by qRT-PCR using iScript™ Reverse Transcription Supermix and SsoAdvanced™ SYBR Green Supermix (Bio-Rad) in Bio-Rad CFX96 following manufacturer’s protocols. GAPDH was used as internal control. Primers are listed in Supplemental Materials.
Luciferase reporter assay
Luciferase reporters were transfected or transduced into cells. Luciferase activity was determined by Luciferase Assay System following manufacturer’s protocols (Promega).
Chromatin-immunoprecipitation (ChIP) assay
ChIP assays were performed as described before (9) or following dual-crosslinking protocol (for CHD7) (46).
RNase A treatment and DDR focus rescue experiments
RNase A treatment and DDR focus rescue were performed as described(18).
Immuno-staining for DNA damage foci
DDR foci were determined as described(18).
Detection of DDRNA by Northern blotting
DDRNA was detected by Northern blot as described using a probe near the I-SceI site(20). In brief, 30 μg total RNA was separated on an agarose gel and subsequently transferred to nylon membrane. 50 ng of a fragment digested from the I-SceI-GPF reporter was labeled with [α-32P]dATP and [α-32P]dCTP by random priming, purified by using MicroSpin G25 Columns (Amersham), and used as probe to detect DDRNA generated near the I-SceI site. The U6 RNA levels were detected by an oligonucleotide probe from U6 (5′-TATGTGCTGCCGAAGCGAGCAC-3′) and used as loading control. Signals were detected by a Typhoon phospho-imager.
HR-Repair assay
4x105/well of U20S-HR-reporter cell line (35) was plated into 6-well plates and transduced with lentiviruses encoding miR-30 or shRNAs for TNRC6A or Dicer or a vector control virus, and selected with puromycin. All the cell lines were then transduced with an I-SceI-encoding lentivirus (pLentivirus-MCS-IRES-Bsd-I-SceI) or a vector control and selected with 5 μg/ml of blasticidin. 7–8 days after transduction, cells were collected, and the percentage of GFP positive cells was determined by flow cytometry. Each experiment was performed in triplicates.
BrdU incorporation assay
3–4x105/well of BJ cells were seeded in 6-well plates, and after overnight incubation, treated with proper dosage of ionizing radiation and incubated at 37°C for 24h. 1 7mu;M BrdU (Sigma) was added into medium and incubated with the cells for 1h at 37°C. BrdU incorporating cells were stained with FITC Anti-BrdU (BD, catalog number 347583) according to manufacturer’s protocol and analyzed by flow cytometry. Each experiment was performed in triplicates.
Apoptosis assay
Apoptosis was determined by Annexin-V kit (eBioscience).
Analysis of tumor formation and senescence in mouse models
miR-30c-1 transgenic mice were generated by targeting miR-30c-1-coding sequence into Rosa26 locus as described(20), and crossed to KRT14-Cre and Pdx1-Cre mice to generate tissue-specific transgenic lines. Cancer development was analyzed as described (23;47). Sample sizes are chosen based on similar published studies. Animals with desired genotypes are randomly chosen. Quantification of IHC results were performed by blinded investigators who do not know the nature of the samples. Animal protocols are approved by the IACUC committee of The Scripps Research Institute and Wake Forest University Medical Center.
Chromatin remodeling
Chromatin remodeling was determined by NaCl solubility assay as described(19).
Supplementary Material
Acknowledgments
This study was supported by Overseas Collaboration Fund from National NSF of China 31428013 (PS, RX), NIH/NCI grants CA106768, CA131231, CA172115 (PS), P30CA012197, and Thomas K. Hearn Brain Tumor Center. WS is supported by International Postdoctoral Exchange Fellowship Program by Office of China Postdoctoral Council (20140027).
We thank Dr. Joanna Wysocka for CHD7 cDNA, and Tumor Tissue and Pathology, Biostatistics and Bioinformatics, Cell & Viral Vector Laboratory and Cellular Imaging Shared Resources of WFBCCC. This study was supported by Overseas Collaboration Fund from National NSF of China 31428013 (PS, RX), NIH/NCI grants CA106768, CA131231, CA172115 (PS), P30CA012197, and Thomas K. Hearn Brain Tumor Center. WS is supported by International Postdoctoral Exchange Fellowship Program by Office of China Postdoctoral Council (20140027).
Footnotes
CONFLICT OF INTEREST
Authors declare no competing financial interests in relation to the work described.
References
- 1.Fabbri M, Croce CM, Calin GA. MicroRNAs. Cancer J. 2008;14:1–6. doi: 10.1097/PPO.0b013e318164145e. [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landthaler M, Gaidatzis D, Rothballer A, Chen PY, Soll SJ, Dinic L, et al. Molecular characterization of human Argonaute-containing ribonucleoprotein complexes and their bound target mRNAs. RNA. 2008;14:2580–96. doi: 10.1261/rna.1351608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Courtois-Cox S, Jones SL, Cichowski K. Many roads lead to oncogene-induced senescence. Oncogene. 2008;27:2801–9. doi: 10.1038/sj.onc.1210950. [DOI] [PubMed] [Google Scholar]
- 5.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–7. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 6.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 7.Wang W, Chen JX, Liao R, Deng Q, Zhou JJ, Huang S, et al. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol Cell Biol. 2002;22:3389–403. doi: 10.1128/MCB.22.10.3389-3403.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwong J, Hong L, Liao R, Deng Q, Han J, Sun P. p38alpha and p38gamma mediate oncogenic ras-induced senescence through differential mechanisms. J Biol Chem. 2009;284:11237–46. doi: 10.1074/jbc.M808327200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwong J, Chen M, Lv D, Luo N, Su W, Xiang R, et al. Induction of p38delta Expression Plays an Essential Role in Oncogenic ras-Induced Senescence. Mol Cell Biol. 2013;33:3780–94. doi: 10.1128/MCB.00784-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferbeyre G, de Stanchina E, Lin AW, Querido E, McCurrach ME, Hannon GJ, et al. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol. 2002;22:3497–508. doi: 10.1128/MCB.22.10.3497-3508.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Narita M, Lowe SW. Senescence comes of age. Nat Med. 2005;11:920–2. doi: 10.1038/nm0905-920. [DOI] [PubMed] [Google Scholar]
- 12.Sun P, Yoshizuka N, New L, Moser BA, Li Y, Liao R, et al. PRAK Is Essential for ras-Induced Senescence and Tumor Suppression. Cell. 2007;128:295–308. doi: 10.1016/j.cell.2006.11.050. [DOI] [PubMed] [Google Scholar]
- 13.Zhao T, Li J, Chen AF. MicroRNA-34a induces endothelial progenitor cell senescence and impedes its angiogenesis via suppressing silent information regulator 1. Am J Physiol Endocrinol Metab. 2010;299:E110–E116. doi: 10.1152/ajpendo.00192.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faraonio R, Salerno P, Passaro F, Sedia C, Iaccio A, Bellelli R, et al. A set of miRNAs participates in the cellular senescence program in human diploid fibroblasts. Cell Death Differ. 2011 doi: 10.1038/cdd.2011.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srikantan S, Gorospe M, Abdelmohsen K. Senescence-associated microRNAs linked to tumorigenesis. Cell Cycle. 2011;10:3211–2. doi: 10.4161/cc.10.19.17050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hong L, Lai M, Chen M, Xie C, Liao R, Kang YJ, et al. The miR-17-92 cluster of microRNAs confers tumorigenicity by inhibiting oncogene-induced senescence. Cancer Res. 2010;70:8547–57. doi: 10.1158/0008-5472.CAN-10-1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei W, Ba Z, Gao M, Wu Y, Ma Y, Amiard S, et al. A role for small RNAs in DNA double-strand break repair. Cell. 2012;149:101–12. doi: 10.1016/j.cell.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 18.Francia S, Michelini F, Saxena A, Tang D, de HM, Anelli V, et al. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature. 2012;488:231–5. doi: 10.1038/nature11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang Q, Goldstein M. Small RNAs Recruit Chromatin-Modifying Enzymes MMSET and Tip60 to Reconfigure Damaged DNA upon Double-Strand Break and Facilitate Repair. Cancer Res. 2016;76:1904–15. doi: 10.1158/0008-5472.CAN-15-2334. [DOI] [PubMed] [Google Scholar]
- 20.Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, et al. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell. 2007;131:146–59. doi: 10.1016/j.cell.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 21.DiGiovanni J. Modification of multistage skin carcinogenesis in mice. Prog Exp Tumor Res. 1991;33:192–229. doi: 10.1159/000419252. [DOI] [PubMed] [Google Scholar]
- 22.DiGiovanni J. Multistage carcinogenesis in mouse skin. Pharmacol Ther. 1992;54:63–128. doi: 10.1016/0163-7258(92)90051-z. [DOI] [PubMed] [Google Scholar]
- 23.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–50. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 24.Caldwell ME, DeNicola GM, Martins CP, Jacobetz MA, Maitra A, Hruban RH, et al. Cellular features of senescence during the evolution of human and murine ductal pancreatic cancer. Oncogene. 2012;31:1599–608. doi: 10.1038/onc.2011.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim Y, et al. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–5. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- 26.Schnetz MP, Bartels CF, Shastri K, Balasubramanian D, Zentner GE, Balaji R, et al. Genomic distribution of CHD7 on chromatin tracks H3K4 methylation patterns. Genome Res. 2009;19:590–601. doi: 10.1101/gr.086983.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schnetz MP, Handoko L, Akhtar-Zaidi B, Bartels CF, Pereira CF, Fisher AG, et al. CHD7 targets active gene enhancer elements to modulate ES cell-specific gene expression. PLoS Genet. 2010;6:e1001023. doi: 10.1371/journal.pgen.1001023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, et al. CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature. 2010;463:958–62. doi: 10.1038/nature08733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engelen E, Akinci U, Bryne JC, Hou J, Gontan C, Moen M, et al. Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat Genet. 2011;43:607–11. doi: 10.1038/ng.825. [DOI] [PubMed] [Google Scholar]
- 30.Kim MS, Oh JE, Kim YR, Park SW, Kang MR, Kim SS, et al. Somatic mutations and losses of expression of microRNA regulation-related genes AGO2 and TNRC6A in gastric and colorectal cancers. J Pathol. 2010;221:139–46. doi: 10.1002/path.2683. [DOI] [PubMed] [Google Scholar]
- 31.Hill DA, Ivanovich J, Priest JR, Gurnett CA, Dehner LP, Desruisseau D, et al. DICER1 mutations in familial pleuropulmonary blastoma. Science. 2009;325:965. doi: 10.1126/science.1174334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar MS, Pester RE, Chen CY, Lane K, Chin C, Lu J, et al. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev. 2009;23:2700–4. doi: 10.1101/gad.1848209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–7. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- 34.d’Adda di FF. A direct role for small non-coding RNAs in DNA damage response. Trends Cell Biol. 2014;24:171–8. doi: 10.1016/j.tcb.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 35.Wang H, Shao Z, Shi LZ, Hwang PY, Truong LN, Berns MW, et al. CtIP protein dimerization is critical for its recruitment to chromosomal DNA double-stranded breaks. J Biol Chem. 2012;287:21471–80. doi: 10.1074/jbc.M112.355354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu Y, Ayrapetov MK, Xu C, Gursoy-Yuzugullu O, Hu Y, Price BD. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol Cell. 2012;48:723–33. doi: 10.1016/j.molcel.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Price BD, D’Andrea AD. Chromatin remodeling at DNA double-strand breaks. Cell. 2013;152:1344–54. doi: 10.1016/j.cell.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berkovich E, Monnat RJ, Jr, Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007;9:683–90. doi: 10.1038/ncb1599. [DOI] [PubMed] [Google Scholar]
- 39.Hurd EA, Capers PL, Blauwkamp MN, Adams ME, Raphael Y, Poucher HK, et al. Loss of Chd7 function in gene-trapped reporter mice is embryonic lethal and associated with severe defects in multiple developing tissues. Mamm Genome. 2007;18:94–104. doi: 10.1007/s00335-006-0107-6. [DOI] [PubMed] [Google Scholar]
- 40.Jongmans MC, Admiraal RJ, van der Donk KP, Vissers LE, Baas AF, Kapusta L, et al. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet. 2006;43:306–14. doi: 10.1136/jmg.2005.036061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eystathioy T, Chan EK, Tenenbaum SA, Keene JD, Griffith K, Fritzler MJ. A phosphorylated cytoplasmic autoantigen, GW182, associates with a unique population of human mRNAs within novel cytoplasmic speckles. Mol Biol Cell. 2002;13:1338–51. doi: 10.1091/mbc.01-11-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meister G, Landthaler M, Peters L, Chen PY, Urlaub H, Luhrmann R, et al. Identification of novel argonaute-associated proteins. Curr Biol. 2005;15:2149–55. doi: 10.1016/j.cub.2005.10.048. [DOI] [PubMed] [Google Scholar]
- 43.Meister G. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet. 2013;14:447–59. doi: 10.1038/nrg3462. [DOI] [PubMed] [Google Scholar]
- 44.Zamudio JR, Kelly TJ, Sharp PA. Argonaute-bound small RNAs from promoter-proximal RNA polymerase II. Cell. 2014;156:920–34. doi: 10.1016/j.cell.2014.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun P, Dong P, Dai K, Hannon GJ, Beach D. p53-independent role of MDM2 in TGF-beta1 resistance. Science. 1998;282:2270–2. doi: 10.1126/science.282.5397.2270. [DOI] [PubMed] [Google Scholar]
- 46.Zeng PY, Vakoc CR, Chen ZC, Blobel GA, Berger SL. In vivo dual cross-linking for identification of indirect DNA-associated proteins by chromatin immunoprecipitation. Biotechniques. 2006;41:694, 696, 698. doi: 10.2144/000112297. [DOI] [PubMed] [Google Scholar]
- 47.Yoshizuka N, Chen RM, Xu Z, Liao R, Hong L, Hu WY, et al. A Novel Function of p38-Regulated/Activated Kinase in Endothelial Cell Migration and Tumor Angiogenesis. Mol Cell Biol. 2012;32:606–18. doi: 10.1128/MCB.06301-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mukhopadhyay A, Deplancke B, Walhout AJ, Tissenbaum HA. Chromatin immunoprecipitation (ChIP) coupled to detection by quantitative real-time PCR to study transcription factor binding to DNA in Caenorhabditis elegans. Nat Protoc. 2008;3:698–709. doi: 10.1038/nprot.2008.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gertsenstein M, Nutter LM, Reid T, Pereira M, Stanford WL, Rossant J, et al. Efficient generation of germ line transmitting chimeras from C57BL/6N ES cells by aggregation with outbred host embryos. PLoS One. 2010;5:e11260. doi: 10.1371/journal.pone.0011260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.