

Abstract
Kaposi’s Sarcoma-associated Herpesvirus (KSHV) is the causative agent of several human cancers such as Kaposi’s Sarcoma (KS), which represents the most common AIDS-associated malignancy that lacks effective treatment options. Despite its clear role in AIDS-malignancies, the fact that only a small set of KSHV-infected patients will eventually develop these tumors implies that additional co-factors are required for the development of KSHV-related cancers. In the current study, we demonstrate for the first time that KSHV de novo infection or viral latent proteins are able to transactivate Human Endogenous Retrovirus K (HERV-K) through a variety of cellular signaling pathways and transcriptional factors. Moreover, we found that HERV-K transactivation, particularly activation of its encoded oncogenic NP9 protein, plays an important role in KSHV pathogenesis and tumorigenesis in vitro and in vivo. Our data provide innovative insights into the mechanisms of HERV-K transactivation contributing to viral oncogenesis, which may represent a promising target for KS treatment.
Keywords: HERV, KSHV, Kaposi’s Sarcoma, viral oncogenesis
INTRODUCTION
Approximately 20% of human cancers have been found related to viral infections, including Kaposi’s Sarcoma-associated Herpesvirus (KSHV, also named as Human Herpesvirus 8, HHV-8).1 KSHV is the causative agent of several cancers arising in patients with compromised immune systems, including Kaposi’s Sarcoma (KS) and Primary Effusion Lymphoma (PEL).2,3 Despite the reduced incidence of KS since the invention of Highly Active Antiretroviral Therapy (HAART) for Human Immunodeficiency Virus (HIV), KS remains the most common Acquired Immunodeficiency Syndrome (AIDS)-associated tumor.4,5 The prevalence of KSHV in the US HIV infected population remains high and incidence of new infections has increased in the HAART era.6 A longitudinal study of solid organ transplant recipients in the United States reported 15% of KSHV seropositivity in this specific subpopulation.7 Transplant recipients who develop primary KSHV infection after the transplantation, will have a relatively high probability of developing these KSHV-related malignancies, especially KS.8, 9 Since its discovery about 25 years ago, KSHV has now become a model pathogen for viral oncogenesis research, but many key questions regarding its mechanisms of pathogenesis and oncogenesis still remain unclear, hindering the identification of rational targets or the development of effective therapeutic strategies against these malignancies. Although KSHV has been closely linked to several human malignancies, only a small portion of KSHV-infected patients will eventually develop these tumors,1 implying that additional host or environmental co-factors such as co-infecting pathogens are required for the development of KSHV-related malignancies.
Human Endogenous Retrovirus (HERV) sequences occupy ~6% - 8% of the human genome, and have resided in our genome for several million years.10,11 Due to the accumulation of multiple nonsense mutations, the majority of HERVs are dysfunctional, however, some are still active and may play a role in human disease, in particular the HERV type K (HML-2) family.12–14 HERV-K transactivation has been observed in a variety of human cancers, such as leukemia,15 lymphoma,16 breast cancer17,18 and melanoma.19 For instance, the expression of the HERV-K envelope (env) protein in malignant breast cancer cell lines have been found higher than non-malignant breast cells, and some anti-HERV-K-specific monoclonal antibodies effectively inhibited breast cancer cells growth and induced their apoptosis of in vitro and in vivo.17 Interestingly, several herpesviruses have been reported to induce HERV-K transactivation. For instance, HERV-K18 can be transactivated as a superantigen (SAg) by Epstein–Barr virus (EBV) infection, and subsequently activates TCRVB13 T cells through MHC-II which plays a central role in EBV infection and pathogenesis.20–22 However, currently there are no data describing the role of HERV-K transactivation in viral oncogenesis, especially KSHV-related malignancies. In the current study, we demonstrate for the first time that KSHV de novo infection or viral latent proteins are able to transactivate HERV-K through a complex of mechanisms. Moreover, HERV-K transactivation (in particular activation of its oncogenic NP9 protein) are required for KSHV pathogenesis and tumorigenesis in vitro and in vivo.
RESULTS
KSHV de novo infection or encoded latent proteins transactivate HERV-K in vitro and in vivo
During an infection time course analysis, we found that KSHV de novo infection gradually increased HERV-K envelope gene (env) transcripts from primary human umbilical vein endothelial cells (HUVEC) when compared to the UV-inactivated KSHV infected cells by using qRT-PCR (Fig. 1A). Currently, the qRT-PCR based detection of HERV-K env transcripts is the most common and reliable method to evaluate the level of HERV-K transactivation in host cells.15–17 Our qRT-PCR primers were designed to measure the total levels of HERV-K env transcripts, including type 1 and 2 proviruses. Interestingly, our data indicate that KSHV+ PEL tumor cell lines (BC-1, BC-3, BCP-1 and BCBL-1) also have significantly higher levels of HERV-K env transcripts when compared with the virus-negative lymphoma cell line, BL-41 (Fig. S1). To understand the clinical relevance of HERV-K transactivation in KSHV-infected HIV+ patients, we examined the levels of HERV-K env transcripts in peripheral blood mononuclear cells (PBMCs) samples collected from a cohort of HIV+ patients prior to undergoing the HAART. KSHV infection status have been determined by measuring the titers of anti-KSHV-encoded LANA and K8.1 circulating IgG as described previously.26,27 Our results indicated a higher level of HERV-K env transcripts in the KSHV+ group (n=11) than those in the KSHV- group (n=10, Fig. 1B). Since there are no significant differences in HIV viral loads and CD4 counts between these two groups (data not shown), we think that KSHV infection may be responsible for the HERV-K transactivation in these patients.
Figure 1. KSHV de novo infection or viral latent proteins transactivate HERV-K in vitro and in vivo.
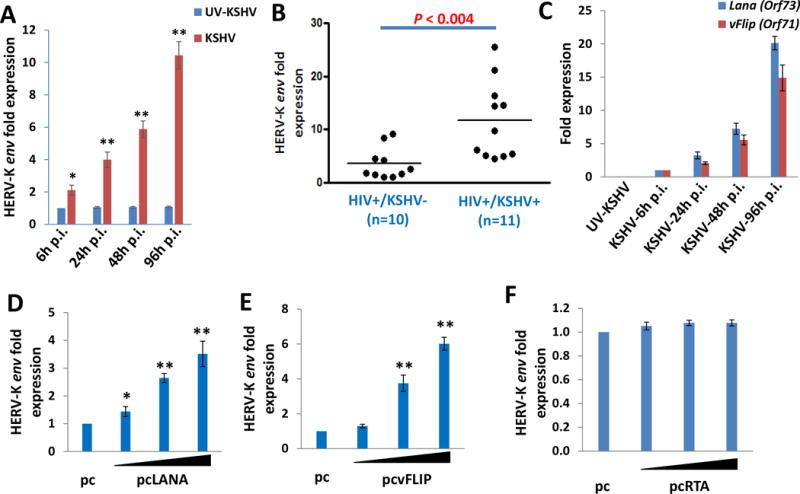
(A) Human umbilical vein endothelial cells (HUVEC) were infected with purified KSHV (MOI~10) or UV-inactivated KSHV for 2h, then the induction of HERV-K reactivation at indicated time-points post-infection (p.i.) was measured and compared to UV-inactivated KSHV infected cells control by qRT-PCR with the specific primers for HERV-K env gene. (B) The levels of HERV-K transactivation within peripheral blood mononuclear cells (PBMCs) from HIV+ patients with or without KSHV co-infection were quantified using qRT-PCR. KSHV infection status was identified using ELISA as described in the Methods. (C) HUVEC were infected by purified KSHV as described above, then the transcripts of viral latent genes Lana (Orf73) and vFlip (Orf71) at indicated time-points p.i. were measured and compared to control mock cells by using qRT-PCR. (D-F) HUVEC were transfected with control vector (pc) or vectors encoding LANA (pcLANA), vFLIP (pcvFLIP) or RTA (pcRTA) at 0.2, 1.0 or 2.5 μg, respectively, for 48 h, then the induction of HERV-K transactivation was quantified by using qRT-PCR. Error bars represent the S.D. from 3 independent experiments. * = p<0.05, ** = p<0.01.
KSHV has two infection-phases: a latent phase with only a limited number of viral genes expressed and a lytic phase in which most viral genes are expressed that ultimately produces infectious virions.31 In most KSHV-infected host cells (>90%), the virus exists in the latency stage,32 suggesting that some virus encoded latent proteins are potentially responsible for HERV-K transactivation. We detected the expression of two major KSHV-encoded latent genes, Latency-associated nuclear antigen (Lana, Orf73)33 and viral FADD-like interferon converting enzyme (FLICE) inhibitory protein (vFlip, Orf71)34 during KSHV de novo infection. Notably, we found that the expression of these two latent genes displayed an increase in expression that was relatively concordant with HERV-K env expression during the time course of KSHV infection (Fig. 1C). To further determine whether these latent genes are indeed responsible for KSHV-induced HERV-K transactivation, we transfected HUVEC with a recombinant LANA or vFLIP construct,35,36 respectively. We found that ectopic expression of LANA or vFLIP significantly increased HERV-K env transcripts from HUVEC in a dose-dependent manner (Fig. 1D-E). As a comparison, we found that ectopic expression of RTA, a viral lytic protein which initially controlling KSHV “latent to lytic” switch,59 almost does not induce HERV-K env expression (Fig. 1F).
Identification of cellular mechanisms for KSHV latent proteins induction of HERV-K expression
We next sought to understand the underlying mechanisms for LANA or vFLIP induced HERV-K transactivation in primary endothelial cells. We and others have reported that KSHV latent proteins are capable of activating several intracellular signaling pathways, e.g., LANA can activate the MAPK pathway37 and vFLIP can activate the NF-κB pathway.34 Our data here confirmed that ectopic expression of LANA or vFLIP induced the phosphorylation of MAPK-ERK or NF-κB p65, respectively, from transfected HUVEC (Fig. 2A-B). Next, we found that only inhibition of MAPK by U0126 effectively reduced HERV-K env transcripts from LANA-transfected cells, while inhibition of NF-κB by Bay11-7082 had no effects (Fig. 2C & E). In contrast, only inhibition of NF-κB but not MAPK effectively reduced HERV-K env transcripts from vFLIP-transfected cells (Fig. 2D & E). Furthermore, inhibition of either MAPK or NF-κB can partially reduce HERV-K env transcripts from KSHV-infected cells, and dual inhibition of these pathways has synergistic effects on reduction of HERV-K env transcripts (Fig. 2F). These data demonstrate that the MAPK and/or NF-κB pathways are indeed required for KSHV or viral latent proteins induced HERV-K transactivation.
Figure 2. Activation of intracellular signaling pathways is involved in HERV-K transactivation by KSHV.
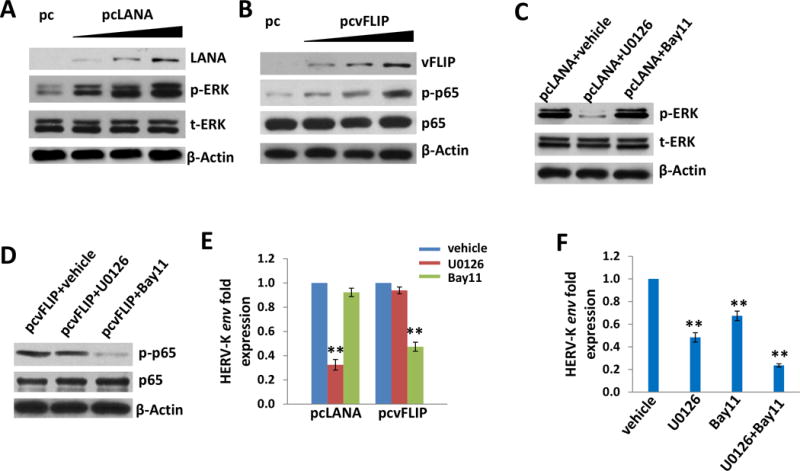
(A-B) HUVEC were transfected with control vector (pc) or vectors encoding LANA (pcLANA) or vFLIP (pcvFLIP) at 0.2, 1.0 or 2.5 μg, respectively, for 48 h, then protein expression was analyzed by using immunoblots. (C-F) HUVEC were first incubated with either vehicle or MEK inhibitor (10 μM of U0126) or NF-κB inhibitor (10 μM of Bay11-7082) for 1 h, then transfected or infected as described above. The induction of HERV-K transactivation was quantified by using qRT-PCR and protein expression was detected by immunoblots. Error bars represent the S.D. from 3 independent experiments. ** = p<0.01.
In fact, HERV-K transactivation largely depends on the transcriptional regulatory elements within its retroviral long terminal repeats (LTRs), which have potential binding sited for both viral and cellular transcriptional factors (TRs).38 Currently, there are a few TRs which have been experimentally shown to modulate HERV-K LTR activities, including Sp1 and YY1 proteins.39,40 A previous study has shown that LANA directly interacts with Sp1 in the nucleus of KSHV+ lymphoma cells.41 Here we confirmed the interaction of LANA and Sp1 in KSHV-infected HUVEC by using immunofluorescence and co-immunoprecipitation assays (Fig. S2A-B). Moreover, knock-down of Sp1 by RNAi partially reduced HERV-K env transcripts from LANA-transfected cells (Fig. S2C). These data provide additional mechanistic insights into viral latent protein mediated induction of HERV-K expression through interaction with cellular TRs.
HERV-K transactivation is closely related to KSHV-induced primary endothelial cell invasiveness
One hallmark of KSHV-infected endothelial cells is displaying a migratory or invasive phenotype, with can facilitate viral dissemination and angiogenesis during KS development.41 Our data indicated that knock-down of HERV-K env by RNAi significantly blocked the invasiveness of KSHV-infected HUVEC by using the transwell assays (Fig. 3A-C). This reduction is independent of cell growth, since we do not observe silencing of HERV-K env affecting HUVEC cell growth (data not shown). Our previous study has demonstrated that VEGF is one of the major pro-angiogenic cytokines responsible for KSHV-induced primary endothelial cell invasiveness.42 Here we found that silencing of HERV-K env significantly reduced the VEGF production and the expression of VEGF receptor 1 (VEGFR1) but not the VEGF receptor 2 (VEGFR2) from KSHV-infected HUVEC (Fig. 3D-E). Together, these data indicate that HERV-K transactivation is closely related to KSHV-induced primary endothelial cell malignant behaviors.
Figure 3. Targeting HERV-K transactivation significantly reduces KSHV-induced primary endothelial cell invasiveness.
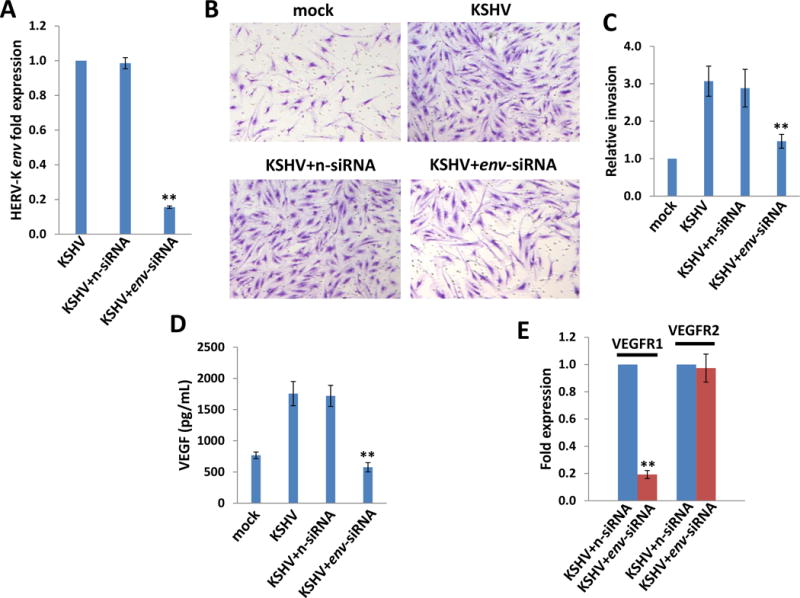
(A-C) HUVEC cells were incubated with purified KSHV (MOI~10) for 2h, then transfected with non-target control siRNA (n-siRNA) or HERV-K env-siRNA for additional 48 h. The transwell assays were performed to determine relative invasiveness as described in the Methods. (D) The concentrations of VEGF in culture supernatants were determined using ELISA. (E) The gene transcripts were quantified by using qRT-PCR. Error bars represent the S.D. from 3 independent experiments. ** = p<0.01.
KSHV infection activates HERV-K encoded oncogenic NP9 expression which enhances viral pathogenesis in endothelial cells
Among HERV-K (HML-2) elements, there are two major types of proviruses (type 1 and 2). Unlike type 2 proviruses, type 1 elements share a 292-nt fragment deletion in the env region, which gives rise to a difference between two isoforms of regulatory proteins encoded by the double-spliced transcripts. Type 2 proviral transcripts, 1.8 kb long, code the 15-kDa accessory protein Rec,63 which is a functional homologue of Rex and Rev from other retroviruses.64 Type 1 specific double-spliced RNA product, NP9, is a 9-kDa protein which shares only the N-terminal 15 amino acid residues with Rec.18,44,45 Furthermore, the NP9 protein has been found as a oncogenic protein and present in a variety of tumors and transformed cells.18,44,45 Our data here indicate that KSHV de novo infection induced a gradient increase of Np9 transcripts from HUVEC using qRT-PCR with Np9-specific primers,65 which was subsequently confirmed by immuoblots with a NP9 polyclonal antibody (kindly provided by Dr. Friedrich A. Grasser from Universitatsklinikum des Saarlandes, Germany)28 (Fig. 4A-B). Notably, NP9 protein is only expressed in KSHV-infected cells, while none in the uninfected cells. In contrast, we found that KSHV de novo infection slightly induced the increase of Rec transcripts (with no statistical significance) using qRT-PCR with Rec-specific primers65 (Fig. S3). Next, we observed the strong expression of NP9 within AIDS-KS tumor tissues while only low levels of expression in adjacent normal tissues from two HIV+ patients without any HAART treatment (Fig. 4C). Additionally, we found that NP9 was exclusively expressed in the nucleus by transfecting HUVEC with the pDsred-NP9 construct28 (Fig. 4D), although we observed some cytoplasmic staining of NP9 in AIDS-KS tissues (Fig. 4C). Since this is a self-made polyclonal antibody which has not been tested for immunohistochemistry staining, we cannot exclude the existence of some non-specific staining in the immunohistochemistry assays. The results from co-immunoprecipitation assays in both directions revealed the protein interaction between NP9 and LANA in KSHV-infected HUVEC (Fig. 4E). Moreover, ectopic expression of NP9 from the recombinant construct pSG5-NP928 significantly increased HUVEC invasion and anchorage-independent growth (Fig. 4F-H). Interestingly, we found that ectopic expression of NP9 greatly up-regulated the expression of one cellular glycoprotein, CD147 (also named as Emmprin), and its downstream proteins, ADAMTS1 (A disintegrin and metalloproteinase with thrombospondin motifs 1) and ADAMTS9 (A disintegrin and metalloproteinase with thrombospondin motifs 9) (Fig. 4F). Both high and low molecular weight (∼65 and ~35 kDa, respectively) CD147 glycoforms were elevated in NP9-transfected cells, in particular the mature high molecular weight glycoform related to biological activities.46 qRT-PCR analysis indicated that ectopic expression of NP9 also increased the transcripts of these genes (Fig. S4). Our previous studies reported that KSHV infection or ectopic expression of LANA induced CD147 expression, which enhances primary endothelial cell invasiveness.35 Our recent transcriptomic analysis has determined that ADAMTS1 and ADAMTS9 are two novel CD147-regulated downstream proteins, and they are all highly expressed in AIDS-KS tissues.30 Moreover, silencing of CD147, ADAMTS1 or ADAMTS9 by RNAi significantly reduced KSHV-induced primary endothelial cell invasiveness.30,35 Here we also found that silencing of CD147, ADAMTS1 or ADAMTS9 by RNAi significantly blocked the NP9-induced HUVEC invasion (Fig. S5), indicating that CD147-ADAMTS1/ADAMTS9 axis is indeed contributed to NP9-mediated cellular functions.
Figure 4. KSHV infection induces HERV-K encoded oncogenic NP9 expression which enhancing primary endothelial cells invasion and colony formation.
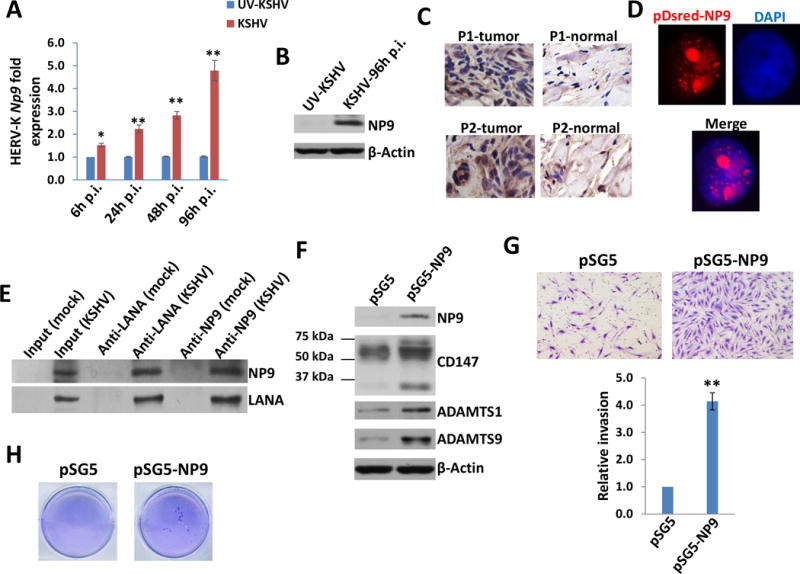
(A-B) HUVEC were infected with purified KSHV (MOI~10) or UV-inactivated KSHV for 2h, then the induction of HERV-K NP9 at indicated time-points post-infection (p.i.) was measured and compared to UV-inactivated KSHV infected cells control using qRT-PCR and immunoblots. (C) The strong expression of NP9 protein in KS tissues from our cohort of two AIDS-KS patients without any treatment by immunohistochemistry staining. (D-E) HUVEC were transfected with the pDsred-NP9 vector for 48 h, then protein expression was detected by immunofluorescence and nuclear was shown by DAPI. Immunoprecipitation assays in both directions were performed using Catch and Release Immunoprecipitation Kit (Millipore) with anti-LANA or anti-NP9 antibodies, respectively. (F-H) HUVEC were transfected with pSG5 control vector or pSG5-NP9 for 48 h, then protein expression was detected by immunoblots. Cell invasiveness was determined using the transwell assays. Anchorage-independent growth ability was determined using the soft agar assays. Error bars represent the S.D. from 3 independent experiments. * = p<0.05, ** = p<0.01.
Since KSHV-infected primary endothelial cells (e.g., HUVEC) usually are not able to form tumors in mice,31 we recently established a KS-like xenograft model using a KSHV long-term-infected telomerase-immortalized human umbilical vein endothelial (TIVE-LTC) cell line, which stably supports KSHV latency (kindly provided by Dr. Rolf Renne at University of Florida).24,30 Our data indicated that TIVE-LTC have much higher levels of NP9, CD147 and downstream protein expression than the parental non-infected TIVE cells (Fig. 5A). Co-immunoprecipitation assays confirmed the interaction of LANA and NP9 in TIVE-LTC (Fig. 5B). We previously showed that TIVE-LTC displayed much stronger abilities of cell invasiveness and anchorage-independent growth than its parental TIVE cells, the latter almost cannot form colonies in soft agar assays.30 To further study the functional role of NP9 in TIVE-LTC, we first directly silenced it by using lentiviral vector containing shRNA specifically targeting Np9 (Np9-shRNA) to obtain stably “knock-down” cells. A non-silencing (n)-shRNA was used as a negative control, and we do not observe silencing of Np9 affecting TIVE-LTC cell growth (data not shown). Here we demonstrated that silencing of Np9 by RNAi dramatically reduced TIVE-LTC invasion and anchorage-independent growth abilities (Fig. 5C-E).
Figure 5. The HERV-K NP9 protein is involved in the pathogenesis of KSHV long-term-infected endothelial cells.
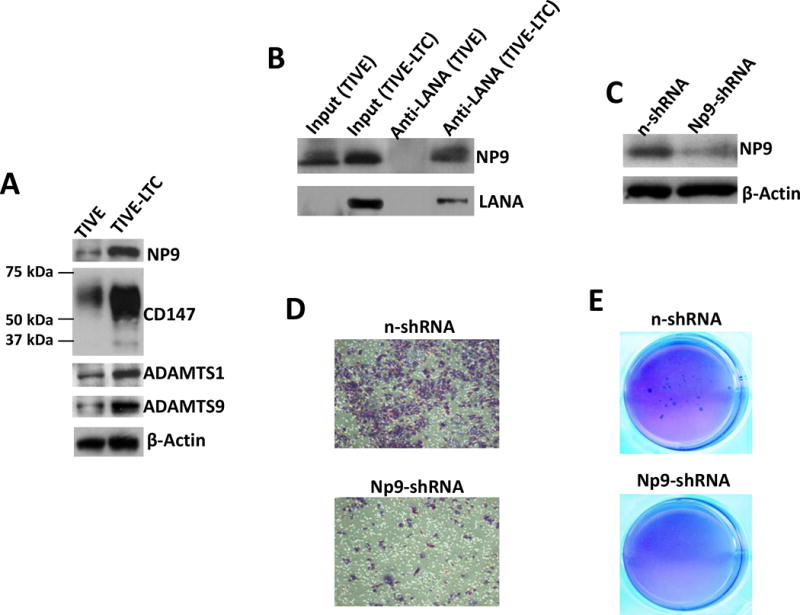
(A) The protein expression in KSHV long-term-infected telomerase-immortalized human umbilical vein endothelial (TIVE-LTC) and parental uninfected TIVE cells was detected and compared by immunoblots. (B) Immunoprecipitation assays were performed with anti-LANA antibody as described previously. (C-E) The stably “knock-down” of Np9 in TIVE-LTC were established by using lentiviral vector containing shRNA specifically targeting Np9 (Np9-shRNA) as described in the Methods. A non-silencing (n)-shRNA was used as a negative control. The protein expression, cell invasiveness and anchorage-independent growth abilities were measured as described above.
Targeting HERV-K NP9 significantly suppresses KSHV-induced tumorigenesis in vivo
We next seek to determine the role of HERV-K NP9 in KSHV-induced tumorigenesis in vivo by using the established KS-like xenograft model.30 We injected the Np9 stably “knock-down” TIVE-LTC or control cells subcutaneously into the two sides of flanks of nude mice, respectively. These mice were checked and measured every 2~3 days for the presence of palpable tumors for 30 days. Our results indicate that silencing of Np9 significantly repressed KSHV-induced tumorigenesis in nude mice. Mice injected with Np9 stably “knock-down” cells formed much smaller tumors, when compared to mice injected with control “n-shRNA” cells at 30 days (Fig. 6A-C). H & E staining confirmed that there were significantly fewer tumor cells or tumor biomass with more immune cell infiltrated in the tumor tissues from mice injected with Np9 stably “knock-down” cells (Fig. 6D). Of note, we also observed dramatically reduced LANA expression in tumor tissues from mice injected with Np9 stably “knock-down” cells, although the underlying mechanisms remain unclear and we do not observe the similar phenotype in vitro cultures (data not shown). Immunoblot results confirmed the reduced levels of NP9, CD147, ADAMTS1 and ADAMTS9 expression in tumor lysates from mice injected with Np9 stably “knock-down” cells (Fig. 6E). Taken together, these data strongly support the important role of HERV-K transactivation (in particular activation of NP9 and related signaling) as the cellular co-factors for KSHV-induced tumorigenesis in this in vivo model.
Figure 6. Targeting HERV-K NP9 significantly suppresses KSHV-induced tumorigenesis in vivo.
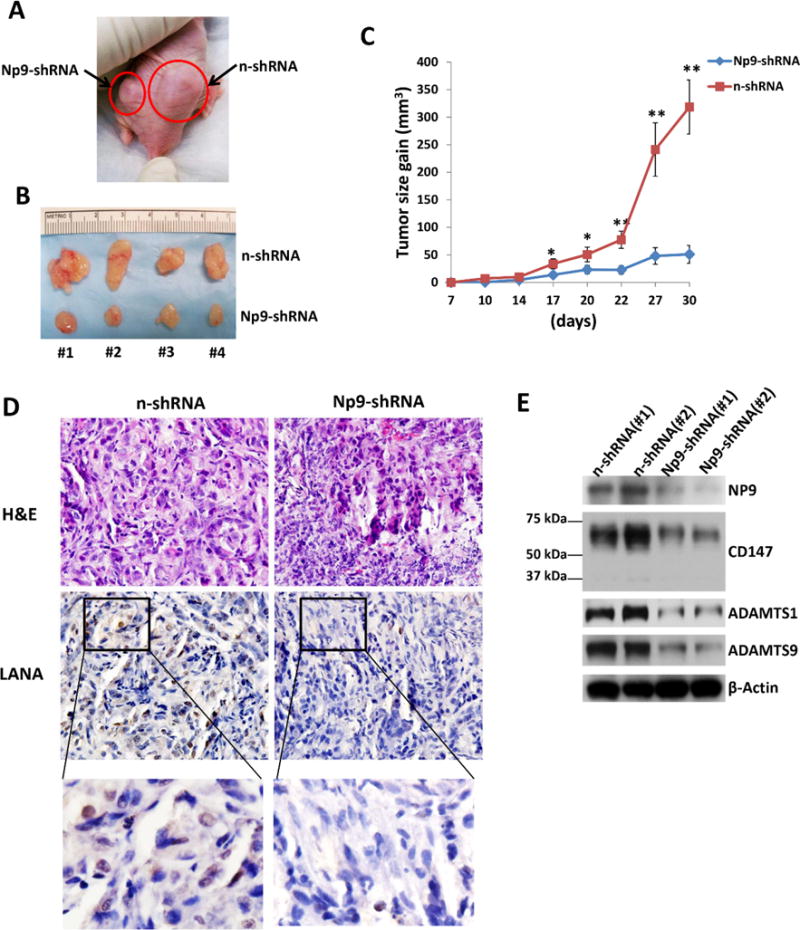
(A-C) The Np9 stably “knock-down” TIVE-LTC or control cells (approximately 5 × 105 cells were mixed at a ratio of 1:1 with growth factor-depleted Matrigel) were injected subcutaneously into the right and left flanks of nude mice, respectively. The mice were observed and measured every 2~3 d for the presence of palpable tumors for 30 d. Error bars represent the S.D. from 2 independent experiments. * = p<0.05, ** = p<0.01. (D-E) Protein expression within tumor tissues from representative injected mice was measured by using immunohistochemistry or immunoblots, respectively.
DISCUSSION
In the current study, we demonstrate for the first time that the KSHV-encoded latent proteins LANA and vFLIP can induce HERV-K transactivation through both intracellular signaling pathways (e.g., MAPK and NF-κB) and cellular transactional factors (TRs, e.g., Sp1), resulting in enhanced cell invasion, anchorage-independent growth and KSHV-induced tumorigenesis (summarized in Fig. 7). In silico analysis of HERV-K 5′ LTR regions has found more than 40 cellular TRs with putative binding sites, including Sp1.38 Future work will explore which TRs are indeed responsible for KSHV-induced HERV-K transactivation. Besides TRs interacting with HERV-K LTRs, the expression of HERV-K can also be regulated by some epigenetic mechanisms including DNA methylation and histone modification.47 Interestingly, LANA has been found to interact with or regulate a variety of epigenetic factors such as EZH2, KDM3a and DNMT3a.48–51 Therefore, it will be interesting to determine whether these epigenetic factors are also involved in KSHV-induced HERV-K transactivation. One recent study reports that the expression of HERV-K correlates with the expression of genes in retinoblastoma (Rb) pathway including p16INK4A-CDK4 in melanoma cells.52 In fact, LANA can interact with Rb and regulate the Rb/E2F pathway, protecting lymphoid cells from p16 INK4A induced cell cycle arrest and inducing S-phase entry.53,54 Therefore, it will be important to determine the potential involvement of Rb/E2F pathway in KSHV/LANA-induced HERV-K transactivation.
Figure 7. Schematic diagram of potential mechanisms for HERV-K transactivation promoting KSHV-induced tumorigenesis.
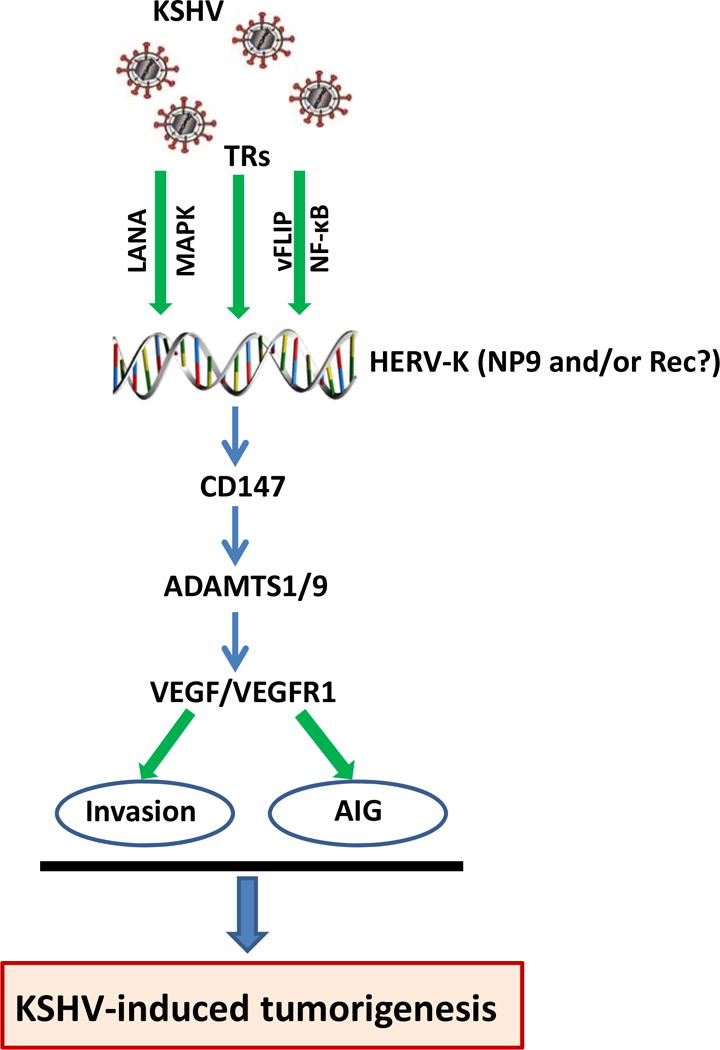
AIG: anchorage-independent growth. TRs: transcriptional factors.
Based on the types of proviruses, two different isoforms of regulatory proteins are encoded by the double-spliced transcripts of HERV-K env region, Rec and NP9, both of which have been reported to link with cancer development.66, 67 In the current study, we found that KSHV de novo infection prominently increases the expression of NP9, which is also highly expressed in AIDS-KS tumor tissues. Interestingly, NP9 has been found to not only activate the Akt, ERK and Notch1 pathways but also to up-regulate β-catenin, which is essential for survival of leukemia stem cells.45 More importantly, all of these pathways are closely related to KSHV pathogenesis and/or KS development.55–58 However, it still requires to understand the role of Rec in the KSHV-related tumorigenesis in future studies, since we found that our RNAi silencing of HERV-K env caused the reduction of both Rec and Np9 transcripts (data not shown).
We here report that “knock-down” of Np9 by RNAi effectively suppresses KSHV-induced tumorigenesis in vivo. Dr. Anil Sood at the MD Anderson Cancer Center has validated the use of 1,2-dioleoyl-snglycero-3-phosphatidylcholine (DOPC) for the efficient systemic delivery of EphA2-specific siRNA (EphA2-siRNA-DOPC) in an established xenograft model for intraperitoneal ovarian cancer.60 This work has revealed a significant reduction in intra-abdominal tumor expression of EphA2 48 h after intravenous injection of EphA2-siRNA-DOPC, and twice-weekly dosing results in sustained target knock-down and significant anti-tumor efficacy using EphA2-siRNA-DOPC alone or in combination with paclitaxel. Therefore, the use of siRNA-DOPC or siRNA-nano-particles targeting oncogenic NP9 protein may represent a novel and clinically feasible approach for the treatment of KSHV-associated tumors.
Our H & E staining images indicated that there is more immune cells infiltration in the tumor tissues from mice injected with Np9 “knock-down” cells when compared to control mice (Fig. 6D), although the underlying mechanisms need further investigation. Actually, HERVs can promote an immunosuppressive response that may lead to cancer formation and spreading.61 For instance, HERV Env protein contains an immunosuppressive domain, which was confirmed in animal models as a cause of tumor growth for tumor cells harboring the insertion of Moloney MLV and in env knockdown in B16 melanoma cells and Neuro-2a neuroblastoma cell lines.62 Therefore, whether targeting HERV-K transactivation can be part of immunotherapy for KSHV-related malignancies may represent an interesting direction.
MATERIALS AND METHODS
Cell culture and reagents
KSHV+ PEL cell line, BCBL-1 as well as a Burkitt’s lymphoma cell line, BL-41 were kindly provided by Dr. Dean Kedes (University of Virginia), which are cultured as described previously.23 The other PEL cell lines including BC-1, BC-3 and BCP-1 were purchased from American Type Culture Collection (ATCC) and cultured as recommended by the manufacturer. KSHV long-term-infected telomerase-immortalized human umbilical vein endothelial cells (TIVE-LTC) and the parental non-infected TIVE and cells were cultured as previously described.24 All the cells were cultured at the conditions of 37°C with 5% CO2.
KSHV purification and infection
BCBL-1 cells were incubated with valproic acid (0.6 mM) for 5 days, and KSHV virions in the culture supernatants was purified using ultracentrifugation as described previously.25 HUVEC were incubated with purified virus for 2 h at 37°C. The concentration of viral particles (MOI) was calculated as described previously.25
Patients and ethics statement
The study was approved by the Institutional Review Boards (IRB) for Human Research at Louisiana State University Health Science Center – New Orleans (No. 8079). All subjects have been provided the written informed consent. A total of 21 HIV+ patients with HAART treatment in our HIV Outpatient (HOP) Clinic are involved. There are 8 females and 13 males, the average age is 50.2 y (range 23-65 y). The average CD4 T cell counts of these patients are 544/mL (range 33-1775/mL), and the average viral loads of HIV is 5904 copies/mL (range 30-63367 copies/mL).
Plasma and PBMC preparation
Whole blood from HIV+ patients was collected and stored heparin-coated tubes, then peripheral blood mononuclear cells (PBMCs) were isolated using a Ficoll-Hypaque cushion. Plasma was obtained through the centrifugation. The KSHV infection status is determined by using the quantitative ELISAs as described previously.26,27
Immunoblotting and immunoprecipitation
The following antibodies (100-200 μg/mL) were used in immunoblotting: p-ERK/t-ERK, p-p65/t-p65, ADAMTS1 (Cell Signaling, Cat. #4370, #4695, #3033, #8242, #12897), ADAMTS9 (Thermo, Cat. #PA1-1760), CD147 (BD, Cat. #555961), LANA (ABI, Cat. #13-210-100), vFLIP (Ximbio, Cat. #151778) and HERV-K NP9 (kindly provided by Dr. Friedrich A. Grasser from Universitatsklinikum des Saarlandes, Germany).28 The antibody detecting β-Actin (Cell Signaling, Cat. #4970) was used as the loading control. Immunoprecipitation assays were carried out using the Catch and Release Immunoprecipitation Kit (Milipore).
Plasmid transfection and RNA interference
HUVEC were transfected with control vectors, pcDNA3.1-LANA (pcLANA), pcDNA3.1-vFLIP (pcvFLIP), pcDNA3.1-RTA (pcRTA), pDsred-NP9, pSG5-NP9 (both are kindly provided by Dr. Friedrich A. Grasser),28 in 12-well plates using Lipofectamine 3000 (Invitrogen). Transfection efficiency was determined as described previously.29 For RNAi assays, ON-TARGET plus SMART pool siRNA for HERV-K Env, Sp1 (Dharmacon), or the negative control siRNA, were delivered by using the DharmaFECT transfection reagent. To establish stable HERV-K knockdown cells, we used Dharmacon SMARTvector Lentiviral Np9-shRNA and a non-silencing (n)-shRNA as a negative control.
qRT-PCR
Total cellular RNA was isolated and purified using the RNeasy Mini kit (QIAGEN). cDNA was synthesized using SuperScript III First-Strand Synthesis SuperMix Kit (Invitrogen). Primers used for amplification of target genes were listed in Table. S1. Amplification was performed on an iCycler IQ Real-Time PCR Detection System, and analyzed as described previously.23
Transwell invasion and soft agar assays
Transwell invasion assays were performed using Matrigel Invasion Chambers (BD) and the relative invasion was calculated as describe previously.25 The anchorage-independent growth abilities were assessed using soft agar assays as described previously.25
KS-like Nude mouse model
5 × 105 TIVE-LTC cells in 50 μL PBS plus 50 μL growth factor-depleted Matrigel (BD Biosciences) were together injected subcutaneously into the flanks of nude mice, 6-8 week-old, male (Jackson Laboratory), 4 mice for each group. At the end of experiment, the tumors were excised for immunoblots and immunohistochemistry analyses. All protocols were approved by the LSUHSC Institutional Animal Care and Use Committee (IACUC) in accordance with the national guidelines.
Statistical analyses
Significance for differences among the experimental groups was calculated and determined using the two-tailed Student’s t-test (Excel 2016). p values <0.05 or <0.01 were considered significant or highly significant, respectively.
Supplementary Material
Acknowledgments
We thank Dr. Rolf Renne at University of Florida for his kind gifts of TIVE-LTC and TIVE cells, and Dr. Friedrich A. Grasser from Universitatsklinikum des Saarlandes, Germany for kindly providing HERV-K NP9 plasmids and antibody. This work was supported by grants from a DOD Career Development Award to (CA140437 to Z. Qin), a Louisiana Clinical and Translational Science Center Pilot grant (U54GM104940 from NIH), a LSU LIFT2 funding and NIH P20-GM121288-01 (PI: Krzysztof Reiss) subproject to Z. Qin, NIH RO1-AI101046, R01-AI106676, and P01CA214091 and Department of Defense W81XWH-16-1-0318 to E.K. Flemington, the federal funds from the National Cancer Institute, NIH, under Contract No. HHSN261200800001E to D. Whitby, and the awards from the National Natural Science Foundation of China (81472547, 81672924 to Z. Qin and 81400164, 81772930 to L. Dai). Funding sources had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Contribution: L.D. and Z.Q. designed and performed experiments, analyzed results, wrote the manuscript, and Z.Q. is the corresponding author. L.D.V. and W.M. performed experiments. D.W. A.C.O and E.K.F performed statistical analysis and provided critical input.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Mesri EA, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host Microbe. 2014;15:266–282. doi: 10.1016/j.chom.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 3.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 4.Engels EA, Biggar RJ, Hall HI, Cross H, Crutchfield A, Finch JL, et al. Cancer risk in people infected with human immunodeficiency virus in the United States. Int J Cancer. 2008;123:187–194. doi: 10.1002/ijc.23487. [DOI] [PubMed] [Google Scholar]
- 5.Bonnet F, Lewden C, May T, Heripret L, Jougla E, Bevilacqua S, et al. Malignancy-related causes of death in human immunodeficiency virus-infected patients in the era of highly active antiretroviral therapy. Cancer. 2004;101:317–324. doi: 10.1002/cncr.20354. [DOI] [PubMed] [Google Scholar]
- 6.Labo N, Miley W, Benson CA, Campbell TB, Whitby D. Epidemiology of Kaposi’s sarcoma-associated herpesvirus in HIV-1-infected US persons in the era of combination antiretroviral therapy. AIDS. 2015;29:1217–1225. doi: 10.1097/QAD.0000000000000682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jenkins FJ, Hoffman LJ, Liegey-Dougall A. Reactivation of and primary infection with human herpesvirus 8 among solid-organ transplant recipients. J Infect Dis. 2002;185:1238–1243. doi: 10.1086/340237. [DOI] [PubMed] [Google Scholar]
- 8.Luppi M, Barozzi P, Santagostino G, Trovato R, Schulz TF, Marasca R, et al. Molecular evidence of organ-related transmission of Kaposi sarcoma-associated herpesvirus or human herpesvirus-8 in transplant patients. Blood. 2000;96:3279–3281. [PubMed] [Google Scholar]
- 9.Ariza-Heredia EJ, Razonable RR. Human herpes virus 8 in solid organ transplantation. Transplantation. 2011;92:837–844. doi: 10.1097/TP.0b013e31823104ec. [DOI] [PubMed] [Google Scholar]
- 10.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 11.Hughes JF, Coffin JM. Evidence for genomic rearrangements mediated by human endogenous retroviruses during primate evolution. Nat Genet. 2001;29:487–489. doi: 10.1038/ng775. [DOI] [PubMed] [Google Scholar]
- 12.Dewannieux M, Harper F, Richaud A, Letzelter C, Ribet D, Pierron G, et al. Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome Res. 2006;16:1548–1556. doi: 10.1101/gr.5565706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee YN, Bieniasz PD. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog. 2007;3:e10. doi: 10.1371/journal.ppat.0030010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kraus B, Boller K, Reuter A, Schnierle BS. Characterization of the human endogenous retrovirus K Gag protein: identification of protease cleavage sites. Retrovirology. 2011;8:21. doi: 10.1186/1742-4690-8-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Depil S, Roche C, Dussart P, Prin L. Expression of a human endogenous retrovirus, HERV-K, in the blood cells of leukemia patients. Leukemia. 2002;16:254–259. doi: 10.1038/sj.leu.2402355. [DOI] [PubMed] [Google Scholar]
- 16.Contreras-Galindo R, Kaplan MH, Leissner P, Verjat T, Ferlenghi I, Bagnoli F, et al. Human endogenous retrovirus K (HML-2) elements in the plasma of people with lymphoma and breast cancer. J Virol. 2008;82:9329–9336. doi: 10.1128/JVI.00646-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang-Johanning F, Rycaj K, Plummer JB, Li M, Yin B, Frerich K, et al. Immunotherapeutic potential of anti-human endogenous retrovirus-K envelope protein antibodies in targeting breast tumors. J Natl Cancer Inst. 2012;104:189–210. doi: 10.1093/jnci/djr540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Armbruester V, Sauter M, Krautkraemer E, Meese E, Kleiman A, Best B, et al. A novel gene from the human endogenous retrovirus K expressed in transformed cells. Clin Cancer Res. 2002;8:1800–1807. [PubMed] [Google Scholar]
- 19.Buscher K, Trefzer U, Hofmann M, Sterry W, Kurth R, Denner J. Expression of human endogenous retrovirus K in melanomas and melanoma cell lines. Cancer Res. 2005;65:4172–4180. doi: 10.1158/0008-5472.CAN-04-2983. [DOI] [PubMed] [Google Scholar]
- 20.Sutkowski N, Conrad B, Thorley-Lawson DA, Huber BT. Epstein-Barr virus transactivates the human endogenous retrovirus HERV-K18 that encodes a superantigen. Immunity. 2001;15:579–589. doi: 10.1016/s1074-7613(01)00210-2. [DOI] [PubMed] [Google Scholar]
- 21.Sutkowski N, Chen G, Calderon G, Huber BT. Epstein-Barr virus latent membrane protein LMP-2A is sufficient for transactivation of the human endogenous retrovirus HERV-K18 superantigen. J Virol. 2004;78:7852–7860. doi: 10.1128/JVI.78.14.7852-7860.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsiao FC, Lin M, Tai A, Chen G, Huber BT. Cutting edge: Epstein-Barr virus transactivates the HERV-K18 superantigen by docking to the human complement receptor 2 (CD21) on primary B cells. J Immunol. 2006;177:2056–2060. doi: 10.4049/jimmunol.177.4.2056. [DOI] [PubMed] [Google Scholar]
- 23.Dai L, Trillo-Tinoco J, Cao Y, Bonstaff K, Doyle L, Del Valle L, et al. Targeting HGF/c-MET induces cell cycle arrest, DNA damage, and apoptosis for primary effusion lymphoma. Blood. 2015;126:2821–2831. doi: 10.1182/blood-2015-07-658823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.An FQ, Folarin HM, Compitello N, Roth J, Gerson SL, McCrae KR, et al. Long-term-infected telomerase-immortalized endothelial cells: a model for Kaposi’s sarcoma-associated herpesvirus latency in vitro and in vivo. J Virol. 2006;80:4833–4846. doi: 10.1128/JVI.80.10.4833-4846.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qin Z, Freitas E, Sullivan R, Mohan S, Bacelieri R, Branch D, et al. Upregulation of xCT by KSHV-encoded microRNAs facilitates KSHV dissemination and persistence in an environment of oxidative stress. PLoS Pathog. 2010;6:e1000742. doi: 10.1371/journal.ppat.1000742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mbisa GL, Miley W, Gamache CJ, Gillette WK, Esposito D, Hopkins R, et al. Detection of antibodies to Kaposi’s sarcoma-associated herpesvirus: a new approach using K8.1 ELISA and a newly developed recombinant LANA ELISA. J Immunol Methods. 2010;356:39–46. doi: 10.1016/j.jim.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benavente Y, Mbisa G, Labo N, Casabonne D, Becker N, Maynadie M, et al. Antibodies against lytic and latent Kaposi’s sarcoma-associated herpes virus antigens and lymphoma in the European EpiLymph case-control study. Br J Cancer. 2011;105:1768–1771. doi: 10.1038/bjc.2011.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gross H, Barth S, Pfuhl T, Willnecker V, Spurk A, Gurtsevitch V, et al. The NP9 protein encoded by the human endogenous retrovirus HERV-K(HML-2) negatively regulates gene activation of the Epstein-Barr virus nuclear antigen 2 (EBNA2) Int J Cancer. 2011;129:1105–1115. doi: 10.1002/ijc.25760. [DOI] [PubMed] [Google Scholar]
- 29.Qin Z, Dai L, Defee M, Findlay VJ, Watson DK, Toole BP, et al. Kaposi’s sarcoma-associated herpesvirus suppression of DUSP1 facilitates cellular pathogenesis following de novo infection. J Virol. 2013;87:621–635. doi: 10.1128/JVI.01441-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dai L, Qiao J, Nguyen D, Struckhoff AP, Doyle L, Bonstaff K, et al. Role of heme oxygenase-1 in the pathogenesis and tumorigenicity of Kaposi’s sarcoma-associated herpesvirus. Oncotarget. 2016;7:10459–10471. doi: 10.18632/oncotarget.7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mesri EA, Cesarman E, Boshoff C. Kaposi’s sarcoma and its associated herpesvirus. Nat Rev Cancer. 2010;10:707–719. doi: 10.1038/nrc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ye F, Lei X, Gao SJ. Mechanisms of Kaposi’s Sarcoma-Associated Herpesvirus Latency and Reactivation. Adv Virol 2011. 2011 doi: 10.1155/2011/193860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kellam P, Boshoff C, Whitby D, Matthews S, Weiss RA, Talbot SJ. Identification of a major latent nuclear antigen, LNA-1, in the human herpesvirus 8 genome. J Hum Virol. 1997;1:19–29. [PubMed] [Google Scholar]
- 34.Grossmann C, Podgrabinska S, Skobe M, Ganem D. Activation of NF-kappaB by the latent vFLIP gene of Kaposi’s sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype. J Virol. 2006;80:7179–7185. doi: 10.1128/JVI.01603-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qin Z, Dai L, Slomiany MG, Toole BP, Parsons C. Direct activation of emmprin and associated pathogenesis by an oncogenic herpesvirus. Cancer Res. 2010;70:3884–3889. doi: 10.1158/0008-5472.CAN-09-4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu L, Eby MT, Rathore N, Sinha SK, Kumar A, Chaudhary PM. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Ikappa B kinase complex. J Biol Chem. 2002;277:13745–13751. doi: 10.1074/jbc.M110480200. [DOI] [PubMed] [Google Scholar]
- 37.Dai L, Chen Y, Toole B, Parsons C, Qin Z. Induction of hyaluronan production by oncogenic KSHV and the contribution to viral pathogenesis in AIDS patients. Cancer Lett. 2015;362:158–166. doi: 10.1016/j.canlet.2015.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manghera M, Douville RN. Endogenous retrovirus-K promoter: a landing strip for inflammatory transcription factors? Retrovirology. 2013;10:16. doi: 10.1186/1742-4690-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuchs NV, Kraft M, Tondera C, Hanschmann KM, Lower J, Lower R. Expression of the human endogenous retrovirus (HERV) group HML-2/HERV-K does not depend on canonical promoter elements but is regulated by transcription factors Sp1 and Sp3. J Virol. 2011;85:3436–3448. doi: 10.1128/JVI.02539-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim JD, Yu S, Kim J. YY1 is autoregulated through its own DNA-binding sites. BMC Mol Biol. 2009;10:85. doi: 10.1186/1471-2199-10-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verma SC, Borah S, Robertson ES. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J Virol. 2004;78:10348–10359. doi: 10.1128/JVI.78.19.10348-10359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qian LW, Xie J, Ye F, Gao SJ. Kaposi’s sarcoma-associated herpesvirus infection promotes invasion of primary human umbilical vein endothelial cells by inducing matrix metalloproteinases. J Virol. 2007;81:7001–7010. doi: 10.1128/JVI.00016-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dai L, Bratoeva M, Toole BP, Qin Z, Parsons C. KSHV activation of VEGF secretion and invasion for endothelial cells is mediated through viral upregulation of emmprin-induced signal transduction. Int J Cancer. 2012;131:834–843. doi: 10.1002/ijc.26428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buscher K, Hahn S, Hofmann M, Trefzer U, Ozel M, Sterry W, et al. Expression of the human endogenous retrovirus-K transmembrane envelope, Rec and Np9 proteins in melanomas and melanoma cell lines. Melanoma Res. 2006;16:223–234. doi: 10.1097/01.cmr.0000215031.07941.ca. [DOI] [PubMed] [Google Scholar]
- 45.Chen T, Meng Z, Gan Y, Wang X, Xu F, Gu Y, et al. The viral oncogene Np9 acts as a critical molecular switch for co-activating beta-catenin, ERK, Akt and Notch1 and promoting the growth of human leukemia stem/progenitor cells. Leukemia. 2013;27:1469–1478. doi: 10.1038/leu.2013.8. [DOI] [PubMed] [Google Scholar]
- 46.Tang W, Chang SB, Hemler ME. Links between CD147 function, glycosylation, and caveolin-1. Mol Biol Cell. 2004;15:4043–4050. doi: 10.1091/mbc.E04-05-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maksakova IA, Mager DL, Reiss D. Keeping active endogenous retroviral-like elements in check: the epigenetic perspective. Cell Mol Life Sci. 2008;65:3329–3347. doi: 10.1007/s00018-008-8494-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He M, Zhang W, Bakken T, Schutten M, Toth Z, Jung JU, et al. Cancer angiogenesis induced by Kaposi sarcoma-associated herpesvirus is mediated by EZH2. Cancer Res. 2012;72:3582–3592. doi: 10.1158/0008-5472.CAN-11-2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim KY, Huerta SB, Izumiya C, Wang DH, Martinez A, Shevchenko B, et al. Kaposi’s sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen regulates the KSHV epigenome by association with the histone demethylase KDM3A. J Virol. 2013;87:6782–6793. doi: 10.1128/JVI.00011-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Di Bartolo DL, Cannon M, Liu YF, Renne R, Chadburn A, Boshoff C, et al. KSHV LANA inhibits TGF-beta signaling through epigenetic silencing of the TGF-beta type II receptor. Blood. 2008;111:4731–4740. doi: 10.1182/blood-2007-09-110544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shamay M, Krithivas A, Zhang J, Hayward SD. Recruitment of the de novo DNA methyltransferase Dnmt3a by Kaposi’s sarcoma-associated herpesvirus LANA. Proc Natl Acad Sci U S A. 2006;103:14554–14559. doi: 10.1073/pnas.0604469103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li Z, Sheng T, Wan X, Liu T, Wu H, Dong J. Expression of HERV-K correlates with status of MEK-ERK and p16INK4A-CDK4 pathways in melanoma cells. Cancer Invest. 2010;28:1031–1037. doi: 10.3109/07357907.2010.512604. [DOI] [PubMed] [Google Scholar]
- 53.Friborg J, Jr, Kong W, Hottiger MO, Nabel GJ. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature. 1999;402:889–894. doi: 10.1038/47266. [DOI] [PubMed] [Google Scholar]
- 54.An FQ, Compitello N, Horwitz E, Sramkoski M, Knudsen ES, Renne R. The latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus modulates cellular gene expression and protects lymphoid cells from p16 INK4A-induced cell cycle arrest. J Biol Chem. 2005;280:3862–3874. doi: 10.1074/jbc.M407435200. [DOI] [PubMed] [Google Scholar]
- 55.Wong JP, Damania B. Modulation of oncogenic signaling networks by Kaposi’s sarcoma-associated herpesvirus. Biol Chem. 2017;398:911–918. doi: 10.1515/hsz-2017-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li W, Jia X, Shen C, Zhang M, Xu J, Shang Y, et al. A KSHV microRNA enhances viral latency and induces angiogenesis by targeting GRK2 to activate the CXCR2/AKT pathway. Oncotarget. 2016;7:32286–32305. doi: 10.18632/oncotarget.8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li S, Hu H, He Z, Liang D, Sun R, Lan K. Fine-Tuning of the Kaposi’s Sarcoma-Associated Herpesvirus Life Cycle in Neighboring Cells through the RTA-JAG1-Notch Pathway. PLoS Pathog. 2016;12:e1005900. doi: 10.1371/journal.ppat.1005900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Angelova M, Ferris M, Swan KF, McFerrin HE, Pridjian G, Morris CA, et al. Kaposi’s sarcoma-associated herpesvirus G-protein coupled receptor activates the canonical Wnt/beta-catenin signaling pathway. Virol J. 2014;11:218. doi: 10.1186/s12985-014-0218-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A. 1998;95:10866–10871. doi: 10.1073/pnas.95.18.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Landen CN, Jr, Chavez-Reyes A, Bucana C, Schmandt R, Deavers MT, Lopez-Berestein G, et al. Therapeutic EphA2 gene targeting in vivo using neutral liposomal small interfering RNA delivery. Cancer Res. 2005;65:6910–6918. doi: 10.1158/0008-5472.CAN-05-0530. [DOI] [PubMed] [Google Scholar]
- 61.Gonzalez-Cao M, Iduma P, Karachaliou N, Santarpia M, Blanco J, Rosell R. Human endogenous retroviruses and cancer. Cancer Biol Med. 2016;13:483–488. doi: 10.20892/j.issn.2095-3941.2016.0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mangeney M, Heidmann T. Tumor cells expressing a retroviral envelope escape immune rejection in vivo. Proc Natl Acad Sci U S A. 1998;95:14920–14925. doi: 10.1073/pnas.95.25.14920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lower R, Tonjes RR, Korbmacher C, Kurth R, Lower J. Identification of a Rev-related protein by analysis of spliced transcripts of the human endogenous retroviruses HTDV/HERV-K. J Virol. 1995;69:141–149. doi: 10.1128/jvi.69.1.141-149.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Magin C, Lower R, Lower J. cORF and RcRE, the Rev/Rex and RRE/RxRE homologues of the human endogenous retrovirus family HTDV/HERV-K. J Virol. 1999;73:9496–9507. doi: 10.1128/jvi.73.11.9496-9507.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gonzalez-Hernandez MJ, Swanson MD, Contreras-Galindo R, Cookinham S, King SR, Noel RJ, Jr, et al. Expression of human endogenous retrovirus type K (HML-2) is activated by the Tat protein of HIV-1. J Virol. 2012;86:7790–7805. doi: 10.1128/JVI.07215-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hohn O, Hanke K, Bannert N. HERV-K(HML-2), the Best Preserved Family of HERVs: Endogenization, Expression, and Implications in Health and Disease. Frontiers in oncology. 2013;3:246. doi: 10.3389/fonc.2013.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Downey RF, Sullivan FJ, Wang-Johanning F, Ambs S, Giles FJ, Glynn SA. Human endogenous retrovirus K and cancer: Innocent bystander or tumorigenic accomplice? Int J Cancer. 2015;137:1249–1257. doi: 10.1002/ijc.29003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.