

Abstract
Solar ultraviolet (sUV) irradiation is a major environmental carcinogen that can cause inflammation and skin cancer. The costs and morbidity associated with skin cancer are increasing, and therefore identifying molecules that can help prevent skin carcinogenesis is important. In this study, we identified the p53-related protein kinase (PRPK) as a novel oncogenic protein that is phosphorylated by the T-LAK cell-originated protein kinase (TOPK). Knockdown of TOPK inhibited PRPK phosphorylation and conferred resistance to solar simulated light (SSL)-induced skin carcinogenesis in mouse models. In the clinic, acute SSL irradiation significantly increased epidermal thickness as well as total protein and phosphorylation levels of TOPK and PRPK in human skin tissues. We identified two PRPK inhibitors, FDA-approved rocuronium bromide (Zemuron®) or betamethasone 17-valerate (Betaderm®) that could attenuate TOPK-dependent PRPK signaling. Importantly, topical application of either rocuronium bromide or betamethasone decreased SSL-induced epidermal hyperplasia, neovascularization and cutaneous squamous cell carcinoma (cSCC) development in SKH1 (Crl: SKH1-Hrhr) hairless mice by inhibiting PRPK activation, and also reduced expression of the proliferation and oncogenesis markers, COX-2, cyclin D1 and MMP-9. This study is the first to demonstrate that targeting PRPK could be useful against sUV-induced cSCC development.
Keywords: TOPK, PRPK, solar ultraviolet, inflammation, cutaneous squamous cell carcinoma
INTRODUCTION
Skin cancer has an estimated annual incidence of 700,000 cases in the United States (1–3). In particular, the incidence of cutaneous squamous cell carcinoma (cSCC), a type of non-melanoma skin cancer (NMSC), is increasing dramatically every year. Surgery is an effective treatment against early-stage NMSC. Nonetheless, cSCC accounts for approximately 20% of all cutaneous malignancies; and 4% of cSCC can reoccur locally or metastasize to regional lymph nodes or distant organs resulting in death, with a five-year survival rate that is less than 20% for patients with cSCC metastases (4–7). The costs and morbidity associated with skin cancer are rising every year and therefore, effective preventive or therapeutic strategies are needed. Solar ultraviolet (sUV) irradiation is a major environmental carcinogen that causes inflammation and skin cancer. Long-term exposure of solar ultraviolet (sUV) irradiation to skin induces inflammatory responses, oxidative stress, DNA damage, gene mutations and immunosuppressive responses, which altogether have been implicated in various skin diseases, including an increased risk of skin cancers. Actinic keratosis (AK), induced by sUV irradiation, acts as precursor to cSCC and is a pre-malignant skin lesion characterized with an abnormal proliferation of atypical keratinocytes confined to the epidermis of the skin (8). Therefore, to intervene at a premalignant stage with chemopreventive strategies before development of cSCC, as well as preventing metastasis of cSCC, seems reasonable.
Metastasis of cSCC is characterized by rapid growth that is associated with increased angiogenesis, invasion, colonization and eventually proliferation to other organs (9). Chronic UV irradiation increases the expression of cyclooxygenase-2 (COX-2), which is the primary source to elevate the production of prostaglandin E2 (PGE2) in the skin, and this prostaglandin plays a key role in skin carcinogenesis by contributing to the uncontrolled proliferation of damaged cells that eventually form tumors (10, 11). The important proliferation marker cyclin D1, which contributes to malignant progression in most human cancers, is overexpressed in SCC and basal cell carcinoma (BCC) (12, 13). Another important marker for cancer progression is the matrix metalloproteinase (MMP) family of proteins, which are essential for degradation of the extracellular matrix (ECM) allowing tumor cells to invade other organs. Furthermore, Notch 1 is down-regulated in solar keratoses and invasive SCCs arising on sUV-exposed skin suggest a tumor suppressor effect for the Notch signaling pathway in UV-related squamous cell photocarcinogenesis (14).
Identifying the major signaling molecules in cSCC development is therefore a critically important task. We previously reported that the T-LAK cell-originated protein kinase (TOPK) is a member of the MAPK protein family and a potential oncogene in the transformation of colorectal cancer (15). TOPK is also highly overexpressed in many types of human cancers, including colon, lung and breast cancers (16–18).
Here, we identified the p53-related protein kinase (PRPK) as a novel kinase downstream of TOPK that promotes skin carcinogenesis. PRPK was originally discovered and named by Abe, et al. (19), but the authors eventually concluded that PRPK might not be directly associated with p53-dependent cell proliferation (20). Furthermore, Peterson, et al. suggested that p53 phosphorylation is not the major role of PRPK (21).
We show that PRPK is a novel molecular driver and critical player in skin carcinogenesis, and that it is directly phosphorylated and activated by TOPK. Targeting PRPK with the FDA approved drugs, rocuronium bromide (Zemuron®) or betamethasone 17-valerate (Betaderm®), suppressed chronic solar simulated light (SSL)-induced cSCC development and decreased expression of COX-2, cyclin D1 and MMP-9 in SKH1 (Crl: SKH1-Hrhr) hairless mice. Thus, molecular targeting of PRPK could be an effective approach in the clinic to treat or prevent cSCC.
RESULTS
Knockout of TOPK completely blocks solar simulated light (SSL)-induced skin cancer
To investigate the role of TOPK in skin carcinogenesis, we examined the status of TOPK in human skin samples. We detected high levels of total and phosphorylated TOPK (Thr9) protein expression in SCC and BCC (Figure 1A), and phosphorylated TOPK (Thr9) levels were also markedly higher in AKs and SCCs from patients compared to matched normal skin (Figure 1B).
Figure 1. Expression of phosphorylated TOPK in AKs and SCCs from human skin.
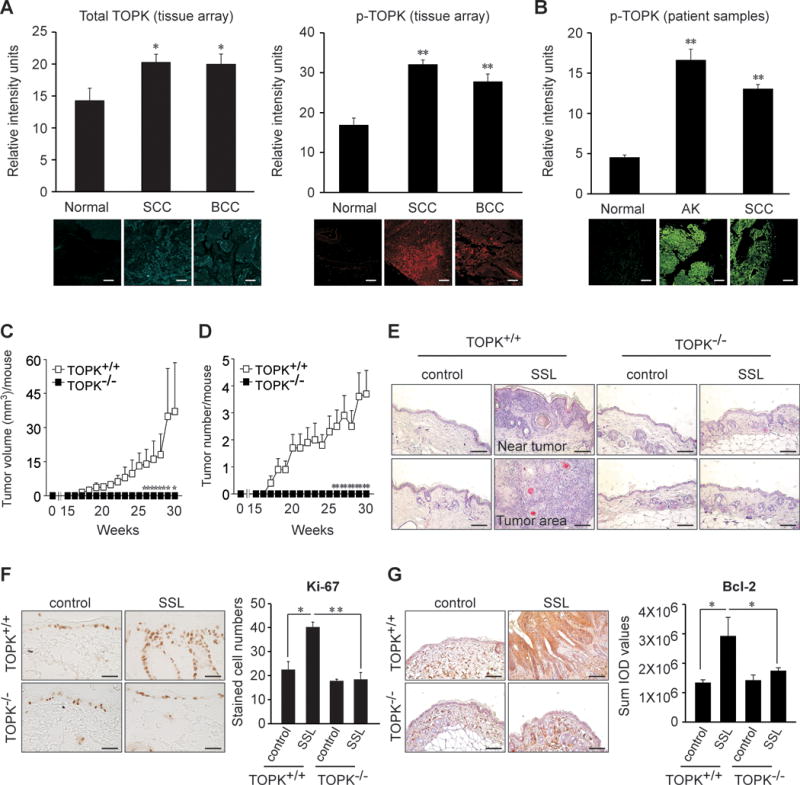
A human skin tissue array was analyzed by immunofluorescence staining to assess the expression of (A, left panel) total TOPK and (A, right panel) phosphorylated TOPK (Thr9) in SCCs (40 tissues) and BCCs (14 tissues) compared to normal skin (10 tissues). (B) The expression of phosphorylated TOPK (Thr9) was assessed in normal skin (30 samples from 6 human subjects), AKs (25 samples from 5 human subjects) and SCCs (40 samples from 8 human subjects). (C, D) Mice were divided into 4 groups that included SSL-untreated control groups or SSL-treated groups of SKH1 (Crl: SKH1-Hrhr) wildtype (TOPK+/+) or SKH1 TOPK knockout (TOPK−/−) mice (20 mice per each group). Average (C) tumor volume and (D) tumor number induced by chronic SSL in SKH1 TOPK+/+ or TOPK−/− mice. For Figure 1A–D, the asterisks (*, **) indicate a significant (p < 0.05 or p < 0.01, respectively) difference compared to the normal tissue group. (E) H&E staining of SKH1 TOPK+/+ or TOPK−/− mouse skin after chronic exposure to SSL. (F) Ki-67 proliferation marker levels in TOPK−/− mouse skin treated with chronic SSL. (G) Anti-apoptotic Bcl-2 protein levels in TOPK−/− mouse skin treated with chronic SSL. Bcl-2 levels are presented as the sum of integrated optical density (IOD) values. The asterisks (*, **) indicate a significant (p < 0.05 or p < 0.01, respectively) difference compared to SSL-treated TOPK+/+ mice. Scale bar = 100 μm.
Next, we examined chronic SSL-induced skin carcinogenesis in TOPK knockout (TOPK−/−) and wild-type (TOPK+/+) control SKH1 (Crl: SKH1-Hrhr) hairless mice. Interestingly, TOPK−/− mice showed no tumors on SSL-exposed skin tissues (Figure 1C, D). In contrast, TOPK+/+ mice treated with SSL showed increased tumor volume (Figure 1C) and number (Figure 1D). Furthermore, TOPK−/− mice showed base levels of epidermal thickness similar to the SSL-unexposed control group, but SSL-treated TOPK+/+ mice showed markedly thicker epidermal changes (Figure 1E). Moreover, the Ki-67 proliferation marker was decreased in SSL-exposed TOPK−/− mouse skin compared to SKH1 TOPK+/+ mice (Figure 1F). Notably, levels of the anti-apoptotic protein Bcl-2 were decreased in TOPK−/− mice, compared to SSL-treated TOPK+/+ mice (Figure 1G). These results suggest that TOPK is important in the development of skin carcinogenesis and exerts anti-apoptotic effects in SSL-exposed mouse skin.
We also examined phosphorylated and total TOPK protein levels in human primary SCCs and SCCs metastasized to skin or lymph nodes. Our results indicated that phosphorylated and total TOPK protein levels in SCC metastasized to skin or lymph nodes were significantly higher compared to human SCC primary tumors. Results of Pearson’s correlation coefficient (σ) analysis showed a positive correlation between tumor stage and tumors metastasized to skin or lymph nodes with values of 0.6405 for phosphorylated TOPK (Supplementary Figure 1A) and 0.7529 for total TOPK (Supplementary Figure 1B). These results indicated that phosphorylated and total TOPK proteins are associated with both SCC development and metastasis.
PRPK is a novel oncogenic kinase phosphorylated by TOPK in skin carcinogenesis
Our results so far suggest that TOPK is involved in skin carcinogenesis; however, whether TOPK or one of its downstream targets is directly involved is unclear. To identify the downstream phosphorylation targets of TOPK, we performed an in vitro kinase assay and screened several potential protein substrates. Results indicated that TOPK phosphorylated PRPK in vitro. The phosphorylation at Ser250 is known to be important for PRPK catalytic activity (22), and in vitro kinase assay results confirmed that TOPK strongly phosphorylated PRPK at Ser250 (Figure 2A). Furthermore, our results showed that PRPK could bind to the endogenous TOPK protein in SCC cell lines A431, SCC13 and human precancerous HaCaT keratinocytes (Figure 2B). However, in normal human epidermal keratinocytes (NHEKs), PRPK and the endogenous TOPK protein had only a low binding affinity (Figure 2B). These results indicate that PPRK is a downstream target of TOPK and that PRPK likely plays an important role in sUV-induced skin carcinogenesis.
Figure 2. PRPK is a novel downstream phosphorylation target of TOPK.
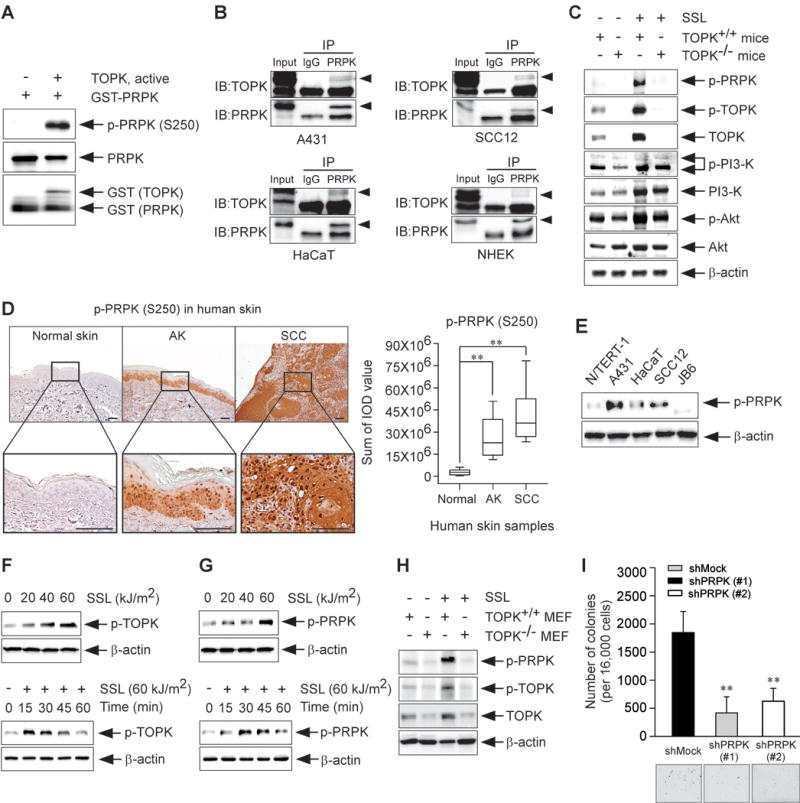
(A) Kinase assay results of active TOPK and a purified GST-tagged PRPK protein were obtained by Western blot analysis. (B) Endogenous binding activity between TOPK and PRPK in human skin epidermoid carcinoma A431 cells, human skin squamous cell carcinoma SCC12, human precancerous HaCaT keratinocytes and normal human epidermal keratinocytes (NHEKs), respectively. Cell lysates were incubated with a PRPK antibody for 24 h at 4°C and pulled down with A/G agarose beads. After immunoprecipitation, protein-bead mixtures were washed 3 times with 1 × PBS and then TOPK/or PRPK proteins were detected by Western blot analysis. (C) Phosphorylated PRPK (Ser250), TOPK (Thr9), PI3-K p85 (Tyr458)/p55 (Tyr199) and Akt (Ser473) levels in chronic SSL-treated TOPK−/− mouse skin. (D) Immunohistochemical staining of normal human skin tissues (30 samples from 6 human subjects), AKs (25 samples from 5 human subjects) and SCCs (40 samples from 8 human subjects) was used to detect expression of phosphorylated PRPK (Ser250). The phosphorylated PRPK levels are presented as the sum of IOD values. The asterisks (**) indicate a significant (p < 0.01) difference compared to the normal skin tissue group. Scale bar = 100 μm. (E) Phosphorylated PRPK levels were detected by Western blot analysis in normal skin (N/TERT-1, JB6), human precancerous keratinocytes (HaCaT) and human SCC (A431, SCC12) cell lines. (F) Phosphorylated TOPK and (G) phosphorylated PRPK levels in HaCaT cells are stimulated by SSL in a dose- (upper panel) or time-dependent (bottom panel) manner. (H) Phosphorylated PRPK levels in TOPK−/− MEFs treated with SSL. (I) Effect of PRPK knockdown on anchorage-independent growth of human skin epidermoid carcinoma A431 cells and data represents mean values ± S.D. of 3 independent experiments. The asterisks (**) indicate a significant (p < 0.01) difference compared to the shMock group.
Phosphorylated PRPK levels were absent in chronic SSL-exposed TOPK−/− mice, as compared with SKH1 TOPK+/+ mice (Figure 2C, Supplementary Figure 2A) as well as in TOPK−/− mice exposed to acute SSL irradiation (Supplementary Figure 2B). As previously reported, TOPK promotes cell migration through the mediation of the PI3-K and Akt pathways in lung cancer (23). Therefore, we examined whether PI3-K, Akt or PRPK were affected by TOPK status and also examined whether PRPK was directly affected by the PI3-K/Akt pathway. Results indicated that SSL-stimulated phosphorylation of PI3-K and Akt was not significantly different between TOPK+/+ and TOPK−/− mouse skin (Figure 2C). In contrast, phosphorylated PRPK levels were dramatically decreased in TOPK−/− mouse skin compared with TOPK+/+ mouse skin (Figure 2C). The presence of the PI3-K inhibitor, LY294002, did not affect PRPK phosphorylation in A431 cells (Supplementary Figure 3A), indicating that the PI3-K pathway is not associated with PRPK phosphorylation. To determine the role of PRPK in skin carcinogenesis, we examined human skin samples, and detected highly phosphorylated PRPK expression in AKs and SCCs compared to normal skin (Figure 2D). Furthermore, phosphorylated PRPK was overexpressed in SCC cell lines, including A431 and SCC12 (Figure 2E). TOPK and PRPK phosphorylation in HaCaT cells was also increased by SSL irradiation in a dose- and time-dependent manner (Figure 2F, G). When TOPK expression was knocked down, SSL-induced PRPK phosphorylation also disappeared (Figure 2H). Next, we investigated the role of PRPK in malignant growth of A431 cells and observed that knocking down PRPK expression (shPRPK), significantly decreased colony formation, as compared with shMock-transfected cells (Figure 2I). Our results suggest that PRPK is a novel molecular driver associated with the TOPK pathway in skin carcinogenesis.
Discovery of rocuronium bromide and betamethasone 17-valerate as PRPK inhibitors for skin cancer prevention
To identify PRPK inhibitors that protect against cSCC development and move closer to clinical trials, we used computer-modeling analysis to screen drug candidates already approved by the Food and Drug Administration (FDA) for other applications. We identified two candidates, rocuronium bromide (abbreviation; rocuronium) and betamethasone 17-valerate (abbreviation; betamethasone), as potential PRPK inhibitors.
We constructed a computer model to dock each of these drugs to the PRPK protein. Rocuronium fit nicely into the ATP binding site of PRPK, with close contacts at the Val47, Val58, Ile116 and Leu169 residues (Figure 3A). A hydrogen bond is formed between rocuronium and Ile116. Similarly, betamethasone also docked into the ATP binding site of PRPK with close contacts at the Val47, Val58, Ile116, Arg123 and Ile182 residues (Figure 3B). Betamethasone forms hydrogen bonds with Ile116 and Arg123. The docking results suggest that each compound favorably binds to PRPK. However, based on the docking score, betamethasone (-6.0 kcal/mol) might bind more tightly than rocuronium (-3.8 kcal/mol) with PRPK.
Figure 3. The FDA approved drugs, rocuronium bromide and betamethasone 17-valerate, are identified as PRPK inhibitors.
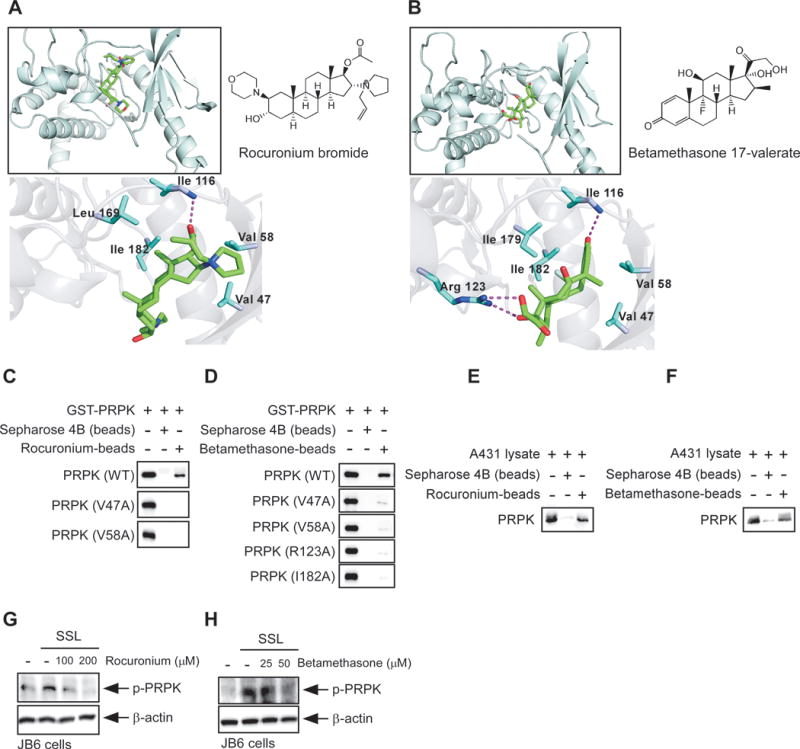
Binding modes of (A) rocuronium bromide and (B) betamethasone 17-valerate with PRPK. The binding of (C) rocuronium bromide or (D) betamethasone 17-valerate with PRPK was confirmed by an in vitro pull-down assay as described in Materials and Methods. A431 cell lysates were incubated with Sepharose 4B-conjugated (E) rocuronium bromide or (F) betamethasone 17-valerate and a pull-down assay was performed. Effects of (G) rocuronium bromide or (H) betamethasone 17-valerate on SSL-induced PRPK phosphorylation in JB6 P+ mouse epidermal cells.
We next examined the binding activity of rocuronium and betamethasone with wild-type or mutant PRPK proteins in which Val47, Val58, Arg123 or Ile182 was replaced with alanine. Rocuronium or betamethasone could interact with wild-type PRPK, and endogenous PRPK proteins isolated from the human A431 SCC cell line, but could not bind with any PRPK mutant protein (Figure 3C–F). Thus, these results suggest that these sites are important for binding with PRPK. Notably, rocuronium or betamethasone inhibited SSL-induced phosphorylation of PRPK in JB6 P+ mouse epidermal cells (Figure 3G, H). In vitro kinase assays using rocuronium or betamethasone (50 μM) and PRPK, indicated that PRPK activity was inhibited by 64% or 41%, respectively. We also performed a kinase assay to determine whether the PRPK inhibitors directly affected PI3-K or Akt activity and results indicated that neither rocuronium nor betamethasone affected PI3-K or Akt activity (Supplementary Figure 3B, C). Furthermore, neither rocuronium nor betamethasone affected TOPK kinase activity (Supplementary Figure 3D). Rocuronium or betamethasone did not absorb in either the UVB or UVA (280 to 400 nm) range and results indicated that the preventive effects of these compounds are not associated with UV absorption (i.e., a sunblock effect) in skin carcinogenesis (Supplementary Figure 4). Thus, our data showed that PI3-K and Akt are likely not involved in PRPK signaling. PRPK is a novel downstream substrate of TOPK and rocuronium or betamethasone is a specific PRPK inhibitor.
Anti-inflammatory effects induced by PRPK inhibition on SSL-induced human or mouse skin
Inflammation is known to contribute to tumor development (24) and we examined the effect of a 2-minimal erythema dose (MED) of SSL exposure in clinical human skin samples. For this study, participants were recruited from a pool of test subjects who had agreed to be re-contacted for future studies and had been previously screened and/or participated in previous skin chemoprevention trials. The eligibility criteria for participants included an age of 18 years or older and the Fitzpatrick skin type II (burns easily, tans poorly) or III (burns moderately, tans gradually). Our results indicated that acute SSL irradiation increased epidermal thickness as a feature of inflammation and also enhanced total protein and phosphorylation levels of TOPK and PRPK in human skin in a time-dependent manner (Figure 4A–D).
Figure 4. Effects of PRPK inhibitors on acute SSL-induced inflammation in human or mouse skin.
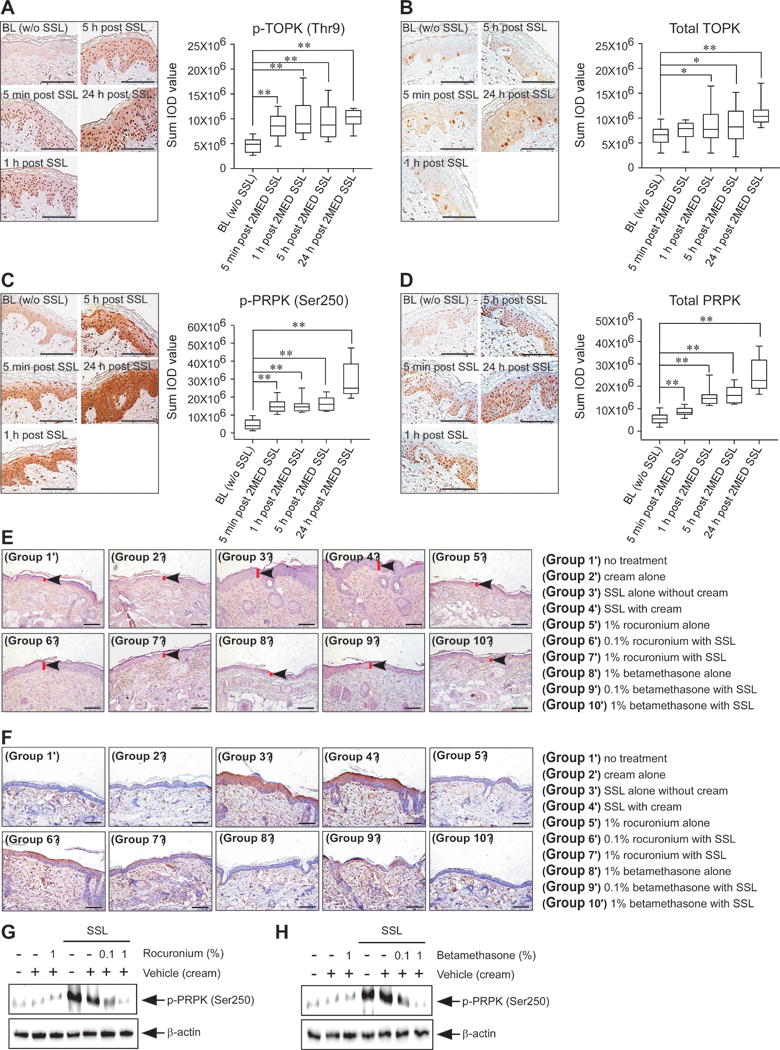
For clinical human tissues, the minimal erythemic dose (MED) was defined as the smallest dose of energy necessary to produce confluent erythema with four distinct borders at 24 h post-exposure. MED was determined on a buttock area previously unexposed to sunlight. Each test area was subdivided into six sub-sites (each 1 cm2) corresponding to the liquid light guide pattern on the solar simulator. The solar simulator was calibrated prior to each use and a series of six increasing SSL irradiation exposures were administered to each sub-site area. Following exposure, the test sites were covered until evaluations were completed. After determination of the MED for each individual, the contralateral buttock was exposed to twice the determined MED. A 6-mm skin punch biopsy sample was collected from one buttock at baseline prior to SSL exposure and additional 4 mm punch biopsies were removed at 5 min, 1, 5, and 24 hours post-SSL irradiation. IHC analysis of normal human skin (BL) without SSL exposure (30 photos from 10 human subjects), 5 min post 2 MED SSL exposure (30 photos from 10 human subjects), 1 h post 2 MED SSL exposure (30 photos from 10 human subjects), 5 h post 2 MED SSL exposure (30 photos from human 10 subjects) and 24 h post 2 MED SSL exposure (30 photos from 10 human subjects) to detect expression of (A) phosphorylated TOPK (Thr9), (B) total TOPK, (C) phosphorylated PRPK (Ser250) or (D) total PRPK. Phosphorylated TOPK, total TOPK, phosphorylated PRPK and total PRPK levels are presented as sum of IOD values (right panel in each figure). Left panel in each figure shows representative staining (brown color). The asterisks (*, **) indicate a significant (p < 0.05 or p < 0.01, respectively) difference compared to the normal human skin (BL). (E) At 24 h after SSL irradiation, mouse skin tissues were prepared for H&E staining to compare the epidermal thickness. (F) IHC and (G, H) Western blot analyses were conducted to examine the effect of rocuronium bromide and betamethasone 17-valerate on phosphorylated PRPK levels in acute SSL-stimulated SKH1 hairless mouse skin. Groups were divided for Figure 4E–H as below; Group 1′ = no treatment; Group 2′ = cream alone; Group 3′ = SSL alone without cream; Group 4′ = SSL with cream; Group 5′ = 1% rocuronium bromide alone; Group 6′ = 0.1% rocuronium bromide alone; Group 7′ = 1% rocuronium bromide with SSL; Group 8′ = 1% betamethasone 17-valerate alone; Group 9′ = 0.1% betamethasone 17-valerate with SSL; and Group 10′ = 1% betamethasone 17-valerate with SSL. Scale bars are 100 μm for Figure 4A–F.
The significance of PRPK phosphorylation induced by skin inflammation has not been reported. Our results showed that mouse skin exhibited increased epidermal thickness, and this increase was attenuated by the application of rocuronium or betamethasone in acute SSL-exposed SKH1 hairless mice (Figure 4E). Furthermore, topical treatment with rocuronium or betamethasone also decreased PRPK phosphorylation in SSL-exposed SKH1 hairless mice (Figure 4F–H). These results indicate that PRPK phosphorylation is an important marker in the acute SSL-induced inflammatory response. We also examined cyclobutane pyrimidine dimer (CPD) formation because UV-induced DNA damage characterized by CPD formation is known to be an important molecular initiator for UV-induced immunosuppression and carcinogenesis in skin tissues (25, 26). Our results indicated that acute SSL increased CPD production that was reduced by PRPK inhibitors (Supplementary Figure 5A and B).
Potential therapeutic or preventive value of PRPK inhibitors in skin carcinogenesis
Next, to evaluate the therapeutic or preventive value of rocuronium or betamethasone against skin carcinogenesis, we used cSCC mouse models, including an early-stage prevention model (Supplementary Figure 6A; Groups 1–7, Figure 5), and a late-stage prevention model (Supplementary Figure 6B; Groups 8-16, Figure 6), as described in Materials and Methods. Histological features of SCC include atypical keratinocytes, dermal invasion, abundant production of large keratin pearls, an increased ratio of nucleus/cytoplasm, and acantholysis (27, 28). Hematoxylin and eosin (H&E) staining results revealed that chronic SSL exposure induced both papilloma formation and invasive cSCC (Figures 5A, 6A). Furthermore, large epidermal changes, dermal invasion (Figure 5A, 6A) and the formation of abundant large keratin pearls were observed in the SSL-exposed group (red arrows, Figures 5A, 6A).
Figure 5. Preventive efficacy of a PRPK inhibitor against cutaneous papillomas and SCC in an early-stage prevention model.
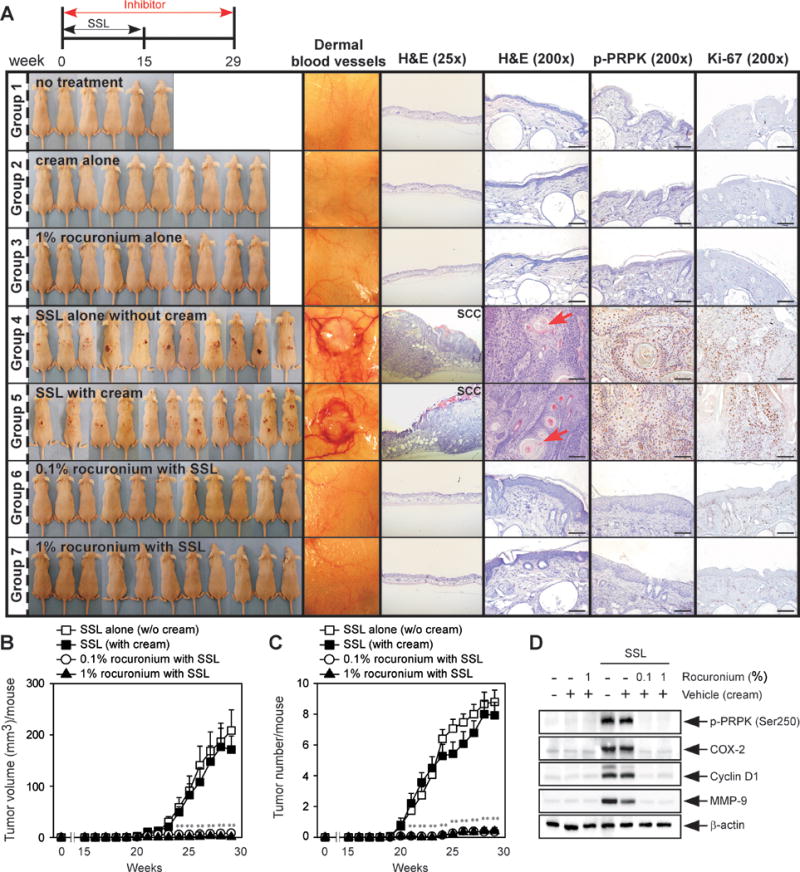
(A) At week 29, tissues were collected and dermal blood vessels were photographed, and tissues subjected to H&E staining and IHC to detect phosphorylated PRPK (Ser250) and Ki-67. Scale bars = 100 μm. (B) Tumor volume and (C) number were measured for 29 weeks. Data represent the mean values ± S.D. Significant differences were determined by one-way ANOVA and the asterisk (**) indicates a significant (p < 0.01) difference compared to the group treated with only SSL (Group 6). (D) Western blot analysis to detect phosphorylated PRPK (Ser250), cyclin D1, COX-2 and MMP-9 in chronic SSL-induced cutaneous papillomas and SCC. Mice were divided into groups as follows: Group 1 = no treatment (n = 6); Group 2 = cream alone (n = 10); Group 3 = 1% rocuronium bromide alone (n = 10); Group 4 = SSL alone without cream (n = 15); Group 5 = SSL with cream (n = 14); Group 6 = 0.1% rocuronium bromide with SSL (n = 15); and Group 7 = 1% rocuronium bromide with SSL (n = 15).
Figure 6. Preventive efficacy of PRPK inhibitors against cutaneous papillomas and SCC in a late-stage prevention model.

(A) At week 30, dermal blood vessels were photographed, and tissues were collected for H&E staining and IHC to detect phosphorylated PRPK (Ser250) and Ki-67. Scale bars = 100 μm. (B, C) Tumor volume and (D, E) number were measured for 30 weeks. Data represent the mean values ± S.D., and significant differences were determined by one-way ANOVA. The asterisks (**) indicate a significant (p < 0.01) difference compared to the group treated with only SSL (Group 12). (F, G) Western blot analysis to detect phosphorylated PRPK (Ser250), cyclin D1, COX-2 and MMP-9 in chronic SSL-induced cutaneous papillomas and SCCs. Mice were divided into groups as follows: Group 8 = no treatment (n = 5); Group 9 = cream alone (n = 10); Group 10 = 1% rocuronium bromide alone (n = 10); Group 11 = 1% betamethasone 17-valerate alone (n = 10); Group 12 = SSL with cream (n = 15); Group 13 = 0.1% rocuronium bromide with SSL (n = 15); Group 14 = 1% rocuronium bromide with SSL (n = 15); Group 15 = 0.1% betamethasone 17-valerate with SSL (n = 15); and Group 16 = 1% betamethasone 17-valerate with SSL (n = 15).
Rocuronium or betamethasone application decreased tumor volume and number in chronic SSL-exposed mouse skin in both the early-stage prevention model (Figure 5B, C) and the late-stage prevention model (Figure 6B–E), and also inhibited PRPK phosphorylation and Ki-67 expression (Figures 5A, 6A). Angiogenesis is an important feature of tumor development (29), and we observed an increased number of large blood vessels in the dermis of the SSL-treated group. Rocuronium or betamethasone clearly inhibited SSL-induced neovascularization in tumor areas (dermal blood vessels, Figure 5A, 6A). Next, we examined the effects of rocuronium or betamethasone on proteins associated with the malignant progression of cSCC, including COX-2, cyclin D1 and MMP-9, in chronic SSL-exposed mouse skin. Rocuronium or betamethasone suppressed SSL-induced expression of each of these proteins in SKH1 hairless mouse skin (Figure 5D, 6F–G).
A high-risk feature of cSCC includes immunosuppression (30). Thus, we examined the level of immune system markers, including interferon-γ (IFN-γ), interleukin-12 (IL-12) and prostaglandin E2 (PGE2), and the T-cell markers, CD3 and CD4, in mouse serum and skin tissue lysates. Down-regulated T-cell receptor (TCR)-related signaling mediated by tumor microenvironment induces the suppression of T-cell function and antitumor immunity in the tumor-bearing host. Very few T-cells are observed in the epidermis and dermis of SSL-induced SCCs (SSL only groups in Supplementary Figure 7A, B). Upon treatment with rocuronium or betamethasone, dense T-cell infiltrates in mouse skin were observed as compared to untreated controls (Supplementary Figure 7A, B). We hypothesized that the PRPK inhibitors, rocuronium and betamethasone, are associated with activation of T-cell function in the dermis and epidermis of skin. SSL-induced immunosuppressive responses are associated with production of Th2-related cytokines (IL-10) and disturbance of antigen presentation to Th1 cells producing IFN-γ (31). Therefore, SSL-induced immunosuppression could be prevented by the production of the Th1 cytokine, IFN-γ. We observed that SSL suppressed production of IFN-γ and that rocuronium or betamethasone treatment enhanced IFN-γ levels (Supplementary Figure 7C, D). Thus, the PRPK inhibitors, rocuronium or betamethasone could attenuate SSL-increased immunosuppression by up-regulating IFN-γ. IL-12 is an immunoregulatory cytokine to reverse UV-induced DNA damage and immunosuppression, and also regulates the function and development of Th1 type cells by stimulating IFN-γ production (32, 33). Furthermore, treatment with recombinant IL-12 reversed the UV-induced immunosuppression (34). Additionally, PGE2 plays a key role as an activator of UV-induced immunosuppression through its down-regulation of the levels of IL-12 (35). Thus, the effects of PRPK inhibitors on SSL-induced changes on the immunoregulatory cytokines, IL-12 and PGE2, were investigated. PRPK inhibitors increased the production of IL-12 in mouse serum (Supplementary Figure 7E, F) and mouse skin (Supplementary Figure 7G, H), and decreased PGE2 levels in the skin (Supplementary Figure 7I, J). Importantly, PRPK inhibitors attenuated the levels of COX-2 expression and PGE2 production in SSL-exposed skin, which might play a crucial role in the up-regulation of IL-12 production. Thus, the stimulation of IFN-γ and IL-12 by PRPK inhibitors could have a pivotal role in prevention of sUV-induced immunosuppression.
Mutations of the p53 gene are usually detected in cSCC (36, 37). The p53 mutations mainly comprise C→T substitutions located on dipyrimidine sites that are specific UV targets (38). We found mutations of the p53 gene in human cSCC (Supplementary Table 1) and SSL-exposed TOPK+/+ mice (Supplementary Table 2, used for Figure 1C–G). Furthermore, mutations of the p53 gene were detected in exon 7 (Supplementary Table 3) and exon 8 (Supplementary Table 4) in SSL-induced cSCC from both the early-stage (Figure 5) and the late-stage prevention mouse models (Figure 6). One hundred percent of the mutations in codon 267 were C to T substitutions. We also observed a high frequency of mutations in codons 270, 277 and 284 of the p53 gene in both human and mouse cSCC (Supplementary Tables 1–4). Importantly, mutations of the p53 gene were not detected in SSL-treated skin from TOPK−/− mice or from mice treated with PRPK inhibitors. Notably, these mice did not develop SCCs.
DISCUSSION
TOPK is an active form of MEK1 (15), and is closely associated with the development and progression of cancers, possibly acting by continuous phosphorylation of oncogenic substrates (39, 40). However, the mechanisms of action for TOPK and its various substrates in skin carcinogenesis are still unclear. Chronic skin inflammation induced by solar UV (sUV) irradiation contributes to cutaneous squamous cell carcinoma (cSCC) development (24). Furthermore, metastasis often occurs in chronic inflammatory skin conditions and increased epidermal thickness is a characteristic of sUV-induced skin inflammation (41). According to Park et al., TOPK is associated with the inflammatory signaling pathway, including endotoxin lipopolysaccharide (LPS)-induced NF-κB activation and iNos gene expression (42). Recently, the FDA-approved antibiotic, cephalosporin (43), and the synthetic compound, ADA-07 (44), have been shown to suppress skin carcinogenesis by blocking TOPK activity.
Our study is the first to show that PRPK phosphorylation is induced by stimulation of skin inflammation. Our clinical data with acute exposure of human skin to SSL supports the importance of the TOPK/PRPK signaling pathway in solar UV-mediated inflammatory responses. Furthermore, skin inflammation was decreased by the PRPK inhibitors, rocuronium or betamethasone. Rocuronium is a muscle relaxant used during surgery (32) and betamethasone is generally used for the relief of the inflammatory and pruritic manifestations of corticosteroid-responsive dermatoses (45–48). Both are included on the List of Essential Medicines of the World Health Organization (WHO).
PRPK (also known as TP53RK) was previously reported to phosphorylate p53 on Ser15, activating its pro-apoptotic activity, which suggested that PRPK-depleted cells would have a higher survival rate. However, the p53 protein phosphorylated on Ser15 still remains even after PRPK depletion, suggesting that this is not the major role of PRPK in p53-related proliferating cells. This is probably because the p53 protein is also phosphorylated by ATR and ATM on Ser15 to provide redundancy of this activity (49). Importantly, PRPK depletion caused a large increase of apoptosis with antimitotic agents. Knockdown of PRPK increased paclitaxel-induced apoptosis, suggesting that taxane treatment could be sensitized by PRPK inhibition to treat cancers (21). Thus, further studies with a combination of PRPK inhibitors and taxanes could provide an approach to resolve drug resistance.
The presence of p53 mutations has been associated with an increased risk of tumor progression to malignancy and the high incidence of p53 mutations increases with histologic progression from mild dysplasia to invasive carcinoma (50, 51). We detected no p53 mutations in the SSL-treated skin from TOPK−/− mice or from SKH1 hairless mice treated with PRPK inhibitors. These mice did not develop cSCCs, suggesting that the role of the TOPK/PRPK pathway in tumorigenesis might rely mainly on mutant p53 gain-of-function properties. Our data imply that the combination of TOPK/PRPK overexpression and p53 mutation could play a pivotal role and be a prognostic factor in poor clinical outcome, and the TOPK/PRPK/mutant p53 axis might be exploited as a diagnostic or therapeutic tool. Further studies are needed to elucidate the mechanisms of the TOPK/PRPK/mutant p53 axis to investigate whether the TOPK/PRPK pathway affects or amplifies mutant p53 oncogenic functions by stimulating gain-of-function activities of mutant p53.
In conclusion, our study indicates that PRPK is phosphorylated by TOPK, and presents a new therapeutic target against sUV-induced skin carcinogenesis. This is also the first report to demonstrate the therapeutic efficacy of FDA-approved rocuronium and betamethasone as PRPK inhibitors and could be available immediately for clinical trial evaluation for testing the effectiveness of PRPK inhibitors against cutaneous papillomas and invasive cSCC.
MATERIALS AND METHODS
Human skin tissues
Normal, AK, SCC, BCC and solar simulated light (SSL)-exposed skin tissues (2-minimal erythemic dose, MED) in humans were supplied by the University of Arizona Cancer Center (Tucson, AZ). The University of Arizona Institutional Review Board approved the study (the University of Arizona Human Subjects Protection Program, FWA00004218/IRB00000291) and written informed consent was obtained from all study participants. Methods were conducted in accordance with relevant regulations and guidelines from the University of Arizona Cancer Center Scientific Review Committee. For the clinical human study with acute SSL exposure, the Multiport UV Solar Simulator Model 600 (Solar Light Co., Philadelphia, PA), corroborated by using a reflectance spectrophotometer (Minolta Chroma Meter Model CR-200; Minolta Corporation, Osaka, Japan), were used to determine the MED of SSL for each individual. The SSL light source comprised 91.3% UVA and 8.7% UVB, and the emission dosages were accurately regulated to be limited to the UVA and UVB spectra.
A human skin cancer tissue array (SK801) and human primary SCCs and SCCs metastasized to skin or lymph node samples were purchased from US Biomax (Rockville, MD).
SSL irradiation system
The SSL source consisted of UVA-340 lamps that emit both UVA (94.5%) and UVB (5.5%) irradiation that was purchased from Q-Lab Corporation (Cleveland, OH). The UVA-340 lamps provide the natural sunlight that includes both UVA and UVB in the critical short wavelength region from 365 nm down to the solar cutoff of 295 nm, with a peak emission at 340 nm.
Comparison of SKH1 TOPK wild-type and knockout mice
SKH1 (Crl: SKH1-Hrhr) TOPK knockout (TOPK−/−) hairless mice were generated as described in Supplementary Materials and Methods. These mice were managed and maintained in the animal facilities of The Hormel Institute following the University of Minnesota Animal Care and Use Committee (IACUC) guidelines. These mice were randomly grouped by age and body weight, and 20 mice per group are typically used and treated for criteria achievement of statistical significance without investigator blinding. For the chronic SSL-treated groups, SKH1 TOPK+/+ and SKH1 TOPK−/− mice aged 6 weeks at the beginning of the study were initially exposed to an SSL dose of 37 kJ/m2 UVA and 1.8 kJ/m2 UVB 3 times per a week. The UV irradiation dose was increased by 10% each week until the 6th week, at which time the UV dose reached 60 kJ/m2 UVA and 2.9 kJ/m2 UVB. This dose was maintained until week 15, at which time the SSL irradiation was discontinued. Tumor growth was monitored for an additional 20 weeks. For acute SSL exposure, the dorsal skins of TOPK+/+ and TOPK−/− mice were exposed to 1 dose of SSL (149 kJ UVA/m2/7.2 kJ UVB/m2) and at 24 h mice were euthanized and skin tissues were collected.
Chronic SSL-induced cutaneous squamous cell carcinoma (cSCC) mouse models
Female SKH1 (Crl: SKH1-Hrhr) hairless mice (5-6 weeks old) were purchased from Charles River (Burlington, MA) and mice were maintained in The Hormel Institute vivarium following the University of Minnesota Animal Care and Use Committee (IACUC) guidelines. In order to establish the criteria for achieving statistical significance, fifteen mice per group were randomly grouped. We created an oil-in-water emulsion cream (patent pending) containing 0.1% or 1% (w/w) each of rocuronium bromide (rocuronium) or betamethasone 17-valerate (betamethasone), and then treated mice with the cream with or without the respective compounds 3 times per a week. The cream was applied to the dorsal area of the SKH1 (Crl: SKH1-Hrhr) hairless mouse skin without investigator blinding, 1 h before SSL irradiation exposure on the same day. At week 1, the SSL dose was 37 kJ/m2 UVA and 1.8 kJ/m2 UVB applied 3 times per week and the dose of SSL irradiation was gradually increased 10 percent every week. At week 6, the SSL dose was 60 kJ/m2 UVA and 2.9 kJ/m2 UVB and this dose was maintained until week 15. We used two separate animal models, including an early-stage prevention (Groups 1-7; Supplementary Figure 6A) and a late-stage prevention (Group 8-16; Supplementary Figure 6B) model. For the early-stage prevention model, rocuronium cor betamethasone cream was applied for 29 weeks and SSL irradiation was for 15 weeks. Mice for the late-stage prevention model received SSL irradiation for 15 weeks and then SSL irradiation was discontinued. At that time, mice began receiving topical application of rocuronium or betamethasone 3 times per a week until week 30.
Acute SSL-induced inflammation in SKH1 hairless mice
Cream only or cream containing 0.1% or 1% (w/w) of rocuronium or betamethasone was topically applied onto the dorsal area of SKH1 hairless mice. After 1 h, SSL at a dose of 149 kJ/m2 UVA and 7.2 kJ/m2 UVB was applied to the mouse dorsal skin. Mice were divided into groups as described in the legend of Figure 4. At 24 h after SSL irradiation, mouse tissues were collected and analyzed.
Computational docking and modeling
The Modeler 9.10 program (Ben Webb at UCSF; San Francisco, CA) was used to build the PRPK structure based on the crystal structure of Bud32 (PDB entry 2VWB). Rocuronium and betamethasone were docked to PRPK using the docking program Glide 5.9 (Schrödinger LLC; New York, NY). For docking analysis, the ATP-binding site-based receptor grid was generated, and rocuronium and betamethasone were prepared by the LigPrep program with default parameters (Schrödinger). Hydrogen atoms were added consistent with a pH 7 and the docking of rocuronium or betamethasone with PRPK was achieved with default parameters in the extra precision (XP) mode.
In vitro pull-down assay
CNBr-activated Sepharose 4B beads (0.3 g) were added in 1 mM HCl and compound (rocuronium bromide or betamethasone 17-valerate) was mixed with solution (pH 8.3, 0.1 M NaHCO3, 0.5 M NaCl). The compound-bead mixtures were incubated for 24 h on a 4°C rotator and then transferred to 0.1 M Tris–HCl buffer (pH 8.0). Acetate buffer (0.1 M, pH 4.0) containing 0.5 M NaCl was used as the washing buffer. Then the compound-bead mixtures were washed with 0.1 M Tris–HCl (pH 8.0) containing 0.5 M NaCl. For the pull-down assay, purified GST-PRPK or A431 cell lysates were incubated overnight on a 4°C rotator with compound-bead mixtures (or control beads) in reaction buffer (50 mM Tris pH 7.5, 150 mM NaCl, 5 mM EDTA, 1 mM dithiothreitol, 2 μg/mL bovine serum albumin, 0.01% Nonidet P-40, and 0.02 mM PMSF). After washing the compound-bead mixtures, proteins bound to the compound-bead mixtures were visualized by Western blot analysis.
In vitro kinase assay
An active TOPK, or Akt1 protein with the substrate for each kinase and with or without inhibitor was incubated for 30 min in a 30°C water bath. Proteins were incubated with 10 μCi [γ-32P] ATP and isotope-unlabeled ATP in kinase buffer containing 20 mM HEPES (pH 7.4), 1 mM dithiothreitol, 10 mM MgCl2, and 10 mM MnCl2. The incorporated radioactivity was measured by autoradiography; and for the non-radioactive kinase assay, the phosphorylated substrate was detected by Western blot analysis. The in vitro PI3-K kinase assay was performed as described previously (52) with PRPK inhibitors or LY294002 (a PI3-K inhibitor).
Anchorage-independent cell growth assay
To generate knockdown PRPK cells, pLKO.1-shPRPK or pLKO.1-mock lentivirus plasmids were co-transfected with psPAX2 and pMD2.0G into HEK 293T cells using the jetPEI poly transfection reagent (Polyplus-transfection SA; New York, NY). The human shPRPK full hairpin sequence is #1, 5′-CCGGGAGATTATATTCAGTCCACTACTCGAGTAGTGGACTGAATATAATCTCTTTTTG-3′, and #2, 5′-CCGGCATAGACTTTGGGCTGAGTTTCTCGAGAAACTCAGCCCAAAGTCTATGTTTTTG-3′. The viral particles containing 8 μg/mL polybrene were infected into A431 cells overnight and the medium was replaced with fresh complete growth medium. After 24 h, A431 cells were selected with 2 μg/mL of puromycin. The shMock or shPRPK-transfected A431 cells were incubated for 10 days as described previously (53). Colony formation was observed and analyzed using a LEICA DM IRB microscope (Nikon Corporation) and the ImagePro Plus software (v.6.1) program (Media Cybernetics Inc., Rockville, MD).
Immunofluorescence (IF) staining and confocal microscopy analysis
After fixation of skin tissues with 4% formaldehyde, tissues were embedded in paraffin blocks, cut and mounted on glass slides, slides were deparaffinized and hydrated, and then permeabilized in 300 μl of 0.5% Triton X-100. For immunostaining, the primary antibody was added and then the Alexa Fluor 488-labeled secondary antibody was added. The 4,6-diamidino-2-phenylindole (DAPI) solution was added for nuclei staining of the cells on the skin tissues. Fluorescence-labeled proteins were analyzed using the Nikon Eclipse TE2000-E Confocal microscope with EZ-C1 software (Nikon Corporation, Minato-ku, Tokyo, JPN).
Western blot analysis
Western blot analyses were performed as described in our previous study (54).
Statistical analyses
The GraphPad prism 5.0 software (GraphPad Software; La Jolla, CA) was used for all the statistical analyses. To compare statistical significance between 3 or more groups, one-way ANOVA was used and Student’s t-test was used to compare between two groups. Statistical p-values (*p < 0.05 or **p < 0.01) were considered to be statistically significant between groups. To measure the degree of association between tumor stages, the Pearson’s linear correlation coefficient was calculated using MATLAB (Natick, MA).
Supplementary Material
Acknowledgments
This work was supported by The Hormel Foundation and National of Institutes of Health grants CA027502, CA166011, CA187027 and CA196639 (Zigang Dong). We thank Dr. Lorenzo A. Pinna (Universita di Padova, Italy) for the pQE-81L-PRPK plasmid, Alyssa Langfald for confocal microscopy analysis and Dr. Tia Rai and Nicki Brickman for assistance with manuscript submission.
Funding Support: This work was supported by The Hormel Foundation and National of Institutes of Health grants CA027502, CA166011, CA187027 and CA196639 (Zigang Dong).
Footnotes
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
E.R. and Z.D. designed the research. E.R. performed experiments and analyzed results. E.R. and A.M.B. wrote the manuscript. E.R. and M.H.L. performed animal studies. T.A.Z., H.G.K., K.B., F.Z., and Y.Y.C. assisted in performing experiments. J.N. and Y.L. performed computer modeling. A.M.B., C.C., J.E. and S.E.D. organized and supplied human patient tissues from University of Arizona Cancer Center.
Supplementary Information accompanies the paper on the Oncogene website.
Data Availability Statement
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
References
- 1.Thompson AK, Kelley BF, Prokop LJ, Murad MH, Baum CL. Risk Factors for Cutaneous Squamous Cell Carcinoma Recurrence, Metastasis, and Disease-Specific Death: A Systematic Review and Meta-analysis. JAMA Dermatol. 2016:1–10. doi: 10.1001/jamadermatol.2015.4994. Epub 2016/01/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guy GP, Jr, Machlin SR, Ekwueme DU, Yabroff KR. Prevalence and costs of skin cancer treatment in the U.S., 2002-2006 and 2007-2011. American Journal of Preventive Medicine. 2015;48(2):183–7. doi: 10.1016/j.amepre.2014.08.036. Epub 2014/12/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rogers HW, Weinstock MA, Feldman SR, Coldiron BM. Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the US Population, 2012. Jama Dermatol. 2015;151(10):1081–6. doi: 10.1001/jamadermatol.2015.1187. [DOI] [PubMed] [Google Scholar]
- 4.Brantsch KD, Meisner C, Schonfisch B, Trilling B, Wehner-Caroli J, Rocken M, et al. Analysis of risk factors determining prognosis of cutaneous squamous-cell carcinoma: a prospective study. Lancet Oncol. 2008;9(8):713–20. doi: 10.1016/S1470-2045(08)70178-5. Epub 2008/07/12. [DOI] [PubMed] [Google Scholar]
- 5.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA: A Cancer Journal for Clinicians. 2010;60(5):277–300. doi: 10.3322/caac.20073. Epub 2010/07/09. [DOI] [PubMed] [Google Scholar]
- 6.Farasat S, Yu SS, Neel VA, Nehal KS, Lardaro T, Mihm MC, et al. A new American Joint Committee on Cancer staging system for cutaneous squamous cell carcinoma: creation and rationale for inclusion of tumor (T) characteristics. Journal of the American Academy of Dermatology. 2011;64(6):1051–9. doi: 10.1016/j.jaad.2010.08.033. Epub 2011/01/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marcil I, Stern RS. Risk of developing a subsequent nonmelanoma skin cancer in patients with a history of nonmelanoma skin cancer: a critical review of the literature and meta-analysis. Archives of Dermatology. 2000;136(12):1524–30. doi: 10.1001/archderm.136.12.1524. Epub 2000/12/15. [DOI] [PubMed] [Google Scholar]
- 8.Ratushny V, Gober MD, Hick R, Ridky TW, Seykora JT. From keratinocyte to cancer: the pathogenesis and modeling of cutaneous squamous cell carcinoma. Journal of Clinical Investigation. 2012;122(2):464–72. doi: 10.1172/JCI57415. Epub 2012/02/02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3(6):453–8. doi: 10.1038/nrc1098. Epub 2003/06/05. [DOI] [PubMed] [Google Scholar]
- 10.Muller-Decker K. Cyclooxygenase-dependent signaling is causally linked to non-melanoma skin carcinogenesis: pharmacological, genetic, and clinical evidence. Cancer and Metastasis Reviews. 2011;30(3–4):343–61. doi: 10.1007/s10555-011-9306-z. Epub 2011/11/01. [DOI] [PubMed] [Google Scholar]
- 11.Trifan OC, Hla T. Cyclooxygenase-2 modulates cellular growth and promotes tumorigenesis. J Cell Mol Med. 2003;7(3):207–22. doi: 10.1111/j.1582-4934.2003.tb00222.x. Epub 2003/11/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shen Y, Xu J, Jin J, Tang H, Liang J. Cyclin D1 expression in Bowen’s disease and cutaneous squamous cell carcinoma. Mol Clin Oncol. 2014;2(4):545–8. doi: 10.3892/mco.2014.273. Epub 2014/06/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang K, Huang C, Shan K, Chen J, Li H. Significance of PC cell-derived growth factor and cyclin D1 expression in cutaneous squamous cell carcinoma. Clinical and Experimental Dermatology. 2012;37(4):411–7. doi: 10.1111/j.1365-2230.2011.04275.x. Epub 2012/03/17. [DOI] [PubMed] [Google Scholar]
- 14.Panelos J, Tarantini F, Paglierani M, Di Serio C, Maio V, Pellerito S, et al. Photoexposition discriminates Notch 1 expression in human cutaneous squamous cell carcinoma. Modern Pathology. 2008;21(3):316–25. doi: 10.1038/modpathol.3801007. Epub 2008/01/15. [DOI] [PubMed] [Google Scholar]
- 15.Zhu F, Zykova TA, Kang BS, Wang Z, Ebeling MC, Abe Y, et al. Bidirectional signals transduced by TOPK-ERK interaction increase tumorigenesis of HCT116 colorectal cancer cells. Gastroenterology. 2007;133(1):219–31. doi: 10.1053/j.gastro.2007.04.048. Epub 2007/07/17. [DOI] [PubMed] [Google Scholar]
- 16.Oh SM, Zhu F, Cho YY, Lee KW, Kang BS, Kim HG, et al. T-lymphokine-activated killer cell-originated protein kinase functions as a positive regulator of c-Jun-NH2-kinase 1 signaling and H-Ras-induced cell transformation. Cancer research. 2007;67(11):5186–94. doi: 10.1158/0008-5472.CAN-06-4506. Epub 2007/06/05. [DOI] [PubMed] [Google Scholar]
- 17.Chang CF, Chen SL, Sung WW, Hsieh MJ, Hsu HT, Chen LH, et al. PBK/TOPK Expression Predicts Prognosis in Oral Cancer. International journal of molecular sciences. 2016;17(7) doi: 10.3390/ijms17071007. Epub 2016/06/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Joel M, Mughal AA, Grieg Z, Murrell W, Palmero S, Mikkelsen B, et al. Targeting PBK/TOPK decreases growth and survival of glioma initiating cells in vitro and attenuates tumor growth in vivo. Mol Cancer. 2015;14:121. doi: 10.1186/s12943-015-0398-x. Epub 2015/06/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abe Y, Matsumoto S, Wei S, Nezu K, Miyoshi A, Kito K, et al. Cloning and characterization of a p53-related protein kinase expressed in interleukin-2-activated cytotoxic T-cells, epithelial tumor cell lines, and the testes. Journal of Biological Chemistry. 2001;276(47):44003–11. doi: 10.1074/jbc.M105669200. Epub 2001/09/08. [DOI] [PubMed] [Google Scholar]
- 20.Abe Y, Takeuchi T, Imai Y, Murase R, Kamei Y, Fujibuchi T, et al. A Small Ras-like protein Ray/Rab1c modulates the p53-regulating activity of PRPK. Biochemical and biophysical research communications. 2006;344(1):377–85. doi: 10.1016/j.bbrc.2006.03.071. Epub 2006/04/08. [DOI] [PubMed] [Google Scholar]
- 21.Peterson D, Lee J, Lei XC, Forrest WF, Davis DP, Jackson PK, et al. A chemosensitization screen identifies TP53RK, a kinase that restrains apoptosis after mitotic stress. Cancer research. 2010;70(15):6325–35. doi: 10.1158/0008-5472.CAN-10-0015. Epub 2010/07/22. [DOI] [PubMed] [Google Scholar]
- 22.Facchin S, Ruzzene M, Peggion C, Sartori G, Carignani G, Marin O, et al. Phosphorylation and activation of the atypical kinase p53-related protein kinase (PRPK) by Akt/PKB. Cellular and Molecular Life Sciences. 2007;64(19–20):2680–9. doi: 10.1007/s00018-007-7179-7. Epub 2007/08/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shih MC, Chen JY, Wu YC, Jan YH, Yang BM, Lu PJ, et al. TOPK/PBK promotes cell migration via modulation of the PI3K/PTEN/AKT pathway and is associated with poor prognosis in lung cancer. Oncogene. 2012;31(19):2389–400. doi: 10.1038/onc.2011.419. Epub 2011/10/15. [DOI] [PubMed] [Google Scholar]
- 24.Bode AM, Dong Z. Molecular and cellular targets. Molecular Carcinogenesis. 2006;45(6):422–30. doi: 10.1002/mc.20222. Epub 2006/05/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Runger TM. How different wavelengths of the ultraviolet spectrum contribute to skin carcinogenesis: the role of cellular damage responses. Journal of Investigative Dermatology. 2007;127(9):2103–5. doi: 10.1038/sj.jid.5700988. Epub 2007/08/19. [DOI] [PubMed] [Google Scholar]
- 26.Ridley AJ, Whiteside JR, McMillan TJ, Allinson SL. Cellular and sub-cellular responses to UVA in relation to carcinogenesis. International journal of radiation biology. 2009;85(3):177–95. doi: 10.1080/09553000902740150. Epub 2009/03/20. [DOI] [PubMed] [Google Scholar]
- 27.Lohmann CM, Solomon AR. Clinicopathologic variants of cutaneous squamous cell carcinoma. Advances in Anatomic Pathology. 2001;8(1):27–36. doi: 10.1097/00125480-200101000-00005. Epub 2001/01/11. [DOI] [PubMed] [Google Scholar]
- 28.Cherpelis BS, Marcusen C, Lang PG. Prognostic factors for metastasis in squamous cell carcinoma of the skin. Dermatol Surg. 2002;28(3):268–73. doi: 10.1046/j.1524-4725.2002.01169.x. Epub 2002/03/19. [DOI] [PubMed] [Google Scholar]
- 29.Yano K, Kajiya K, Ishiwata M, Hong YK, Miyakawa T, Detmar M. Ultraviolet B-induced skin angiogenesis is associated with a switch in the balance of vascular endothelial growth factor and thrombospondin-1 expression. Journal of Investigative Dermatology. 2004;122(1):201–8. doi: 10.1046/j.0022-202X.2003.22101.x. Epub 2004/02/14. [DOI] [PubMed] [Google Scholar]
- 30.Burton KA, Ashack KA, Khachemoune A. Cutaneous Squamous Cell Carcinoma: A Review of High-Risk and Metastatic Disease. Am J Clin Dermatol. 2016;17(5):491–508. doi: 10.1007/s40257-016-0207-3. Epub 2016/07/01. [DOI] [PubMed] [Google Scholar]
- 31.Yokogawa M, Takaishi M, Nakajima K, Kamijima R, Digiovanni J, Sano S. Imiquimod attenuates the growth of UVB-induced SCC in mice through Th1/Th17 cells. Molecular Carcinogenesis. 2013;52(10):760–9. doi: 10.1002/mc.21901. Epub 2012/03/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Allanson M, Reeve VE. Carbon monoxide signalling reduces photocarcinogenesis in the hairless mouse. Cancer immunology, immunotherapy : CII. 2007;56(11):1807–15. doi: 10.1007/s00262-007-0324-1. Epub 2007/04/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Katiyar SK. Interleukin-12 and photocarcinogenesis. Toxicology and applied pharmacology. 2007;224(3):220–7. doi: 10.1016/j.taap.2006.11.017. Epub 2007/01/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwarz A, Maeda A, Kernebeck K, van Steeg H, Beissert S, Schwarz T. Prevention of UV radiation-induced immunosuppression by IL-12 is dependent on DNA repair. J Exp Med. 2005;201(2):173–9. doi: 10.1084/jem.20041212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katiyar SK. Proanthocyanidins from Grape Seeds Inhibit UV-Radiation-Induced Immune Suppression in Mice: Detection and Analysis of Molecular and Cellular Targets. Photochem Photobiol. 2015;91(1):156–62. doi: 10.1111/php.12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edlund K, Larsson O, Ameur A, Bunikis I, Gyllensten U, Leroy B, et al. Data-driven unbiased curation of the TP53 tumor suppressor gene mutation database and validation by ultradeep sequencing of human tumors. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(24):9551–6. doi: 10.1073/pnas.1200019109. Epub 2012/05/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Loyo M, Li RJ, Bettegowda C, Pickering CR, Frederick MJ, Myers JN, et al. Lessons learned from next-generation sequencing in head and neck cancer. Head & neck. 2013;35(3):454–63. doi: 10.1002/hed.23100. Epub 2012/08/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Missero C, Antonini D. Crosstalk among p53 family members in cutaneous carcinoma. Experimental Dermatology. 2014;23(3):143–6. doi: 10.1111/exd.12320. Epub 2014/01/15. [DOI] [PubMed] [Google Scholar]
- 39.Matsuo Y, Park JH, Miyamoto T, Yamamoto S, Hisada S, Alachkar H, et al. TOPK inhibitor induces complete tumor regression in xenograft models of human cancer through inhibition of cytokinesis. Sci Transl Med. 2014;6(259):259ra145. doi: 10.1126/scitranslmed.3010277. Epub 2014/10/24. [DOI] [PubMed] [Google Scholar]
- 40.Li Y, Yang Z, Li W, Xu S, Wang T, Niu M, et al. TOPK promotes lung cancer resistance to EGFR tyrosine kinase inhibitors by phosphorylating and activating c-Jun. Oncotarget. 2016 doi: 10.18632/oncotarget.6826. Epub 2016/01/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hensler S, Mueller MM. Inflammation and skin cancer: old pals telling new stories. Cancer J. 2013;19(6):517–24. doi: 10.1097/PPO.0000000000000010. Epub 2013/11/26. [DOI] [PubMed] [Google Scholar]
- 42.Park JH, Jeong YJ, Won HK, Choi SY, Oh SM. Activation of TOPK by lipopolysaccharide promotes induction of inducible nitric oxide synthase through NF-kappaB activity in leukemia cells. Cellular Signalling. 2014;26(5):849–56. doi: 10.1016/j.cellsig.2014.01.004. Epub 2014/01/21. [DOI] [PubMed] [Google Scholar]
- 43.Fan X, Duan Q, Ke C, Zhang G, Xiao J, Wu D, et al. Cefradine blocks solar-ultraviolet induced skin inflammation through direct inhibition of T-LAK cell-originated protein kinase. Oncotarget. 2016;7(17):24633–45. doi: 10.18632/oncotarget.8260. Epub 2016/03/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao G, Zhang T, Wang Q, Reddy K, Chen H, Yao K, et al. ADA-07 Suppresses Solar Ultraviolet-Induced Skin Carcinogenesis by Directly Inhibiting TOPK. Molecular cancer therapeutics. 2017 doi: 10.1158/1535-7163.MCT-17-0212. Epub 2017/06/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saraceno R, Chiricozzi A, Nistico SP, Tiberti S, Chimenti S. An occlusive dressing containing betamethasone valerate 0.1% for the treatment of prurigo nodularis. The Journal of dermatological treatment. 2010;21(6):363–6. doi: 10.3109/09546630903386606. Epub 2010/06/12. [DOI] [PubMed] [Google Scholar]
- 46.Jensen JM, Scherer A, Wanke C, Brautigam M, Bongiovanni S, Letzkus M, et al. Gene expression is differently affected by pimecrolimus and betamethasone in lesional skin of atopic dermatitis. Allergy. 2012;67(3):413–23. doi: 10.1111/j.1398-9995.2011.02747.x. Epub 2011/12/07. [DOI] [PubMed] [Google Scholar]
- 47.Dobrev H. Evaluation of the inhibitory activity of topical indomethacin, betamethasone valerate and emollients on UVL-induced inflammation by means of non-invasive measurements of the skin elasticity. Photodermatology, Photoimmunology and Photomedicine. 2001;17(4):184–8. doi: 10.1034/j.1600-0781.2001.170408.x. Epub 2001/08/14. [DOI] [PubMed] [Google Scholar]
- 48.Atzmony L, Reiter O, Hodak E, Gdalevich M, Mimouni D. Treatments for Cutaneous Lichen Planus: A Systematic Review and Meta-Analysis. American journal of clinical dermatology. 2016;17(1):11–22. doi: 10.1007/s40257-015-0160-6. Epub 2015/10/29. [DOI] [PubMed] [Google Scholar]
- 49.Nakagawa K, Taya Y, Tamai K, Yamaizumi M. Requirement of ATM in phosphorylation of the human p53 protein at serine 15 following DNA double-strand breaks. Mol Cell Biol. 1999;19(4):2828–34. doi: 10.1128/mcb.19.4.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Girardini JE, Napoli M, Piazza S, Rustighi A, Marotta C, Radaelli E, et al. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer cell. 2011;20(1):79–91. doi: 10.1016/j.ccr.2011.06.004. Epub 2011/07/12. [DOI] [PubMed] [Google Scholar]
- 51.Boyle JO, Hakim J, Koch W, van der Riet P, Hruban RH, Roa RA, et al. The incidence of p53 mutations increases with progression of head and neck cancer. Cancer research. 1993;53(19):4477–80. Epub 1993/10/01. [PubMed] [Google Scholar]
- 52.Kwon JY, Lee KW, Kim JE, Jung SK, Kang NJ, Hwang MK, et al. Delphinidin suppresses ultraviolet B-induced cyclooxygenases-2 expression through inhibition of MAPKK4 and PI-3 kinase. Carcinogenesis. 2009;30(11):1932–40. doi: 10.1093/carcin/bgp216. Epub 2009/09/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim JE, Kim JH, Lee Y, Yang H, Heo YS, Bode AM, et al. Bakuchiol suppresses proliferation of skin cancer cells by directly targeting Hck, Blk, and p38 MAP kinase. Oncotarget. 2016;7(12):14616–27. doi: 10.18632/oncotarget.7524. Epub 2016/02/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim JE, Roh E, Lee MH, Yu DH, Kim DJ, Lim TG, et al. Fyn is a redox sensor involved in solar ultraviolet light-induced signal transduction in skin carcinogenesis. Oncogene. 2016;35(31):4091–101. doi: 10.1038/onc.2015.471. Epub 2015/12/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.