

Abstract
Anaplastic thyroid cancer (ATC) is among the most lethal malignancies. The mitotic kinase PLK1 is overexpressed in the majority of ATCs and PLK1 inhibitors have shown preclinical efficacy. However, they also cause mitotic slippage and endoreduplication, leading to the generation of tetraploid, genetically unstable cell populations.
We hypothesized that PI3K activity may facilitate mitotic slippage upon PLK1 inhibition, and thus tested the effect of combining PLK1 and PI3K inhibitors in ATC models, in vitro and in vivo. Treatment with BI6727 and BKM120 resulted in a significant synergistic effect in ATC cells, independent of the levels of AKT activity. Combination of the two drugs enhanced growth suppression at doses for which the single drugs showed no effect, and led to a massive reduction of the tetraploid cells population. Furthermore, combined treatment in PI3Khigh cell lines showed a significant induction of apoptosis.
Finally, combined inhibition of PI3K and PLK1 was extremely effective in vivo, in an immunocompetent allograft model of ATC.
Our results demonstrate a clear therapeutic potential of combining PLK1 and PI3K inhibitors in anaplastic thyroid tumors.
Keywords: Mitotic slippage, Thyroid, Mouse models
Introduction
Anaplastic thyroid carcinoma (ATCs) is an extremely aggressive tumor that most often presents at diagnosis with local invasion of the neck soft tissues, airway compromise, lymph node involvement, and distant metastasis [1, 2]. These features usually hinder surgical resection, making ATC one of the most lethal tumor types, with a median survival of less than 6 months [3]. No effective therapeutic regimens exist for ATC. Cytotoxic chemotherapy (doxorubicin and paclitaxel) is generally unable to prolong survival of ATC patients [4-6], while radiation therapy, alone or in conjunction with doxorubicin, has not shown any significant improvement in overall survival [4], although recent data seem to suggest a better outcome with intensity-modulated radiotherapy [7].
The poor outcome of these traditional therapeutic approaches is the result of several key features of ATC, such as elevated levels and activity of multidrug resistance proteins [8], strong activation of pro-survival pathways, and frequent chromosomal instability and aneuploidy [9].
The growing availability of targeted kinase inhibitors and immunomodulatory molecules, together with easier access to detailed tumor molecular characterization, have fostered the development and testing of combinatorial approaches that in several cases have shown promising results [10-12].
Up to 70% of human ATCs display loss or inactivation of TP53, while in up to 40% the PI3K cascade is constitutively activated through mechanisms that include PTEN loss and PIK3CA amplification or mutation [13, 14]. Additional common driver oncogenic mutations include BRAF [15] and RAS activating mutations [16].
We have previously described a genetically engineered mouse model of ATC, which faithfully recapitulates the features of human ATC [17]. Comparative analysis between mouse and human ATC expression datasets highlighted several common deregulated genes and pathways, including a “mitosis network” centered around PLK1 [17].
PLK1 is a mitotic regulator overexpressed in breast, colorectal, endometrial, ovarian, and pancreatic cancer [18]. It is also found overexpressed in the majority of ATCs [9, 19]. PLK1 participates in multiple stages of mitosis, including mitotic entry, centrosome maturation, bipolar spindle formation, chromosome segregation, cytokinesis and mitotic exit [20]. A number of PLK1 inhibitors have been tested in the preclinical setting, with promising results [18, 21, 22]. However, clinical trials have found that therapeutic activity often occurs at or above the maximum tolerated doses, due to hematologic toxicity (neutropenia) [23-26]. We have previously reported that a PLK1 inhibitor, GSK461364A [18], is remarkably effective against mouse and human ATC cell lines characterized by different driver mutations, both in vitro and in an allograft model [27]. Interestingly, we also found that, in several cell lines, GSK461364A induced a polyploid cell population, which we hypothesized is the result of mitotic slippage and endoreduplication, the processes through which cells escape mitotic arrest and enter interphase without undergoing chromosome segregation and cytokinesis, thus producing tetraploid multinucleated cells [28, 29].
This effect may contribute to limiting the clinical efficacy of PLK1 inhibition by generating genetically unstable, therapy-resistant cell sub-populations [30, 31]. In this manuscript, we build on these data to test the hypothesis that the mitotic slippage induced by PLK1 inhibition is at least in part enabled by PI3K activation, as previously proposed in paclitaxel-treated [32] and irradiated [33] cells, and that pharmacological inhibition of PI3K might enhance the efficacy and reduce the effective dose (and thus the toxicity) of a PLK1 inhibitor.
Materials and Methods
Cells lines
All cell lines used in this study were maintained at 37°C with 5% CO2 in the culture media indicated in Table 1. Mouse cell lines were established from ATCs developed by genetically engineered mice [17, 34]. Cell line identity was validated by STS profiling as well as by amplifying and sequencing genomic fragments encompassing their known mutations.
Table 1.
| Cell line | Species of origin | Mutations | Culture Medium |
|---|---|---|---|
| T4888M | Mouse | Pten−/−/Tp53−/− | DMEM |
| T3533 | Mouse | Pten−/−/Tp53−/−- | DMEM |
| 3531L | Mouse | Pten−/−/KrasG12D | DMEM |
| T1903 | Mouse | Pten−/−/Tp53−/− | DMEM |
| A274 | Mouse | Pik3caH1047R/Tp53−/− | DMEM |
| A275 | Mouse | Pik3caH1047R/Tp53−/− | DMEM |
| A17 | Mouse | Pik3caH1047R/KrasG12D | DMEM |
| A22 | Mouse | Pik3caH1047R/KrasG12D | DMEM |
| THJ16T | Human | PIK3CAE545R/TP53P72R/R273H | RPMI |
| CAL62 | Human | KRASG12R/TP53A161D | DMEM |
| C643 | Human | HRASG13R/TP53R248Q | RPMI |
| OCUT2 | Human | PIK3CAH1047R/BRAFV600E/TP53−/− | RPMI |
| T238 | Human | PIK3CAE542K/BRAFV600E/TP53S183X | RPMI |
| 8505c | Human | BRAFV600E/TP53R248G | RPMI |
| SW1736 | Human | BRAFV600E/TP53-/−3 | RPMI |
| BHT101 | Human | BRAFV600E/TP53I251T | RPMI |
TP53 transcriptionally silenced [58]
Drug treatments and cell proliferation assay
The PLK1 inhibitor Volasertib (BI6727), and the PI3K inhibitors BKM120 and GDC0941 were purchased from Selleck Chemicals and dissolved in DMSO. For drug sensitivity experiments all the inhibitors were added 12 hours after plating. Alamar Blue was directly added to the culture medium of treated and control cells after 72 hours of treatment. Fluorescence was measured using a plate reader (excitation 530nm, emission 590nm). Statistical analysis and EC50 value calculation were done using GraphPad Prism (GraphPad Software).
Compounds were diluted so that the DMSO concentration was kept at 0.1% in all samples. Volasertib (BI6727) was used between 1nM and 100nM, BKM120 was used between 200nM and 2μM, and GDC0941 was used between 200nM and 5μM. Molar ratios between the drugs used in the combinations were established based on the ratios between the EC50 concentrations for the single drugs.
Statistical analysis of drug synergy was done using the Chou-Talaly method [35] and the Calcusyn Software (Biosoft). To determine synergy between two drugs, the software uses a median-effect method that determines if the drug combination produces greater effects together than expected from the summation of their individual effects. The combination index (CI) values are calculated for the different dose-effect plots (for each of the serial dilutions) based on the parameters derived from the median-effect plots of the individual drugs or drug combinations at the fixed ratios. The CI was calculated based on the assumption of mutually nonexclusive drug interactions. Following the analysis proposed by Bijnsdorp et al. [36], we have considered CI values above 1.1 antagonistic, between 0.9 and 1.1 additive, between 0.7 and 0.9 moderately synergistic, between 0.3 and 0.7 synergistic, and below 0.3 strongly synergistic. The Dose Reduction Index (DRI) denotes how many folds of dose reduction are allowed for each drug due to synergism when compared with the dose of each drug alone.
Cell cycle analysis
For cell cycle analysis, cell lines were treated with DMSO (control), volasertib (EC50) and BKM120 (1μM).
Cells were harvested by trypsin treatment and fixed in 75% ethanol in ice for 30 min. After treatment with RNase (Genentech/Roche) for 5 min at room temperature, cells were stained with propidium iodide (BioSure) overnight, and DNA content was measured using a BD FACSCantoTM II system (BD Biosciences).
Apoptosis analysis
Cells were treated with DMSO (control), volasertib at EC50, and 1μM BKM120 for 24-72 hours. At the end of treatment, cells were harvested by trypsinization. The supernatant was also collected. Cells were stained with Annexin V FITC and propidium iodide (BD Biosciences) for 15 minutes at room temperature in the dark. Samples were analyzed by flow cytometry within 1 hour using a BD FACSCantoTM II system (BD Biosciences). Flow cytometry analysis was performed on the FloJo platform.
Western blot analysis
24 hours after plating at 70% confluence, cells were treated with volasertib (EC70) and BKM120 (1μM), alone and in combination. After 24 hours, cells were homogenized on ice in radioimmunoprecipitation assay (RIPA) buffer supplemented with Halt Protease and Phosphatase Inhibitor cocktail (ThermoFisher Scientific). Protein concentration was determined using the Pierce BCA protein Kit (ThermoFisher Scientific). Western blot analysis was conducted using 50 μg of proteins on ExpressPlus precast gels (Genescript). Proteins were blotted onto polyvinylidene difluoride membranes (Millipore). The membranes were probed with the following antibodies: pAKTS473, phospho-Histone H3 (pHH3Ser10), phospho-Histone H2A.X (pH2AXSer139), poly (ADP-ribose) polymerase (PARP) (Cell Signaling). All the primary antibodies used at a dilution of 1:1000 in 5% BSA in TBS-T. Signals were detected with HRP-conjugated secondary antibodies (ThermoFisher Scientific) and the chemiluminescence substrate Luminata Crescendo (EMD Millipore). Equivalent loading was confirmed with anti-β-tubulin (Sigma-Aldrich).
Colony formation assay
For washout experiments, THJ16T cells were treated for 72 with volasertib (EC50) and BKM120 (1μM), alone and in combination. After pre-treatment, live cells were counted, seeded at 400 cells/well, and cultured under routine conditions for 10 days. Medium was replaced as needed.
For continuous exposure experiments, 400 cells per well were seeded. 12 hours after seeding, cells were treated as above. Medium and drugs were replaced every 3 days. Colonies were fixed with 0.1%methanol and stained with 0.05%crystal violet (Sigma-Aldrich).
Analysis of cell nuclei
Wheat germ agglutinin Alexa Fluor® 594 (WGA) (ThermoFisher Scientific) and 4’, 6-diamidino-2-phenylindole, dihydrochloride (DAPI) (Sigma) were used to stain the membranes and nuclei of cells, respectively.
Cells were fixed and stained according to the manufacturer’s instructions. Binucleated cells were counted. Imaging was conducted using the Nikon STORM/SIM/TIRF microscope.
In vivo experiments
6-8 week-old 129S6/SvE mice were injected subcutaneously with 5 × 106 T4888M cells. When tumors reached a size of 200 mm3, mice were randomized to control, volasertib, BKM120, and combination treatment groups. Volasertib and BKM120 were resuspended in 0.5%Methylcellulose/0.5%Tween80 and administered via oral gavage at 8mg/Kg/day and 24mg/Kg/day, respectively. Tumor volume was calculated from two-dimensional measurements using the equation: tumor volume = (length × width2 ) × 0.5, every two days. Tumor weight was measured at the end of the experiment. Data were plotted and analyzed using GraphPad Prism.
All experiments were performed in accordance with institutional policies on animal welfare.
Immunohistochemistry
Tissues were paraffin embedded and sectioned at 5μm. To assess the proliferation status of the tumor, Ki67 staining was performed. Immunostaining of apoptotic cells was carried out using the TUNEL assay. Representative microphotographs were imported into ImageJ for quantitation of proliferating and apoptotic cells.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism. The results were expressed as the mean ± standard deviation (SD) of the individual experiments. Results were compared using the unpaired, two-tailed Student t-test; p-values of <0.05 were considered significant.
Results
PLK1 inhibition leads to endoreduplication and mitotic slippage
We have utilized a panel of 16 mouse and human anaplastic thyroid cancer (ATC)-derived cell lines carrying driver alterations representative of the whole mutation spectrum observed in the ATC patient population [14] to assess the growth suppressive efficacy of volasertib (BI6727), a well-known orally available PLK1 inhibitor [37].
Similar to what we have previously reported using a different PLK1 inhibitor, GSK461364A [27], all ATC cell lines were extremely sensitive to the growth suppressive activity of volasertib, with EC50’s in the low nanomolar range. Notably, sensitivity to volasertib was completely independent of the nature of the driving genetic alteration (Fig. 1A,B and Suppl. Fig. 1). In general, human cell lines were slightly more sensitive than mouse cell lines.
Figure 1.
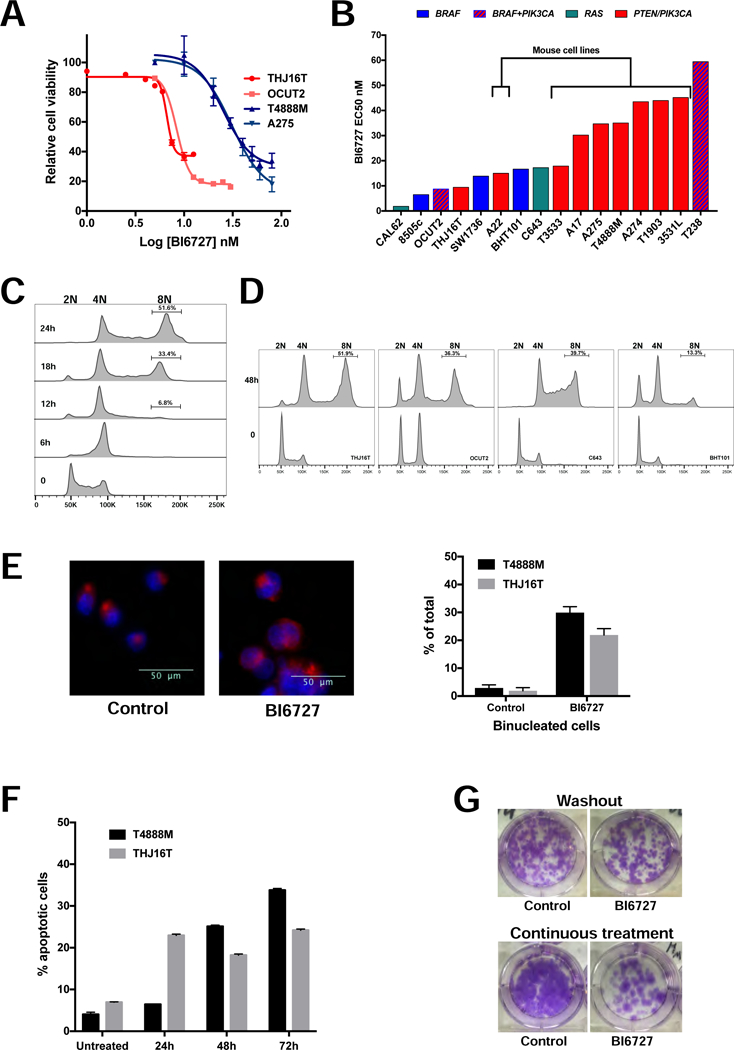
Volasertib (BI6727) is effective in ATC cells. A, dose-response assay in two mouse (blue lines) and two human (red lines) ATC cell lines. B, distribution of Volasertib (BI6727) EC50 values in a panel of mouse and human cell lines, labeled by driver mutation. C, time course of the effect of EC70 Volasertib (BI6727) on the cell cycle profile of T4888M cells. D, effect of EC50 Volasertib (BI6727) on the cell cycle profile of human ATC cell lines. E, left, nuclear (DAPI) and membrane (WGA) staining of THJ16T cells treated with EC70 Volasertib (BI6727) for 48 hours. Arrows indicate binucleated cells. Right, frequency of binucleated cells upon EC70 Volasertib (BI6727) treatment for 24 (T4888M) and 48 (THJ16T) hours. F, time course of apoptotic response to EC70 Volasertib (BI6727) treatment. G, colony forming assay showing the effect of ten-day continuous treatment, or 72h treatment followed by washout and replating for twelve days, on THJ16T cells’ ability to proliferate.
Time course analysis of the effect of volasertib (EC70) on the cell cycle profile of the mouse ATC cell line T4888M showed a rapid arrest in G2/M, within 6 hours from treatment, followed by progressive accumulation of an 8N population, between 18 and 24 hours after treatment (Fig. 1C). Again, this phenotype was common to all cell lines, independent of the driver mutation, and was also observed at lower drug concentrations corresponding to the EC50 for these lines (Fig. 1D). Human cell lines accumulated 8N cells with a slightly slower kinetics compared to mouse cells (Fig. 1D).
DAPI staining of mouse and human ATC cells treated with EC70 volasertib for 24 and 48h respectively, showed that 20% of the human THJ16T and 30% of the mouse T4888M cells contained two nuclei, suggesting that the 8N population observed in the cell cycle analysis is the result of mitotic slippage and endoreduplication (Fig. 1E).
Finally, volasertib (BI6727) treatment at EC70 induced a time-dependent but relatively modest apoptotic response in both T4888M and THJ16T cells (Fig. 1F). The accumulation of an 8N population in cells treated with volasertib alone raises the possibility that these cells, which have escaped the mitotic arrest and subsequent cell death by catastrophe, might give rise to a genomically unstable cell subset associated with increased tumor aggressiveness. To test the longterm effect of volasertib treatment, we performed colony forming assays using the THJ16T cells. Cells were treated with vehicle or EC50 volasertib for three days, and then the same number of live control or treated cells were plated at low density in drug-free medium and allowed to form colonies over a ten days period. As shown in Fig. 1G (top), the cell growth arrest elicited by volasertib is fully reversible, since volasertib-treated cells formed approximately the same number of colonies as the vehicle-treated cells. In a second approach, cells were plated at low density and exposed to volasertib (EC50) continuously for twelve days. Strikingly, despite continuous drug exposure, a relevant percentage of cells was able to escape growth arrest and/or apoptosis, forming large colonies (Fig. 1G, bottom).
Thus, ATC cells evade volasertib-induced cell cycle arrest and death via mitotic slippage.
PI3K inhibition synergizes with PLK1 inhibition
The mitotic slippage and aberrant cell cycle progression of volasertib-treated ATC cells could be associated with the TP53 mutant or null status of the vast majority of ATCs (Table 1). In addition, we hypothesized that the high activation level of the PI3K signaling cascade observed in most ATCs might contribute to mitotic slippage and endoreduplication by accelerating cell cycle progression through G2 and/or by increasing pro-survival signals and impairing mitotic catastrophe [38, 39].
To test this hypothesis, we treated a panel of mouse and human ATC cell lines with a combination of volasertib and the pan-PI3K inhibitor, BKM120. We observed a striking reduction in cell viability in almost all cell lines tested (Fig. 2A,B). Synergy analysis according to the mass-action law-based theory [35] was conducted by calculating the drugs Combination Index at both EC75 and EC90. Synergistic interaction was demonstrated for 12 of 14 cell lines analyzed (Fig. 2C). The combination also showed that drug reduction index (DRI) value was always above 1 at both EC75 and EC90 in the cell lines showing synergism, indicating that dose reduction is attainable in the clinical setting (Table 2).
Figure 2.
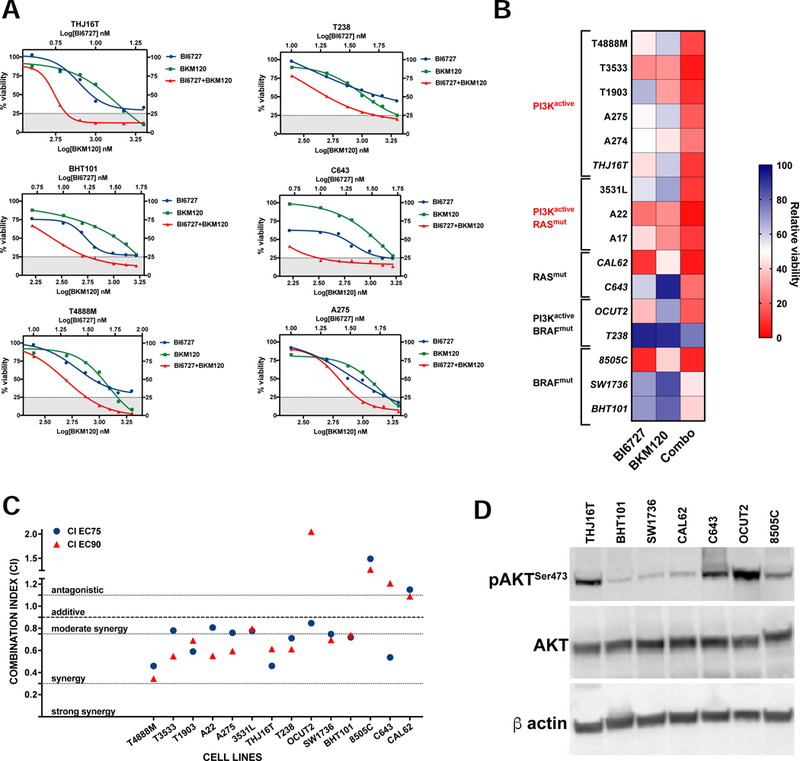
PI3K inhibition synergizes with Volasertib (BI6727). A, dose-response assays showing increased responses of ATC cell lines to combined PI3K and PLK1 inhibition. B, heat map showing the effect of combining 10nM Volasertib (BI6727) with the corresponding BKM120 dose (from the experiments in A). C, distribution of the Combination Indices at EC75 and EC90 showing synergy in the majority of cell lines analyzed. D, western blot analysis of AKT activation status in the human cell lines utilized.
Table 2.
: Dose Reduction Index (DRI) of volasertib (BI6727) and BKM120 combination in ATC cell lines.
| DRI EC75 | DRI EC90 | |||
|---|---|---|---|---|
| BI6727 | BKM120 | BI6727 | BKM120 | |
| T4888M | 2.44 | 20.44 | 3.05 | 58.63 |
| T3533 | 2.58 | 2.33 | 3.11 | 2.6 |
| T1903 | 4.28 | 2.81 | 3.76 | 2.33 |
| A22 | 2.27 | 2.73 | 3.51 | 3.75 |
| A275 | 1.68 | 6.03 | 1.92 | 13.83 |
| 3531L | 2.62 | 2.53 | 2.68 | 2.36 |
| THJ16T | 2.6 | 13.21 | 1.84 | 14.16 |
| T238 | 4.05 | 2.15 | 6.04 | 2.24 |
| OCUT2 | 1.93 | 3.05 | 0.76 | 1.35 |
| SW1736 | 1.55 | 9.62 | 1.56 | 18.91 |
| BHT101 | 2.01 | 4.51 | 1.9 | 4.66 |
| 8505C | 1.01 | 1.98 | 1.08 | 2.9 |
| C643 | 3.43 | 4.06 | 2.5 | 1.24 |
| CAL62 | 0.92 | 15.32 | 0.94 | 31.53 |
Response to the combined treatment was not synergistic only in those cell lines that were already extremely sensitive to volasertib treatment as single agent (Fig. 2B,C). While cell lines with mutations activating the PI3K pathway were clearly more sensitive to the combined treatment, synergy was also detected in several cell lines with unrelated driver mutations (BRAF, RAS). Interestingly, in these lines, there was only a weak correlation between AKT phosphorylation level and Combination Index, suggesting that even basal PI3K activity might be sufficient to reduce arrest in G2/M and/or to confer protection from mitotic catastrophe (Fig. 2D).
PI3K inhibition prevents mitotic slippage and endoreduplication
In order to define the mechanisms responsible for the synergy between BKM120 and volasertib, we analyzed the cell cycle profile of T4888M ATC cells treated with volasertib or volasertib + BKM120 for 6, 12, 18, and 24 hours. PI3K inhibition resulted in a striking reduction of the 8N population, which practically disappeared from the cell cycle profile. The same result was evident after cotreating the human THJ16T ATC cells for 24 hours (Fig. 3A). Of note, PI3K inhibition alone resulted, as expected, in G1 arrest (Fig. 3A, bottom).
Figure 3.

PI3K inhibition prevents mitotic slippage and endoreduplication. A, top, time course of the effect of EC50 Volasertib (BI6727) and 1μM BKM120 on the cell cycle profile of T4888M cells; bottom, effect of EC50 Volasertib (BI6727) and 1μM BKM120 on the cell cycle profile of THJ16T cells. B, apoptotic response to single (EC50 Volasertib and 1μM BKM120) and combined treatments for 72h in three ATC cell lines. C, left, nuclear (DAPI) and membrane (WGA) staining of THJ16T and T4888M cells treated with single and combined drugs for 24 (T4888M) and 48 (THJ16T) hours. Arrows indicate binucleated cells. Right, frequency of binucleated cells upon single and combined treatment. D, colony forming assay showing the effect of twelve-day continuous treatments or 72h treatment followed by washout and replating for ten days, on THJ16T cells’ ability to proliferate.
Most importantly, the combined treatment resulted in a dramatic increase in the induction of apoptosis in all the cell lines we analyzed (Fig. 3B).
As expected, combined volasertib/BKM120 treatment significantly reduced the number of T4888M and THJ16T binucleated cells at 24 and 48 hours, respectively (Fig. 3C).
To assess the long-term effect of co-targeting PLK1 and PI3K, we performed colony forming assays as described above, using single and combined treatment of THJ16T cells.
Strikingly, 72-hour combined volasertib/BKM120 treatment dramatically reduced the number of colonies growing in regular growth medium after drug washout (Fig. 3D, top). In addition, continuous treatment for twelve days completely suppressed colony formation (Fig. 3D, bottom). Identical results were obtained with the T4888M cell line (not shown).
Taken together, these results strongly suggest that combined inhibition of PLK1 and PI3K exerts significant long-term growth suppressive effects on ATC cells.
BKM120 effect depend on on-target activity
Recent data have clearly demonstrated that, at concentrations above 1μM, BKM120 inhibits microtubule dynamics by directly binding to tubulin [40]. Although in all our experiments 1μM was the highest BKM120 concentration used, we repeated selected dose-response and cell cycle profiling assays using a more specific pan-PI3K inhibitor, GDC0941 [41].
When we tested the combination of volasertib and GDC0941 in two mouse ATC cell lines, T4888M (Pten−/−) and A275 (Pik3caH1047R), we observed the same strongly synergistic interaction previously observed using BKM120 (Fig. 4A). Furthermore, GDC0941 was as effective as BKM120 in preventing mitotic slippage and endoreduplication, as shown by the almost complete disappearance of the 8N peak after cell cycle profiling (Fig.4B).
Figure 4.
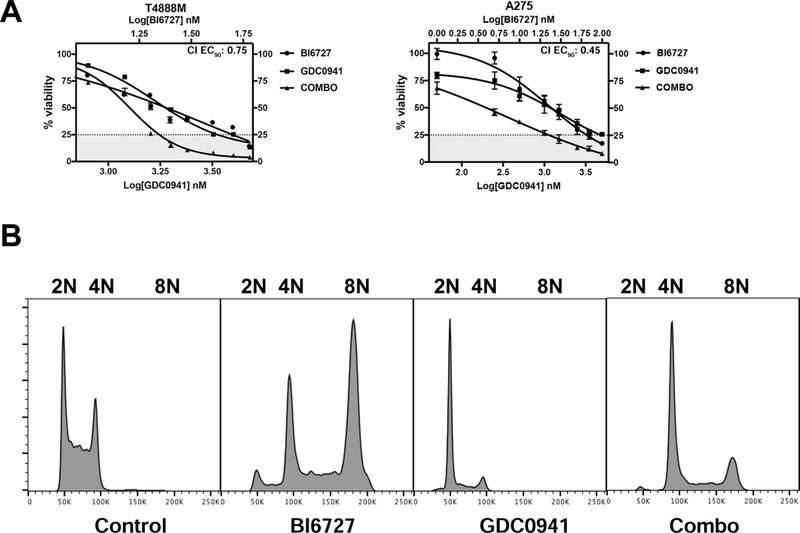
The pan-PI3K inhibitor GDC0941 has overlapping effect with BKM120. A, dose-response assays showing synergy between GDC0941 and Volasertib (BI6727) in two ATC cell lines. B, GDC0941 suppresses the 8N population induced by Volasertib (BI6727).
Thus, the observed effect of BKM120 on volasertib-induced mitotic slippage is due to on-target PI3K inhibition.
Western blotting analysis of biomarkers of BKM120 and volasertib action, using protein extracts from THJ16T and T4888M cells treated for 24 hours, confirmed on target compound activity at the concentration used in the cell cycle assays presented above, as shown by the inhibition of AKT phosphorylation (for BKM120) and the induction of histone H3 and histone H2AX phosphorylation (for volasertib) (Fig. 5). Furthermore, PARP cleavage, a marker of apoptosis, was increased in protein extracts from cells co-treated with the two inhibitors.
Figure 5.
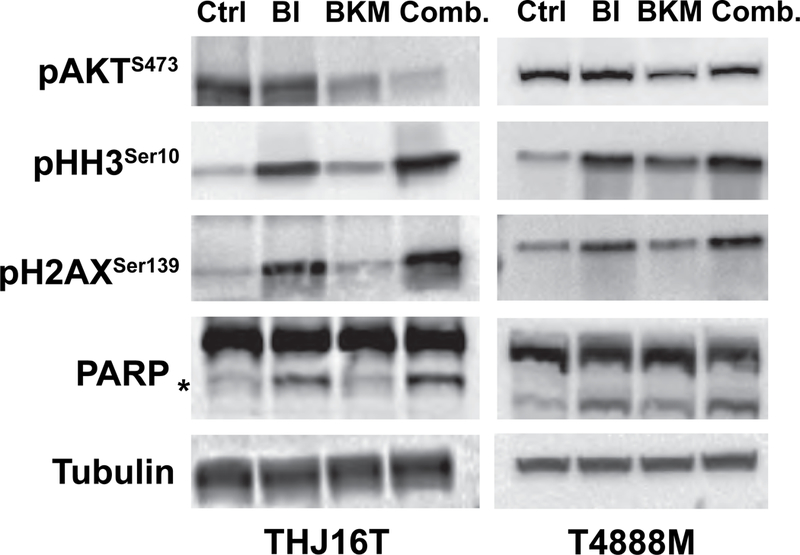
Western blot analysis on extracts from THJ16T and T4888M cells treated for 24 hours with EC70 Volasertib (BI6727) and/or 1μM BKM120, showing on target activity of both BKM120 and volasertib.
Combined PI3K and PLK1 inhibition is strongly effective in vivo
Based on these promising in vitro data suggesting that PI3K inhibition strongly synergizes with PLK1 inhibition, we tested the effect of this combination on an immunocompetent, syngeneic ATC allograft model.
T4888M mouse ATC cells were implanted in the flank of 129S6/SvEv mice and treatment was started when tumors reached 200mm3.
PLK1 inhibitors have shown strong dose-limiting hematological toxicity in clinical trials [42]. In addition, our pilot experiments have shown that combined treatment with 30mg/kg/day BKM120 and 10mg/kg/day volasertib, the most commonly used doses for in vivo experiments [43-46], was not tolerated by our mouse model.
Thus, we decided to reduce by 20% the dose of both BKM120 and volasertib and to modify the treatment schedule introducing a 2-day treatment interruption every week.
Even at these reduced doses, both BKM120 and volasertib significantly inhibited in vivo tumor growth, although volasertib treatment appeared to partially lose efficacy after about two weeks. Combined treatment, however, exerted a strikingly profound and sustained tumor suppressive effect, as evidenced by both tumor volume analysis over time (Fig. 6A) and tumor weight analysis at time of sacrifice (Fig. 6B). Drug-induced toxicity, as measured by body weight analysis and blood cell counts, was limited and did not result in any deaths among the treated mice (not shown).
Figure 6.
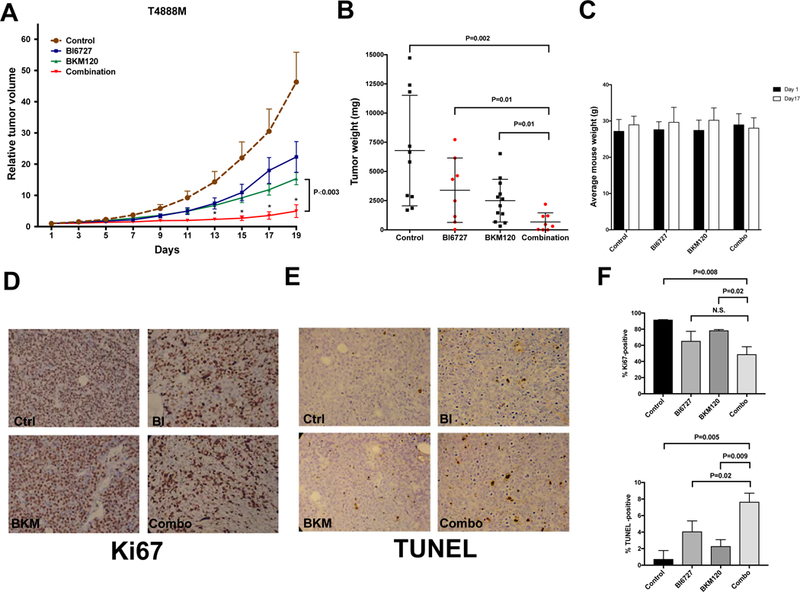
In vivo efficacy of a combination of BKM120 and volasertib (BI6727). A, relative tumor growth (±SEM) of T4888M allografts treated with vehicle, single drugs, and combination. Values are normalized to the tumor volume at the beginning of treatment. P<0.003 from day 13 onwards when comparing BKM120 to the combined treatment. B, tumor weights at endpoint. C, Average mouse weight at the beginning and at day 17 of treatment. D,E immunohistochemical detection of proliferation (D) and apoptosis (E) in sections from tumors collected at endpoint. F, quantification of Ki67 and TUNEL positive cells.
Finally, immunohistochemical analysis of proliferation and apoptosis markers in the four treatments cohorts evidenced a drastic reduction of proliferation in both the volasertib and the combination-treated groups, and a significantly increased apoptotic index in the combination group.
Thus, combined in vivo inhibition of PLK1 and PI3K is well tolerated and effective in inhibiting tumor growth in a clinically relevant immunocompetent mouse model of ATC.
Discussion
Treatment options for anaplastic thyroid cancer are extremely limited and usually only palliative in nature. The increasing understanding of the driver alterations characterizing these tumors has allowed the identification of numerous actionable targets, often leading to encouraging clinical results [12].
Furthermore, additional recent preclinical data in cell lines and xenograft models have shown promising results combining classic cytotoxic therapy with targeted inhibitors [47, 48] or lipid metabolism inhibitors [49].
Although the results of a number of clinical trials in other tumor types have dampened the interest for PLK1 inhibitors due to limited activity observed at tolerable doses [23-26], our previous [27] and current data clearly show that PLK1 is still a viable therapeutic target in ATC.
However, we also found that in ATC cells a substantial cell population escapes G2/M arrest via mitotic slippage and endoreduplication. This phenomenon is reminiscent of what was observed with Aurora kinases inhibitors [50], and is likely favored by the absence of TP53 in most ATCs. The fate of these endoreduplicated cells is not clear: while most of these cells died shortly after slippage in the Aurora kinases study [50], our clonogenic assay data show that a substantial number of volasertib-treated cells survives and resumes regular proliferation, possibly after “depolyploidization” [29], with obvious possible detrimental consequences on therapeutic efficacy. We have so far been unsuccessful in our attempts to isolate and follow up the fate of this 8N population, thus this speculation remains to be experimentally substantiated.
In an effort to counter the development of this cell population, we hypothesized that increased PI3K signaling, a common feature of ATC cells, may contribute to the escape from mitotic blockage by increasing mitotic exit speed [51, 52], favoring survival after slippage [53], and directly protecting cells from mitotic catastrophe [38, 54, 55].
This notion represented the rationale for combining, in this study, volasertib with a pan-PI3K inhibitor. This is the first report, to our knowledge, of such a combinatorial approach. Pilot experiments (Suppl. Fig. 2) were performed to identify the optimal combination strategy, and showed that simultaneous treatment was clearly superior to sequential drug administration.
Our data, obtained from a large panel of mouse and human ATC cell lines, clearly show that PI3K inhibition prevents the slippage and endoreduplication processes induced by PLK1 inhibition, and strikingly synergizes with volasertib in inducing apoptotic cell death. The correlation between the disappearance of the 8N peak and the dramatic increase in apoptosis (although tested only with one combination ratio) strongly suggests that when ATC cells are prevented (by PI3K inhibition) to undergo slippage, they readily enter apoptosis.
It is also important to point that the combined treatment virtually annihilated the long-term clonogenic capacity of ATC cells upon continuous treatment, a significant finding from a therapeutic perspective.
Previous studies have shown that a slow rate of degradation of Cyclin B1 is a critical promoter of mitotic slippage [28]. Furthermore, it has been proposed that MCL1 dynamics influences both mitotic slippage and cell death after mitotic block [56, 57]. However, we have found no evidence of alterations in MCL1 or cyclin B1 levels upon PI3K/PLK1 inhibition (not shown).
Notably, several cell lines without activating mutations in the PI3K pathway benefited from the combined treatment, especially when volasertib alone was not overly effective as single agent, strongly suggesting that basal PI3K activity is sufficient to drive slippage and endoreduplication. This notion would be extremely valuable in the clinical setting, since about 60% of ATC patients do not harbor PI3K-activating mutations.
Importantly, our data translated extremely well into the preclinical setting, using our immunocompetent allograft model of ATC. As expected from the clinical trials data, dose-limiting toxicity was observed when volasertib and BKM120 were combined at full doses (doses regularly used in published studies). However, a 20% dose reduction applied to both compounds, together with a therapy vacation during weekends, reduced toxicity to tolerable levels while resulting in a striking therapeutic activity, associated with inhibition of proliferation and increased apoptosis in the tumors.
In summary our in vivo data show for the first time that otherwise incurable ATC may represent a tumor type uniquely sensitive to combinations of PLK1 and PI3K inhibitors. These findings warrant consideration of this combinatorial approach for rapid translation into the clinical investigation setting.
Supplementary Material
ATC is one of the most lethal tumor types, with a median survival of less than 6 months from diagnosis.
PLK1 is a mitotic regulator overexpressed in ATC, and the PI3K cascade is constitutively activated in 40% of ATCs.
ATC cells are sensitive to the PLK1 inhibitor, volasertib, but often evade cell cycle arrest and death via mitotic slippage and endoreduplication.
PI3K inhibition synergizes with volasertib in vitro, preventing slippage and inducing apoptosis, and potentiates volasertib activity in vivo.
Acknowledgements
We acknowledge the Animal Housing, Analytical Imaging, and Flow Cytometry Core Facilities of Albert Einstein College of Medicine, which are partially supported by the NIH Cancer Center Support Grant to the Albert Einstein Cancer Center (P30CA013330). This work utilized a High-Speed/Resolution Whole Slide Scanner that was purchased with funding from a National Institutes of Health SIG grant 1S10OD019961-01. Research reported in this publication was supported by NIH grants CA172012, CA128943, and CA167839 to A. Di Cristofano.
Footnotes
Declarations of interest: none
Conflict of interests : None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Smallridge RC, Approach to the patient with anaplastic thyroid carcinoma, J Clin Endocrinol Metab, 97 (2012) 2566–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Patel KN, Shaha AR, Poorly differentiated and anaplastic thyroid cancer, Cancer Control, 13 (2006) 119–128. [DOI] [PubMed] [Google Scholar]
- [3].Smallridge RC, Marlow LA, Copland JA, Anaplastic thyroid cancer: molecular pathogenesis and emerging therapies, Endocr Relat Cancer, 16 (2009) 17–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sherman EJ, Lim SH, Ho AL, Ghossein RA, Fury MG, Shaha AR, Rivera M, Lin O, Wolden S, Lee NY, Pfister DG, Concurrent doxorubicin and radiotherapy for anaplastic thyroid cancer: a critical re-evaluation including uniform pathologic review, Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology, 101 (2011) 425–430. [DOI] [PubMed] [Google Scholar]
- [5].Higashiyama T, Ito Y, Hirokawa M, Fukushima M, Uruno T, Miya A, Matsuzuka F, Miyauchi A, Induction chemotherapy with weekly paclitaxel administration for anaplastic thyroid carcinoma, Thyroid, 20 (2010) 7–14. [DOI] [PubMed] [Google Scholar]
- [6].Ain KB, Egorin MJ, DeSimone PA, Treatment of anaplastic thyroid carcinoma with paclitaxel: phase 2 trial using ninety-six-hour infusion. Collaborative Anaplastic Thyroid Cancer Health Intervention Trials (CATCHIT) Group, Thyroid, 10 (2000) 587–594. [DOI] [PubMed] [Google Scholar]
- [7].Park JW, Choi SH, Yoon HI, Lee J, Kim TH, Kim JW, Lee IJ, Treatment outcomes of radiotherapy for anaplastic thyroid cancer, Radiat Oncol J, 36 (2018) 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zheng X, Cui D, Xu S, Brabant G, Derwahl M, Doxorubicin fails to eradicate cancer stem cells derived from anaplastic thyroid carcinoma cells: characterization of resistant cells, Int J Oncol, 37 (2010) 307–315. [DOI] [PubMed] [Google Scholar]
- [9].Salvatore G, Nappi TC, Salerno P, Jiang Y, Garbi C, Ugolini C, Miccoli P, Basolo F, Castellone MD, Cirafici AM, Melillo RM, Fusco A, Bittner ML, Santoro M, A cell proliferation and chromosomal instability signature in anaplastic thyroid carcinoma, Cancer Res, 67 (2007) 10148–10158. [DOI] [PubMed] [Google Scholar]
- [10].Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, Wen PY, Zielinski C, Cabanillas ME, Urbanowitz G, Mookerjee B, Wang D, Rangwala F, Keam B, Dabrafenib and Trametinib Treatment in Patients With Locally Advanced or Metastatic BRAF V600-Mutant Anaplastic Thyroid Cancer, J Clin Oncol, 36 (2018) 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Iyer PC, Dadu R, Gule-Monroe M, Busaidy NL, Ferrarotto R, Habra MA, Zafereo M, Williams MD, Gunn GB, Grosu H, Skinner HD, Sturgis EM, Gross N, Cabanillas ME, Salvage pembrolizumab added to kinase inhibitor therapy for the treatment of anaplastic thyroid carcinoma, Journal for immunotherapy of cancer, 6 (2018) 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Iyer PC, Dadu R, Ferrarotto R, Busaidy NL, Habra MA, Zafereo M, Gross N, Hess KR, Gule-Monroe M, Williams MD, Cabanillas ME, Real-World Experience with Targeted Therapy for the Treatment of Anaplastic Thyroid Carcinoma, Thyroid, 28 (2018) 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bonhomme B, Godbert Y, Perot G, Al Ghuzlan A, Bardet S, Belleannee G, Criniere L, Do Cao C, Fouilloux G, Guyetant S, Kelly A, Leboulleux S, Buffet C, Leteurtre E, Michels JJ, Tissier F, Toubert ME, Wassef M, Pinard C, Hostein I, Soubeyran I, Molecular Pathology of Anaplastic Thyroid Carcinomas: A Retrospective Study of 144 Cases, Thyroid, 27 (2017) 682–692. [DOI] [PubMed] [Google Scholar]
- [14].Landa Inigo, Ibrahimpasic Tihana, Boucai Laura, Sinha Rileen, Knauf, A. Jeffrey, Shah, H. Ronak, Dogan, Snjezana, F. Ricarte, C. Julio, Krishnamoorthy, P. Gnana, Xu, Bin, Schultz, Nikolaus, Berger, F. Michael, Sander, Chris, Taylor, S. Barry, Ghossein, Ronald, Ganly, Ian, Fagin, A. James, Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers, Journal of Clinical Investigation, 126 (2016) 1052–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Quiros RM, Ding HG, Gattuso P, Prinz RA, Xu X, Evidence that one subset of anaplastic thyroid carcinomas are derived from papillary carcinomas due to BRAF and p53 mutations, Cancer, 103 (2005) 2261–2268. [DOI] [PubMed] [Google Scholar]
- [16].Nikiforov YE, Genetic alterations involved in the transition from welldifferentiated to poorly differentiated and anaplastic thyroid carcinomas, Endocr Pathol, 15 (2004) 319–327. [DOI] [PubMed] [Google Scholar]
- [17].Antico Arciuch VG, Russo MA, Dima M, Kang KS, Dasrath F, Liao XH, Refetoff S, Montagna C, Di Cristofano A, Thyrocyte-specific inactivation of p53 and Pten results in anaplastic thyroid carcinomas faithfully recapitulating human tumors, Oncotarget, 2 (2011) 1109–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Degenhardt Y, Greshock J, Laquerre S, Gilmartin AG, Jing J, Richter M, Zhang X, Bleam M, Halsey W, Hughes A, Moy C, Liu-Sullivan N, Powers S, Bachman K, Jackson J, Weber B, Wooster R, Sensitivity of cancer cells to Plk1 inhibitor GSK461364A is associated with loss of p53 function and chromosome instability, Mol Cancer Ther, 9 (2010) 2079–2089. [DOI] [PubMed] [Google Scholar]
- [19].Zhang XG, Lu XF, Jiao XM, Chen B, Wu JX, PLK1 gene suppresses cell invasion of undifferentiated thyroid carcinoma through the inhibition of CD44v6, MMP-2 and MMP-9, Exp Ther Med, 4 (2012) 1005–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Strebhardt K, Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy, Nature reviews. Drug discovery, 9 (2010) 643–660. [DOI] [PubMed] [Google Scholar]
- [21].Hikichi Y, Honda K, Hikami K, Miyashita H, Kaieda I, Murai S, Uchiyama N, Hasegawa M, Kawamoto T, Sato T, Ichikawa T, Cao S, Nie Z, Zhang L, Yang J, Kuida K, Kupperman E, TAK-960, a novel, orally available, selective inhibitor of Pololike kinase 1, shows broad-spectrum preclinical antitumor activity in multiple dosing regimens, Mol Cancer Ther, (2011). [DOI] [PubMed] [Google Scholar]
- [22].Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, Gurtler U, Garin-Chesa P, Lieb S, Quant J, Grauert M, Adolf GR, Kraut N, Peters JM, Rettig WJ, BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo, Curr Biol, 17 (2007) 316–322. [DOI] [PubMed] [Google Scholar]
- [23].Mross K, Frost A, Steinbild S, Hedbom S, Rentschler J, Kaiser R, Rouyrre N, Trommeshauser D, Hoesl CE, Munzert G, Phase I dose escalation and pharmacokinetic study of BI 2536, a novel Polo-like kinase 1 inhibitor, in patients with advanced solid tumors, J Clin Oncol, 26 (2008) 5511–5517. [DOI] [PubMed] [Google Scholar]
- [24].Hofheinz RD, Al-Batran SE, Hochhaus A, Jager E, Reichardt VL, Fritsch H, Trommeshauser D, Munzert G, An open-label, phase I study of the polo-like kinase-1 inhibitor, BI 2536, in patients with advanced solid tumors, Clin Cancer Res, 16 (2010) 4666–4674. [DOI] [PubMed] [Google Scholar]
- [25].Sebastian M, Reck M, Waller CF, Kortsik C, Frickhofen N, Schuler M, Fritsch H, Gaschler-Markefski B, Hanft G, Munzert G, von Pawel J, The efficacy and safety of BI 2536, a novel Plk-1 inhibitor, in patients with stage IIIB/IV non-small cell lung cancer who had relapsed after, or failed, chemotherapy: results from an open-label, randomized phase II clinical trial, Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer, 5 (2010) 1060–1067. [DOI] [PubMed] [Google Scholar]
- [26].Olmos D, Barker D, Sharma R, Brunetto AT, Yap TA, Taegtmeyer AB, Barriuso J, Medani H, Degenhardt YY, Allred AJ, Smith DA, Murray SC, Lampkin TA, Dar MM, Wilson R, de Bono JS, Blagden SP, Phase I study of GSK461364, a specific and competitive Polo-like kinase 1 inhibitor, in patients with advanced solid malignancies, Clin Cancer Res, 17 (2011) 3420–3430. [DOI] [PubMed] [Google Scholar]
- [27].Russo MA, Kang KS, Di Cristofano A, The PLK1 Inhibitor GSK461364A Is Effective in Poorly Differentiated and Anaplastic Thyroid Carcinoma Cells, Independent of the Nature of Their Driver Mutations, Thyroid, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Raab M, Kramer A, Hehlgans S, Sanhaji M, Kurunci-Csacsko E, Dotsch C, Bug G, Ottmann O, Becker S, Pachl F, Kuster B, Strebhardt K, Mitotic arrest and slippage induced by pharmacological inhibition of Polo-like kinase 1, Molecular oncology, 9 (2015) 140–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Puig PE, Guilly MN, Bouchot A, Droin N, Cathelin D, Bouyer F, Favier L, Ghiringhelli F, Kroemer G, Solary E, Martin F, Chauffert B, Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy, Cell Biol Int, 32 (2008) 1031–1043. [DOI] [PubMed] [Google Scholar]
- [30].Visconti R, Grieco D, Fighting tubulin-targeting anticancer drug toxicity and resistance, Endocr Relat Cancer, 24 (2017) T107–T117. [DOI] [PubMed] [Google Scholar]
- [31].Ganem NJ, Storchova Z, Pellman D, Tetraploidy, aneuploidy and cancer, Curr Opin Genet Dev, 17 (2007) 157–162. [DOI] [PubMed] [Google Scholar]
- [32].Swanton C, Marani M, Pardo O, Warne PH, Kelly G, Sahai E, Elustondo F, Chang J, Temple J, Ahmed AA, Brenton JD, Downward J, Nicke B, Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs, Cancer Cell, 11 (2007) 498–512. [DOI] [PubMed] [Google Scholar]
- [33].Kandel ES, Skeen J, Majewski N, Di Cristofano A, Pandolfi PP, Feliciano CS, Gartel A, Hay N, Activation of Akt/protein kinase B overcomes a G(2)/m cell cycle checkpoint induced by DNA damage, Mol Cell Biol, 22 (2002) 7831–7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Dima M, Miller KA, Antico-Arciuch VG, Di Cristofano A, Establishment and characterization of cell lines from a novel mouse model of poorly differentiated thyroid carcinoma: powerful tools for basic and preclinical research, Thyroid, 21 (2011) 1001–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chou TC, Talalay P, Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors, Adv Enzyme Regul, 22 (1984) 27–55. [DOI] [PubMed] [Google Scholar]
- [36].Bijnsdorp IV, Giovannetti E, Peters GJ, Analysis of drug interactions, Methods Mol Biol, 731 (2011) 421–434. [DOI] [PubMed] [Google Scholar]
- [37].Rudolph D, Steegmaier M, Hoffmann M, Grauert M, Baum A, Quant J, Haslinger C, Garin-Chesa P, Adolf GR, BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity, Clin Cancer Res, 15 (2009) 3094–3102. [DOI] [PubMed] [Google Scholar]
- [38].Koul D, Fu J, Shen R, Lafortune TA, Wang S, Tiao N, Kim YW, Liu JL, Ramnarian D, Yuan Y, Garcia-Echevrria C, Maira SM, Yung WK, Antitumor Activity of NVP-BKM120--A Selective Pan Class I PI3 Kinase Inhibitor Showed Differential Forms of Cell Death Based on p53 Status of Glioma Cells, Clin Cancer Res, 18 (2012) 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Muller A, Gillissen B, Richter A, Richter A, Chumduri C, Daniel PT, Scholz CW, Pan-class I PI3-kinase inhibitor BKM120 induces MEK1/2-dependent mitotic catastrophe in non- Hodgkin lymphoma leading to apoptosis or polyploidy determined by Bax/Bak and p53, Cell death & disease, 9 (2018) 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Brachmann SM, Kleylein-Sohn J, Gaulis S, Kauffmann A, Blommers MJ, Kazic-Legueux M, Laborde L, Hattenberger M, Stauffer F, Vaxelaire J, Romanet V, Henry C, Murakami M, Guthy DA, Sterker D, Bergling S, Wilson C, Brummendorf T, Fritsch C, Garcia-Echeverria C, Sellers WR, Hofmann F, Maira SM, Characterization of the mechanism of action of the pan class I PI3K inhibitor NVPBKM120 across a broad range of concentrations, Mol Cancer Ther, 11 (2012) 17471757. [DOI] [PubMed] [Google Scholar]
- [41].Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, Friedman LS, Hayes A, Hancox TC, Kugendradas A, Lensun L, Moore P, Olivero AG, Pang J, Patel S, Pergl-Wilson GH, Raynaud FI, Robson A, Saghir N, Salphati L, Sohal S, Ultsch MH, Valenti M, Wallweber HJ, Wan NC, Wiesmann C, Workman P, Zhyvoloup A, Zvelebil MJ, Shuttleworth SJ, The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonylpiperazin-1-ylmethyl)-4-morpholin-4-ylt hieno[3,2d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer, J Med Chem, 51 (2008) 5522–5532. [DOI] [PubMed] [Google Scholar]
- [42].Degenhardt Y, Lampkin T, Targeting Polo-like kinase in cancer therapy, Clin Cancer Res, 16 (2010) 384–389. [DOI] [PubMed] [Google Scholar]
- [43].Xiao D, Yue M, Su H, Ren P, Jiang J, Li F, Hu Y, Du H, Liu H, Qing G, Polo-like Kinase-1 Regulates Myc Stabilization and Activates a Feedforward Circuit Promoting Tumor Cell Survival, Mol Cell, 64 (2016) 493–506. [DOI] [PubMed] [Google Scholar]
- [44].Bhola NE, Jansen VM, Bafna S, Giltnane JM, Balko JM, Estrada MV, Meszoely I, Mayer I, Abramson V, Ye F, Sanders M, Dugger TC, Allen EV, Arteaga CL, Kinome-wide functional screen identifies role of PLK1 in hormoneindependent, ER-positive breast cancer, Cancer Res, 75 (2015) 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ, Witt H, Korshunov A, Read TA, Sun JL, Schmitt EM, Miller CR, Buckley AF, McLendon RE, Westbrook TF, Northcott PA, Taylor MD, Pfister SM, Febbo PG, WechslerReya RJ, An animal model of MYC-driven medulloblastoma, Cancer Cell, 21 (2012) 155167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, Schnell C, Guthy D, Nagel T, Wiesmann M, Brachmann S, Fritsch C, Dorsch M, Chene P, Shoemaker K, De Pover A, Menezes D, Martiny-Baron G, Fabbro D, Wilson CJ, Schlegel R, Hofmann F, Garcia-Echeverria C, Sellers WR, Voliva CF, Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor, Mol Cancer Ther, 11 (2012) 317–328. [DOI] [PubMed] [Google Scholar]
- [47].Milosevic Z, Bankovic J, Dinic J, Tsimplouli C, Sereti E, Dragoj M, Paunovic V, Milovanovic Z, Stepanovic M, Tanic N, Dimas K, Pesic M, Potential of the dual mTOR kinase inhibitor AZD2014 to overcome paclitaxel resistance in anaplastic thyroid carcinoma, Cell Oncol (Dordr), (2018). [DOI] [PubMed] [Google Scholar]
- [48].Di Desidero T, Antonelli A, Orlandi P, Ferrari SM, Fioravanti A, Ali G, Fontanini G, Basolo F, Francia G, Bocci G, Synergistic efficacy of irinotecan and sunitinib combination in preclinical models of anaplastic thyroid cancer, Cancer Lett, 411 (2017) 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhong WB, Tsai YC, Chin LH, Tseng JH, Tang LW, Horng S, Fan YC, Hsu SP, A Synergistic Anti-Cancer Effect of Troglitazone and Lovastatin in a Human Anaplastic Thyroid Cancer Cell Line and in a Mouse Xenograft Model, Int J Mol Sci, 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Marxer M, Ma HT, Man WY, Poon RY, p53 deficiency enhances mitotic arrest and slippage induced by pharmacological inhibition of Aurora kinases, Oncogene, 33 (2014) 3550–3560. [DOI] [PubMed] [Google Scholar]
- [51].Lu J, Tan M, Huang WC, Li P, Guo H, Tseng LM, Su XH, Yang WT, Treekitkarnmongkol W, Andreeff M, Symmans F, Yu D, Mitotic deregulation by survivin in ErbB2-overexpressing breast cancer cells contributes to Taxol resistance, Clin Cancer Res, 15 (2009) 1326–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Stiles B, Gilman V, Khanzenzon N, Lesche R, Li A, Qiao R, Liu X, Wu H, Essential role of AKT-1/protein kinase B alpha in PTEN-controlled tumorigenesis, Mol Cell Biol, 22 (2002) 3842–3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bellacosa A, Kumar C, Di Cristofano A, Testa JR, Activation of AKT Kinases in Cancer: Implications for Therapeutic Targeting, Adv. Cancer Res, 94 (2005) 29–86. [DOI] [PubMed] [Google Scholar]
- [54].Hirose Y, Katayama M, Mirzoeva OK, Berger MS, Pieper RO, Akt activation suppresses Chk2-mediated, methylating agent-induced G2 arrest and protects from temozolomide-induced mitotic catastrophe and cellular senescence, Cancer Res, 65 (2005) 4861–4869. [DOI] [PubMed] [Google Scholar]
- [55].Hemstrom TH, Sandstrom M, Zhivotovsky B, Inhibitors of the PI3-kinase/Akt pathway induce mitotic catastrophe in non-small cell lung cancer cells, International journal of cancer. Journal international du cancer, 119 (2006) 1028–1038. [DOI] [PubMed] [Google Scholar]
- [56].Dikovskaya D, Cole JJ, Mason SM, Nixon C, Karim SA, McGarry L, Clark W, Hewitt RN, Sammons MA, Zhu J, Athineos D, Leach JD, Marchesi F, van Tuyn J, Tait SW, Brock C, Morton JP, Wu H, Berger SL, Blyth K, Adams PD, Mitotic Stress Is an Integral Part of the Oncogene-Induced Senescence Program that Promotes Multinucleation and Cell Cycle Arrest, Cell Rep, 12 (2015) 1483–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sloss O, Topham C, Diez M, Taylor S, Mcl-1 dynamics influence mitotic slippage and death in mitosis, Oncotarget, 7 (2016) 5176–5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Blagosklonny MV, Giannakakou P, Wojtowicz M, Romanova LY, Ain KB, Bates SE, Fojo T, Effects of p53-expressing adenovirus on the chemosensitivity and differentiation of anaplastic thyroid cancer cells, J Clin Endocrinol Metab, 83 (1998) 2516–2522. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.